

SUMMARY
The sparse activity of hippocampal dentate gyrus (DG) granule cells (GCs) is thought to be critical for cognition and behavior, whereas excessive DG activity may contribute to disorders such as temporal lobe epilepsy (TLE). Glutamatergic mossy cells (MCs) of the DG are potentially critical to normal and pathological functions of the DG because they can regulate GC activity through innervation of GCs or indirectly through GABAergic neurons. Here, we test the hypothesis that MC excitation of GCs is normally weak, but under pathological conditions, MC excitation of GCs is dramatically strengthened. We show that selectively inhibiting MCs during severe seizures reduced manifestations of those seizures, hippocampal injury, and chronic epilepsy. In contrast, selectively activating MCs was pro-convulsant. Mechanistic in vitro studies using optogenetics further demonstrated the unanticipated ability of MC axons to excite GCs under pathological conditions. These results demonstrate an excitatory and epileptogenic effect of MCs in the DG.
In Brief
Dentate gyrus (DG) mossy cells (MCs) have glutamatergic synapses on the DG principal cells, granule cells (GCs), but many studies suggest that MCs inhibit GCs by exciting local GABAergic interneurons. Botterill et al. show that MC excitation of GCs is robust during status epilepticus, which contributes to excitotoxicity and epileptogenesis.
Graphical Abstract
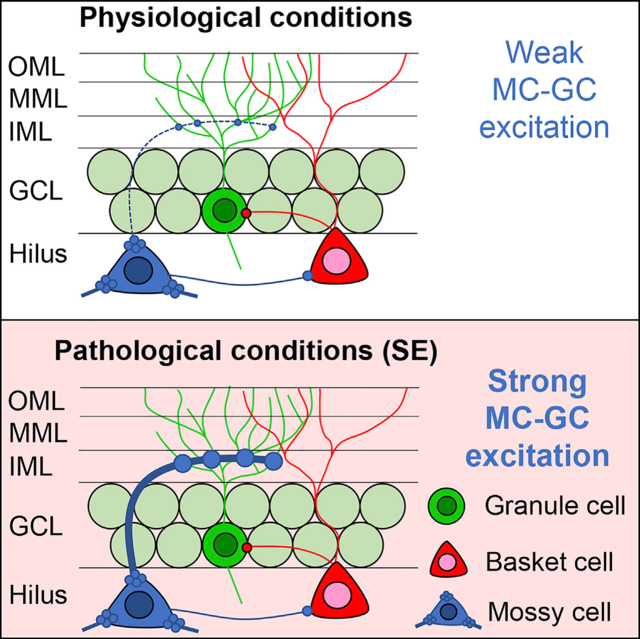
INTRODUCTION
The hippocampal dentate gyrus (DG) is critical to normal spatial memory and other cognitive functions and to regulation of mood, anxiety, and other conditions (Scharfman, 2007). Furthermore, the DG is implicated in diverse neurological diseases and psychiatric disorders, one of which is epilepsy. Specifically, the DG is involved in acquired temporal lobe epilepsy (TLE), a type of epilepsy acquired after brain injury where seizures often involve the hippocampus (Dengler and Coulter, 2016; Pun et al., 2012; Scharfman, 2007). Based on the broad importance of the DG, it is critical to understand how the neuronal subtypes and pathways allow the DG to perform its normal functions and how these are disrupted in disease. One aspect of the DG has been particularly puzzling: the role of a specific cell type called mossy cells (MCs; Scharfman, 2016; Scharfman and Myers, 2013). Here, we present data that demonstrate an excitatory and epileptogenic role of MCs in a mouse model of TLE.
The DG is primarily composed of glutamatergic neurons called granule cells (GCs), which receive strong entorhinal cortical excitatory input and form the DG output to the hippocampus (Amaral et al., 2007). There are additional pathways from the entorhinal cortex to the hippocampus, but the position of the DG between the entorhinal cortex and the hippocampus has led to the common conception that the DG forms a gateway to the hippocampus (Dengler and Coulter, 2016; Dengler et al., 2017; Heinemann et al., 1992; Krook-Magnuson et al., 2015; Lothman et al., 1992; Pun et al., 2012). In the hippocampus, GCs innervate area CA3 and make powerful excitatory “detonator” synapses on the principal cells (pyramidal cells; Henze et al., 2002). Remarkably, GC firing is normally sparse (Diamantaki et al., 2016; Jung and McNaughton, 1993; Neunuebel and Knierim, 2012). Notably, the detonator synapse of GCs can release substantial quantities of glutamate, and in pathological conditions, GC hyperactivity has been suggested to be excitotoxic to hippocampal neurons (Scharfman and MacLusky, 2014a; Sloviter et al., 2003). The regulation of GC activity is therefore paramount for normal hippocampal function. The GCs are controlled by many processes, including ligand and voltage-gated ion channels, innervation by local GABAergic neurons, and MCs.
MCs are located adjacent to the GCs in the hilar region and project extensively to GCs and GABAergic neurons in the DG (Amaral et al., 2007). The major axon arborization is far from the MC soma, throughout the ipsilateral and contralateral DG, and both GCs and DG GABAergic neurons are innervated (Scharfman and Myers, 2013). MCs are important to GC activity because the MC axon terminals are strategically located to regulate GCs, in the proximal third of the GC dendrites (the DG inner molecular layer [IML]; Scharfman, 2016). However, evidence that MCs robustly excite GCs in vivo is surprisingly weak. Instead, several studies suggest that MCs inhibit GCs by activating DG GABAergic neurons that synapse on GCs (Bui et al., 2018; Hsu et al., 2016; Jinde et al., 2012; Sloviter, 1991; Yeh et al., 2018). The net effect of MCs on GCs has been controversial because experimental data suggesting inhibitory effects of MCs on GCs seem discordant with the anatomical data showing numerous excitatory synapses of MCs on GCs in the IML (Buckmaster et al., 1996).
Clarifying this paradox is critical to understanding the normal regulation of GC activity. It also is important in understanding diseases like TLE where MCs are typically damaged and there is abnormal GC activity (Danzer, 2018; Dengler and Coulter, 2016; Pun et al., 2012; Scharfman and Pedley, 2006). It has been suggested that the loss of numerous MCs leads to the hyperactivity in GCs because DG GABAergic neurons called “basket cells” lose MC afferent input, causing disinhibition of GCs (the “dormant basket cell” hypothesis; Sloviter, 1991, 1994). An alternative hypothesis also exists that suggests that surviving MCs become very active in TLE, and this overactivity promotes GC hyperexcitability (the “irritable” MC hypothesis; Ratzliff et al., 2002). These contrasting ideas have created a major debate (Bernard et al., 1998; Jinde et al., 2012; Ratzliff et al., 2004; Scharfman, 2016). Here, we present the view that both hypotheses are partly correct. In normal conditions, MCs appear to support GC inhibition, as suggested by the dormant basket cell hypothesis. However, if certain conditions are met, MC excitation of GCs can strengthen greatly. The pathological conditions that occur during severe seizures (status epilepticus [SE]) are a situation when MC excitation of GCs is surprisingly powerful. This finding has strong implications for TLE because SE is a trigger for epileptogenesis. Thus, inhibiting MCs during SE is antiepileptogenic.
RESULTS
MCs Influence SE In Vivo
To evaluate the function of MCs during SE, dopamine receptor D2 (DrD2-Cre) mice with preferential expression of Cre recombinase in MCs were used as previously described (Gangarossa et al., 2012; Senzai and Buzsáki, 2017) and confirmed (Figure S1). For in vivo manipulations of MC function, we utilized inhibitory Designer Receptors Exclusively Activated by Designer Drugs (iDREADDs). An adeno-associated virus (AAV) containing a modified designer receptor (hM4D) coupled to an inhibitory signaling cascade (Gi) was injected stereotaxically into the DG to target a large number of MCs (Figures 1A–1C). DrD2-Cre+/− mice with viral expression are referred to as iDREADDs mice below. This viral approach infected sufficient MCs so that their axons in the IML were well labeled throughout the DG septotemporal axis bilaterally (Figure S1). DrD2-Cre−/− littermates were treated the same way, but viral expression was undetectable (see below). Immediately after viral injection, we implanted four subdural screw electrodes over the left frontal cortex (FC), the left and right dorsal hippocampus (HC), and the right occipital cortex (OC; Figure 1D). After 2–4 weeks for expression to occur, mice were connected to a video EEG (vEEG) recording system for baseline recording followed by induction of SE, which leads to hippocampal neuronal loss within days and chronic spontaneous convulsive seizures (epilepsy) within 4 weeks. SE was induced using a common method, the convulsant pilocarpine (Figure 1E). To determine the effects of MC inhibition on SE, mice were injected with the iDREADDs activator clozapine N-oxide (CNO) 30 min before pilocarpine administration. CNO was also administered to Cre−/− mice to control for potential off-target effects (see Method Details).
Figure 1. Inhibiting MCs with iDREADDs Attenuates Pilocarpine-Induced SE.

(A1) Timeline. Mice underwent surgery for viral injection and implant of an EEG recording assembly. SE was induced 2–4 weeks later with the convulsant pilocarpine (day 1). EEG was conducted 24 h later (day 2; see Figure 3), neuronal injury was assessed in a subset of mice on day 3 (see Figure 4), and the remaining mice were recorded by vEEG for 2 weeks to quantify chronic seizures (see Figure 5).
(A2) Expanded day 1 timeline shows the sequence of procedures in more detail.
(B) The Cre-dependent viral construct used in iDREADDs experiments was AAV-hSyn-DIO-hM4D(Gi)-mCherry.
(C1) Schematic of the hippocampus.
(C2) Virus was injected into the dorsal and ventral hippocampus, bilaterally.
(D) Subdural screw electrodes were positioned over the left frontal cortex (FC), the left and right dorsal hippocampus (HC), and the right occipital cortex (OC) for vEEG recordings.
(E) Representative 6-h EEG recording showing SE onset (the first 30 min of SE). After a ~1-h baseline period, all mice were injected with CNO, followed by pilocarpine 30 min later. Seizures began within the next 60 min and lasted several hours.
(F1) There were no group differences in the latency to the first electrographic seizure.
(F2) The latency to the first convulsive seizure was significantly delayed in iDREADDs compared to Cre−/− mice.
(G1) There were no group differences in baseline EEG power.
(G2) MC inhibition selectively increased theta power during the CNO period.
(G3) MC inhibition reduced theta, alpha, beta, and gamma power during SE onset.
(H1 and H2) Representative 2-h EEG record and corresponding spectrograms showing the pilocarpine injection and SE onset. Note that EEG power was reduced in iDREADDs relative to Cre−/− mice during SE onset. Data are represented as mean ± SEM. *p < 0.05.
See also Figures S1, S2, and S6.
A two-way ANOVA revealed a main effect of group (Cre−/− versus iDREADDs) on seizure latency (F(1,56) = 9.270, p = 0.003). Although the latency to the first electrographic seizure (for definition, see Method Details) in Cre−/− (35.80 ± 2.72 min) did not significantly differ from iDREADDs mice (44.20 ± 3.38 min; p = 0.146; Figure 1F1), the latency to the first convulsive seizure occurred significantly faster in Cre−/− mice (41.27 ± 2.74 min) than in iDREADDs mice (52.67 ± 3.99 min; p = 0.032; Figure 1F2). These data suggested a potential effect of MCs early in SE, so we evaluated whether there was a difference in the total number of convulsive seizures during a longer period of time during SE, the first 2 h. The total number of convulsive seizures in Cre−/− (4.60 ± 0.54) did not differ from iDREADDs mice (3.53 ± 0.48; t(28) = 1.465, p = 0.154).
Because the data suggested an effect early in SE, we analyzed spectral power during the baseline period, the period when CNO was injected until pilocarpine injection, and the first 30 min of SE (“SE onset”) using major frequency bands: delta (0–3 Hz), theta (4–8 Hz), alpha (9–13 Hz), beta (14–30 Hz), and gamma (31–100 Hz; Kadam et al., 2017; Figure 1G). A two-way ANOVA found no significant differences between Cre−/− and iDREADDs mice in baseline power (F(1,140) = 0.704, p = 0.402; Figure 1G1). Next, we analyzed the period after CNO was injected until the time of pilocarpine injection and found a significant effect of group (Cre−/− versus iDREADDs) in power (F(1,140) = 7.830, p = 0.005). Interestingly, theta power was significantly greater in iDREADDs (4.67 ± 0.24 μV2) compared to Cre−/− mice (3.97 ± 0.33 μV2; p = 0.018; Figure 1G2), but no other frequencies (all p values > 0.154). We also confirmed that CNO did not significantly alter EEG power in absence of iDREADD, as there was no difference in power between the baseline and CNO period in Cre−/− mice (F(1,140) = 2.613, p = 0.108; two-way ANOVA).
We then analyzed power during SE onset and found a significant main effect of group (Cre−/− versus iDREADDs; two-way ANOVA; (F(1,140) = 25.39, p < 0.001). EEG power of iDREADDs mice significantly decreased in theta (Cre−/− 101.8 ± 6.67 μV2 versus iDREADDs 68.54 ± 7.10 μV2; t(28) = 3.413, p = 0.002), alpha (Cre−/− 64.26 ± 6.19 μV2 versus iDREADDs 40.28 ± 3.97 μV2; t(28) = 3.261, p = 0.002), beta (Cre−/− 24.78 ± 2.56 μV2 versus iDREADDs 14.92 ± 1.74 μV2; t(28) = 3.178, p = 0.003), and gamma frequencies (Cre−/− 1.46 ± 0.14 μV2 versus iDREADDs 0.89 ± 0.11 μV2; t(28) = 3.142, p = 0.003; Figure 1G3). Delta frequency did not differ between groups (t(28) = 1.340, p = 0.190). The average power through the entire frequency range (0–100 Hz) during SE onset was significantly decreased in iDREADDs mice (Cre−/− 17.50 ± 1.87 μV2 versus iDREADDs 11.12 ± 1.16 μV2; t(28) = 2.887, p = 0.007), suggesting that iDREADDs have a broad suppressant effect on the EEG during SE onset (Figures 1H1 and 1H2). There were no significant power differences between Cre−/− and iDREADDs mice afterward (30–60, 60–0, or 90–120 min after SE; all p values > 0.150).
To confirm the results with additional analyses, we evaluated the rapid spike-like transients dominating the EEG during SE onset (Figure 2). Specifically, we asked whether there would be a reduction in spike number, frequency, or amplitude (see Method Details). Notably, Cre−/− had significantly more spikes (2660 ± 123 spikes) than iDREADDs mice (1876 ± 147 spikes; t(28) = 4.084, p < 0.001; Figure 2B). The interspike interval was significantly shorter in Cre−/− (232.4 ± 10.6 ms) than in iDREADDs mice (321.9 ± 26.1 ms; t(28) = 3.168, p = 0.003; Figure 2C). There was no difference in the peak amplitude of spikes (t(28) = 0.652, p = 0.519; Figure 2D), suggesting that MCs affect the initiation or “drive” on spikes but the network underlying spikes was just as robust. In summary, spike analysis supported the finding that merely inhibiting MCs reduced several measures of SE onset (Figure 2E), a remarkable effect for a relatively small number of neurons.
Figure 2. Inhibiting MCs Reduces Spike Number and Frequency of the Hippocampal EEG during SE Onset.

(A) Timeline of the entire study showing the timing of day 1, when data in the figure were taken.
(B) The number of spikes during the first 10 min of SE was significantly greater in Cre−/− than in iDREADDs mice.
(C) The interval between spikes (interspike interval) was significantly greater in iDREADDs than in Cre−/− mice.
(D) Mean spike amplitude did not differ between groups.
(E1 and E2) Representative Cre−/− and iDREADDs recordings show the onset of SE following pilocarpine injection. A threshold of 7 SD ≥ the RMS of the baseline EEG amplitude was set as the criterion for spike detection. No events met this criterion during the baseline recordings. After SE began, events termed spikes (arrows) that exceeded the ≥7 SD threshold criterion were analyzed. Data are represented as mean ± SEM. *p < 0.05.
See also Figure S2.
To further evaluate the contribution of MCs to SE, a subset of mice (n = 5) underwent the same procedures but were injected with the excitatory DREADDs (eDREADDs) construct AAV-hSyn-DIO-hM3D(Gq)-mCherry instead (Figures S2A and S2B). Remarkably, iDREADDs and eDREADDs produced opposing effects on seizure latencies and EEG power. Activating MCs in eDREADDs mice 30 min before pilocarpine injection dramatically accelerated seizure-onset latencies (Figures S2C and S2D). Power analysis revealed that activating MCs reduced EEG power during the CNO period (Figure S2E1) but significantly increased EEG power during SE onset (Figure S2E2). Spike analysis further demonstrated that MC activation influenced spiking at SE onset (Figures S2F and S2G; see the legend for statistical comparisons). The opposing effects of iDREADDs and eDREADDs suggest off-target effects of CNO were limited.
MC Inhibition during SE Reduces Intermittent Seizures the Following Day
On the day after pilocarpine injection (24–48 h after pilocarpine; day 2), we observed that the vEEG record of Cre−/− and iDREADDs mice showed intermittent epileptiform-like activity and even seizures (Figure 3). The fact that spontaneous seizures occurred on day 2 in controls (Cre−/− mice) is consistent with prior studies (Bumanglag and Sloviter, 2008; Mazzuferi et al., 2012; Smith et al., 2018). Notably, the seizures on day 2 are not likely to be due to residual pilocarpine, since it is cleared within 24 h (Mazzuferi et al., 2012).
Figure 3. Inhibiting MCs during SE Reduces Seizure Burden the Following Day.

(A) Timeline of the entire study showing the timing of day 2, when data in the figure were taken.
(B1) The number of day 2 convulsive seizures from 24 to 48 h after SE was significantly reduced in iDREADDs compared to Cre−/− mice.
(B2) The number of day 2 non-convulsive seizures was significantly reduced in iDREADDs compared to Cre−/− mice.
(B3) The total number of convulsive and non-convulsive seizures was significantly reduced in iDREADDs compared to Cre−/− mice.
(C1a and C2a) Representative spectrogram from the left and right dorsal hippocampus in Cre−/− and iDREADDs mice. Inset: 1-min-long spectrogram and corresponding EEG showing spikes in the Cre−/− EEG record with high power at high frequencies (>20 Hz) relative to the iDREADDs record.
(C1b and C2b) EEG record corresponding to the spectrograms.
(C1c and C2c) Insets show seizures (1, 3, 6) or bursts of spikes (2, 4, 5, 7, 8) that were more frequent in Cre−/− mice. Data are represented as mean ± SEM. *p < 0.05.
We first quantified convulsive behaviors (stage 3 or higher) and non-convulsive seizures (stages 1 and 2; Racine, 1972). Cre−/− mice had significantly more convulsive seizures (11.60 ± 1.45 seizures) than did iDREADDs mice (4.06 ± 1.33 seizures; Mann-Whitney U = 30.5, p < 0.001; Figure 3B1). There were approximately twice as many non-convulsive seizures in Cre−/− (7.53 ± 1.82 non-convulsive seizures) as iDREADDs mice (3.53 ± 1.43 non-convulsive seizures; Mann-Whitney U = 57.5, p = 0.020; Figure 3B2). When all seizures were pooled, Cre−/− mice (19.13 ± 2.75) had a significantly higher number of seizures than iDREADDs mice (7.60 ± 2.57; Mann-Whitney U = 41.0, p = 0.002; Figure 3B3). Taken together, the results indicated that iDREADDs mice had fewer seizures (Figures 3C1 and 3C2).
MCInhibition during SE Reduces Hippocampal Neuronal Injury
To determine whether inhibition of MCs during SE affected SE-induced hippocampal neuronal injury, a randomly selected subset of Cre−/− (n = 7) and iDREADDs mice (n = 8) were sacrificed 72 h after pilocarpine (day 3; Figure 4A). Brains were hemisected, and the left hemisphere was sectioned in the coronal plane to evaluate the dorsal hippocampus, whereas the right hemisphere was processed in the horizontal plane to best evaluate the ventral hippocampus (Figures 4B and S3; see Method Details). The sections were then processed with the neurodegenerative marker FluoroJade B to evaluate the areas (hilus and area CA1–3) that show neuronal injury after brain insults (Scharfman, 1999; Scharfman and Pedley, 2006; Figures 4C and 4D). A two-way ANOVA found a significant main effect of group (Cre−/− versus iDREADDs) on hilar FluoroJade cell number (F(1,26) = 17.78, p < 0.001), with a greater number of hilar FluoroJade-stained cells in dorsal sections in Cre−/− (43.96 ± 3.29 cells) than iDREADDs mice (21.52 ± 4.18; p = 0.004; Figure 4E1a). Similarly, the number of hilar FluoroJade-stained cells in ventral sections was significantly greater in Cre−/− (44.56 ± 4.69 cells) than in iDREADDs mice (27.50 ± 5.82 cells; p = 0.032; Figure 4E2a). Thus, neuronal injury after SE appeared to be protected by inhibition of MCs during SE.
Figure 4. Inhibiting MCs during SE Reduces Neuronal Injury.

(A) Timeline of the entire study showing the timing of day 3, when data in the figure were taken.
(B) Schematic of the hippocampus and sectioning planes. The dorsal hippocampus was evaluated with coronal sections, whereas the ventral hippocampus was evaluated using horizontal sections.
(C and D) Representative dorsal (C) and ventral (D) hM4D(Gi)-mCherry and FluoroJade photomicrographs in Cre−/− and iDREADDs mice 3 days after SE. Note the viral expression in the IML (arrow) and hilus in iDREADDs mice, but not Cre−/−. Viral expression coincided with reduced FluoroJade staining, suggesting that there was less hilar and CA3 neurodegeneration when MCs were inhibited during SE. Scale bars: 200 μm, 500 μm, and 50 μm (insets).
(E1a and E2a) Quantification of iDREADDs mouse sections revealed significantly fewer hilar FluoroJade-stained cells compared to Cre−/−.
(E1b and E2b) There was a smaller area of the CA3 cell layer stained by FluoroJade (arrows) in iDREADDs compared to Cre−/− mice.
(E1c and E2c) FluoroJade staining did not significantly differ between Cre−/− and iDREADDs mice in CA1. d. FluoroJade staining in the GCL was minimal in both groups. Data are represented as mean ± SEM. *p < 0.05.
See also Figure S3.
Next, we asked if other vulnerable regions within the hippocampus were also protected. Area fraction measurements were used to evaluate FluoroJade staining in CA3, CA1, and the GCL because the staining could be so intense that counting individual cells was difficult (Figure S3B). A two-way ANOVA revealed a main effect of group (Cre−/− versus iDREADDs) on area fraction (F(1,78) = 67.79; p < 0.001), with greater FluoroJade staining in dorsal CA3 of Cre−/− (18.07% ± 1.00%), compared with iDREADDs mice (4.37% ± 1.99%; p < 0.001; Figure 4E1b). A similar result was observed within ventral CA3 sections, with Cre−/− (18.23% ± 2.03%) showing greater FluoroJade staining than iDREADDs mice (4.42% ± 1.58%; p < 0.001; Figure 4E2b), but there were no significant group differences in CA1 (all p values >0.077; Figures 4E1c–4E2c). There was minimal FluoroJade staining in the GCL, consistent with its relative resistance to neuronal injury (Scharfman, 1999; Scharfman and Pedley, 2006), and no significant group differences were found (all p values > 0.999; Figures 4E1d–4E2d). Notably, FluoroJade staining was undetectable in Cre+/− or Cre−/− mice that were injected with hM4D(Gi)-mCherry but did not undergo SE (n = 3 per group).
To determine the cell types in the hilus that were damaged, adjacent sections were processed with markers of the two main vulnerable cell types: MCs (GluR2/3) and hilar cells projecting to the perforant path terminal zone ([HIPP] cells, a GABAergic neuron subtype expressing somatostatin; Freund and Buzsáki, 1996). A two-way ANOVA revealed a main effect of group (Cre−/− versus iDREADDs) on hilar GluR2/3 cells per section (F(1,26) = 19.34, p < 0.001), with Cre−/− mice (8.53 ± 2.76 cells) showing significantly fewer GluR2/3 cells per section in the dorsal hippocampus than the iDREADDs mice (25.52 ± 4.24 cells; p = 0.001; Figure S3C3a), as well as in ventral sections of Cre−/− (19.94 ± 2.96 cells), compared to iDREADDs mice (31.26 ± 2.35 cells; p = 0.039; Figure S3C3b). Adjacent sections processed for somatostatin revealed no group differences (F(1,26) = 0.218, p = 0.644; two-way ANOVA; Figure S3D). Taken together, these results suggest that iDREADDs specifically protected MCs but not somatostatin cells.
MCInhibition during SE Reduces the Severity of Chronic Seizures
Next, we determined whether the neuroprotective effects conferred by the inhibition of MCs during SE decreased the severity of chronic seizures (i.e., epilepsy). A randomly chosen cohort of Cre−/− (n = 8) and iDREADDs mice (n = 7) were used for chronic seizure monitoring 4 weeks after pilocarpine-induced SE (Figure 5A), when spontaneous seizures occurred in Cre−/− mice (Figure 5B). The total number of convulsive seizures in 2 weeks of continuous vEEG monitoring showed more than twice the number of seizures in Cre−/− (25.0 ± 5.23 seizures) than in iDREADDs mice (9.71 ± 2.98 seizures; t(13) = 2.44, p = 0.029; Figure 5B1). Seizure frequency was more than two times greater in Cre−/− (1.78 ± 0.37 seizures per day) than in iDREADDs mice (0.69 ± 0.21 seizures per day; t(13) = 2.44, p = 0.029; Figure 5B2). Next, we measured seizure burden, defined as the number of days out of the 2-week-long recording period when a mouse had at least one seizure. Seizure burden was significantly greater in Cre−/− (6.75 ± 0.86 days) than in iDREADDs mice (3.42 ± 0.97 days; t(13) = 2.56, p = 0.023; Figure 5B3). Cre−/− also had more consecutive days with seizures (4.62 ± 0.59 days) than did iDREADDs mice (2.57 ± 0.75 days; t(13) = 2.16, p = 0.049; Figure 5B4). Finally, Cre−/− had significantly longer seizures (54.75 ± 0.88 s) than did iDREADDs mice (34.29 ± 1.01 s; t(266) = 12.56, p < 0.001; Figures 5B5 and 5C). The results indicate that the inhibition of MCs during SE reduced the severity of epilepsy.
Figure 5. Inhibiting MCs during SE Reduces the Severity of Chronic Seizures (Epilepsy).

(A) Timeline highlighting the chronic period of epilepsy, when the data in the figure were taken. At 4 weeks after SE, mice were recorded with vEEG for 2 consecutive weeks to quantify spontaneous convulsive seizures.
(B1) The total number of seizures was lower in iDREADDs mice compared to Cre−/−.
(B2) Seizure frequency (seizures/day) was lower in iDREADDs mice.
(B3) The number of days where at least one seizure was detected was lower in iDREADDs mice.
(B4) The number of consecutive days with a seizure was lower in iDREADDs mice.
(B5) The duration of convulsive seizures in iDREADDs mice was significantly shorter than Cre−/− mice.
(C1a and C2a) Representative EEG recording.
(C1b and C2b) Spectrogram corresponding to the EEG shows a convulsive seizure in a Cre−/− mouse compared to a convulsive seizure in an iDREADDs mouse. Note that the convulsive seizure in the Cre−/− mouse is considerably longer than the iDREADDs mouse (arrows). Data are represented as mean ± SEM. *p < 0.05.
During Slice Simulation of SE, MC Activation Triggers Seizure-like Activity in GCs and Downstream Hippocampus
Next, we addressed the potential mechanism by which MCs affect SE. We hypothesized that the excitatory MC→GC pathway is normally weak but can be strong under unusual conditions like SE (see Simulating SE in the Method Details for details and rationale). To address the MC excitation of GCs during simulated SE, hippocampal slices were made after mice were injected at one site in the DG with a virus encoding channelrhodopsin and eYFP (ChR2-eYFP; Figure 6A). Slices distal to the injection site that expressed ChR2-eYFP in MC axons were used for recordings (Figures 6B and 6C). Distal slices were used because activating the somata of MCs in a slice near the injection site of the virus will not activate the majority of their terminals, since most MC terminals are distal to their somata (Amaral et al., 2007; Buckmaster et al., 1996; Scharfman and Myers, 2013). ChR2-expressing MC axons were activated by a brief pulse (<3 ms) of 473 nm of blue light aimed at the IML while simultaneously recording from patched GCs (Figure 6D). Responses to brief light pulses were recorded during standard recording conditions and then pharmacologically simulated SE. We found that a single light pulse elicited short-latency (<4 ms) excitatory postsynaptic potentials (EPSPs) or EPSPs followed by inhibitory PSPs (IPSPs) in patched GCs at resting membrane potential (RMP; n = 15 GCs tested). Under standard recording conditions, EPSPs were small in amplitude, consistent with a limited excitatory effect of the MC/GC monosynaptic pathway (Figure 6E1). In six cells of four mice, simulated SE was also tested. During the simulation of SE, the same light pulses that initially elicited weak EPSPs changed gradually and ultimately evoked prolonged complex EPSPs (n = 6/6 cells; Figure 6E2). Notably, the amplitude of the EPSP was increased >5-fold between standard recording conditions (1.09 ± 0.24 mV) and simulated SE (5.95 ± 0.88 mV; paired t test, t(5) = 6.14, p = 0.002), which indicates a substantial strengthening of the MC→GC synapse. However, the latency from the start of the light pulse to the onset of the EPSP did not differ between standard recording conditions (2.29 ± 0.20 ms) and simulated SE (2.86 ± 0.29 ms; paired t test, t(5) = 2.18, p = 0.082).
Figure 6. Optogenetic Activation of MC Axons Reveals a Much Larger Excitatory Effect under Conditions Simulating SE Onset.

(A) Timeline.
(B) DrD2-Cre+/− mice were injected with AAV2-DIO-hChR2(H134R)-eYFP in the left anterior or posterior hippocampus. Slices with MC axons distal to the injection site (ipsilateral or commissural; red dashes) were used to test the effects of brief light pulses on patched GCs.
(C) Representative viral expression of commissurally projecting MC axons located in the IML of the contralateral hemisphere (arrow). Scale bar: 200 μm.
(D1) Representative schematic of the DG. Note the MC axon terminating in the IML (red arrow).
(D2) 473 nm of light was aimed at the IML, the location of intense viral expression, while patching nearby GCs.
(E1) A brief pulse of light (2 ms; blue arrow) produced a weak depolarization under baseline conditions (black arrow).
(E2) After pharmacological simulation of SE began, the depolarization evoked by light became larger and prolonged (black arrow).
(E3) Within 25 min of simulating SE, the same light pulse triggered a paroxysmal depolarizing shift in the GC (PDS; black arrow).
(F) The intrinsic properties of the cell in (E) identified it as a GC (Scharfman, 1992, 1995a), including a triphasic afterhyperpolarization potential (F1) and current pulses that evoked responses with a short time constant and linear I-V relationship (F2 and F3).
See also Figure S4.
Over time, these prolonged EPSPs developed many action potentials (APs) on large and long-lasting EPSPs like paroxysmal depolarization shifts (PDSs; n = 6/6 cells; Figure 6E3). Further support for MC excitation of GCs was shown in a subset of GCs that were patched after the simulation of SE had begun, and all showed PDSs in response to light (n = 4/4 cells in two mice). Intrinsic properties indicated the patched cells were GCs (Figure 6F). Importantly, light directed to areas outside of the IML, such as the hilus or outer molecular layer, reduced the light responses >90% (n = 15/15 slices tested), consistent with the specific activation of MC axons when light was directed to the IML. In summary, optogenetic activation of MC axons failed to evoke PDSs in GCs under standard recording conditions (n = 21 cells), whereas PDSs were evoked in all GCs (n = 10 cells) after simulated SE had begun.
The results from patched GCs provided support for the “pathological” excitation of GCs by MCs. However, whether the PDSs in GCs were sufficient to trigger epileptiform activity in downstream targets (i.e., the hippocampus) was not addressed. To address that question, simultaneous recordings were made of the DG and the hippocampus (Figures 7A and 7B). After a robust light response was recorded in the DG, there was epileptiform activity in CA3, CA1, and the subiculum (Figure 7C). As one would predict from the known projection from GCs to CA3, the onset of CA3 epileptiform discharge that was triggered by light began within ms of the onset of the DG light response (DG: 2.83 ± 0.28 ms; CA3: 9.58 ± 0.48 ms; t(10) = 12.06, p < 0.001, n = 6 slices in four mice). These data suggest that MC axons activated GCs monosynaptically, which in turn activated the CA3 pyramidal cell population by the GC axons, the mossy fibers. In support of this hypothesis, CA3 epileptiform activity evoked by optical stimulation of the IML was blocked by DCG-IV, a selective antagonist of mossy fiber transmission (Yoshino et al., 1996; Figures 7D1–7D3).
Figure 7. Optogenetic Activation of MC Axons during Simulation of SE Triggers Epileptiform Activity that Propagates throughout the Hippocampus.

(A) Timeline.
(B1) Schematic of the hippocampal trisynaptic circuit showing the potential pathways for the spread of seizure activity (dashed arrows).
(B2) 473 nm of light was aimed at the IML, and extracellular field potentials were recorded in the IML, CA3, CA1, and subiculum cell layers (Xs).
(C1) A brief light pulse (2 ms; blue arrow) evoked a field EPSP in the IML (black arrow). Epileptiform discharges were recorded at increasingly longer delays in CA3, CA1, and the subiculum (see black arrows and C2), suggesting propagation along the trisynaptic circuit.
(D1) Epileptiform activity in CA3 and the subiculum was blocked by DCG-IV (2 μM), an antagonist of mossy fiber transmission (D2), which was reversed after 30 min in DCG-IV-free conditions (D3).
See also Figures S4 and S5.
Recordings in the DG and the hippocampus also showed that a single light pulse to the IML could trigger spreading depolarization (SD), a severe type of seizure activity (Figure S4; Rogawski, 2012). We hypothesized a role of NMDA receptors because of the prior demonstration that NMDA receptor activation contributes to SE (Naylor et al., 2013; Yen et al., 2004). This hypothesis was supported because the NMDA receptor antagonist DL-APV blocked epileptiform activity and SD using extracellular (Figure S4C) or patch recordings of GCs (n = 4 GCs, 4 slices in 3 mice; Figure S4D). Further evidence for a critical role of NMDA receptors in light-evoked epileptiform activity was seen in additional experiments that did not use 4-AP or GABA receptor blockade (n = 4 slices in two mice). In these experiments, simultaneously patched GC and CA3 PCL extracellular recordings were made in 0 mM Mg2+ and 5 mM K+. Notably, the IML light pulse did not produce a prolonged PDS, as shown in Figure 6E3. Rather, the light pulse triggered one or multiple APs in GCs (n = 1 cell/slice in the four slices mentioned above) and a subsequent CA3 burst discharge. These data suggest the following: (1) NMDA receptors play a critical role in the ability of MCs to drive GCs above their threshold and ultimately induce a CA3 burst discharge, and (2) optical activation of MC axons can evoke APs in GCs and epileptiform-like activity in the hippocampus even when GABAergic inhibition is not blocked.
One question that emerged in these recordings was whether the burst discharges were triggered by light or were spontaneous burst discharges, which occur in hippocampal pyramidal cells exposed to GABA receptor blockade (Miles et al., 1988; Scharfman, 1994b; Swann and Brady, 1984), 4-AP (Perreault and Avoli, 1991), or low [Mg2+]o (Scharfman, 1994a). Therefore, it was important to confirm that light-triggered epileptiform activity was not a coincidence of a light pulse occurring serendipitously before a spontaneous epileptiform event. To address this possibility, we recorded the interburst intervals of spontaneous epileptiform events and showed the interburst interval was long (>1 s, n = 3 or 4 slices in three mice) relative to the interval between an optical stimulus and a burst discharge (<100 ms; Figure S5). DCG-IV blocked the light-evoked CA3 burst discharges (consistent with burst generation in the DG), but spontaneous discharges in CA3 persisted (consistent with burst generation in CA3).
To confirm that MCs expressing hM4D(Gi)-mCherry were hyperpolarized by CNO in vitro, we injected the virus and made slices near the injection site 2–4 weeks later (Figures S6A1–S6A3). The RMP of hilar MCs without hM4D(Gi)-expression was not affected by CNO (n = 6 cells in four mice; Figure S6A4). However, consistent with an effect of iDREADDs, bath application of CNO caused mCherry-labeled MCs (n = 6 cells in four mice) to hyperpolarize 8.00 ± 1.67 mV (range, 4.5–16 mV; t(10) = 4.59, p<0.001; Figure S6A5). All hilar cells tested had the physiological characteristics of MCs (Figure S6A6; Scharfman and Myers, 2013). Notably, different doses of CNO have been suggested to influence recordings (Yeh et al., 2018), so we chose a dose (10 mM) that is widely used in experiments of this kind (Gallo et al., 2018; Kätzel et al., 2014). An additional control experiment showed that CNO reduced field EPSPs of GCs evoked by electrical stimulation of MC axons (by electrically stimulating the IML; see Method Details) in slices from iDREADDs mice (reduction in fEPSP slope, 50.29% ± 9.26%; n = 3 slices in three mice; Figure S6B).
To test the hypothesis that CNO reduced effects of pilocarpine-induced SE by inhibiting MCs during SE, we recorded from MCs and simulated SE in slices from iDREADDs mice. MCs were patched during treatment with vehicle (n = 3 cells in three mice) or CNO (n = 3 cells in three mice), and then SE was simulated. MCs depolarized and discharged repetitively during simulated SE (Figure S6C), similar to prior studies of MCs in slices during simulation of repetitive seizures (Scharfman and Schwartzkroin, 1989, 1990a, 1990b). MCs in vehicle-treated slices often did not recover from simulated SE, consistent with prior studies (Scharfman and Schwartzkroin, 1989, 1990a, 1990b). However, CNO-treated MCs recovered better (Figure S6C).
DISCUSSION
Our results suggest a remarkable contribution of MCs to the regulation of GC activity, which is relevant to epileptogenesis and TLE. Specifically, selective inhibition of MCs reduced manifestations of pilocarpine-induced SE and the sequelae that typically follow SE, such as neuronal injury and chronic epilepsy. Therefore, MC inhibition appears to have a rapid protective effect on epileptogenesis after SE. Slice electrophysiology experiments suggested that MC inhibition was effective because it curtailed a strengthening of MC→GC synapses during SE. Notably, selective optogenetic activation of MC axons produced a weak excitation of GCs under normal conditions, but once SE was simulated, the MC axons could trigger PDSs in GCs, and this caused epileptiform bursts that propagated through the hippocampal trisynaptic circuit. Severe seizure-like events such as SD were also triggered. The capacity for MCs to exert strong excitatory effects on GCs is supported by the anatomical demonstration of numerous MC→GC synapses proximal to GC somata (Buckmaster and Schwartzkroin, 1994; Buckmaster et al., 1996).
Remarkably, studies until now have mainly argued that MCs prevent rather than contribute to epilepsy (Bui et al., 2018; Jinde et al., 2012; Sloviter, 1991, 1994). Interestingly, Bui et al. (2018) studied MC activation during chronic convulsive seizures, and their results suggest that MC activation curtails them, which seems inconsistent with our results. However, in our experiments, MCs were inhibited in the normal brain, whereas Bui et al. (2018) studied chronic epilepsy. This is a critical difference in our view, because there are many changes in cells and circuitry in the DG in chronic epilepsy. Also, there were technical differences. For example, Bui et al. (2018) primarily studied dorsal MCs and targeted MCs differently.
Effects of MCs on SE
MCs produced effects during SE that are remarkable for such a small cell population. It is also notable that effects may have been greater if every MC expressed DREADDs, or if we had tested different doses or numbers of CNO injections (Yeh et al., 2018). This is useful to consider because the effects of MCs were mainly found during SE onset, suggesting that MCs play a role in the initiation of SE. But with broader targeting of MCs or manipulating other factors, the effects might have been greater. On the other hand, effects of MCs may not persist throughout SE because multiple brain areas become recruited and help sustain SE. Collectively, data from both iDREADDs and eDREADDs approaches suggest that inhibiting MCs during SE is protective and antiepileptogenic, whereas stimulating MCs during SE is pro-convulsant.
Interestingly, although iDREADDs decreased EEG power during SE onset (Figure S2), eDREADDs did not. One explanation arose from recordings of MCs during simulated SE. Consistent with prior studies (Scharfman and Schwartzkroin, 1989, 1990a, 1990b), normal MCs appear to be activated very strongly during the onset of SE or seizure activity. In this situation, iDREADDs and eDREADDs may differ because MC inhibition could have protective effects, but MC excitation may not be able to increase MC activity more than it is already. However, eDREADDs did have opposing effects relative to iDREADDs in other measures of SE onset. Therefore, all effects of DREADDs may not be solely on somatic firing. Instead, iDREADDs may inhibit glutamate release from MC axons, and eDREADDs may lead to more glutamate release. If this occurs independently from the soma, in some cases iDREADDs and eDREADDs will produce opposing effects, but in other cases there may not be opposite effects. It has been shown that DREADDs also act on terminals to modify transmitter release (Zhu and Roth, 2014). This finding is further supported by our slice studies of iDREADD-expressing MC axons, where the MCcell body is removed in slices that are distal to the DREADD-expressing MCs. In this case, CNO inhibited MC→GC excitation (Figure S6B), presumably through actions on the MC axon terminals.
Notably, mice treated with iDREADDs exhibited a selective increase in theta power before pilocarpine injection (Figure 1G2). These results are relevant to the finding that MCs can discharge at theta rhythm (Henze and Buzsaki, 2007; Soltesz et al., 1993). They are also consistent with the report that MC ablation led to increased theta power, which was explained by a reduction in MC→GABAergic neuron→GC inhibition (Jinde et al., 2012). The results are also consistent with a recent study showing that an opposing strategy—MC excitation–increased calcium signals in GABAergic neurons (Yeh et al., 2018). It is noteworthy that manipulating the activity of MCs had a significant effect on theta oscillations. One possible explanation for this effect is that MCs influence GC firing, which could subsequently influence the activity of downstream regions of the hippocampus involved in theta rhythm (e.g., CA3 and CA1) because of the trisynaptic pathway. Nonetheless, it is notable that our approach used subdural screw electrodes instead of depth electrodes to minimize hilar damage. The ability to detect hippocampal theta in cortical screws may reflect volume conduction, given the relatively short distance between the cortical screw electrode and the dorsal hippocampus. Alternatively, several studies have suggested that hippocampal theta can entrain or couple with cortical theta rhythms (Alexander et al., 2018; Vinck et al., 2016; Young and McNaughton, 2009) and may explain why theta changes were detected without the use of depth electrodes.
MC Inhibition during SE Blunts Epileptogenesis: Decreased Seizures on Day 2
In SE models of TLE, it had been commonly believed that the initial brain insult (e.g., SE) is followed by a seizure-free period, often called the latent period, that precedes the development of spontaneously recurring seizures (i.e., chronic epilepsy; Cavalheiro et al., 1991; Löscher et al., 2015; Sloviter, 2008). However, vEEG recordings have shown that in mice, there is epileptiform activity and seizures (convulsive and non-convulsive) in the days after pilocarpine-induced SE (Mazzuferi et al., 2012; Smith et al., 2018), suggesting that epileptogenesis begins much earlier than the onset of convulsive seizures, an idea supported by rat data (Williams et al., 2009). Therefore, it was not surprising that Cre−/− mice exhibited epileptiform activity and seizures on day 2 (Figure 3). However, it was surprising that iDREADDs mice exhibited few seizures the day after SE (day 2). The day 2 results support the idea that MC inhibition during SE protected against an early phase of epileptogenesis.
MC Inhibition during SE Blunts Epileptogenesis: Decreased Hippocampal Neuronal Injury
Hippocampal neuronal injury is a common characteristic of human TLE and rodent models of TLE, particularly acquired TLE, where a brain insult is thought to be causal (Ben-Ari and Dudek, 2010; Dudek and Staley, 2012; Mathern et al., 2002). The pattern of hippocampal neuronal loss is often called mesial temporal lobe sclerosis (MTS) and refers to the reduction of neurons in the hilus, including MCs, DG GABAergic cells, and the pyramidal cells in areas CA1 and CA3 (Scharfman and Pedley, 2006; Wieser and ILAE Commission on Neurosurgery of Epilepsy, 2004). Historically, it has been suggested that neuronal loss contributes to the development of chronic epilepsy, although many other effects of brain insults are also likely to be important (Ben-Ari and Dudek, 2010; Goldberg and Coulter, 2013; Pitkänen and Lukasiuk, 2011).
Here, we showed that MC inhibition during SE decreased MC and CA3 neuronal loss but not loss of hilar somatostatin neurons or CA1 PCs. The neuroprotective effect of iDREADDs on MCs and PCs is consistent with the idea that MCs and CA3 PCs, but not hilar somatostatin cells or CA1 PCs, are innervated by the massive boutons of GCs (i.e., the “detonator synapses”). The massive boutons have many more glutamatergic vesicles than other hippocampal boutons and have been linked to excitotoxicity of hilar and CA3 neurons (Sloviter et al., 2003). Additional factors are likely to contribute to vulnerability such as striatal enriched tyrosine phosphatase (STEP), which is very strongly expressed in hilar somatostatin cells with weaker expression in MCs (Choi et al., 2007). CA1 neuronal loss also may have more than one mechanism besides afferent drive from CA3, such as strong afferent drive from the temporoammonic pathway (Ang et al., 2006).
MC Inhibition during SE Blunts Chronic Seizures
Our analyses revealed that MC inhibition during SE dramatically reduced chronic seizures. Thus, inhibition of MCs during SE conferred significant long-term benefits against chronic epilepsy. This idea supports the hypothesis that hilar neuronal loss is a precipitating factor in TLE (Scharfman and Pedley, 2006; Sloviter, 1991). However, until now, many clinical and animal studies that have addressed this hypothesis are correlative (e.g., Hester and Danzer, 2013; Margerison and Corsellis, 1966). Why MC protection is important is underscored by prior studies indicating that surviving MCs inhibit chronic seizures, probably by the MC→GABAergic neuron→GC pathway (Bui et al., 2018).
Strengthening the MC→GC Synapse
Why MCs normally have excitatory effects on GCs that greatly strengthen during SE can be explained based on our recordings during simulated SE and prior studies. Our recordings during simulated SE showed that MCs are likely to be very strongly activated at this time, consistent with previous studies of MCs during repetitive perforant path stimulation in slices (Scharfman and Schwartzkroin, 1989, 1990a, 1990b). MC activation persisted, and ultimately MCs appeared to remain in a depolarized state without much ability to discharge (Scharfman and Schwartzkroin, 1989,1990a, 1990b) similar to our data (Figure S6). These findings are consistent with the report that hilar cells are labeled with markers of neurodegeneration (e.g., FluoroJade) as soon as 4 h after pilocarpine-induced SE in mice (Wang et al., 2008).
A likely result of the strong activation of MCs early in SE is that MCs release more glutamate, which would increase MC→GC excitation. However, it is also likely that the MC→GABAergic neuron pathway also increases, so it is relevant that GABA release may become depleted at the onset of a seizure (Soukupova et al., 2014; Zhang et al., 2012) and, if so, probably the onset of SE as well. If GABA release is not depleted, GABAergic inhibition may still be abnormal because GABA can become depolarizing during seizures (Magloire et al., 2019; Staley, 2004; Zhang et al., 2011). Other studies suggest an alternative, which is that the MC→GC synapse is potentiated preferentially when the MCs discharge at high frequency (Hashimotodani et al., 2017). Therefore, for many reasons, there may be greater MC→GC excitation than MC→GABAergic neuron/GC inhibition during SE onset. Other factors are likely to be important as well. For example, principal cells depolarize during a seizure (Avoli et al., 2016; Zhang et al., 2011), and this includes GCs (based on simulated seizures in vitro; Scharfman and Schwartzkroin, 1990a, 1990b). The depolarization of GCs increases the strength of the MC→GC synapse (Scharfman, 1995b), which appears to be due to a greater input resistance in GCs (Scharfman, 1994a) and NMDA receptor activation (Scharfman, 1994a; Scharfman and Schwartzkroin, 1990a, 1990b).
Once MCs strongly activate GCs, the ability for the GCs to detonate the cells they innervate—specifically, the target cells of the detonator synapses (MC and CA3 PCs)—leads to strong, persistent MC and CA3 PC excitation. This excitation has important implications because CA3 PCs have recurrent excitatory synapses that recruit other CA3 PCs into epileptiform activity (Miles et al., 1988). This may be why CA3 firing became prolonged and synchronized and led to burst discharges in the downstream targets of CA3. Notably, if the “detonation” is persistent, MC and PC overactivity leads to their death (i.e., excitotoxicity; Scharfman and MacLusky, 2014a; Sloviter et al., 2003), providing an explanation for the MC and CA3 PC FluoroJade data (Figures 4 and S3). Together, the results and the findings of prior studies suggest a series of factors that provide an explanation of the dramatic strengthening of excitation of the MC→GC synapse during SE onset and its consequences.
Summary and Conclusions
Collectively, we provide evidence of a critical role of MCs in TLE using a common and validated animal model (Löscher, 2002). Surprisingly, our results are not what would be predicted from the existing literature. Instead, we report an excitatory role of MCs, which was revealed by our distinct experimental design and use of selective methods to manipulate MCs in vivo and in vitro. The findings support an alternative framework for considering the role of MCs in the normal DG and in TLE.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Helen Scharfman (helen.scharfman@nki.rfmh.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study design
All experimental procedures were done in accordance with the National Institutes of Health (NIH) guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) at the Nathan Kline Institute. We used DrD2-Cre mice to selectively target and manipulate MCs in vivo and in vitro. For in vivo studies, we used DREADDs to inhibit or excite MCs prior to experimentally-induced seizures elicited by the convulsant pilocarpine. For in vitro studies, we used optogenetics to selectively activate MC axons under standard recording conditions and under recording conditions that pharmacologically simulated SE.
Animals & genotyping
DrD2-Cre transgenic mice were kindly provided by Dr. Emmanuel Valjent (France). Hemizygous DrD2-Cre males were bred in-house to C57BL/6N females (Stock No. 027, Charles River Laboratories). Breeding pairs were fed Purina 5008 rodent chow (W.F. Fisher) and provided 2”×2” nestlets (W.F. Fisher). Mice were weaned at postnatal day 25–30 and housed with same-sex siblings (2–4 per cage) in standard laboratory cages with corn cob bedding and a 12 hr light-dark cycle. Standard rodent chow (Purina 5001, W.F. Fisher) and water were available ad libitum. Genotypes were confirmed for all mice (Genotyping Core Laboratory, New York University Langone Medical Center). We used adult male mice for all studies (experiments starting at ~8 wks of age), since pilocarpine-induced SE is rare in female rodents and if it occurs, the mice do not recover well (Scharfman and MacLusky, 2014b).
METHOD DETAILS
Stereotaxic Surgery
For chemogenetic inhibition of MCs and their axons, we injected the Cre-dependent AAV2-hSyn-DIO-hM4D(Gi)-mCherry (iDREADDs; 4×1012 vg/mL, Addgene) into the dorsal and ventral hippocampus, bilaterally (i.e., 4 injections) of DrD2-Cre+/− mice (n = 15). The viral construct contains a modified designer receptor (hM4D) coupled to inhibitory (Gi) signaling. In presence of CNO, hM4D(Gi) activates inward rectifying potassium channels that hyperpolarize hM4D(Gi)-expressing cells and also reduces presynaptic glutamate release from their axon terminals (Zhu and Roth, 2014). DrD2-Cre−/− (n = 15) were injected with the same virus and showed no viral expression (Figures 4C and 4D).
At approximately 8 wks of age, mice underwent stereotaxic surgery for viral injections and implantation of EEG electrodes. Mice were initially anesthetized with 5% isoflurane (Aerrane, Henry Schein). The mice were then immediately secured in a rodent stereotaxic apparatus (Model #502063, World Precision Instruments). A homeothermic blanket system maintained body temperature at 37°C (Harvard Apparatus). Isoflurane (1%–2%) was mixed with oxygen and delivered through a nose cone attached to the stereotaxic apparatus. Buprenex (Buprenorphine, 0.1 mg/kg, s.c.) was delivered prior to any surgical manipulations to reduce discomfort. The scalp of each mouse was then shaved and swabbed with Betadine (Purdue Products). Lubricating gel was applied to the eyes to prevent dehydration (Patterson Veterinary).
After a midline scalp incision, a surgical drill (Model C300, Grobert) was used to make four craniotomies for viral injections. All stereotaxic coordinates for craniotomies are described in anterior-posterior (AP) and medial-lateral (ML) coordinates (in reference to bregma). Craniotomies were made over the dorsal (−1.9 mm AP, ± 1.2 mm ML) and ventral (−3.1 mm AP, ± 2.4 mm ML) hippocampus bilaterally. A 33-gauge infusion needle (#C315I-SPC, Plastics One) attached to a 0.5 μl Hamilton syringe was lowered from the skull surface 2.0 mm into each relatively anterior site in the hippocampus to target dorsal areas and 2.8 mm into each posterior site to target ventral hippocampus. Each site was injected with 160 nL of virus at a rate of 40 nL/min. The needle remained in place for an additional 5 min after each injection to allow for diffusion of the virus and then it was slowly removed.
Next, subdural screw electrodes (0.10” length stainless steel jeweler’s screws #8209, Pinnacle Technology) were secured in the two anterior craniotomies positioned over the left and right dorsal hippocampus. A subdural screw electrode was also secured in the right posterior craniotomy that we used for viral injections and is referred to as the right occipital cortex for all recordings. In addition, a subdural screw electrode was secured in a craniotomy made over the left frontal cortex (0.0 mm AP, −2.6 mm ML). Last, screw electrodes were secured in craniotomies made over the right olfactory bulb (+2.3 mm AP, +1.8 mm ML) which served as a ground and the cerebellum (−5.7 mm AP, −0.5 mm ML) which served as a reference electrode. The subdural screw electrodes were attached to an 8-pin connector that was centered over the skull and secured with dental cement. Mice were transferred to a clean cage at the end of surgery and placed on a heating blanket (37°C) until fully ambulatory.
Continuous vEEG monitoring
All mice were given a minimum 2–4 wks surgical recovery period prior to vEEG recordings. During this time, each mouse was housed individually in a standard laboratory cage within a room where the vEEG equipment was housed so they would acclimate to the recording environment. After the recovery period, each mouse was individually placed into a 21 cm × 19 cm transparent cage comparable to a standard laboratory cage. A pre-amplifier was inserted into the 8-pin headcap and connected to a multichannel commutator (Pinnacle Technology). This tethered EEG system allowed for free range of movement throughout the entire recording cage. EEG signals were acquired at 500 Hz and bandpass filtered at 1–100 Hz in Sirenia Acquisition software (Pinnacle Technology). Simultaneous video recordings synchronized with the EEG record were captured using an infrared LED camera (#AP-DCS100W, Apex CCTV) for offline analyses.
Induction of SE
Induction of SE with the convulsant pilocarpine was done in cohorts of 2–4 mice while blinded to the experimental group. Baseline vEEG recordings were acquired for at least 1 hr to capture a wide range of EEG signals associated with various behavioral states (e.g., exploration, grooming, and rest). Following the baseline period, mice were injected with the peripheral muscarinic antagonist scopolamine methyl nitrate (1 mg/kg s.c.; #S2250, Sigma Aldrich) to reduce the peripheral effects of pilocarpine. The β2-adrenergic agonist terbutaline hemisulfate (1 mg/kg s.c.; #T2528, Sigma Aldrich) was also administered to support respiration. Ethosuximide (150 mg/kg s.c.; #E7138, Sigma Aldrich) was administered to reduce the occurrence of brainstem seizures which can lead to mortality (Iyengar et al., 2015). For inhibition of hM4D(Gi)-expressing MCs and their axons during SE, mice were injected with CNO (10 mg/kg, s.c.; #BML-NS105–0005, Enzo Life Sciences). We selected a 10 mg/kg dose of CNO because this dose has been widely used in the literature to inhibit diverse cell types throughout the brain with minimal to no off-target effects (Mahler and Aston-Jones, 2018; Smith et al., 2016). Importantly, we controlled for potential off-target effects of CNO by also injecting Cre−/− mice with the same dose of CNO as iDREADDs mice (MacLaren et al., 2016). Previous studies have shown that blood plasma levels of CNO peak within 30 min of injection (Manvich et al., 2018), but the observed brain and behavioral effects last for hr (Whissell et al., 2016). For this reason, and also because numerous injections would cause behavioral stress, one injection of CNO was used to inhibit MCs for the first hr of SE. SE was induced by injecting pilocarpine hydrochloride (250 mg/kg, s.c., #P6503, Sigma Aldrich) 30 min after the CNO injection, at the time when CNO plasma levels are peaking (as noted above). All mice were injected with diazepam (5 mg/kg, s.c.; NDC# 0409–3213-12, Hospira) 2 hr after the pilocarpine injection to reduce the severity of SE, which appears to prevent morbidity and mortality after SE (Goodkin and Kapur, 2009; Iyengar et al., 2015). Mice were injected with 1 mL (s.c.) of lactated Ringer’s solution (Aspen Veterinary Resources) at this time to support hydration. Our previous studies suggest that SE is most intense for several hr after the pilocarpine injection, but there is continued spiking in the EEG overnight (VonDran et al., 2014). To ensure the continued inhibition of MCs during this time, a second injection of CNO (10 mg/kg, s.c.) was given to Cre−/− and iDREADDs mice 6 hr after pilocarpine was injected. For all in vivo experiments, stock solutions were freshly prepared in 0.9% NaCl in dH2O (saline; i.e., CNO, scopolamine, terbutaline) or phosphate buffered saline (i.e., ethosuximide).
Quantification of video EEG
All vEEG recordings were analyzed offline using Sirenia Seizure Pro (v. 1.7.9, Pinnacle Technology). Seizures were defined as rapid and rhythmic (> 3 Hz) deflections in all EEG channels that lasted > 5 s (Cho et al., 2015) and were at least 3 standard deviations above the baseline root mean square (RMS) amplitude (Iyengar et al., 2015). Seizures were considered convulsive if the video record showed behaviors consistent with stages 3–5 on the Racine scale (stage 3, unilateral forelimb clonus; stage 4, bilateral forelimb clonus with rearing; stage 5, stage 4 followed by loss of posture; Racine, 1972). Seizures were considered non-convulsive if the EEG parameters above were met, but no stage 3–5 behaviors were detected in the video record (Racine, 1972). Using these criteria, we compared the latency to the first electrographic seizure (i.e., convulsive or non-convulsive) and convulsive seizure after injection of pilocarpine. Furthermore, we measured SE, defined as the first seizures that were severe (large amplitude EEG deflections appearing in all 4 electrodes simultaneously) and persisted continuously for ≥ 5 min, a standard definition (Goodkin and Kapur, 2009; Iyengar et al., 2015).
Power spectrum analysis
EEG power was analyzed offline using Spike2 software (Cambridge Electronic Design; Kam et al., 2016). We used Spike2 to convert the EEG into a power spectrum using the Fast Fourier Transform (FFT) algorithm. The block size of the FFT was set at 512 points (1.024 s) to produce 256 bins, each with a resolution of 0.98 Hz using Hanning windows. For each subject, power was calculated using the EEG signal that corresponded to the left dorsal hippocampus. We analyzed power using frequency ranges that are typical for EEG studies of this kind (Kadam et al., 2017): delta (0 to 3 Hz), theta (4–8 Hz), alpha (9–13 Hz), beta (14–30 Hz) and gamma (31–100 Hz). For the “Baseline” and “CNO” power analyses, we analyzed the last 20 min of each period. We analyzed “SE” in 30 min increments from the onset of electrographic SE.
Spike analysis
EEG signals acquired in Sirenia Acquisition were exported for analysis with Clampfit software (v. 10.7, Molecular Devices). As in the power analysis, we selected the EEG signal that corresponded to the left dorsal hippocampus for spike analysis. We calculated baseline RMS amplitude for each subject during a 5 min artifact-free period of the baseline recording. In Clampfit software, we used the ‘Event Detection’ feature to detect spikes that were 7 standard deviations (SD) above the RMS amplitude, which is a more conservative criteria than used previously (Chauvière et al., 2012; Ziemann et al., 2008). For each subject, spike analysis was conducted on a 10 min segment of the baseline period and the first 10 min of electrographic SE. The primary measure of interest was the number of spikes that exceeded the spike threshold (i.e., ≥ 7 SDs), but the interval between spikes (interspike interval) and peak amplitude of each spike was also recorded.
Anatomy
Perfusions
Mice were initially anesthetized with isoflurane, followed by urethane (2.5 g/kg; i.p.). After deep anesthesia, the abdominal cavity was opened and the subject was transcardially perfused with ~10 mLof room temperature saline, followed by ~20 mL of cold 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4) using a peristaltic pump (Minipuls2, Gilson). The brains were extracted and stored overnight at 4°C in 4% paraformaldehyde in 0.1 M PB. The brains were then hemisected and sections (50 μm) were made using a vibratome (Vibratome 3000, Ted Pella). For each mouse, the left hemisphere was cut in the coronal plane and the right hemisphere was cut in the horizontal plane. Brains were sectioned horizontally as well as in the standard coronal plane because layers of the DG and CA3 are difficult to interpret in ventral coronal sections, whereas the layer borders are easily detected in ventral horizontal sections (Duffy et al., 2013). Serial sections (1 in 12 series; sections 600 mm apart) were stored in 24-well tissue culture plates containing cryoprotectant (25% glycerol, 30% ethylene glycol, 45% 0.1 M phosphate buffer, pH 6.7) at 4°C (Botterill et al., 2017).
Expression
Pilocarpine-treated mice were sacrificed at 3 days after SE, or after the chronic recording period (e.g., 6–8 wks after SE). The expression of hM4D(Gi) in DrD2-Cre+/− mice was confirmed by the mCherry tag. Notably, Cre−/− mice did not have any mCherry fluorescence (see Figure 4).
Our viral strategy was designed to target the majority of MCs with iDREADDs (Figure S1A). Sections were processed as described previously (Duffy et al., 2013). Briefly, sections were incubated in rabbit polyclonal anti-mCherry primary antibody (1:2000; Abcam #ab167453), followed by Alexa 568 goat-anti rabbit secondary antibody (1:500; Invitrogen #A11036). The sections were counterstained with Hoechst 33342 (1:60000; Thermo Fisher Scientific #62249) and coverslipped with Citifluor antifade mounting medium (Electron Microscopy Sciences; Botterill et al., 2015). Photomicrographs were captured on an Olympus BX61 microscope equipped with a CCD camera (Qlmaging Retiga 2000R).
The robust nature of MC labeling in the DrD2-Cre line is described elsewhere (Gangarossa et al., 2012; Senzai and Buzsaki, 2017). To demonstrate that virally-infected cells in DrD2-Cre+/− were putative MCs, a cohort of mice (n = 5) were injected in the dorsal and ventral hippocampus bilaterally with the AAV2-DIO-eYFP (4.6×1012 vg/mL; University of North Carolina (UNC) Vector Core, 160 nL per injection). These mice were used for colocalization with the glutamatergic marker GluR2/3 (Figure S1B). Sections were incubated in primary antibodies for anti-chicken GFP (1:2000; Abcam #ab13970) and anti-rabbit GluR2/3 (1:100; Millipore#AB1506) for 48 hr at 4°C, followed by Alexa 488 goat anti-chicken (1:1000; Invitrogen #A11039) and Alexa 568 goat anti-rabbit (1:500; Invitrogen #A11036) secondary antibodies. Images were acquired on a LSM510 laser scanning confocal microscope (Zeiss). In sections near the injection site, we counted the number of hilar eYFP fluorescent cells that colocalized with GluR2/3 (see Figure S1).
FluoroJade B
The degeneration of hippocampal neurons following pilocarpine-induced SE was evaluated with the anionic fluorescein derivative FluoroJade B (#1FJB, Histo-Chem). The sections were processed following the manufacturer’s instructions using a 1 in 12 series (600 μm between sections). We analyzed a minimum of 3 sections per subject at 20x magnification. Hilar FluoroJade-stained cells were counted throughout the thickness of the slice, excluding the cut edges, and the average number of cells per section was calculated for each subject.
Because the CA pyramidal cell layers are tightly packed, it is difficult to count individual FluoroJade cells. Instead, we evaluated the percent area fraction of FluoroJade staining within the borders of the GCL, CA3, and CA1 cell layers as described previously (Jain et al., 2019). Using this technique, images were converted to grayscale and thresholded against background staining (ImageJ, v. 1.44p, National Institutes of Health). We then traced the border of each cell layer and measured the area of FluoroJade staining within the traced area (Figure S3B3). Area fraction was defined as the total area within the layer that was above threshold divided by the total area of the layer. Adjacent Nissl-stained sections confirmed that the observed patterns of FluoroJade staining corresponded to regions with significant neuronal loss (Figure S3B4).
GluR2/3 and Somatostatin
To determine which hilar cells were damaged after SE, we processed sections for markers of the two most vulnerable hilar cells: MCs (GluR2/3) and HIPP cells (somatostatin). Free floating sections were washed in 0.1 M Tris Buffer (TB), followed by 5 min in 1% H202 (GluR2/3) or incubated in sodium citrate buffer (pH = 6.0) at 85°C for 30 min (somatostatin). The sections were washed with 0.1 M TB (3 × 5 min) and then blocked in 5% goat serum (Vector), 0.25% (v/v) Triton X-100, and 1% (w/v) bovine serum albumin diluted in 0.1 M TB for 30 min. Primary antibodies for anti-rabbit GluR2/3 (1:100; Millipore) or anti-rabbit somatostatin (1:1000; Peninsula Laboratories) were diluted in blocking solution and incubated overnight at 4°C. On the following day, sections were washed in 0.1 M TB (3 × 5 min) and then incubated in biotinylated goat anti-rabbit secondary antibody (1:500; Vector) for 2 hr. The sections were then rinsed (2 × 5 min) in 0.1 M TB and then incubated in avidin-biotin complex (1:500; Vector) for 1 hr. Sections were reacted in a solution containing 0.5 μg/mL 3, 3′-diaminobenzidine (Sigma), 40 μg/mL ammonium chloride (Sigma), 25 μg/mL (D+)-glucose (Sigma), and 3 μg/mL glucose oxidase (Sigma) in 0.1 M TB. Sections were washed in 0.1 M TB (3 × 5 min) and then mounted onto gelatin-coated slides and dried overnight at room temperature. On the following day, sections were dehydrated with increasing concentrations of ethanol (70%, 95%, 100%), cleared in Xylene, and coverslipped with Permount (Electron Microscopy Sciences).
A minimum of 3 dorsal and 3 ventral sections were used for cell counting, with a minimum of 600 μm between sections. GluR2/3- or somatostatin-immunoreactive cells in the hilus were counted using the same methods used for FluoroJade described above. The average number of cells per section was calculated by dividing the total number of cells counted by the number of sections analyzed.
Slice electrophysiology
Viral injections
Using the same methods described above for injection of virus in iDREADDs or eYFP, AAV2-EF1α-DIO-hChR2(H134R)-eYFP (3.4×1012 vg/mL; UNC Core, 160 nL per injection) was injected at one site in the DG (either the left anterior or left posterior hippocampal coordinates) for optogenetic studies. Slices were prepared 2–4 wks later, a time when ChR2-eYFP expression in MC axons within the IML was robust (Figure 6C).
Slice preparation
Mice were deeply anesthetized by isoflurane inhalation, decapitated, and the brain was removed and placed into 4°C sucrose-based artificial cerebrospinal fluid (ACSF, in mM: 90 sucrose, 2.5 KCl, 1.25 NaH2PO4,4.5 MgSO4, 25.0 NaHCO3,10.0 D-glucose, 80.0 NaCl, and 0.5 CaCl2; pH 7.4). All ACSF solutions here and below were oxygenated (95% O2, 5% CO2, All-Weld Products Corporation). The slices were cut horizontally in 4°C sucrose-based ACSF with a vibratome (350–400 μm thick; HMV450, Micron Instruments).
Recordings
For patch clamp recordings, slices were allowed to recover for 45 min at 35°C in sucrose ACSF and kept in sucrose ACSF at room temperature until being transferred to a recording chamber (RC-27LD, Warner) maintained at 32°C with a temperature controller (TC-324B, Warner) and in-line heater (SH-27B, Warner) and perfused with NaCl ACSF (in mM: 130 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4,25.0 NaHCO3,10.0 D-glucose, and 2.4 CaCl2) at 6 mL/min with a peristaltic pump (Masterflex C/L, Cole-Parmer). For patch recordings, borosilicate glass (1.5 mm outside diameter; 0.86 inner diameter, Sutter Instruments) was pulled horizontally (P-97, Sutter) to 7–10 MΩ. The internal solution of the recording electrode was (in mM): 130.0 K-gluconate, 4.0 KCl, 2.0 NaCl, 10.0 HEPES, 0.2 EGTA, 4.0 Mg-ATP, 0.3 Na2-GTP, 14.0 Tris-phosphocreatine, and 0.5% biocytin (pH of 7.25; 302 ± 5 mOsm). For extracellular recordings, electrodes were filled with NaCl ACSF. Recordings were filtered at 10 kHz (Digidata 1440A, Molecular Devices) and acquired by a MultiClamp 700B amplifier and pClamp software (v. 10.7, Molecular Devices).
Recordings in Figures 7, S4, S5, and S6 used an interface recording chamber and the preparation differed from what is described above in the following ways: the sucrose ASCF contained (in mM): 252.0 sucrose, 3.5 KCl, 1.25 NaH2PO4, 2.0 MgSO4, 26.0 NaHCO3, 10.0 D-glucose, and 2.4 CaCl2. Slices recovered in the interface chamber for 30 min at 31–32°C. After recovery, slices were exposed to NaCl ACSF and recordings began 30 min later. The interface chamber was based on a standard design (Scharfman, 1995b) and slices were perfused at 1–2 mL/min. Data were acquired with an Axoclamp2B amplifier (Molecular Devices), but otherwise was the same.
Optogenetic stimulation of MC axons
A thin optic fiber was placed in a micromanipulator and focused on the IML to activate MC axons in the IML. The optic fiber was made as follows: a 200 mm fiber patch cable (M86L01, ThorLabs) was unsheathed for 2 cm and polished to a thickness of 150 μm. The modified optic fiber was secured in a glass Pasteur pipette by a micromanipulator and the tip was positioned immediately above the slice surface. For patch experiments, the tip was positioned ~200–400 mm lateral to and angled at the IML proximal to the patched cell. The response was greatly reduced if the optic fiber was moved from the IML toward the hilus or fissure. The optic fiber was connected to a 473 nm diode-pumped solid-state (DPSS) laser (LRS-0473, Laserglow Technologies) that was calibrated to 10 mW at the start of each experiment using a power meter (PM100D, ThorLabs). Individual light pulses (0.5 to 2.5 msec duration) were triggered using pClamp software, with an interstimulus interval of 1 min, unless noted otherwise.
Electrical stimulation of MC axons
Slices were used that had been injected with virus encoding iDREADDs as described above. Slices were selected from areas of the DG that were distal to the injection site, so expression was primarily in the IML (i.e., ventral slices that were ipsilateral to a dorsal injection site or contralateral to the injection site). An extracellular recording electrode was positioned in the IML and an electrical stimulus (100 μA, 10–50 μsec; AMPI stimulus isolation unit) was delivered using a monopolar electrode (stainless steel Teflon-coated wire, 25 mm wide, without the Teflon; A-M Systems). Recording sites were > 250 μm from the stimulation site and at a depth that exhibited the maximal response. Experiments were conducted when there was a robust IML response with a much smaller or no response to the same stimulus in the adjacent GC layer or middle molecular layer. After slice selection, a baseline period was used to determine responses to stimulation were stable and then CNO was added to the ACSF and responses were monitored at the same frequency of stimulation (0.016 Hz). Responses (field EPSPs; fEPSPs) were measured by analyzing the maximal slope or amplitude (baseline to peak) and the results were the same (Figure S6B). CNO was dissolved in saline to make a stock solution that was stored in aliquots at −20°C until the day of recording when an aliquot was diluted in ACSF to reach 10 μM. Control experiments were conducted the same way but recordings were made for the same period of time without CNO.
Simulating SE
We hypothesized that MC excitation would strengthen during SE because of prior recordings of monosynaptically-coupled MCs and GCs, where the MC innervated the GC (Scharfman, 1995b). MC excitation of the GC was only evident when GABAa receptors were blocked and increased when the GC was depolarized (Scharfman, 1995b). In addition, the literature suggests that strong inhibition of GCs by MCs (the MC→GABAergic neuron→GC pathway) occurs under most conditions in vivo (Bui et al., 2018; Hsu et al., 2016; Jinde et al., 2012), and MC excitation of GCs in vitro is typically subthreshold (Chancey et al., 2014; Hsu et al., 2016; Jackson and Scharfman, 1996; Jinde et al., 2012; Scharfman, 1995b).
The methods to simulate SE were based on combining these factors (GABA receptor antagonism, depolarization) together to produce a pathological set of conditions that would elicit slice activity analogous to SE, which we defined as spontaneous seizure-like events lasting for hours, often with postictal periods (which occurred after some seizure activity and all instances of SD). Therefore, to simulate SE we combined factors (GABA receptor antagonists, elevated [K+]o; Table S1; Figure 6). Because NMDA receptors are relieved of their voltage-dependent block at depolarized potentials, and have been implicated in SE (Naylor et al., 2013; Yen et al., 2004), [Mg2+]o was reduced also (Table S1).
Notably, GABA receptor antagonism and principal cell depolarization are consistent with changes that have been suggested to occur during seizures. For example, reduced GABAergic inhibition, depletion of GABAergic vesicles, and/or depolarizing effects of GABA have been suggested to occur during the transition to seizures or during seizure activity (Magloire et al., 2019; Soukupova et al., 2014; Staley, 2004; Zhang et al., 2011). Principal cells become depolarized due to increased glutamate release and increased extracellular potassium (de Curtis and Gnatkovsky, 2009; Weiss et al., 2013; Zhang et al., 2011). Neuronal depolarization would lead to increased discharge and therefore increased transmitter release; the increase in release was simulated in some experiments with 4-AP (Table S1).
Latency and amplitude measurements of light-evoked EPSPs were made for 6 GCs where recordings were made before and after simulated SE had begun. Specifically, we selected the time during simulated SE when the EPSP had increased to a large degree, but had not yet evoked an AP, which is a time when EPSP amplitude is hard to distinguish from the rising phase of the AP. This approach allowed for a quantitative comparison of EPSPs in standard recording and simulated SE conditions, although it underestimates the strengthening of excitation. The number of APs and duration of the light response continues to increase.
Evaluating CNO effects in patched MCs during simulated SE
After a baseline period in normal ACSF, pharmacology and changes in the ACSF were used to simulate SE like other slice experiments (Table S1). However, for the MC experiments we initially only used 3 changes for 20 min and then all changes for another 20 min. The intention was to increase the severity of simulated SE more slowly than other experiments to allow us to better observe changes in MCs (which were quite fast). These changes were made first: we added: GABAa and GABAb receptor antagonists and used an ACSF containing 5 mM K+ and 0 mM Mg2+. For the second 20 min-period, 4-AP was added. Seizure activity was monitored with a simultaneous extracellular recording in the PC layer.
Two experiments were compared: 1) CNO was present as the slice was put into the chamber and continued throughout the procedure, and 2) CNO was not present. In each case, a MC was patched within approximately 30 min after the slice was put into the chamber, and after a baseline period to ensure the MC was stable, changes were made as described in the paragraph above, and then ACSF used in the baseline period was initiated (“wash”). For AP measurements, one cell was not firing in the baseline period and in this case, an AP was elicited by a current pulse sufficient to reach threshold.
This experimental design was based on prior recordings from MCs during 30 s cycles of perforant path stimulus trains (Scharfman and Schwartzkroin, 1989,1990a, 1990b). MCs rapidly depolarized during stimulation and discharged at high frequency. After extensive stimulation APs became stunted and the RMP became depolarized, which persisted after stimulation. In these experiments, spontaneous short bursts occurred in area CA3 long after stimulation (Scharfman and Schwartzkroin, 1990a, 1990b). In the present experiments, MCs exhibited similar effects and the effects were reduced by CNO-treatment (Figure S6).
For the experiments with patched MCs, the stock solutions of CNO were made in saline and stored at −20°C until use. On the day of the experiment, an aliquot of the stock solution was diluted in ACSF to reach working concentrations.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were carried out using Prism 8.0 (GraphPad). Results are presented as the mean ± standard error of the mean (SEM). Comparisons of parametric data from two groups were made using unpaired two-tailed Student’s t test. For non-parametric data, two-tailed Mann-Whitney were used when the data was not normally distributed, as determined by a D’Agostino-Pearson omnibus normality test (Prism). For parametric data with multiple comparisons, two-way ANOVAs followed by Bonferroni’s multiple comparisons post hoc tests were used. For Figure 1G3, we evaluated each frequency (delta through gamma) using planned comparison tests. In Figure S6, we compared vehicle-versus CNO-treated MCs across time (baseline, SE, recovery) using a repeated-measures two-way ANOVA. For all analyses, statistical significance was achieved if the p value was < 0.05 (denoted on all graphs by an asterisk).
Sample sizes were determined on the basis of power analysis (G*Power software). We determined that for a two-tailed analysis with statistical significance set at α = 0.05 and power > 80%, we required approximately 7–8 subjects per group in our acute and long-term EEG studies. Researchers were blind to experimental conditions during data acquisition and analysis. Littermates were randomly selected for inclusion in the study from the breeding colony.
DATA AND CODE AVAILABILITY
The published article includes all datasets generated or analyzed during this study.
Supplementary Material
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-mCherry (#AB167453) | Abcam | RRID:AB_2571870 |
| Rabbit anti-GluR2/3 (#AB1506) | Millipore | RRID:AB_90710 |
| Chicken anti-GFP (#AB13970) | Abcam | RRID:AB_300798 |
| Rabbit anti-somatostatin (#T4103) | Peninsula Laboratories | RRID:AB_518614 |
| Alexa 568 Goat Anti-Rabbit (#A11036) | Invitrogen | RRID:AB_10563566 |
| Alexa 488 Goat Anti-Chicken (#A11039) | Invitrogen | RRID:AB_142924 |
| FluoroJade B | Histo-Chem | Cat#1 FJB |
| Hoechst 33442 Trihydrochloride, Trihydrate | Thermofisher | Cat#H 1399 |
| Bacterial and Virus Strains | ||
| AAV2-hSyn-DIO-hM4D(Gi)-mCherry | Addgene | Cat#44362; RRID:Addgene_44362 |
| AAV2-EF1 α-DIO-hChR2(H134R)-eYFP | University of North Carolina Vector Core | N/A |
| AAV2-EF1 α-DIO-eYFP | University of North Caroline Vector Core | N/A |
| AAV2-hSyn-DIO-hM3D(Gq)-mCherry | Addgene | Cat#44361; RRID:Addgene_44361 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pilocarpine Hydrochloride | Sigma | Cat#P6503 |
| Scopolamine Methyl Nitrate | Sigma | Cat#S2250 |
| Terbutaline Hemisulfate | Sigma | Cat#T2528 |
| Ethosuximide | Sigma | Cat#E7138 |
| Diazepam | Hospira | NDC #0409–3213-12 |
| Bicuculline Methiodide (BMI) | Sigma | Cat#14343 |
| CGP52432 | Sigma | Cat#SML0593 |
| 4-Aminopyridine (4-AP) | Sigma | Cat#A0152 |
| Clozapine N-Oxide (CNO) | Enzo Life Sciences | Cat#BML-NS105–0005 |
| DL-APV | Sigma | Cat#A5282 |
| DCG-IV | Tocris | Cat#0975 |
| DNQX | Tocris | Cat#0189 |
| Deposited Data | ||
| Raw and analyzed data | This paper; Mendeley Data | http://dx.doi.org/10.17632/m8jpfctxmy.2 |
| Experimental Models: Organisms/Strains | ||
| Mouse: DrD2-Cre (C57BL/6N) | Dr. Emmanuel Valjent | N/A |
| Mouse: C57BL/6N (stock #027) | Charles River | RRID:IMSR_CRL:027 |
| Software and Algorithms | ||
| Sirenia Acquisition | Pinnacle Technology | RRID:SCR_016183 |
| Sirenia Seizure | Pinnacle Technology | RRID:SCR_016184 |
| Spike2 | Cambridge Electronic Design | RRID:SCR_00903 |
| Clampfit | Molecular Devices | RRID:SCR_011323 |
| pClamp | Molecular Devices | RRID:SCR_011323 |
| Prism | GraphPad | RRID:SCR_002798 |
| G*Power | G*Power | RRID:SCR_013726 |
| ImageJ | NIH | RRID:SCR_003070 |
| Adobe Photoshop | Adobe | RRID:SCR_014199 |
| Inkscape | Inkscape | RRID:SCR 014479 |
Highlights.
MC excitation of GCs increases during pathological conditions that cause epilepsy
Pathological excitation of GCs by MCs leads to excitotoxicity of the targets of GCs
GABA and NMDA receptors regulate pathological excitation of GCs by MCs
By increasing excitotoxicity early in epileptogenesis, MCs contribute to epilepsy
ACKNOWLEDGMENTS
We thank members of the Scharfman lab and Dr. Neil MacLusky for advice on the project. This work was supported by the New York State Office of Mental Health and NIH R01 MH-109305, 106983 (H.E.S.) and a postdoctoral fellowship from the Natural Sciences and Engineering Research Council of Canada (J.J.B.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.100.
REFERENCES
- Alexander AS, Rangel LM, Tingley D, and Nitz DA (2018). Neurophysiological signatures of temporal coordination between retrosplenial cortex and the hippocampal formation. Behav. Neurosci 132, 453–468. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Scharfman HE, and Lavenex P (2007). The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies). Prog. Brain Res 183, 3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang CW, Carlson GC, and Coulter DA (2006). Massive and specific dysregulation of direct cortical input to the hippocampus in temporal lobe epilepsy. J. Neurosci 28, 11850–11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, deCurtis M, Gnatkovsky V, Gotman J, Kohling R, Levesque M, Manseau F, Shiri Z, and Williams S (2016). Specific imbalance of excitatory/inhibitory signaling establishes seizure onset pattern in temporal lobe epilepsy. J. Neurophysiol 115, 3229–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, and Dudek FE (2010). Primary and secondary mechanisms of epileptogenesis in the temporal lobe: there is a before and an after. Epilepsy Curr. 10, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Esclapez M, Hirsch JC, and Ben-Ari Y (1998). Interneurones are not so dormant in temporal lobe epilepsy: a critical reappraisal of the dormant basket cell hypothesis. Epilepsy Res. 32, 93–103. [DOI] [PubMed] [Google Scholar]
- Botterill JJ, Guskjolen AJ, Marks WN, Caruncho HJ, and Kalynchuk LE (2015). Limbic but not non-limbic kindling impairs conditioned fear and promotes plasticity of NPY and its Y2 receptor. Brain Struct. Funct 220, 3641–3655. [DOI] [PubMed] [Google Scholar]
- Botterill JJ, Nogovitsyn N, Caruncho HJ, and Kalynchuk LE (2017). Selective plasticity of hippocampal GABAergic interneuron populations following kindling of different brain regions. J. Comp. Neurol 525, 389–406. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, and Schwartzkroin PA (1994). Hippocampal mossy cell function: a speculative view. Hippocampus 4, 393–402. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Wenzel HJ, Kunkel DD, and Schwartzkroin PA (1996). Axon arbors and synaptic connections of hippocampal mossy cells in the rat in vivo. J. Comp. Neurol 388, 271–292. [DOI] [PubMed] [Google Scholar]
- Bui AD, Nguyen TM, Limouse C, Kim HK, Szabo GG, Felong S, Maroso M, and Soltesz I (2018). Dentate gyrus mossy cells control spontaneous convulsive seizures and spatial memory. Science 359, 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumanglag AV, and Sloviter RS (2008). Minimal latency to hippocampal epileptogenesis and clinical epilepsy after perforant pathway stimulation-induced status epilepticus in awake rats. J. Comp. Neurol 510, 561–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalheiro EA, Leite JP, Bortolotto ZA, Turski WA, Ikonomidou C, and Turski L (1991). Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia 32, 778–782. [DOI] [PubMed] [Google Scholar]
- Chancey JH, Poulsen DJ, Wadiche JI, and Overstreet-Wadiche L (2014). Hilar mossy cells provide the first glutamatergic synapses to adultborn dentate granule cells. J. Neurosci 34, 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvière L, Doublet T, Ghestem A, Siyoucef SS, Wendling F, Huys R, Jirsa V, Bartolomei F, and Bernard C (2012). Changes in interictal spike features precede the onset of temporal lobe epilepsy. Ann. Neurol 71, 805–814. [DOI] [PubMed] [Google Scholar]
- Cho KO, Lybrand ZR, Ito N, Brulet R, Tafacory F, Zhang L, Good L, Ure K, Kernie SG, Birnbaum SG, et al. (2015). Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat. Commun 8, 6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Lin SL, Lee B, Kurup P, Cho HY, Naegele JR, Lombroso PJ, and Obrietan K (2007). Status epilepticus-induced somatostatinergic hilar interneuron degeneration is regulated by striatal enriched protein tyrosine phosphatase. J. Neurosci 27, 2999–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer SC (2018). Contributions of adult-generated granule cells to hippocampal pathology in temporal lobe epilepsy: a neuronal bestiary. Brain Plast. 3, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Curtis M, and Gnatkovsky V (2009). Reevaluating the mechanisms of focal ictogenesis: The role of low-voltage fast activity. Epilepsia 50, 2514–2525. [DOI] [PubMed] [Google Scholar]
- Dengler CG, and Coulter DA (2016). Normal and epilepsy-associated pathologic function of the dentate gyrus. Prog. Brain Res 228, 155–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengler CG, Yue C, Takano H, and Coulter DA (2017). Massively augmented hippocampal dentate granule cell activation accompanies epilepsy development. Sci. Rep 7, 42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamantaki M, Frey M, Berens P, Preston-Ferrer P, and Burgalossi A (2016). Sparse activity of identified dentate granule cells during spatial exploration. eLife 5, e20252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, and Staley KJ (2012). The time course and circuit mechanisms of acquired epileptogenesis In Jasper’s Basic Mechanisms ofthe Epilepsies, Chapter31, Noebels JL, Avoli M, Rogawski MA, Olsen RW, and Delgado-Escueta AV, eds. (Oxford University Press; ). [PubMed] [Google Scholar]
- Duffy AM, Schaner MJ, Chin J, and Scharfman HE (2013). Expression of c-fos in hilar mossy cells of the dentate gyrus in vivo. Hippocampus 23, 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, and Buzsaki G (1996). Interneuronsofthe hippocampus. Hippocampus 8, 347–470. [DOI] [PubMed] [Google Scholar]
- Gallo EF, Meszaros J, Sherman JD, Chohan MO, Teboul E, Choi CS, Moore H, Javitch JA, and Kellendonk C (2018). Accumbens dopamine D2 receptors increase motivation by decreasing inhibitory transmission to the ventral pallidum. Nat. Commun 9, 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangarossa G, Longueville S, De Bundel D, Perroy J, Herve D, Girault JA, and Valjent E (2012). Characterization of dopamine D1 and D2 receptor-expressing neurons in the mouse hippocampus. Hippocampus 22, 2199–2207. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, and Coulter DA (2013). Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat. Rev. Neurosci 14, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, and Kapur J (2009). The impact of diazepam’s discovery on the treatment and understanding of status epilepticus. Epilepsia 50, 2011–2018. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Nasrallah K, Jensen KR, Chavez AE, Carrera D, and Castillo PE (2017). LTP at hilar mossy cell-dentate granule cell synapses modulates dentate gyrus output by increasing excitation/inhibition balance. Neuron 95, 928–943.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, and Zhang CL (1992). The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res. Suppl. 7, 273–280. [PubMed] [Google Scholar]
- Henze DA, and Buzsáki G (2007). Hilar mossy cells: functional identification and activity in vivo. Prog. Brain Res 163, 199–216. [DOI] [PubMed] [Google Scholar]
- Henze DA, Wittner L, and Buzsáki G (2002). Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat. Neurosci 5, 790–795. [DOI] [PubMed] [Google Scholar]
- Hester MS, and Danzer SC (2013). Accumulation of abnormal adult-generated hippocampal granule cells predicts seizure frequency and severity. J. Neurosci 33, 8926–8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TT, Lee CT, Tai MH, and Lien CC (2016). Differential recruitment of dentate gyrus interneuron types by commissural versus perforant pathways. Cereb. Cortex 26, 2715–2727. [DOI] [PubMed] [Google Scholar]
- Iyengar SS, LaFrancois JJ, Friedman D, Drew LJ, Denny CA, Burghardt NS, Wu MV, Hsieh J, Hen R, and Scharfman HE (2015). Suppression of adult neurogenesis increases the acute effects of kainic acid. Exp. Neurol 264, 135–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB, and Scharfman HE (1996). Positive feedback from hilar mossy cells to granule cells in the dentate gyrus revealed by voltage-sensitive dye and microelectrode recording. J. Neurophysiol 76, 601–616. [DOI] [PubMed] [Google Scholar]
- Jain S, LaFrancois JJ, Botterill JJ, Alcantara-Gonzalez D, and Scharfman HE (2019). Adult neurogenesis in the mouse dentate gyrus protects the hippocampus from neuronal injury following severe seizures. Hippocampus 29, 683–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinde S, Zsiros V, Jiang Z, Nakao K, Pickel J, Kohno K, Belforte JE, and Nakazawa K (2012). Hilar mossy cell degeneration causes transient dentate granule cell hyperexcitability and impaired pattern separation. Neuron 76, 1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung MW, and McNaughton BL (1993). Spatial selectivity of unit activity in the hippocampal granular layer. Hippocampus 3, 165–182. [DOI] [PubMed] [Google Scholar]
- Kadam SD, D’Ambrosio R, Duveau V, Roucard C, Garcia-Cairasco N, Ikeda A, de Curtis M, Galanopoulou AS, and Kelly KM (2017). Methodological standards and interpretation of video-electroencephalography in adult control rodents. A TASK1-WG1 report of the AES/ILAE Translational Task Force of the ILAE. Epilepsia 58 (Suppl 4), 10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam K, Duffy AM, Moretto J, LaFrancois JJ, and Scharfman HE (2016). Interictal spikes during sleep are an early defect in the Tg2576 mouse model of β-amyloid neuropathology. Sci. Rep 6, 20119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kätzel D, Nicholson E, Schorge S, Walker MC, and Kullmann DM (2014). Chemical-genetic attenuation of focal neocortical seizures. Nat. Commun 5, 3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Bui A, Lew S, Oijala M, and Soltesz I (2015). In vivo evaluation of the dentate gate theory in epilepsy. J. Physiol 593, 2379–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W (2002). Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 50, 105–123. [DOI] [PubMed] [Google Scholar]
- Löscher W, Hirsch LJ, and Schmidt D (2015). The enigma of the latent period in the development of symptomatic acquired epilepsy - Traditional view versus new concepts. Epilepsy Behav. 52, 78–92. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, and Bertram EH (1992). The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res. Suppl. 7, 301–313. [PubMed] [Google Scholar]
- MacLaren DA, Browne RW, Shaw JK, Krishnan Radhakrishnan S, Khare P, Esparña RA, and Clark SD (2016). Clozapine N-oxide administration produces behavioral effects in Long-Evans rats: implications for designing DREADD experiments. eNeuro 3, ENEURO.0219–16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magloire V, Mercier MS, Kullmann DM, and Pavlov I (2019). GABAergic interneurons in seizures: Investigating causality with optogenetics. Neuroscientist 25, 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, and Aston-Jones G (2018). CNO evil? Considerations for the use of DREADDs in behavioral neuroscience. Neuropsychopharmacology 43, 934–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manvich DF, Webster KA, Foster SL, Farrell MS, Ritchie JC, Porter JH, and Weinshenker D (2018). The DREADD agonist clozapine N-oxide (CNO) is reverse-metabolized to clozapine and produces clozapine-like interoceptive stimulus effects in rats and mice. Sci. Rep 8, 3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margerison JH, and Corsellis JA (1966). Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain 89, 499–530. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Adelson PD, Cahan LD, and Leite JP (2002). Hippocampal neuron damage in human epilepsy: Meyer’s hypothesis revisited. Prog. Brain Res 135, 237–251. [DOI] [PubMed] [Google Scholar]
- Mazzuferi M, Kumar G, Rospo C, and Kaminski RM (2012). Rapid epileptogenesis in the mouse pilocarpine model: video-EEG, pharmacokinetic and histopathological characterization. Exp. Neurol 238, 156–167. [DOI] [PubMed] [Google Scholar]
- Miles R,Traub RD, and Wong RK (1988). Spread of synchronous firing in longitudinal slices from the CA3 region of the hippocampus. J. Neurophysiol 60, 1481–1496. [DOI] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Niquet J, and Wasterlain CG (2013). Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol. Dis 54, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neunuebel JP, and Knierim JJ (2012). Spatial firing correlates of physiologically distinct cell types of the rat dentate gyrus. J. Neurosci 32, 3848–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault P, and Avoli M (1991). Physiology and pharmacology of epileptiform activity induced by 4-aminopyridine in rat hippocampal slices. J. Neurophysiol 65, 771–785. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, and Lukasiuk K (2011). Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 10, 173–186. [DOI] [PubMed] [Google Scholar]
- Pun RY, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, Uhl JD, Schmeltzer SN, Faulkner C, Bronson SL, Murphy BL, et al. (2012). Excessive activation of mTOR in postnatally generated granule cells is sufficientto cause epilepsy. Neuron 75, 1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. I. After-discharge threshold. Electroencephalogr. Clin. Neurophysiol 32, 269–279. [DOI] [PubMed] [Google Scholar]
- Ratzliff Ad., Santhakumar V, Howard A, and Soltesz I (2002). Mossy cells in epilepsy: rigor mortis or vigor mortis? Trends Neurosci. 25, 140–144. [DOI] [PubMed] [Google Scholar]
- Ratzliff Ad., Howard AL, Santhakumar V, Osapay I, and Soltesz I (2004). Rapid deletion of mossy cells does not result in a hyperexcitable dentate gyrus: implications for epileptogenesis. J. Neurosci. 24, 2259–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA (2012). Migraine and epilepsy-shared mechanisms within the family of episodic disorders In Jasper’s Basic Mechanisms of the Epilepsies, Chapter 73, Noebels JL, Avoli M, Rogawski MA, Olsen RW, and Delgado-Escueta AV, eds. (Oxford University Press; ). [Google Scholar]
- Scharfman HE (1992). Differentiation of rat dentate neurons by morphology and electrophysiology in hippocampal slices: granule cells, spiny hilar cells and aspiny ‘fast-spiking’ cells. Epilepsy Res Suppl. 7, 93–109. [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (1994a). EPSPs of dentate gyrus granule cells during epileptiform bursts of dentate hilar “mossy” cells and area CA3 pyramidal cells in dis-inhibited rat hippocampal slices. J. Neurosci 14, 6041–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (1994b). Synchronization ofarea CA3 hippocampal pyramidal cells and non-granule cells of the dentate gyrus in bicuculline-treated rat hippocampal slices. Neuroscience 59, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (1995a). Electrophysiological diversity of pyramidal-shaped neurons at the granule cell layer/hilus border of the rat dentate gyrus recorded in vitro. Hippocampus 5, 287–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (1995b). Electrophysiological evidence that dentate hilar mossy cells are excitatory and innervate both granule cells and interneurons. J. Neurophysiol 74, 179–194. [DOI] [PubMed] [Google Scholar]
- Scharfman HE (1999). The role of nonprincipal cells in dentate gyrus excitability and its relevance to animal models of epilepsy and temporal lobe epilepsy. Adv. Neurol 79, 805–820. [PubMed] [Google Scholar]
- Scharfman HE (2007). The Dentate Gyrus: A Comprehensive Guide to Structure, Function, and Clinical Implications, Volume 163 (Elsevier; ). [Google Scholar]
- Scharfman HE (2016). The enigmatic mossy cell of the dentate gyrus. Nat. Rev. Neurosci 17, 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, and MacLusky NJ (2014a). Differential regulation of BDNF, synaptic plasticity and sprouting in the hippocampal mossy fiber pathway of male and female rats. Neuropharmacology 76, 696–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, and MacLusky NJ (2014b). Sex differences in the neurobiology of epilepsy: a preclinical perspective. Neurobiol. Dis 72, 180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, and Myers CE (2013). Hilar mossy cells of the dentate gyrus: a historical perspective. Front. Neural Circuits 6, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, and Pedley TA (2006). Temporal lobe epilepsy In The Neurobiology of Disease, Gilman A, ed. (Academic Press; ). [Google Scholar]
- Scharfman HE, and Schwartzkroin PA (1989). Protection of dentate hilar cells from prolonged stimulation by intracellular calcium chelation. Science 246, 257–260. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, and Schwartzkroin PA (1990a). Consequences of prolonged afferent stimulation of the rat fascia dentata: epileptiform activity in area CA3 of hippocampus. Neuroscience 35, 505–517. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, and Schwartzkroin PA (1990b). Responses of cells of the rat fascia dentata to prolonged stimulation of the perforant path: sensitivity of hilar cells and changes in granule cell excitability. Neuroscience 35, 491–504. [DOI] [PubMed] [Google Scholar]
- Senzai Y, and Buzsaki G (2017). Physiological properties and behavioral correlates of hippocampal granule cells and mossy cells. Neuron 93, 691–704.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS (1991). Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the “dormant basket cell” hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus 1, 41–66. [DOI] [PubMed] [Google Scholar]
- Sloviter RS (1994). The functional organization ofthe hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann. Neurol 35, 640–654. [DOI] [PubMed] [Google Scholar]
- Sloviter RS (2008). Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis: the importance of the “latent period” and other concepts. Epilepsia 49, 85–92. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Harvey BD, Bumanglag AV, Bender RA, and Frotscher M (2003). “Dormant basket cell” hypothesis revisited: relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J. Comp. Neurol 459, 44–76. [DOI] [PubMed] [Google Scholar]
- Smith KS, Bucci DJ, Luikart BW, and Mahler SV (2016). DREADDS: Use and application in behavioral neuroscience. Behav. Neurosci 130, 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZZ, Benison AM, Bercum FM, Dudek FE, and Barth DS (2018). Progression of convulsive and nonconvulsive seizures during epileptogenesis after pilocarpine-induced status epilepticus. J. Neurophysiol 119,1818–1835. [DOI] [PubMed] [Google Scholar]
- Soltesz I, Bourassa J, and Deschênes M (1993). The behavior of mossy cells of the rat dentate gyrus during theta oscillations in vivo. Neuroscience 57, 555–564. [DOI] [PubMed] [Google Scholar]
- Soukupová M, Binaschi A, Falcicchia C, Zucchini S, Roncon P, Palma E, Magri E, Grandi E, and Simonato M (2014). Impairment of GABA release in the hippocampus at the time ofthe first spontaneous seizure in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol 257, 39–49. [DOI] [PubMed] [Google Scholar]
- Staley KJ (2004). Role ofthe depolarizing GABA response in epilepsy. Adv. Exp. Med. Biol 548, 104–109. [DOI] [PubMed] [Google Scholar]
- Swann JW, and Brady RJ (1984). Penicillin-induced epileptogenesis in immature rat CA3 hippocampal pyramidal cells. Brain Res. 314, 243–254. [DOI] [PubMed] [Google Scholar]
- Vinck M, Bos JJ, Van Mourik-Donga LA, Oplaat KT, Klein GA, Jackson JC, Gentet LJ, and Pennartz CM (2016). Cell-type and state-dependent synchronization among rodent somatosensory, visual, perirhinal cortex, and hippocampus CA1. Front. Syst. Neurosci 9, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonDran MW, LaFrancois J, Padow VA, Friedman WJ, Scharfman HE, Milner TA, and Hempstead BL (2014). p75NTR, but not proNGF, is upregulated following status epilepticus in mice. ASN Neuro 6, 1759091414552185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu YH, Huang YG, and Chen LW (2008). Time-course of neuronal death in the mouse pilocarpine model of chronic epilepsy using Fluoro-Jade C staining. Brain Res. 1241, 157–167. [DOI] [PubMed] [Google Scholar]
- Weiss SA, McKhann G Jr., Goodman R, Emerson RG, Trevelyan A, Bikson M, and Schevon CA (2013). Field effects and ictal synchronization: insights from in homine observations. Front. Hum. Neurosci 7, 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whissell PD, Tohyama S, and Martin LJ (2016). The use of DREADDs to deconstruct behavior. Front. Genet 7, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieser HG; ILAE Commission on Neurosurgery of Epilepsy (2004). ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 45, 695–714. [DOI] [PubMed] [Google Scholar]
- Williams PA, White AM, Clark S, Ferraro DJ, Swiercz W, Staley KJ, and Dudek FE (2009). Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J. Neurosci 29, 2103–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CY, Asrican B, Moss J, Quintanilla LJ, He T, Mao X, Casse F, Gebara E, Bao H, Lu W, et al. (2018). Mossy cells control adult neural stem cell quiescence and maintenance through a dynamic balance between direct and indirect pathways. Neuron 99, 493–510.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen W, Williamson J, Bertram EH, and Kapur J (2004). A comparison of three NMDA receptor antagonists in the treatment of prolonged status epilepticus. Epilepsy Res. 59, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino M, Sawada S, Yamamoto C, and Kamiya H (1996). A metabotropic glutamate receptor agonist DCG-IV suppresses synaptic transmission at mossy fiber pathway ofthe guinea pig hippocampus. Neurosci. Lett 207, 70–72. [DOI] [PubMed] [Google Scholar]
- Young CK, and McNaughton N (2009). Coupling of theta oscillations between anterior and posterior midline cortex and with the hippocampus in freely behaving rats. Cereb. Cortex 19, 24–40. [DOI] [PubMed] [Google Scholar]
- Zhang ZJ, Valiante TA, and Carlen PL (2011). Transition to seizure: from “macro”- to “micro”-mysteries. Epilepsy Res. 97, 290–299. [DOI] [PubMed] [Google Scholar]
- Zhang ZJ, Koifman J, Shin DS, Ye H, Florez CM, Zhang L, Valiante TA, and Carlen PL (2012). Transition to seizure: ictal discharge is preceded by exhausted presynaptic GABA release in the hippocampal CA3 region. J. Neurosci 32, 2499–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, and Roth BL (2014). Silencing synapses with DREADDs. Neuron 82, 723–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemann AE, Schnizler MK, Albert GW, Severson MA, Howard MA 3rd, Welsh MJ, and Wemmie JA (2008). Seizure termination by acidosis depends on ASIC1a. Nat. Neurosci 11,816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.