

Gastrointestinal transcription factors drive lineage-specific programs in organ specification and cancer.
Abstract
Transcription factors (TFs) are spatially and temporally regulated during gut organ specification. Although accumulating evidence shows aberrant reactivation of developmental programs in cancer, little is known about how TFs drive lineage specification in development and cancer. We first defined gastrointestinal tissue–specific chromatin accessibility and gene expression during development, identifying the dynamic epigenetic regulation of SOX family of TFs. We revealed that Sox2 is not only essential for gastric specification, by maintaining chromatin accessibility at forestomach lineage loci, but also sufficient to promote forestomach/esophageal transformation upon Cdx2 deletion. By comparing our gastrointestinal lineage-specific transcriptome to human gastrointestinal cancer data, we found that stomach and intestinal lineage-specific programs are reactivated in Sox2high/Sox9high and Cdx2high cancers, respectively. By analyzing mice deleted for both Sox2 and Sox9, we revealed their potentially redundant roles in both gastric development and cancer, highlighting the importance of developmental lineage programs reactivated by gastrointestinal TFs in cancer.
INTRODUCTION
The gastrointestinal (GI) tract is one long tube consisting of the esophagus, the stomach, the small intestine, and the large intestine (i.e., colon), and its functions are to digest food, absorb nutrients, and excrete waste. Because of genetic inadequacies and/or environmental insults, cancers of the GI tract are among the most common and deadliest in the world (1).
In select GI cancers, epithelial metaplasia or transdifferentiation into adjacent organ identities precedes malignant transformation and is associated with the expression of lineage-specifying factors. For example, a subtype of gastric and esophageal cancers exhibits intestinal metaplasia, characterized by the presence of ectopic intestinal cell types in the proximal organs (2). Moreover, the recent temporal transcriptome and proteomic analyses of the mouse stomach showed that many developmentally regulated factors are abnormally reactivated in gastric cancer (3). Since these factors are likely regulated by transcription factors (TFs), a better understanding of GI lineage-specifying developmental programs directed by TFs would provide important mechanistic insight into GI tumorigenesis.
During GI development, the stomach, the small intestine, and the colon are derived from an elongating tube of endodermal cells, termed the primitive gut tube (PGT). In mice, the PGT is first formed at embryonic day 8.5 (E8.5), when the restricted expression of TFs localizes to distinct regions of the PGT. Sex-determining region Y-box 2 (SOX2), pancreatic and duodenal homeobox 1 (PDX1), and caudal type homeobox 2 (CDX2) are expressed in the foregut, midgut, and hindgut, respectively (4). As development proceeds, the foregut will give rise to the esophagus, the lung, the stomach, and the proximal intestine, while the mid- and hindguts will develop into the pancreas, the liver, the remaining small intestine, and the colon. In more detail, the murine stomach will first regionalize into the squamous forestomach and the glandular hindstomach. Further hindstomach maturation yields two subregions: the corpus, which houses H+ K+ adenosine triphosphatase–positive (ATPase+) parietal cells, and the antrum, which contains PDX1+ epithelial cells (5).
In the pancreas, the lung, and the liver, tissue lineage decisions are initiated by organ-specific master regulators, which activate organ-specific transcriptional programs (6). While CDX2 has been shown to be critical for specifying an intestinal fate through the regulation of intestinal enhancers (7–9), it remains unclear which factors are necessary for gastric specification and maturation (5). Cdx2 deletion led to a notable forestomach/esophageal transformation and ectopic expression of Sox2, suggesting their antagonistic relationship. Previous studies also have provided evidence for the role of Sox2 in gastric development and homeostasis. Analyses of mice with various Sox2 hypomorphic alleles revealed foregut defects and altered expression of GI cell type markers (10). However, the overall gastric morphology and specification in these mice were still maintained. In addition, although Sox2 labels gastric stem cells, it is dispensable during adult homeostasis; unexpectedly, its deletion in Wnt signaling–activated gastric cancer promoted gastric tumorigenesis, implying potential redundancy (11, 12). While these studies have implicated Sox2 in gastric development and cancer, its role in gastric lineage specification and the underlying mechanisms are still unclear.
To define regulation of GI specification mediated by GI TFs, we analyzed enhancers and gene expression during mouse stomach development, identifying the dynamic expression of SOX family TFs in gastric development. Subsequently, we elucidated the critical role of Sox2 in gastric lineage specification through the regulation of forestomach enhancers and defined its genetic relationship with Cdx2. Moreover, by comparing our GI developmental gene expression data to the human GI cancer data, we have shown that stomach and intestinal lineage programs are enriched in Sox2high/Sox9high and Cdx2high GI cancers, respectively. By analyzing mice deleted for both Sox2 and Sox9 in our gastric development and cancer models, we have revealed their potential redundancy. Together, our study highlights the importance of tissue-specific programs reactivated by GI lineage TFs in cancer.
RESULTS
Analyses of chromatin accessibility and gene expression during GI development identify dynamic epigenetic regulation of SOX family of TFs in gastric development
To define changes in the chromatin landscape as GI organs specify into their associated regions, we isolated epithelial cells from E13.5 and E16.5 murine guts and preformed assay for transposase-accessible chromatin sequencing (ATAC-seq). On the basis of the E16.5 forestomach, hindstomach, small intestine, and colon ATAC-seq profiles, we identified peaks enriched within the gastric and intestinal regions and those overlapping between them (Fig. 1A). These peaks primarily localized to promoter and distal intergenic regions, suggesting enrichment of DNA regulatory elements such as enhancers (fig. S1A). We found that region-enriched chromatin became accessible at E16.5 upon completion of regionalization (Fig. 1A). For example, gastric and intestinal lineage-specific genes such as gastrokine-1 (Gkn1) and fatty acid–binding protein-2 (Fabp2) were inaccessible in E13.5 but became accessible in E16.5 (Fig. 1B). In contrast, the chromatin near the FoxA1 (a pan-endodermal marker) locus was accessible in both stomach and intestinal tissues at both stages (Fig. 1B). To identify transcriptionally regulated genes critical for regionalization, we also conducted RNA sequencing (RNA-seq) using epithelium isolated from the same E16.5 GI regions. Comparison of all stomach genes versus all intestinal genes showed unique gene signatures between the two organs, indicating organ-specific transcriptional programs (fig. S1B). Unsupervised hierarchical clustering of the 2000 most differentially regulated genes showed similarity between the hindstomach and the intestinal organs (Fig. 1C), and genes up-regulated in each region were significantly enriched within the matched ATAC-seq profiles, suggesting epigenetic regulation of GI region–specific genes (Fig. 1D). To further define epigenetically active genes, we also analyzed H3K27ac modification, a histone mark signifying active DNA regulatory elements such as enhancers and gene promoters (13, 14), in the four E16.5 GI regions. Peak distribution confirms that most H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq) peaks were found at promoter and distal intergenic regions, conferring regulation via promoters and enhancers (fig. S1C). In addition, we observed that while the chromatin near developmental and region-enriched genes remained open in all GI regions at E16.5, genes marked by H3K27ac conferred region specificity. For example, the chromatin near the Sox4 (an endodermal marker gene) locus was open and epigenetically active in all GI organs, while Sox2 and Cdx2 enhancers were active only in the stomach and intestinal tissues, respectively (Fig. 1E). To identify key transcriptional regulators of GI development, we conducted TF motif analysis of E13.5 and E16.5 region-specific ATAC-seq profiles and identified the SOX TFs as one of the most enriched TF families in both E13.5 stomach peaks and E16.5 forestomach/hindstomach common peaks (Fig. 1F). Immunohistochemistry analysis of E13.5 stomachs showed an enrichment of SOX2 and SOX9 (fig. S2A).
Fig. 1. SOX TFs are enriched in the stomach as the chromatin landscape acquires accessibility during development.
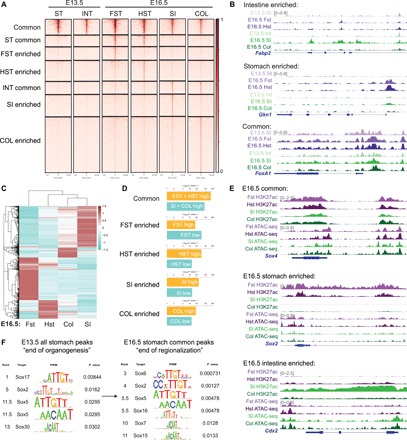
(A) ATAC-seq profiles of E13.5 stomach and intestine compared to E16.5 forestomach, hindstomach, intestine, and colon. Region-enriched peaks are determined on the basis of E16.5 profiles, and their corresponding accessibility is analyzed in the E13.5 tissues. (B) Representative examples of ATAC-seq tracks show acquired chromatin accessibility in E16.5 stomach-specific (Gkn1) and intestinal-specific (Fabp2) loci as the GI organs become regionalized, while common, endodermal loci such as FoxA1 are accessible throughout gut development. (C) Unsupervised hierarchical clustering of E16.5 forestomach, hindstomach, small intestine, and colon transcriptomes based on the top 2000 most variable genes. (D) Binding and Expression Target Analysis (BETA) comparing association of region-specific ATAC-seq peaks to genes differentially regulated in each region at E16.5. (E) Representative examples of H3K27ac ChIP-seq and ATAC-seq peaks of common (Sox4), stomach-specific (Sox2), and intestinal-specific (Cdx2) active DNA regulatory elements such as promoters and enhancers are shown. (F) TF motif analysis of all E13.5 stomach and E16.5 stomach common ATAC-seq peaks shows enrichment of SOX TF motif. Rank indicates the ranking of the motif enrichment in all 637 mouse TF motifs. For ATAC-seq (n = 1 per group), six to eight embryonic guts were pooled. For H3K27ac ChIP-seq (n = 1 per group), 25 to 30 embryonic guts were pooled. For RNA-seq (n = 2 per group), 13 to 15 embryonic guts were pooled. ST, stomach; INT, intestine; FST, forestomach; HST, hindstomach; SI, small intestine; COL, colon.
Sox2 is critical for gastric specification and regionalization
To determine the role for Sox2 in gastric development, we generated Sox2 knockout (KO) mice (Sox2CreERT2/flox) and administered tamoxifen at E8.5 to bypass early embryonic lethality observed in Sox2 KO embryos (15). Upon complete deletion of Sox2, whole-mount images of Sox2 KO mice and hematoxylin and eosin (H&E) analysis revealed a marked reduction in stomach size and loss of squamous epithelium compared to controls (Fig. 2, A and B). Immunohistochemistry analysis of the mutant epithelium demonstrated an absence of transformation-related protein 63 (TP63), a forestomach marker, but properly localized expression of H+ K+ ATPase and PDX1 (i.e., hindstomach markers) (Fig. 2C) and CDX2, an intestinal marker (fig. S2B). These results suggest that Sox2 is critical for gastric growth and forestomach specification, while it is dispensable for hindstomach regionalization. We next examined the lineage trajectory of endodermal progenitors upon loss of Sox2. To conduct cell lineage–tracing experiments, we crossed the Sox2 KO mutants with R26mT/mG reporter mice in which Sox2CreERT2+ and their descendant cells were permanently labeled with green fluorescent protein (GFP) upon tamoxifen induction at E8.5, while Sox2CreERT2− cells expressed TdTomato (Fig. 2D). We observed more GFP+ cells in the distal region of the small intestine in Sox2-deleted mice compared to the controls (Fig. 2E). These Sox2−/GFP+ cells in the mutant intestines were negative for intestinal (i.e., Cdx2 and Tff3) and gastric markers (Tp63 and H+ K+ ATPase), unlike Sox2+/GFP+ cells in the control intestines (fig. S2C). Collectively, these results suggest that Sox2-deleted endodermal progenitors lose the ability to respond to niche signals, and thus, fail to acquire intestinal identity properly.
Fig. 2. Sox2 is essential for gastric specification and regionalization.
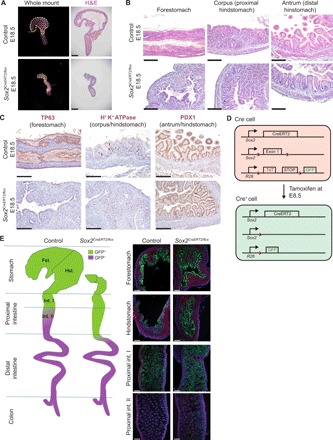
(A) Whole-mount images and subsequent histological analyses using H&E staining of control and Sox2-deleted guts at E18.5 (n ≥ 3 each). Tamoxifen was administered at E8.5. Scale bars, 200 μm. (B) Higher magnification images of gastric regions highlight the loss of squamous, forestomach and epithelium and the maintenance of glandular, hindstomach morphology in Sox2 KO mutants. Scale bars, 100 μm. (C) Immunohistochemistry of regional gastric markers reiterates the loss of TP63+ forestomach epithelium but the proper expression of hindstomach markers, H+ K+ ATPase and PDX1, in Sox2 KO embryos (n ≥ 3 each). Scale bars, 100 μm. (D) Schematic of alleles used to generate conditional Sox2-deleted embryos using tamoxifen-induced Cre-LoxP recombination for cell lineage–tracing experiments is shown. (E) Schematic (left) and fluorescence images (right) of cell lineage tracing of Sox2-deleted endodermal progenitors throughout gut development and their allocation into the different regions of the GI tract (n = 3 each). Scale bars, 100 μm.
Sox9 compensates for the loss of Sox2 and maintains residual gastric development
Residual gastric development maintained upon the loss of Sox2 implies potential redundancy of additional factors responsible for preserving gastric specification (Fig. 2, A to C). Since we identified the expression of Sox9 in the developing stomach (fig. S2A), we examined its expression in the Sox2 KO embryos and found that SOX9 became strongly activated in the proximal mutant stomach (Fig. 3A). Analysis of forestomach and hindstomach ATAC-seq peaks showed that Sox2 and Sox9 are broadly accessible despite their region-specific expression (Fig. 3B). These data suggest that the gastric chromatin is primed and can support either SOX TF expression. To determine their potential redundancy in gastric development, we generated mice conditionally deleted for both Sox2 and Sox9 [double knockout (DKO)] (Sox2CreERT2/flox;Sox9flox/flox) at E8.5, the onset of gut organogenesis. Despite efficient deletion of Sox2, these DKO embryos still maintained gastric specification due to the Cre escapers expressing Sox9 (Fig. 3C and fig. S3A). Accordingly, staining for region-specific markers showed complete loss of the forestomach marker TP63, while the hindstomach/antral marker PDX1 was maintained (fig. S3A). To circumvent the temporally restricted activity of Sox2CreERT2, we used a constitutively active, gut epithelial Cre, ShhCre, to replicate the Sox2;Sox9 DKO experiments. While we observed a drastic reduction in organ size by H&E staining, we similarly observed the presence of Sox9+ gastric epithelial cells in the DKO mutants, masking the true DKO phenotype (fig. S3B).
Fig. 3. Sox9 compensates for the loss of Sox2 and maintains residual gastric development.
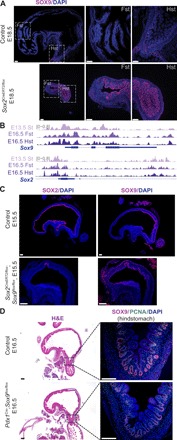
(A) Immunofluorescence staining shows that SOX9 is ectopically expressed in Sox2 deleted proximal stomachs (n ≥ 3 each). Scale bars, 100 μm. DAPI, 4′,6-diamidino-2-phenylindole. (B) ATAC-seq tracks of Sox2 and Sox9 in stomach tissues at E13.5 and E16.5 display broad chromatin accessibility maintained during gut regionalization. (C) SOX9 expression is maintained in Sox2;Sox9 DKO embryos, shown through immunofluorescence staining (n = 3 each). Scale bars, 100 μm. (D) H&E (left) and fluorescence images (right) of Sox9-deleted embryos using an alternative gut epithelial Cre (Pdx1Cre) show normal gastric specification, with maintained SOX9 and proliferating cell nuclear antigen (PCNA) expression in the hindstomach. Dashed boxes on H&E images correspond to regions shown for subsequent immunofluorescence staining (n = 3 each). Scale bars, 100 μm.
In addition, when we generated Sox9 KO embryos using an alternative hindstomach epithelial Cre, Pdx1Cre, we observed the same Cre escapers (Fig. 3D) and maintenance of hindstomach differentiation by H+ K+ ATPase staining (fig. S3C). This result was consistent with the incomplete recombination of the Sox9 floxed allele previously reported during pancreatic development (Pdx1Cre;Sox9flox/flox) (16). Therefore, our findings imply that Sox9 is critical for gastric epithelial survival during development, as the Cre escapers expressing Sox9 have a selective advantage and are able to maintain gastric specification in the absence of Sox2.
SOX2 directly regulates forestomach lineage-specific genes by maintaining chromatin accessibility at their loci
Since our data demonstrated that Sox2 deletion leads to the complete loss of forestomach identity, we hypothesized that it regulates the expression of genes critical for forestomach specification. To address this hypothesis, we performed SOX2 ChIP-seq experiment using E16.5 forestomach epithelium. We compared Sox2 binding sites to our E16.5 forestomach ATAC-seq profile (i.e., the tissue highly expressing Sox2) and the E16.5 small intestine ATAC-seq profile (i.e., the tissue lacking Sox2 expression). We found the enrichment of SOX2+ peaks in the forestomach profile compared to the intestinal profile (Fig. 4A). Approximately 66% of all Sox2 binding sites localized to promoters and distal intergenic regions, suggesting that Sox2-directed regulation occurs at promoters and enhancers, respectively (Fig. 4B and fig. S1D). We then compared the SOX2 targets to the 2100 most up-regulated genes in the forestomach transcriptome. Notably, we found that more than 10% of highly expressed forestomach genes were direct targets of Sox2 (Fig. 4C). These targets were enriched for Gene Ontology (GO) terms such as keratinocyte differentiation and epidermis development, which are features of the squamous epithelium found in the forestomach and the esophagus (Fig. 4D). In addition, co-TF motif analysis of all SOX2+ peaks identified TP63, a critical forestomach stem cell marker, as the top-ranked co-binding TF, suggesting their transcriptional cooperation (Fig. 4E) (17).
Fig. 4. SOX2 directly induces forestomach lineage-specific genes by maintaining chromatin accessibility at their loci.
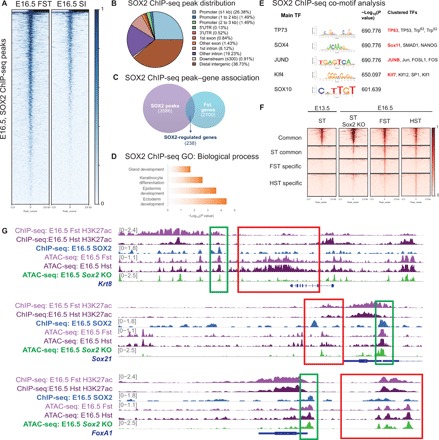
(A) SOX2 ChIP-seq peaks are enriched in the E16.5 forestomach ATAC-seq profile versus the E16.5 intestinal ATAC-seq profile, which lacks Sox2 expression. For SOX2 ChIP-seq (n = 1), 30 embryonic stomachs were pooled. (B) Genomic distribution of SOX2 ChIP-seq peaks. 5′UTR, 5′ untranslated region. (C) Venn diagram outlining the overlap between SOX2 target gene associations and the top 2100 most highly expressed genes in the E16.5 forestomach transcriptome (P < 0.05 and fold change > 1.5 when compared to the E16.5 small intestinal transcriptome). FC, fold change. (D) GO term analysis of SOX2-regulated forestomach genes. (E) Co-TF motif analysis shows enrichment for motifs important in squamous epithelium (TP63), endodermal development (SOX4), and embryonic stem cells [Kruppel-like factor 4 (KLF4)]. (F) Comparison of E16.5 forestomach and hindstomach and E13.5 stomach ATAC-seq heat maps to E16.5 Sox2 KO stomach ATAC-seq data shows loss of forestomach-enriched peaks. The peaks are categorized on the basis of the common and region-enriched peaks defined in Fig. 1. (G) Alignment of E16.5 forestomach and hindstomach ATAC-seq and H3K27ac ChIP-seq tracks with SOX2 ChIP-seq to show examples of SOX2 directly regulated genes (the green box highlights Sox2 binding site in proximity to the gene of interest). E16.5 Sox2 KO ATAC-seq profile demonstrates that forestomach lineage-specific open chromatin regions become inaccessible in Sox2 KO embryos, but the same is not observed for Sox2-regulated endodermal genes (the red box outlines corresponding ATAC-seq signal).
Since Sox2 is known to act as a pioneering factor in reprograming (18), we examined whether Sox2 loss changes the chromatin landscape during gastric development. To address this question, we performed ATAC-seq using E16.5 Sox2-deleted gastric epithelium. Analyzing wild-type E16.5 forestomach, E16.5 hindstomach, and Sox2 KO whole-stomach ATAC-seq profiles, we found the complete loss of forestomach-enriched peaks that were originally identified in Fig. 1A, suggesting lack of chromatin changes associated with regionalization (Fig. 4F). For example, ATAC-seq traces near the locus of Krt8, a SOX2-regulated gene expressed in adult gastric glands (19), showed a drastically reduced level of chromatin accessibility (Fig. 4G). Corroborating these data, regression analysis showed that the E16.5 Sox2 KO whole-stomach ATAC-seq profile is more similar to the E13.5 stomach (i.e., less differentiated organ) ATAC-seq profile than the E16.5 fore- or hindstomach profiles (fig. S1E). However, the common peaks accessible in all of the developing GI organs were still maintained in the Sox2-deleted gut. For instance, the chromatin near the loci of SOX2+ endodermal targets such as Sox21 and Sox4 was still accessible, suggesting that additional co-factors are able to preserve chromatin accessibility despite Sox2 deletion (Fig. 4G). Nevertheless, these results demonstrate the importance of Sox2 in gastric specification by maintaining chromatin accessibility at forestomach lineage-specific loci.
Loss of Sox2 rescues forestomach/esophageal transformation induced by Cdx2 deletion
A previous study showed that the deletion of the intestinal-specific factor Cdx2 in the developing endoderm transformed the intestinal, villous epithelium into gastric epithelium (7). This mutant epithelium ectopically expressed SOX2 and TP63 and acquired a forestomach, squamous cellular morphology. However, the role of these TFs in gastric transformation is still unknown. Given our findings demonstrating the critical role of Sox2 in gastric specification, we hypothesized that the ectopic expression of Sox2 in Cdx2 KO embryos might be sufficient to drive forestomach/esophageal transformation. To address this hypothesis, we generated embryos conditionally deleted for both Sox2 and Cdx2 at E8.5 (R26CreERT2;Cdx2flox/flox;Sox2flox/flox). We observed the partial restoration of villous/glandular-like epithelium in these embryos (Fig. 5A). The ectopic expression of the forestomach marker, TP63, was completely lost, while the pattern of proliferating cell nuclear antigen–positive (PCNA+) cells was partially restored (Fig. 5B). Further characterization of the Sox2;Cdx2 DKO intestinal epithelium revealed the absence of intestinal markers (i.e., TTF3+/Alcian blue+ goblet cells and alkaline phosphatase+ enterocytes) (fig. S4A) and the hindstomach/corpus marker H+ K+ ATPase (fig. S4B). The DKO intestinal epithelium stained positive for gastric-type surface mucous cells, evidenced by periodic acid–Schiff staining and the hindstomach/antral marker PDX1, suggesting antral transformation of DKO intestinal epithelium (fig. S4B). Furthermore, these Cdx2 and Sox2 DKO embryos ectopically activated Sox9 expression in the distal intestinal epithelium (Fig. 5B), recapitulating the Sox2-Sox9 dynamics observed in the Sox2 KO embryos (Fig. 3A). In addition to the villus-like morphology (Fig. 5A) and the intervillous restriction of PCNA+ proliferating cells (Fig. 5B), these results further support an antral/proximal intestine–like identity of Sox2;Cdx2 DKO epithelial cells.
Fig. 5. Loss of Sox2 rescues forestomach/esophageal transformation induced by Cdx2 deletion.
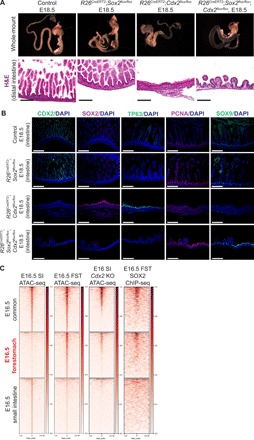
(A) Whole-mount images and subsequent histological analyses using H&E staining of control and Sox2;Cdx2 doubly deleted guts at E18.5 (n ≥ 3 each). Cre-LoxP recombination was induced at E8.5. Scale bars, 100 μm. (B) Immunofluorescence images show CDX2, SOX2, TP63, PCNA, and SOX9 staining in control, Sox2 KO, Cdx2 KO, and Sox2;Cdx2 DKO embryos. Scale bars, 100 μm. (C) Comparison of E16.5 common-, forestomach-, and small intestine–enriched ATAC-seq peaks in publicly available Cdx2 KO ATAC-seq data. Ectopic forestomach-specific peaks in Cdx2 KO embryos are associated with SOX2 binding (SOX2 ChIP-seq). Cdx2KO ATAC-seq was performed in E16 intestine tissue by Banerjee et al. and retrieved from GSM3181688. SOX2 ChIP-Seq was performed in E16.5 forestomach epithelium.
To define the epigenetic regulation of gastric transformation induced by Cdx2 deletion, we compared our E16.5 forestomach ATAC-seq and SOX2 ChIP-seq profiles to E16 Cdx2 KO ATAC-seq data generated by Banerjee et al. (9). We found that Cdx2 KO embryos contained ectopic activation of forestomach-specific ATAC-seq peaks that were partially associated with Sox2 binding (Fig. 5C). Together, our results demonstrate that the ectopic expression of Sox2 is responsible for squamous transformation induced by Cdx2 deletion.
Developmental stomach and intestinal programs are enriched in Sox2high- and Cdx2high-expressing GI cancers, respectively
Multiple GI cancers show an up-regulation of SOX2, SOX9, and CDX2 (20–22). Although the role of Sox2 and its transcriptional mechanisms in squamous cell esophageal carcinomas have been elucidated (17, 23), its role in gastric cancer is still controversial: It has been proposed to function as both a tumor suppressor and an oncogene (24). As our data demonstrated that Sox2 is both essential and sufficient for gastric (forestomach) lineage specification, we hypothesized that these lineage-specifying TFs reactivate GI developmental lineage programs in human cancers. To examine whether E16.5 organ-specific programs become activated in GI cancers, we used The Cancer Genome Atlas (TCGA) GI cancer database to identify Sox2high-, Sox9high-, and Cdx2high-expressing human gastric [stomach adenocarcinoma (STAD)] and colorectal [colon adenocarcinoma (COAD)] cancers (25–28). By performing gene set enrichment analyses, we demonstrated that Sox2high and Cdx2high cancers are enriched for E16.5 stomach and intestinal genes, respectively (Fig. 6, A and B). In addition, Sox9high cancer samples exhibited a gastric program bias (Fig. 6, A and B). These results suggest that the overexpression of these TFs in GI cancers may promote tumorigenesis by activating lineage-specific programs.
Fig. 6. Developmental stomach and intestinal programs are enriched in Sox2high- and Cdx2high-expressing GI cancers, respectively.
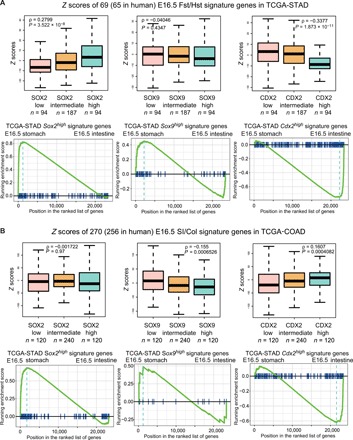
(A) Top: Differential genes expressed in Sox2high, Sox9high, and Cdx2high human stomach cancer (STAD) transcriptomes are compared to our E16.5 GI RNA-seq profiles. Bottom: Gene set enrichment analysis reveals strong associations between Sox2high/Sox9high cancer genes and the E16.5 stomach profiles and between Cdx2high cancer genes and the E16.5 intestinal profiles. (B) Similar analysis conducted using human colon cancer (COAD) transcriptomes from TCGA shows the activation of stomach and intestinal lineage-specific genes in Sox2high/Sox9high and Cdx2high cancers, respectively.
Single deletion of Sox2 or Sox9 in gastric adenoma mice increases cancer severity, whereas DKO rescues tumor severity
To investigate the role of SOX TFs during gastric cancer, we used our previously published gastric adenoma model harboring the activation of Notch signaling in gastric acid–secreting cells (Atp4bCre;R26NICD/+) (29). Since the expression of SOX9 is increased in human gastric cancer (21) and in our adenoma mouse model (Fig. 7A), we hypothesized that Sox9 promotes gastric cancer initiation. To determine its role in gastric cancer in vivo, we conditionally deleted Sox9 in Atp4bCre;R26NICD/+;Sox9flox/flox mice. However, we observed more disorganized glandular epithelia and increased presence of Alcian blue+ mucin cells (i.e., a hallmark of early-stage gastric cancer) in Sox9 KO adenoma mice compared to the controls (Fig. 7B). We detected a notably increased level of Sox2 nuclear staining in the adenomatous glands of Sox9 KO adenoma mice in contrast to staining in the glandular stomach of the control (Fig. 7C). To determine the role of Sox2 in gastric cancer in vivo, we generated Sox2-deleted mice in the adenoma background. Consistent with recently published work addressing the role of Sox2 in Wnt signaling–activated gastric cancer (12), we observed increased tumor severity upon Sox2 deletion (Fig. 7B). We also observed up-regulated Sox9 expression in these mice (Fig. 7C). These results suggest that both factors act in a redundant manner to compensate for the loss of either SOX TF during cancer initiation. Notably, this Sox2-Sox9 dynamic was also observed during gastric embryonic development: The loss of Sox2 up-regulated Sox9 and subsequently maintained residual gastric specification (Fig. 3A). To examine whether Sox2 and Sox9 act in a redundant manner during cancer initiation, we generated Sox2 and Sox9 DKO mice in the adenoma background. These mice exhibited more organized glandular structures and reduced Alcian blue staining in comparison to single-KO adenoma mice (Fig. 7B). Histopathology scoring confirmed significant reductions in disease severity and the presence of dysplastic epithelium when comparing DKO adenoma mice to their single-KO counterparts (fig. S5, A and B). These data demonstrate that both SOX TFs cooperatively regulate genes that are important during cancer initiation, and single-SOX TF deletions are inadequate in preventing gastric cancer initiation.
Fig. 7. Single deletion of Sox2 or Sox9 in gastric adenoma mice increases cancer severity, whereas DKO rescues tumor severity when compared to singly deleted counterparts.
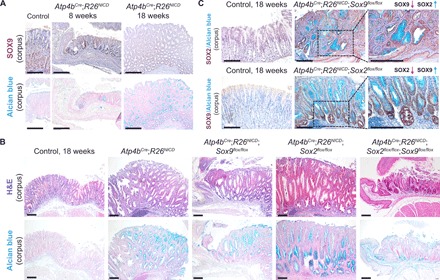
(A) SOX9 immunohistochemistry (top) and Alcian blue staining (bottom) show SOX9 overexpression at sites of adenomatous epithelium in early (8 weeks) and late (18 weeks) adenoma mice (n = 3 each). Scale bars, 100 μm. (B) Corresponding H&E (top) and Alcian blue (bottom) staining shows the aberrant changes in gastric epithelial structure and cell types in the various adenoma models (n ≥ 3 for Atp4bCre;R26NICD, Atp4bCre;R26NICD;Sox2flox/flox, Atp4bCre;R26NICD;Sox9flox/flox, and Atp4bCre;R26NICD;Sox2flox/flox;Sox9flox/flox mice). Scale bars, 100 μm. (C) SOX2 immunohistochemistry in Atp4bCre;R26NICD;Sox9flox/flox mice (top) and SOX9 immunohistochemistry in Atp4bCre;R26NICD;Sox2flox/flox mice (bottom) demonstrate reciprocal activation in the absence of either SOX TF (n = 3 each). Scale bars, 100 μm.
DISCUSSION
As the endoderm and mesoderm assemble into the PGT and gut development proceeds, organ and regional boundaries become specified through the expression of key developmental TFs (4). These factors would likely bind to unique DNA regulatory elements such as H3K27ac-rich promoters and enhancers of fate determining genes, thereby regulating expression in a spatial and temporal manner. To understand global chromatin and gene expression changes during GI development, we analyzed chromatin accessibility and the H3K27ac-active enhancer mark throughout all regions of the GI tract, which include the colon, and found both broadly permissive and region-enriched chromatin regions during gut development. In identifying the dynamic expression of SOX family TFs in gastric development, we focused on the roles of Sox2 and Sox9. We demonstrated that Sox2 is critical for gastric specification and regionalization by directly regulating the expression of forestomach-specific genes and chromatin accessibility at their loci.
In the absence of Sox2, Sox9 was up-regulated and maintained hindstomach specification. Because of inefficient Cre-LoxP excision, Sox9 expression was still maintained in Sox9-deleted embryos, masking the true Sox9 KO, and Sox2;Sox9 DKO phenotypes. The Cre escapers still expressing Sox9 had a selective survival advantage over Sox9−/− cells and were able to maintain residual gastric specification.
A previous study showed that Sox2 overexpression is sufficient to induce gastric cell type markers, but CDX2 expression and villus-like glands were still maintained (30), in contrast to the complete forestomach/esophageal transformation upon Cdx2 deletion at E8.5 (7). The Sox2 overexpression study used Villin-rtTA transgenic mice, in which Sox2 expression begins at E13.5. Given the various phenotypes induced by Cdx2 deletions at different developmental stages due to chromatin-mediated developmental plasticity (8, 9), it would be interesting to examine Sox2 overexpression at earlier stages. Sox2 deletion at E8.5 followed by lineage tracing showed that despite the fact that Sox2-deleted gastric progenitors were properly incorporated into the intestine, they failed to express intestinal genes, suggesting a deficiency in responding to niche signals. Therefore, this study also highlights potentially distinct epigenetic regulation of the stomach and intestine mediated by SOX2 and CDX2, respectively.
Since developmental programs are known to be frequently reactivated in cancer, we also investigated their significance in cancer by comparing our GI developmental and available adult cancer transcriptomes. We found an enrichment of forestomach, hindstomach, and intestinal lineage-specific developmental genes in Sox2high-, Sox9high-, and Cdx2high-expressing gastric and colorectal cancers, respectively. To determine the importance of developmental lineage programs induced by these TFs in GI cancers, we examined the roles of Sox2 and Sox9 in gastric cancer mice. Consistent with the Sox2-Sox9 dynamics observed during development, we found that a single deletion of either TF leads to activation of the reciprocal factor, while the double deletion of Sox2 and Sox9 in the adenoma background ameliorates cancer progression when compared to single-KO adenoma models. This SOX TF redundancy provides new insight into the complex roles of Sox2 proposed in gastric cancer (12, 24).
A recent study has established CDX2 as a prognostic biomarker in stage II and stage III colon cancer, suggesting that a subgroup of patients with stage II colon cancer might benefit from adjuvant chemotherapy based on the lack of CDX2 expression in cancer stem cells (31). Notably, SOX2 expression has been associated with a colon cancer stem cell state and down-regulation of CDX2 (22). This relationship between SOX2 and CDX2 in human colon cancer is consistent with our finding of their genetic relationship during mouse GI specification. Therefore, a better understanding of these developmental lineage-specific TFs and their epigenetic mechanisms would likely provide significant insight into the disease mechanisms of human GI cancers.
MATERIALS AND METHODS
Experimental animals
All animal husbandry was conducted in adherence to the standards proposed by the Canadian Council on Animal Care and approved by the institutional animal care committees of The Centre for Phenogenomics, Toronto. ShhCre-eGFP (32)/R26mT/mG (33)/Pdx1Cre (34), Cdx2floxed (35)/R26NICD (36), R26CreERT2 (37), and Atp4bCre (38) mice were obtained as gifts from C.-c. Hui, R. Shivdasani, J. Rossant, and J. Mills, respectively. Sox2CreERT2, Sox2floxed, and Sox9floxed mice were purchased from the Jackson Laboratory.
Tamoxifen oral gavage to pregnant dams
Murine females aged 15 weeks were used for embryonic tamoxifen administration via oral gavage. On E8.5 and E10.5, 100 μl of tamoxifen (20 mg/ml) and progesterone (10 mg/ml) in sunflower seed oil was administered to the dam on each day. Females were euthanized at the appropriate end point, and embryos were immediately collected via cesarean section for histological and high-throughput experiments.
Embryonic epithelial isolation for ATAC-seq, RNA-seq, and ChIP-seq
For E13.5 mouse experiments, ShhCre;R26YFP/+ embryos were used for downstream high-throughput experiments. After dissection, the stomach and intestinal organs were isolated, separated, washed in cold phosphate-buffered saline (PBS) and digested in 4 ml of a 3:1 solution of trypsin LE: 1× PBS at 37°C. After 10 min, tissue was vigorously pipetted until the organs broke down into a single-cell suspension, which was confirmed using bright-field microscopy. The enzymatic reaction was then inhibited with 10% fetal bovine serum. After washing cells in cold PBS, the single cells were resuspended in 400 μl of 1:5000 SYTOX Blue (Thermo Fisher Scientific, S34857) in cold 1× PBS, filtered through a 30-μm mesh and submitted for sorting. Sorted cells were washed again and processed for ATAC-seq [50,000 cells from three to four yellow fluorescent protein–positive (YFP+) embryos] using the previously published protocol (39).
For E16.5 experiments, whole-organ digestion using trypsin was detrimental to epithelial cell viability; thus, an alternative isolation approach was used. Given that strict cell numbers are required for ATAC-seq, gut epithelium was isolated from ShhCre;R26YFP/+ embryos before single-cell dissociation. In more detail, the E16.5 embryonic stomach, the proximal small intestine, and the colon were dissected and opened longitudinally using a 0.5-cm tungsten needle. The forestomach was separated from the hindstomach along the squamous-glandular border under a dissecting microscope and incubated in dispase solution for 15 min at 37°C. Next, the epithelial layer was manually separated from the mesenchyme and flash-frozen for RNA isolation or collagenase-digested for sorting. The hindstomach, proximal small intestine, and colon were incubated in 10 mM EDTA, 5 mM EDTA, and 5 mM EDTA in 1× PBS, respectively, for 30 to 40 min at 4°C on a shaker. Tissues were then moved to fresh 1× PBS and shaken vigorously for 2 to 3 min or until epithelial glands/villi/crypts were observed in suspension. Isolated epithelial cells were immediately frozen, and RNA isolation was conducted using an RNA isolation kit (QIAGEN, catalog no. 74104). To prepare for sorting for ATAC-seq experiments, the fresh epithelial pellets of the forestomach, hindstomach, small intestine, and colon were then digested in collagenase-based solution for 20 min at 37°C. The resulting single-cell suspension was washed in cold 1× PBS, resuspended in 500 μl of 1:5000 SYTOX Blue, and submitted for sorting. Sorted cells were then used for ATAC-seq using the same protocol as mentioned above.
Chromatin immunoprecipitation sequencing
After isolation of embryonic epithelium using dispase for forestomach and EDTA for the hindstomach, intestine, and colon, epithelial pellets were fixed in 1% paraformaldehyde for 10 min at room temperature, quenched with one-twentieth volume of 2.5 M glycine for 5 min, washed twice with cold 1× PBS, and flash-frozen. When enough sample was acquired (approximately 0.1 g of tissue per region), fixed pellets were pooled, dounced in cold PBS with protease inhibitor, and filtered through a 70-μm mesh. Samples were then incubated in cold cell membrane and nuclear lysis buffers containing nondenaturating detergents for 30 min each on a shaker at 4°C. Washed samples were then pelleted and resuspended in 300 μl of sonication buffer containing 0.1% SDS. Sonication was conducted using the Diagenode Bioruptor (Diagenode, B01020001) at high power for 40 cycles, 30 s ON/OFF. Sonicated samples were cleared with 1:10 dilution of 30% Triton X-100 and added to antibody bound beads (human anti-SOX2: R&D Systems, AF2018-SP; rabbit anti-H3K27ac: Abcam, ab4729). The next day, beads were washed with high- and low-salt buffers and treated with proteinase K overnight at 65°C. DNA was isolated using phenol/chloroform extraction, and 10 ng of immunoprecipitated DNA was used for cDNA library construction using the Rubicon DNA-seq ThruPLEX 48S Kit. Libraries were size-selected and submitted to be sequenced at 50–base pair read length, single-end reads, with 15 million to 20 million reads per library. All of the -omics data can be accessed at GSE134277.
Histology, immunofluorescence, and immunohistochemistry
After dissection of either embryos or adult stomachs, tissue was immediately fixed in fresh 4% PFA in 1× PBS overnight. Whole-mount pictures were taken with a Leica microscope and ImageJ software (National Institutes of Health). After dehydration, tissues were processed, embedded, and cut into 5- to 7-μm sections. After baking overnight at 37°C, these slides were deparaffinized and rehydrated for staining. For simple dyes, Harris’s hematoxylin was used with alcoholic eosin Y (Electron Medical Sciences), Alcian blue with nuclear fast red, periodic acid–Schiff (Sigma-Aldrich, 395B-1KT) with Harris’s hematoxylin, and alkaline phosphatase (Roche, 11093274910) staining with nuclear fast red.
For protein staining, slides were antigen-retrieved using a citrate buffer (pH 6) for 30 min, blocked for 1 hour, and stained with the following antibodies: rabbit anti-Sox2 (Abcam, ab97959; 1:200), rabbit anti-Sox9 (Millipore, AB5535; 1:300), mouse anti-proton pump (H+ K+ ATPase beta subunit) (Cedarlane, D032-3; 1:500), mouse anti-TP63 (Santa Cruz Biotechnology, sc-8431; 1:200), mouse anti-PCNA (Santa Cruz Biotechnology, sc-56; 1:200), mouse anti-PDX1 (Developmental Studies Hybridoma Bank, F109-D12; 1:200), rabbit anti-CDX2 (R&D System, AF3665; 1:50), and chicken anti-GFP (Abcam, ab13970; 1:2000). For immunofluorescence, secondary staining with Alexa Fluor 488– and Alexa Fluor 568–conjugated (1:500; Thermo Fisher Scientific) antibodies was followed by mounting with 4′,6-diamidino-2-phenylindole–containing VECTASHIELD media (Vector Laboratories). For immunohistochemistry, we used the 3,3′-diaminobenzidine (Abcam) protocol followed by hematoxylin staining. Images were acquired using a Nikon microscope DS-Qi1Mc camera.
Histopathology scoring
H&E-stained sections of gastric samples were blindly scored on the basis of seven cancer traits, on a scale from 0 to 4 per trait; the scoring system was adapted from a previously adapted paper, where the trait “Hyalinosis Red refractile droplets and crystals” was disregarded (40). Cumulative scores were calculated, and significance was determined using an unpaired Student’s t test, comparing two groups at a time.
ATAC-seq and ChIP-seq data analysis
After removing adaptors by Cutadapt, pair-end ATAC-seq reads were aligned to the mouse reference genome (GRCm38) using Bowtie2 with the parameter “-X 2000” (41). For ChIP-seq, the filtered single-end reads were aligned to the mouse reference genome (GRCm38) using Bowtie2 with default parameters. ATAC-seq and ChIP-seq peaks were then called by MACS2 -callpeak with parameters “--keep-dup = 1” and “--SPMR” (42). ENCODE mm10 blacklist regions (www.encodeproject.org/annotations/ENCSR636HFF/) were excluded from the called peaks for downstream analysis. Resultant bedGraph files were converted to big wiggle files using the University of California, Santa Cruz (UCSC) bedGraphToBigWig tool (43). Genomic signal within 2 kb of peaks were visualized using deepTools (44). The Binding and Expression Target Analysis (BETA) tool from Cistrome was used to assess the activating or repressive function of selected peaks (45). TF motifs enriched in the selected peaks were identified using the “SeqPos” tool from Cistrome (46) and the R package “PWMEnrich” (www.bioconductor.org/packages//2.13/bioc/html/PWMEnrich.html).
To determine the similarities between different tissues by ATAC-seq data, all the ATAC-seq peaks from different tissues were merged and the ATAC-seq signal of merged peaks were retrieved from .bw files. Pearson correlations were then calculated in a pairwise manner using the ATAC-seq signal.
RNA-seq analysis
The trimmed pair-end reads were aligned to the mouse reference genome (GRCm38) with STAR (version 2.4.2a) (47), and gene expression was then quantified using the reads per kilobase per million mapped reads method. COAD (TCGA-COAD) and STAD (TCGA-STAD) RNA-seq data were downloaded from the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/) (27, 28). Signature genes of a specific feature were selected by the threshold of fold change [fragments per kilobase of transcript, per million mapped reads (FPKM) +1] > 2 and false discovery rate < 0.05. The “getLDS” function from the R package “biomaRt” was used to align gene symbols between mouse and human (48). Enrichment plots of gene signatures were generated using the R package “clusterProfiler” (49).
Supplementary Material
Acknowledgments
We thank the Rossant and Huang Labs for sharing primary and secondary antibodies. Funding: The work was supported by SickKids Foundation startup, Cancer Research Society, and the University of Toronto’s Medicine by Design Initiative/the Canada First Research Excellence Fund (T.H.K.) and the NSERC discovery grant (498706), the CIHR New Investigator Award, and the OMIR Early Researcher Award (H.H.H.). Author contributions: R.F. designed and performed experiments, analyzed data, and wrote the manuscript. H.G. performed experiments; analyzed ATAC-seq, ChIP-seq, and RNA-seq data; and drafted the bioinformatics analytical sections. C.S. performed histopathology scoring. M.A. helped with RNA-seq analysis. T.Y. assisted in RNA-seq experiments. P.B.D. provided Sox2 mouse lines. H.H.H. supervised genomic and epigenomic data analyses. T.-H.K. conceived and supervised the study, analyzed data, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All of the presented -omics data are available at the NCBI GEO (accession number: GSE134277). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaax8898/DC1
Fig. S1. Quality control of genomic datasets.
Fig. S2. Analyses of gut region–specific markers during embryonic development.
Fig. S3. Hindstomach specification maintained by Sox9+ Cre escapers.
Fig. S4. Molecular characterization of Sox2;Cdx2 DKO epithelium.
Fig. S5. Disease quantification of adenoma samples.
REFERENCES AND NOTES
- 1.Ferlay J., Shin H.-R., Bray F., Forman D., Mathers C., Parkin D. M., Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 127, 2893–2917 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Giroux V., Rustgi A. K., Metaplasia: Tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat. Rev. Cancer 17, 594–604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X., Zhang C., Gong T., Ni X., Li J., Zhan D., Liu M., Song L., Ding C., Xu J., Zhen B., Wang Y., Qin J., A time-resolved multi-omic atlas of the developing mouse stomach. Nat. Commun. 9, 4910 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherwood R. I., Chen T.-Y., Melton D. A., Transcriptional dynamics of endodermal organ formation. Dev. Dyn. 238, 29–42 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willet S. G., Mills J. C., Stomach organ and cell lineage differentiation: From embryogenesis to adult homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2, 546–559 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zorn A. M., Wells J. M., Vertebrate endoderm development and organ formation. Annu. Rev. Cell Dev. Biol. 25, 221–251 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao N., White P., Kaestner K. H., Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev. Cell 16, 588–599 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar N., Tsai Y. H., Chen L., Zhou A., Banerjee K. K., Saxena M., Huang S., Toke N. H., Xing J., Shivdasani R. A., Spence J. R., Verzi M. P., The lineage-specific transcription factor CDX2 navigates dynamic chromatin to control distinct stages of intestine development. Development 146, dev172189 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee K. K., Saxena M., Kumar N., Chen L., Cavazza A., Toke N. H., O’Neill N. K., Madha S., Jadhav U., Verzi M. P., Shivdasani R. A., Enhancer, transcriptional, and cell fate plasticity precedes intestinal determination during endoderm development. Genes Dev. 32, 1430–1442 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Que J., Luo X., Schwartz R. J., Hogan B. L., Multiple roles for Sox2 in the developing and adult mouse trachea. Development 136, 1899–1907 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnold K., Sarkar A., Yram M. A., Polo J. M., Bronson R., Sengupta S., Seandel M., Geijsen N., Hochedlinger K., Sox2+ adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9, 317–329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarkar A., Huebner A. J., Sulahian R., Anselmo A., Xu X., Flattery K., Desai N., Sebastian C., Yram M. A., Arnold K., Rivera M., Mostoslavsky R., Bronson R., Bass A. J., Sadreyev R., Shivdasani R. A., Hochedlinger K., Sox2 suppresses gastric tumorigenesis in mice. Cell Rep. 16, 1929–1941 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S. A., Flynn R. A., Wysocka J., A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., Jaenisch R., Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avilion A. A., Nicolis S. K., Pevny L. H., Perez L., Vivian N., Lovell-Badge R., Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 17, 126–140 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seymour P. A., Freude K. K., Dubois C. L., Shih H. P., Patel N. A., Sander M., A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev. Biol. 323, 19–30 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watanabe H., Ma Q., Peng S., Adelmant G., Swain D., Song W., Fox C., Francis J. M., Pedamallu C. S., DeLuca D. S., Brooks A. N., Wang S., Que J., Rustgi A. K., Wong K. K., Ligon K. L., Liu X. S., Marto J. A., Meyerson M., Bass A. J., SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J. Clin. Invest. 124, 1636–1645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soufi A., Donahue G., Zaret K. S., Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Starzyńska T., Cytokeratin expression in human stomach. Immunofluorescence study. Mater. Med. Pol. 23, 273–276 (1991). [PubMed] [Google Scholar]
- 20.Camilo V., Barros R., Celestino R., Castro P., Vieira J., Teixeira M. R., Carneiro F., Pinto-de-Sousa J., David L., Almeida R., Immunohistochemical molecular phenotypes of gastric cancer based on SOX2 and CDX2 predict patient outcome. BMC Cancer 14, 753 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sashikawa Kimura M., Mutoh H., Sugano K., SOX9 is expressed in normal stomach, intestinal metaplasia, and gastric carcinoma in humans. J. Gastroenterol. 46, 1292–1299 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Lundberg I. V., Edin S., Eklöf V., Öberg Å., Palmqvist R., Wikberg M. L., SOX2 expression is associated with a cancer stem cell state and down-regulation of CDX2 in colorectal cancer. BMC Cancer 16, 471 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu K., Jiang M., Lu Y., Chen H., Sun J., Wu S., Ku W. Y., Nakagawa H., Kita Y., Natsugoe S., Peters J. H., Rustgi A., Onaitis M. W., Kiernan A., Chen X., Que J., Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell 12, 304–315 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrasco-Garcia E., Santos J. C., Garcia I., Brianti M., García-Puga M., Pedrazzoli J. Jr., Matheu A., Ribeiro M. L., Paradoxical role of SOX2 in gastric cancer. Am. J. Cancer Res. 6, 701–713 (2016). [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Research Network , Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Network , Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., Schultz N., Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., Schultz N., The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim T.-H., Shivdasani R. A., Notch signaling in stomach epithelial stem cell homeostasis. J. Exp. Med. 208, 677–688 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raghoebir L., Bakker E. R. M., Mills J. C., Swagemakers S., Kempen M. B. V., Munck A. B. D., Driegen S., Meijer D., Grosveld F., Tibboel D., Smits R., Rottier R. J., SOX2 redirects the developmental fate of the intestinal epithelium toward a premature gastric phenotype. J. Mol. Cell Biol. 4, 377–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dalerba P., Sahoo D., Paik S., Guo X., Yothers G., Song N., Wilcox-Fogel N., Forgó E., Rajendran P. S., Miranda S. P., Hisamori S., Hutchison J., Kalisky T., Qian D., Wolmark N., Fisher G. A., van de Rijn M., Clarke M. F., CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N. Engl. J. Med. 374, 211–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harfe B. D., Scherz P. J., Nissim S., Tian H., McMahon A. P., Tabin C. J., Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118, 517–528 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Muzumdar M. D., Tasic B., Miyamichi K., Li L., Luo L., A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Hingorani S. R., Petricoin E. F. III, Maitra A., Rajapakse V., King C., Jacobetz M. A., Ross S., Conrads T. P., Veenstra T. D., Hitt B. A., Kawaguchi Y., Johann D., Liotta L. A., Crawford H. C., Putt M. E., Jacks T., Wright C. V. E., Hruban R. H., Lowy A. M., Tuveson D. A., Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Verzi M. P., Shin H., Ho L.-L., Liu X. S., Shivdasani R. A., Essential and redundant functions of caudal family proteins in activating adult intestinal genes. Mol. Cell. Biol. 31, 2026–2039 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murtaugh L. C., Stanger B. Z., Kwan K. M., Melton D. A., Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 14920–14925 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Badea T. C., Wang Y., Nathans J., A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J. Neurosci. 23, 2314–2322 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Syder A. J., Karam S. M., Mills J. C., Ippolito J. E., Ansari H. R., Farook V., Gordon J. I., A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proc. Natl. Acad. Sci. U.S.A. 101, 4471–4476 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buenrostro J. D., Wu B., Chang H. Y., Greenleaf W. J., ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogers A. B., Taylor N. S., Whary M. T., Stefanich E. D., Wang T. C., Fox J. G., Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer Res. 65, 10709–10715 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kent W. J., Zweig A. S., Barber G., Hinrichs A. S., Karolchik D., BigWig and BigBed: Enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramirez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang S., Sun H., Ma J., Zang C., Wang C., Wang J., Tang Q., Meyer C. A., Zhang Y., Liu X. S., Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 8, 2502–2515 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mei S., Qin Q., Wu Q., Sun H., Zheng R., Zang C., Zhu M., Wu J., Shi X., Taing L., Liu T., Brown M., Meyer C. A., Liu X. S., Cistrome Data Browser: A data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 45, D658–D662 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durinck S., Spellman P. T., Birney E., Huber W., Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu G., Wang L. G., Han Y., He Q. Y., clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaax8898/DC1
Fig. S1. Quality control of genomic datasets.
Fig. S2. Analyses of gut region–specific markers during embryonic development.
Fig. S3. Hindstomach specification maintained by Sox9+ Cre escapers.
Fig. S4. Molecular characterization of Sox2;Cdx2 DKO epithelium.
Fig. S5. Disease quantification of adenoma samples.