

PTPRE-AS1 deficiency aggravates pulmonary inflammation but reduces colitis severity by modulating M2 macrophage activation.
Abstract
Long noncoding RNAs (lncRNAs) are important regulators of diverse biological processes; however, their function in macrophage activation is undefined. We describe a new regulatory mechanism, where an unreported lncRNA, PTPRE-AS1, targets receptor-type tyrosine protein phosphatase ε (PTPRE) to regulate macrophage activation. PTPRE-AS1 was selectively expressed in IL-4–stimulated macrophages, and its knockdown promoted M2 macrophage activation via MAPK/ERK 1/2 pathway. In vivo, PTPRE-AS1 deficiency enhanced IL-4–mediated M2 macrophage activation and accelerated pulmonary allergic inflammation while reducing chemical-induced colitis. Mechanistically, PTPRE-AS1 bound WDR5 directly, modulating H3K4me3 of the PTPRE promoter to regulate PTPRE-dependent signaling during M2 macrophage activation. Further, the expression of PTPRE-AS1 and PTPRE was significantly lower in peripheral blood mononuclear cells from patients with allergic asthma. These results provide evidence supporting the importance of PTPRE-AS1 in controlling macrophage function and the potential utility of PTPRE-AS1 as a target for controlling inflammatory diseases.
INTRODUCTION
Macrophages are essential components of innate immunity and have critical roles in tissue homeostasis. They orchestrate the initiation and resolution phases of both innate and adaptive immunity and significantly affect the protective immunity and immune-mediated tissue injury (1, 2) associated with microbial infection, asthma, inflammatory bowel diseases, tumorigenesis, and autoimmune disease. Macrophages are divided into functionally distinct forms, including classically activated, alternatively activated, and numerous other phenotypically distinct subsets, depending on their microenvironment (3, 4). Interleukin-4 (IL-4)–induced M2 macrophage activation contributes to anti-inflammatory activity, tissue repair, and wound healing (5) and can also increase cell recruitment and mucus secretion, resulting in airway hyperresponsiveness in allergic asthma (6). Hence, improved understanding of the molecular mechanisms underlying macrophage subset activation could assist in the diagnosis and treatment of human inflammatory diseases.
Long noncoding RNAs (lncRNAs) are a class of noncoding transcripts of >200 nucleotides that can regulate gene expression at various levels, including chromatin modification, and transcriptional and posttranscriptional processing (7, 8). While lncRNAs have been studied primarily in the context of genomic imprinting, developmental processes, and cancer, emerging evidence suggests that they have important regulatory roles in both innate and adaptive immune responses. For example, the lncRNA THRIL regulates tumor necrosis factor–α (TNF-α) expression through interaction with hnRNPL during innate activation of THP1 macrophages (9). Another lncRNA, lincRNA-EPS, inhibits macrophage-mediated inflammation by reducing transcription (10); however, the roles of lncRNAs in IL-4–mediated M2 macrophage activation are largely unknown.
In this study, we conducted genome-wide analysis of lncRNA expression profiles in bone marrow–derived macrophages (BMDMs) and identified a previously unknown lncRNA, PTPRE-AS1, which was significantly up-regulated during IL-4–induced M2 macrophage activation. Functionally, PTPRE-AS1 inhibits M2 macrophage–associated gene expression (of IL-10, Arg-1, and CD206, among others) via epigenetic promotion of receptor-type tyrosine protein phosphatase ε (PTPRE) expression. Further, PTPRE-AS1 deficiency in mice resulted in significantly increased cockroach extract (CRE)–induced pulmonary allergic inflammation, while it reduced the severity of dextran sodium sulfate (DSS)–induced acute colitis. The expression levels of PTPRE-AS1 and PTPRE were significantly lower in peripheral blood mononuclear cells (PBMCs) from patients with allergic asthma relative to those from healthy controls. Overall, our study identifies a previously unknown lncRNA, PTPRE-AS1, with a biological, mechanistic, and clinical impact on inflammatory diseases.
RESULTS
LncRNA PTPRE-AS1 is highly induced in macrophages exposed to IL-4
We hypothesized that if lncRNAs are involved in regulating macrophage activation, their expression would likely be tightly controlled following stimulation with lipopolysaccharide (LPS) or IL-4. To test this hypothesis and identify lncRNAs regulated during macrophage activation, we conducted transcriptome microarray and bioinformatic analyses of BMDMs treated with LPS (designated operationally as M1 subsets) and IL-4 (M2). In the discovery phase, LPS and IL-4 were shown to induce transcription of numerous protein-coding genes and lncRNAs in their respective macrophage subsets. We identified 553 unique lncRNAs that were differentially expressed in BMDMs following IL-4 stimulation, among which 52% (289 lncRNAs) were suppressed and 48% (264 lncRNAs) were enhanced. To further narrow down the candidate lncRNAs, we specifically selected the differentially expressed antisense lncRNAs after IL-4 stimulation, and the strongly enhanced antisense lncRNAs in IL-4 stimulation were compared with the LPS stimulation group (Fig. 1A). To validate the microarray data, we analyzed the expression of five antisense lncRNA candidates in BMDMs following IL-4 or LPS stimulation using real-time quantitative polymerase chain reaction (RT-qPCR). Although three of the four lncRNAs had the same pattern of expression upon IL-4 treatment as that determined by microarray analysis, they had no effect on M2 activation (fig. S1).
Fig. 1. PTPRE-AS1 is highly expressed and acts as a repressor in IL-4–induced M2 macrophage activation.
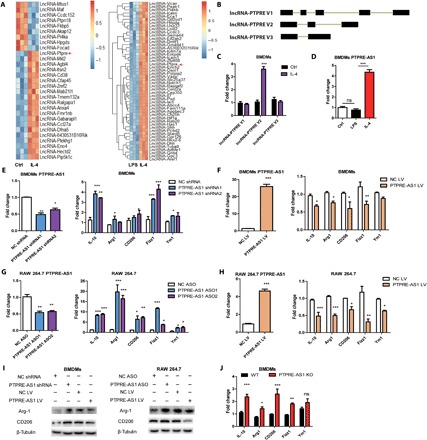
(A) Heatmap of antisense lncRNAs with significantly altered expression upon stimulation of BMDMs with IL-4 and LPS, respectively. (B) lncRNA-PTPRE encodes three splice variants. (C) Evaluation of the expression of three lncRNA-PTPRE splice variants in IL-4–stimulated BMDMs. (D) Expression of PTPRE-AS1 in BMDMs stimulated with IL-4 or LPS. (E) Knockdown of PTPRE-AS1 in BMDMs using two distinct shRNAs (left). (F) Overexpression of PTPRE-AS1 in BMDMs with PTPRE-AS1 LV or NC LV (left); after transfection, M2-associated gene expression in IL-4–stimulated BMDMs was quantified by RT-qPCR analysis (right). NC, negative control. (G) Knockdown of PTPRE-AS1 in RAW 264.7 cells transfected with two distinct ASOs (200 nM) (left). (H) Overexpression of PTPRE-AS1 in RAW 264.7 cells with PTPRE-AS1 LV or NC LV (left), followed by IL-4 stimulation; M2-associated gene expression was quantified by RT-qPCR (right). (I) Western blots of protein levels of Arg-1 and CD206 in BMDMs (left) and RAW 264.7 cells (right) with PTPRE-AS1 knockdown or overexpression, followed by IL-4 stimulation for 24 hours. (J) M2-associated gene expression levels in WT and PTPRE-AS1–deficient mouse BMDMs were detected by RT-qPCR after IL-4 treatment for 24 hours. Data are presented as mean ± SEM from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ns, no significance.
Notably, among these differentially expressed antisense lncRNAs, that of lncRNA-PTPRE (5830432E09RIK) with unrecognized function was robustly enhanced during IL-4–induced M2 macrophage activation. In addition, microarray analysis results demonstrated that its expression was higher among IL-4–induced antisense lncRNAs than those treated with LPS (Fig. 1A). This lncRNA sequence mapped to the reverse strand of the cis gene, tyrosine phosphatase receptor type E (PTPRE), and was therefore designated as lncRNA-PTPRE.
Three lncRNA-PTPRE splice variants were detected (Fig. 1B); using RT-qPCR, we determined that only transcript variant 2 (1494 base pairs; gene accession number: NR_015548) was enhanced in IL-4–stimulated BMDMs, compared with control and LPS-induced M1 macrophages (Fig. 1, C and D), suggesting a potential role for this lncRNA in M2 macrophage activation. We designated the lncRNA-PTPRE variant 2 sequence as PTPRE-AS1.
PTPRE-AS1 acts as a repressor of IL-4–induced M2 macrophage activation by enhancing PTPRE gene expression
To determine whether differential expression of PTPRE-AS1 influences activation of M2 macrophages, we evaluated BMDMs with confirmed PTPRE-AS1 knockdown and found that IL-4–treated cells exhibited significantly elevated levels of M2-associated gene expression, specifically IL-10, Arg-1, CD206, Fizz1, and Ym1 (Fig. 1E). Further, “gain-of-function” studies of BMDMs ectopically expressing PTPRE-AS1 (PTPRE-AS1 LV) demonstrated reduced expression of IL-10, Arg-1, CD206, Fizz1, and Ym1 in these cells following IL-4 stimulation (Fig. 1F). Similarly, enhanced levels of IL-10, Arg-1, CD206, Fizz1, and Ym1 expression were noted in RAW 264.7 cells with PTPRE-AS1 knocked down, whereas their levels were reduced in RAW 264.7 cells overexpressing PTPRE-AS1 (Fig. 1, G and H). Consistently, we found that PTPRE-AS1 affects M2-associated gene expression (Arg-1, CD206) in the same way at protein level as mRNA level in both BMDMs and RAW 264.7 cells following IL-4 stimulation (Fig. 1I). Furthermore, BMDMs from PTPRE-AS1 knockout (KO) mice displayed strongly M2-associated gene expression (Fig. 1J). Together, these findings suggest that PTPRE-AS1 can inhibit IL-4–induced M2 macrophage activation.
As PTPRE-AS1 is located on the opposite strand to the coding gene, PTPRE, and lncRNAs have been reported to exert cis-regulatory effects on nearby genes, we investigated whether the effect of PTPRE-AS1 on M2 macrophage activation was due to its influence on PTPRE expression. The PTPRE protein can localize to either the cell membrane (PTPRE-M) or cytosol (PTPRE-C), while PTPRE-C is constitutively expressed in the spleen and colon and PTPRE-M is primarily expressed in the brain and lung. In addition, PTPRE-C expression was significantly decreased in the lung and colon of the CRE-induced allergic model and DSS-induced colitis model, respectively (fig. S2, A and B). Moreover, our results demonstrated that PTPRE-AS1 knockdown did result in an approximately 40% reduction of PTPRE-C expression in both BMDMs and RAW 264.7 cells, whereas ectopic expression of PTPRE-AS1 significantly increased the levels of PTPRE-C mRNA (Fig. 2, A and B). BMDMs from PTPRE-AS1–null mice also exhibited reduced PTPRE-C expression levels (fig. S2C).These findings confirm that PTPRE-AS1 can positively regulate PTPRE-C expression.
Fig. 2. PTPRE-AS1 suppresses M2 macrophage activation by targeting PTPRE.
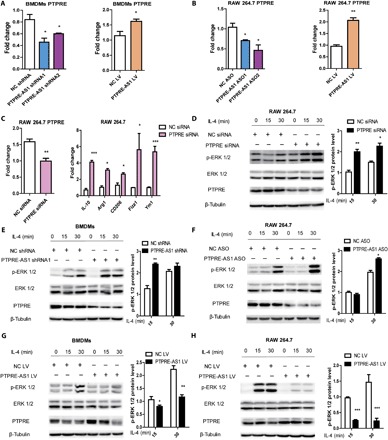
(A) RT-qPCR analysis of PTPRE expression levels in BMDMs with PTPRE-AS1 knockdown by two distinct shRNAs (left) or overexpression using PTPRE-AS1 LV (right). (B) RT-qPCR analysis of PTPRE expression levels in RAW 264.7 cells with PTPRE-AS1 knockdown by two distinct ASOs (left) or overexpression from PTPRE-AS1 LV (right). (C) Analysis of PTPRE in RAW 264.7 cells transfected with PTPRE siRNA (200 nM) or control siRNA (left). Transfected cells were stimulated with IL-4, and M2-associated gene expression levels were detected by RT-qPCR (right). Data are presented as means ± SEM from three independent experiments. Analysis of ERK activation in RAW 264.7 cells transfected with PTPRE or control siRNA (D), in BMDMs with PTPRE-AS1 or NC shRNA (E), in RAW 264.7 cells with PTPRE-AS1 or NC ASO (F), and in PTPRE-AS1–overexpressed BMDMs (G) and RAW 264.7 cells (H), followed by IL-4 stimulation. p-ERK 1/2 protein levels were normalized to those of β-tubulin and quantified using ImageJ software. The means and SEM of relative p-ERK 1/2 levels from three independent experiments are shown. *P < 0.05; **P < 0.01; ***P < 0.001.
Next, to further examine whether the effect of PTPRE-AS1 was mediated through its direct target, PTPRE, IL-4–treated M2 macrophages with confirmed PTPRE knockdown were evaluated and exhibited significantly enhanced levels of M2-associated gene expression, compared with those in negative controls (NC; Fig. 2C). PTPRE, a protein tyrosine phosphatase, is directly or indirectly involved in regulating insulin receptor signaling, Janus kinase–signal transducer and activator of transcription (JAK-STAT) signaling, and the mitogen-activated protein kinase (MAPK)/extracellular signal–regulated kinase (ERK) 1/2 pathways (11, 12). Efficient small interfering RNA (siRNA)–mediated knockdown of PTPRE was accompanied by substantially enhanced levels of IL-4–induced ERK 1/2 phosphorylation (Fig. 2D); however, siPTPRE-treated macrophages exhibited no differences in IL-4–induced phosphorylation levels of STAT6 (fig. S3A). These data indicate that PTPRE potentially represses M2 macrophage activation through the MAPK/ERK 1/2 signaling pathway.
Consistent with the findings in siPTPRE-treated macrophages, ERK 1/2 phosphorylation levels were strongly enhanced in both BMDMs and RAW 264.7 cells, with PTPRE-AS1 knockdown treated with IL-4 (Fig. 2, E and F), whereas they were reduced in these cells overexpressing PTPRE-AS1 (Fig. 2, G and H). Again, there was no apparent change in IL-4–induced phosphorylation levels of STAT6 (fig. S3B). Together, these findings suggest that PTPRE-AS1 can suppress IL-4–mediated M2 macrophage activation by enhancing PTPRE expression, which inhibits IL-4–induced activation of MAPK/ERK 1/2 signaling.
PTPRE-AS1 binds to WDR5 and mediates H3 lysine 4 trimethylation of the PTPRE promoter region, thereby epigenetically activating PTPRE gene expression
Recent studies have suggested that a significant number of lncRNAs exert their activity in cooperation with chromatin-modifying enzymes to promote epigenetic activation or silencing of gene expression. Specifically, lncRNAs can bind to chromatin remodeling complexes, including polycomb repressive complexes and mixed-lineage leukemia (MLL), to modulate downstream gene expression, serving as scaffolds for the histone modification complex (13). To further explore the mechanism underlying PTPRE-AS1–mediated effects, we performed subcellular fractionation, localization, and RNA fluorescence in situ hybridization (FISH) assays. The results showed first that PTPRE-AS1 was localized to both the nucleus and cytoplasm (Fig. 3A). Second, using a bioinformatics algorithm, PTPRE-AS1 was predicted to interact with a panel of chromatin modifiers, including WDR5 (WD repeat domain 5; H3K4me3), LSD1 (H3K4me3), SETDB1 (H3K9me3), DNMT1, and EZH2 (H3K27me3) (http://pridb.gdcb.iastate.edu/RPISeq/references.php) (Fig. 3B). Next, we conducted RNA binding protein immunoprecipitation assays (RIP) using a panel of antibodies specific for each of the above chromatin modifiers and found 17-fold enrichment of PTPRE-AS1 when anti-WDR5 antibodies were used, relative to the use of immunoglobulin G (IgG) control, while no enrichment was observed with the nonspecific control U1 (Fig. 3C). These results thus imply a direct interaction of PTPRE-AS1 and WDR5.
Fig. 3. PTPRE-AS1 binds directly to WDR5 to activate PTPRE.
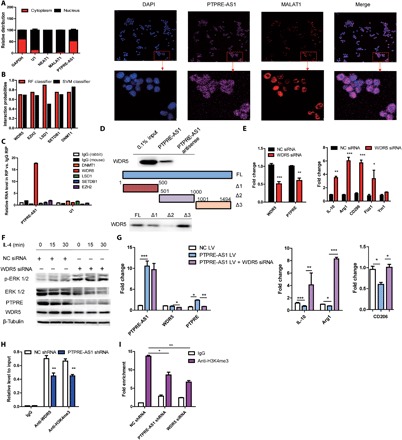
(A) RT-qPCR analysis of PTPRE-AS1 abundance in BMDM nuclear and cytoplasmic fractions (left). GAPDH, cytoplasmic marker. U1, nuclear marker. Subcellular distribution of PTPRE-AS1, visualized by RNA FISH (right). MALAT1, positive control for the nucleus. (B) Bioinformatic prediction of PTPRE-AS1–interacting histone proteins and DNA methylation modifiers. RPISeq predictions were based on random forest (RF) or support vector machine (SVM). (C) RIP analyses of PTPRE-AS1 and its predicted binding partners. (D) Western blotting analyses of WDR5 in samples pulled down by full-length (FL) or truncated (1–500Δ1, 501–1000Δ2, 1001–1494Δ3) PTPRE-AS1. (E) RT-qPCR analyses of WDR5 and PTPRE in RAW 264.7 cells with or without WDR5 knockdown after IL-4 treatment (left), and detection of M2-associated gene expression (right). (F) Western blotting analysis to evaluate levels of p-ERK 1/2, ERK 1/2, and PTPRE in WDR5-knockdown RAW 264.7 cells. (G) RT-qPCR analyses of PTPRE and M2-associated gene expression in RAW 264.7 cells overexpressing PTPRE-AS1, with or without WDR5 knockdown, followed by IL-4 stimulation. (H) ChIP analyses of the binding efficiency of WDR5 and H3K4Me3 histone to the PTPRE gene promoter in RAW 264.7 cells with or without PTPRE-AS1 knockdown. (I) ChIP-qPCR analyses of H3K4 trimethylation levels in the PTPRE gene promoter region in RAW 264.7 cells with or without WDR5 or PTPRE-AS1 knockdown. Data are presented as means ± SEM from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
WDR5 is a core subunit of MLL and SET1, both of which are histone H3 Lys4 (H3K4) methyltransferase complexes and “effectors” of H3K4 methylation during gene transactivation (14). To further identify the WDR5-interacting region of PTPRE-AS1, four biotinylated PTPRE-AS1 fragments (full-length, 1–500Δ1, 501–1000Δ2, and 1001–1494Δ3) were reconstructed and were each transfected into RAW 264.7 cells. RNA pull-down assays using the transfected RAW 264.7 cells revealed that WDR5 was detected in the retrieved RNA pull-down proteins from PTPRE-AS1–overexpressing RAW 264.7 cells by Western blotting. Results showed that both the 1–500Δ1 and 1001–1494Δ3 fragments of PTPRE-AS1 mediated interaction with WDR5 (Fig. 3D).
Next, we explored whether WDR5 is involved in the effect of PTPRE-AS1 on PTPRE expression. The results showed that levels of PTPRE significantly decreased in RAW 264.7 cells with WDR5 knockdown, concomitant with significantly up-regulated expression of M2-associated gene and activation of MAPK/ERK 1/2 signaling in IL-4–treated M2 macrophages (Fig. 3, E and F). These findings were consistent with those noted in cells with PTPRE-AS1 and PTPRE knockdown. In addition, we found that enforced expression of PTPRE-AS1 inhibited IL-4–induced M2-associated gene expression, whereas knockdown of WDR5 abolished the effect of ectopically expressed PTPRE-AS1 (Fig. 3G). Therefore, PTPRE-AS1 exerts its regulatory function in a WDR5-dependent manner.
Moreover, to confirm whether PTPRE-AS1 activated the transcription of PTPRE directly via binding to WDR5 and mediating trimethylation of H3K4, we performed chromatin immunoprecipitation (ChIP) assays in PTPRE-AS1 knockdown cells. The results demonstrated that WDR5 directly bound to the promoter region of PTPRE, and PTPRE-AS1 silencing significantly decreased the binding activity of WDR5 (Fig. 3H), implying its important role in bridging WDR5 and the PTPRE gene promoter region. Further, knockdown of both PTPRE-AS1 and WDR5 significantly decreased the levels of H3K4me3 associated with the promoter region of the PTPRE gene (Fig. 3I). Collectively, these studies strongly suggest that PTPRE-AS1 recruits WDR5 to the PTPRE promoter and activates PTPRE transcription to regulate IL-4–induced M2 macrophage activation.
PTPRE-AS1 deficiency attenuates colitis in an acute DSS model
To assess the role of PTPRE-AS1 in the regulation of inflammatory diseases in vivo, we generated PTPRE-AS1 KO mice using the CRISPR-Cas9 system, which resulted in deletion of exons 2 and 3 of lncRNA PTPRE-AS1 and functional loss of PTPRE-AS1 (fig. S4). No apparent abnormalities of external morphology or body weight were noted in PTPRE-AS1–null mice, relative to wild-type (WT) C57BL/6 mice. PTPRE-AS1–null mice also developed normally, and no spontaneous inflammatory pathologies were detected in various organs.
Then, we evaluated the impact of PTPRE-AS1 on an acute model of DSS-induced colitis (Fig. 4A). We noted that levels of PTPRE-AS1 and its target, PTPRE, were significantly suppressed in colon tissues from mice with DSS-induced colitis (fig. S5, A and B), implying that PTPRE-AS1 and PTPRE may have important roles in the pathogenesis of this condition. While aggravated loss of body weight was observed in WT mice from the fifth day after DSS administration, body weight loss was significantly alleviated in PTPRE-AS1–null mice (Fig. 4B). In addition, after DSS administration, PTPRE-AS1–null mice exhibited decreased cumulative disease activity index (DAI) compared with WT mice (Fig. 4C).
Fig. 4. PTPRE-AS1–deficient mice exhibits increased resistance to DSS-induced colitis.
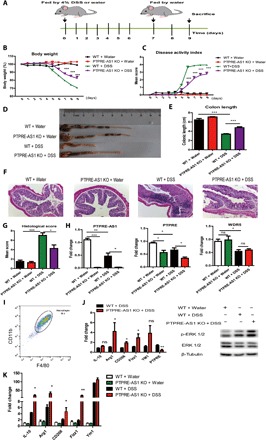
(A) WT and PTPRE-AS1 KO mice were provided with plain water or water containing 4% DSS (eight mice per group), according to the protocol described in Materials and Methods. (B) Mean daily weight change and (C) changes in DAI, scored based on diarrhea, bleeding, and body weight loss. (D and E) On day 9, mice were sacrificed, their colons were removed, and colon lengths were measured and recorded. Photo credit: Yufeng Zhou. (F) Histopathological changes in the colon tissue were examined by H&E staining (magnification, ×100). (G) Histopathological scores. (H) Levels of PTPRE-AS1, PTPRE, and WDR5 in colon tissue were quantified by RT-qPCR. (I) The percentages of macrophages in purified cells from colon tissue were determined by flow cytometry. Representative F4/80 and CD11b plots from at least three independent experiments were shown. (J) The levels of M2-associated genes, PTPRE, and phosphorylated ERK 1/2 protein in IL-4–treated colon macrophages purified from experimental models were quantified. (K) RT-qPCR for M2-associated genes in colon tissue samples. The data shown were from one of three independent experiments. Data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Moreover, colon length, a parameter with very low variability and a marker of intestinal inflammation in the DSS-induced colitis model, significantly reduced after DSS treatment in WT mice compared with controls; however, this parameter was improved in DSS-treated PTPRE-AS1–null mice (Fig. 4, D and E). Histological analysis showed that DSS-treated WT mice developed multiple erosive lesions and significant inflammatory responses, characterized by increased inflammatory cell infiltrations (composed of macrophages, lymphocytes, eosinophils, and occasional neutrophils), goblet cell loss, crypt abscess formation, and submucosal edema. In contrast, DSS-treated PTPRE-AS1–null mice displayed significantly reduced levels of inflammatory lesions compared with those in DSS-treated WT mice (Fig. 4F). Blinded scoring of histological injury in the distal colon from DSS-treated PTPRE-AS1–null mice generated significantly lower scores than those from DSS-treated WT animals (Fig. 4G). Together, these results indicate that PTPRE-AS1 deficiency can attenuate DSS-induced colitis in mice.
The effects of PTPRE-AS1 deficiency on DSS-induced colitis in mice were associated with improved M2-associated gene expression and responses. First, after treatment with water, we observed that colon sections from PTPRE-AS1–null mice showed significantly lower PTPRE than WT mice. Reduced PTPRE levels were also noted in DSS-treated PTPRE-AS1–deficient mice relative to those in DSS-treated WT mice, whereas there was no significant difference in WDR5 expression between DSS-treated WT and PTPRE-AS1–null mice (Fig. 4H), implying the potential involvement of PTPRE in mediating the regulatory effects of PTPRE-AS1 in vivo. We then purified macrophages from colon sections and found that >90% of purified cells expressed CD11b and F4/80 (Fig. 4I). Similar to the in vitro results, we detected significantly increased levels of M2-associated gene and phosphorylated ERK 1/2 protein in IL-4–treated colon macrophages from DSS-treated PTPRE-AS1–deficient mice compared with DSS-treated WT mice (Fig. 4J). In addition, we noted much higher levels of M2-associated gene in colon tissues from DSS-treated PTPRE-AS1–deficient mice than DSS-treated WT mice (Fig. 4K). Collectively, these data suggest that deficiency of PTPRE-AS1 may tip the balance of macrophages toward the M2 subset during DSS-induced colitis, thereby preventing colitis development.
PTPRE-AS1 deficiency exacerbates cockroach allergen–induced lung inflammation
Next, we tested the role of PTPRE-AS1 in a mouse model of M2-associated, CRE allergen–induced pulmonary allergic inflammation (15). The results showed that, compared with control mice, those treated with CRE exhibited significant recruitment of inflammatory cells to the lungs and dense peribronchial infiltrates, concomitant with significantly reduced PTPRE-AS1 and PTPRE levels in lung tissue (fig. S5, C and D). To evaluate whether PTPRE-AS1 directly influences the severity of CRE-induced pulmonary inflammation, we exposed WT and PTPRE-AS1–null mice to CRE (Fig. 5A). Compared with CRE-treated WT mice, CRE-treated PTPRE-AS1–null mice displayed significantly increased numbers of total inflammatory cells in the bronchoalveolar lavage fluid (BALF) (Fig. 5B). Specifically, the data showed that, among all analyzed cell types, the numbers of eosinophils and macrophages were vastly enhanced in CRE-challenged PTPRE-AS1–null mice, as compared with CRE-challenged WT counterparts (Fig. 5C). Moreover, recruitment of inflammatory cells to the lungs was exacerbated in CRE-challenged PTPRE-AS1–null mice, with dense peribronchial infiltrates (Fig. 5D).
Fig. 5. PTPRE-AS1–deficient mice are more susceptible to CRE-induced asthma.
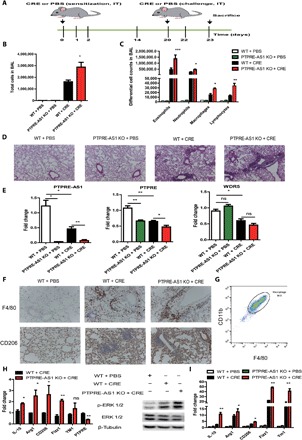
(A) Experimental CRE-induced asthma model including eight mice per group in each of the three independent experiments. (B) Total and (C) differential BALF cell numbers in PBS- and CRE-challenged PTPRE-AS1 KO and WT mice. (D) Representative images of H&E-stained lung tissues from PBS- and CRE-challenged PTPRE-AS1 KO and WT mice (magnification, ×100). (E) Levels of PTPRE-AS1, PTPRE, and WDR5 in lung tissues were detected by RT-qPCR. (F) Representative IHC images of F4/80 and CD206 expression in lung tissues (magnification, ×200). (G) The percentages of macrophages in purified cells from lung tissues were determined by flow cytometry. (H) The levels of M2-associated genes, PTPRE, and phosphorylated ERK 1/2 protein in IL-4–treated lung macrophages purified from experimental models were quantified. (I) RT-qPCR for M2-associated genes in lung tissue samples. The data shown were from one of three independent experiments. Data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Subsequently, we questioned whether PTPRE-AS1 also plays a role in the regulation of PTPRE expression in CRE-induced pulmonary inflammation. We found that lung sections from phosphate-buffered saline (PBS)–treated PTPRE-AS1 KO mice exhibited significantly lower levels of PTPRE expression compared with PBS-treated WT mice. In addition, similar to the DSS-induced colitis model, decreased levels of PTPRE were detected in lung tissues from CRE-treated PTPRE-AS1–null mice, relative to CRE-treated WT mice, whereas WDR5 expression was virtually unchanged (Fig. 5E). Furthermore, compared with CRE-treated WT mice, increased levels of F4/80+ and CD206+ inflammatory cells were observed in lung tissues from CRE-treated PTPRE-AS1–deficient mice (Fig. 5F). We then purified macrophages from lung tissue and found that ~95% of purified cells expressed CD11b and F4/80 (Fig. 5G). Similar to the observations in DSS-induced colitis, the enhanced levels of M2-associated gene and phosphorylated ERK 1/2 protein were detected in IL-4–treated lung macrophages from CRE-treated PTPRE-AS1–null mice compared with CRE-treated WT mice (Fig. 5H). Last, we also observed greater increases in M2-associated gene levels in lung tissues from CRE-treated PTPRE-AS1–null mice than CRE-treated WT mice (Fig. 5I). Thus, these findings suggest that PTPRE-AS1 may have a protective role in M2 macrophage–mediated pathogenesis of pulmonary inflammation by inhibiting M2 macrophage activation.
Reduced levels of PTPRE-AS1 and PTPRE in PBMCs from patients with allergic asthma
To determine the potential importance of PTPRE-AS1 in human inflammatory diseases, we evaluated the expression of PTPRE-AS1 and PTPRE in PBMCs from children with allergic asthma and healthy control subjects. Detailed characteristics of the study subjects are presented in table S1. Similar to the expression patterns in murine models, levels of PTPRE-AS1 and its target, PTPRE, were significantly lower in patients with allergic asthma compared with healthy subjects. WDR5 expression was also decreased in patients with asthma (Fig. 6A). Further, elevated levels of M2-associated genes, including IL-10 and CD206, were detected in patients with asthma (Fig. 6B). To confirm the functional homology of PTPRE-AS1 in human and mouse species, we evaluated the regulatory effects of PTPRE-AS1 in human macrophages. We found that PTPRE-AS1 was also a negative regulator of IL-4–induced M2 macrophage activation and downstream responses in human macrophages (Fig. 6C and fig. S6).
Fig. 6. Expression of PTPRE-AS1 in human allergic asthmatic patients.
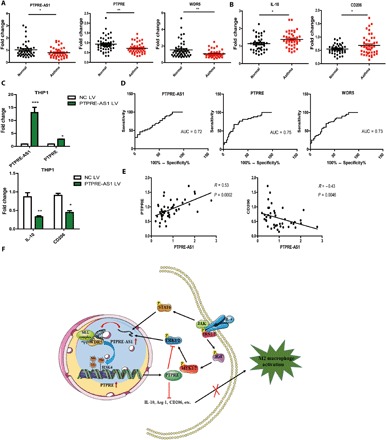
(A) Analyses of the expression levels of PTPRE-AS1, PTPRE, and WDR5 in PBMCs from 40 allergic asthmatic patients and 42 healthy controls. (B) Analyses of the expression of the M2-associated genes in PBMCs from patients with allergic asthma and healthy controls. (C) Macrophages were induced using THP1 cells treated with phorbol 12-myristate 13-acetate (PMA) (500 ng/ml, 48 hours). Overexpression of PTPRE-AS1 in THP1 macrophages transfected with PTPRE-AS1 LV or NC LV. After 72 hours of transfection, M2-associated gene expression in IL-4–stimulated THP1 macrophages was quantified by RT-qPCR analysis. (D) Receiver operating curve (ROC) analysis of PTPRE-AS1, PTPRE, and WDR5 levels in the study population. (E) Correlation analysis of the expression of PTPRE-AS1 and PTPRE, or CD206 levels in patients with allergic asthmatic (Pearson’s correlation). (F) Schematic model of PTPRE-AS1 functions in IL-4–induced M2 macrophage activation. *P < 0.05; **P < 0.01; ***P < 0.001.
The ability of PTPRE-AS1, PTPRE, and WDR5 to differentiate patients with allergic asthma from healthy subjects was assessed by receiver operating curve analysis, which yielded area under the curve (AUC) values of 0.72, 075, and 0.73, respectively (Fig. 6D). The positive correlation between PTPRE-AS1 and PTPRE was validated in human patients with allergic asthma, while a negative correlation between PTPRE-AS1 and CD206 was identified (Fig. 6E). Together, these findings imply that PTPRE-AS1 has potential as a biomarker in childhood allergic asthma.
DISCUSSION
LncRNAs have emerged as important regulators of inflammatory responses and models of inflammatory diseases, and they may have a critical role in differential macrophage activation and function (16, 17). In this study, a new lncRNA, PTPRE-AS1, targeting PTPRE, was identified to selectively express during IL-4–driven M2 macrophage activation. Using both gain- and loss-of-function approaches, we identified PTPRE-AS1 as a negative regulator of IL-4–driven M2 macrophage activation and associated pulmonary inflammation, partly through suppression of MAPK/ERK1/2 activation, while functionally promoting M1-associated colitis. Mechanistically, PTPRE-AS1 controls M2 macrophage activation through epigenetic up-regulation of PTPRE expression by binding to WDR5, leading to chromatin remodeling. Thus, these results suggest a novel regulatory pathway contributing to M2 macrophage activation and its associated inflammatory responses, adding a new dimension to the functional importance of lncRNA regulation and function in inflammatory diseases (Fig. 6F).
Numerous lncRNAs exert their functional effects on neighboring genes exclusively in cis or trans (18, 19). Our results demonstrated that PTPRE-AS1 could activate transcription of the proximal gene PTPRE. The functional importance of PTPRE has been well studied. For example, PTPRE-deficient BMDMs have abnormalities in cytokine production, with reduced TNFα and enhanced IL-10 levels following LPS challenge (20). In addition, the MAPK and JAK-STAT signaling pathways are both involved in IL-4–mediated cellular effect (21, 22). Consistent with the documented role of PTPRE as a phosphatase inhibitor of MAPK/ERK 1/2 and JAK-STAT pathway activation (11, 12, 23), we observed that PTPRE only inhibited IL-4–mediated ERK 1/2 phosphorylation levels, whereas no apparent changes in the STAT6 phosphorylation levels were detected. Moreover, it has been widely assumed that IL-4 induces the proliferation and alternative activation of macrophages (24, 25). In addition, MAPK/ERK 1/2 signaling plays critical roles in macrophage proliferation, cell cycle, and apoptosis (26). We found that the growth rate of lung macrophages from PTPRE-AS1 KO mice significantly improved during IL-4 stimulation compared to WT. The cell cycle stages further showed an increased cell ratio in the S phase for PTPRE-AS1 KO macrophages, whereas the percentage of apoptotic cells significantly decreased (fig. S7), implying that PTPRE-AS1 may affect macrophage proliferation and survival through IL-4–induced MAPK/ERK 1/2 signaling. Collectively, these findings support the possibility that PTPRE-AS1 negatively regulates IL-4–induced M2 macrophage activation by enhancing PTPRE expression, leading to the inhibition of MAPK/ERK 1/2 signaling and, at least in part, reduced macrophage functions.
LncRNAs participate in chromatin remodeling complexes that promote epigenetic activation or silencing of gene expression (27); however, the functions of many individual lncRNAs are unknown. Using a combination of bioinformatics prediction, FISH, and RIP assays, we determined that PTPRE-AS1 is enriched in the nucleus, where it interacts with WDR5. This key component of the histone methyltransferase complex reportedly interacts with lncRNAs and promotes gene expression by mediating H3K4me3 (13, 28, 29). RNA pull-down and ChIP assays further demonstrated that PTPRE-AS1 can mediate H3K4 trimethylation of the PTPRE promoter by directly binding to WDR5, leading to suppression of IL-4–induced M2 macrophage activation.
Recently, dysregulation of the expression and function of lncRNAs is increasingly implicated in various diseases, and related therapeutic strategies are gradually being explored (30, 31). Our data provide further evidence that PTPRE-AS1 is a key contributor to intestinal and lung inflammation. In the acute DSS-induced colitis model, PTPRE-AS1–null mice exhibited increased anti-inflammatory responses compared with WT mice, as evidenced by weight loss, stool consistency, rectal bleeding, and increased colon length. Besides having a critical role in pathogen clearance, colonic macrophages can also regulate inflammatory responses and local homeostasis (32). In particular, M2 macrophages represent an “alternatively” activated phenotype, with anti-inflammatory activities that can promote wound healing (33, 34). Consistent with in vitro studies, we observed increased expression of M2-associated genes and ERK 1/2 phosphorylation in colon macrophages from DSS-treated PTPRE-AS1–null mice relative to those from WT mice, whereas PTPRE levels were significantly reduced. These results imply that deficiency of PTPRE-AS1 may directly reduce PTPRE production to suppress DSS-induced intestinal inflammation by promoting tissue M2 macrophage activation.
Clinical and animal model findings have demonstrated positive correlations between asthma severity and elevated numbers of M2 macrophages, which have active roles in the pathogenesis of allergic asthma (35–38). PTPRE-AS1 deficiency aggravated CRE-induced allergic asthma inflammation, and lung macrophages from CRE-treated PTPRE-AS1-KO mice displayed enhanced M2 activation compared with CRE-treated WT mice. In contrast, PTPRE levels were significantly down-regulated in lung macrophages from CRE-challenged PTPRE-AS1–deficient mice. A recent study suggested PTPRE as a candidate susceptibility locus for allergic asthma (39); however, the role of PTPRE in the pathogenesis of allergic asthma remains unclear. Our findings provide evidence supporting the hypothesis that the enhanced allergen-induced allergic inflammation and M2 macrophage response in the context of PTPRE-AS1 deficiency may be attributable to reduced PTPRE levels, leading to M2 macrophage activation. In addition, our findings indicate a positive association between decreased PTPRE-AS1 expression and human asthma severity. Therefore, the observation that PTPRE-AS1 is critical for M2 macrophage activation in vitro and in vivo strongly suggests that this molecule may be an important regulator of other common inflammatory diseases. Further investigation of PTPRE-AS1 expression and function in human diseases is thus warranted.
In summary, we have used detailed molecular approaches to demonstrate that PTPRE-AS1 negatively regulates IL-4–induced M2 macrophage activation through recruitment of WDR5. Our in vivo results imply that PTPRE-AS1 is crucial in the pathogenesis of inflammatory disease. Together, the insights gained in this study will help to further understand the pathophysiological roles of lncRNAs in inflammatory diseases.
MATERIALS AND METHODS
Mice
PTPRE-AS1–null (PTPRE-AS1-KO) mice were generated using the CRISPR-Cas9 system at the Shanghai Model Organisms Center Inc. Briefly, the CRISPR-Cas9 system was applied in zygotes from C57BL/6 mice, and mutations were introduced by nonhomologous recombination repair, resulting in deletion of exons 2 and 3, and loss of function of lncRNA PTPRE-AS1. PTPRE-AS1–null mouse genotypes were confirmed by DNA sequencing. Primers used to identify genetically modified mice are listed in table S2. All mice were housed, bred, and maintained under specific pathogen–free conditions. All experiments complied with the relevant laws and institutional guidelines, as overseen by the Animal Studies Committee of the Children’s Hospital of Fudan University.
Cell culture, stimulation, and transfection
Mouse BMDMs were isolated and cultured as previously described (15). Briefly, bone marrow was extracted from femurs and, after lysis of erythrocytes, plated in complete medium consisting of Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and macrophage colony-stimulating factor-1 (10 ng/ml; R&D Systems). Cells were allowed to differentiate for 7 to 8 days before use. Isolation of mouse lung and colon macrophages was performed according to published protocols (15, 32), and cells were further purified by positive selection using magnetic anti-mouse CD11b beads (Miltenyi Biotec) and macrophage adherence. The percentages of macrophages in purified cells were detected by fluorescein isothiocyanate (FITC) anti-mouse F4/80 and phycoerythrin (PE) anti-mouse CD11b antibody (eBioscience). The mouse RAW 264.7 cell line was obtained from the Cell Bank, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
For M2 macrophage activation, macrophages were stimulated with IL-4 (20 ng/ml, 24 hours; R&D Systems). Antisense oligonucleotides (ASOs) were synthesized by Sangon Biotech (Shanghai). siRNAs, PTPRE-AS1 short hairpin RNA (shRNA) lentivirus (PTPRE-AS1 shRNA), and PTPRE-AS1–overexpressing lentivirus (PTPRE-AS1 LV) were synthesized by GenePharma (Shanghai). The sequences of siRNAs, ASOs, and controls are provided in table S3. BMDMs and RAW 264.7 cells were transfected with these RNAs using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer’s instructions.
Microarray analysis
BMDMs were stimulated with LPS (200 ng/ml) or IL-4 (20 ng/ml) for 3 and 24 hours, respectively. Total RNA was extracted and purified using the miRNeasy Mini Kit (Qiagen). The lncRNA mRNA Mouse Gene Expression Microarray V 6.0 (Agilent Technologies) was used to investigate differentially expressed lncRNAs in BMDMs after IL-4 or LPS stimulation. Microarray slides were sequenced on an Agilent Microarray Scanner, and the raw data were normalized by Quantile algorithm, GeneSpring Software 12.6.1 (Agilent Technologies) at Shanghai Biotechnology Corporation. To comprehensively analyze the microarray data, filtering criteria were applied (≥2-fold change, P < 0.05).
Real-time quantitative polymerase chain reaction
RNA was extracted from cells using TRIzol Reagent (Invitrogen). Complementary DNA (cDNA) was synthesized using the PrimeScript RT Reagent Kit (Takara Bio). To quantify mRNA expression, cDNAs were amplified by RT-qPCR using the SYBR Premix Ex Taq RT-PCR Kit (Takara Bio). ACTIN and GAPDH expression levels were determined as internal controls for human and mouse, respectively. Fold change in expression level was calculated using the 2−ΔΔCt method. The primer sequences for all genes are provided in table S2.
Western blotting analysis
Lysates were resolved by electrophoresis, transferred to polyvinylidene difluoride membranes, and probed with antibodies directed against phosphorylated (Thr202/Tyr204) ERK 1/2, ERK 1/2, phosphorylated (Tyr641) STAT6, STAT6, WDR5 (Cell Signaling Technology), PTPRE (Abcam), and β-tubulin (Abcam). All results were normalized to those of β-tubulin, which was used as a loading control.
Subcellular fractionation and localization
Nuclear and cytosolic fractions were separated using the Nuclear and Cytoplasmic Extraction Kit (CWBIO, Shanghai). Cells (1 × 107) were harvested, resuspended in 1 ml of Nc-buffer A and 55 μl of Nc-buffer B, and incubated for 20 min on ice. Cells were then centrifuged for 15 min at 12,000g; the resulting supernatants (containing the cytoplasmic component) and nuclear pellets were used for RNA extraction.
RNA FISH assays
The FISH kit was purchased from Stellaris, and experiments were performed according to the manufacturer’s instructions. Hybridization images were visualized using a confocal microscope (Leica). Briefly, cells were seeded, fixed with fixation buffer, and treated with 70% ethanol followed by prehybridization. They were then hybridized with probes (125 nM) for 4 hours. Quasar 670–labeled PTPRE-AS1 and Quasar 570–labeled MALAT1 probes were provided by Stellaris.
RIP assays
RIP was performed using the EZ-Magna RIP Kit (Millipore). Cells (1 × 107) were lysed with RIP lysis buffer. Cell extracts were coimmunoprecipitated with anti-WDR5, EZH2, LSD1, SETDB1 (Cell Signaling Technology), or DNMT1 (Abcam), and the retrieved RNA was subjected to RT-qPCR analysis. For RT-qPCR analysis, U1 was used as a nonspecific control target.
In vitro transcription and RNA pull-down assays
In vitro transcription assays were performed using a MEGAscript kit (Life Technologies, USA), according to the manufacturer’s instructions. PTPRE-AS1 RNAs were labeled by desthiobiotinylation, using the Pierce RNA 3′End Desthiobiotinylation Kit (Thermo Scientific). Then, RNA pull-down assays were performed using the Magnetic RNA-Protein Pull-Down Kit (Thermo Scientific).
ChIP assays
Cells were transfected with mixtures containing equal amounts of PTPRE-AS1 shRNA or WDR5 siRNA. ChIP was conducted using a SimpleChIP Enzymatic Chromatin IP kit (Cell Signaling Technology), according to the manufacturer’s instructions. Anti-WDR5 and anti-H3K4me3 antibodies were purchased from Cell Signaling Technology. Normal rabbit IgG was used as a negative control. Primers for ChIP-qPCR are listed in table S2.
DSS-induced mouse colitis model
Induction of colitis and experimental design
Eight-week-old male mice were divided into the following groups: Water + WT, Water + PTPRE-AS1 KO, DSS + WT, and DSS + PTPRE-AS1 KO. Mice in the DSS + WT and DSS + PTPRE-AS1 KO groups were provided with 4% DSS (36 to 50 kDa, MP Biomedicals) dissolved in sterile, distilled water for the experimental days (1 to 7), followed by 2 days of regular drinking water. Control mice (Water + WT and Water + PTPRE-AS1 KO groups) had access to water (without DSS). Mice were sacrificed on day 9.
Determination of disease activity index
Body weight, gross blood, and stool consistency were analyzed daily. Briefly, no weight loss was registered as 0 points; weight loss of 1 to 5% from baseline was assigned 1 point; 6 to 10%, 2 points; 11 to 15%, 3 points; and >15%, 4 points. For stool consistency, 0 points were assigned for well-formed pellets, 2 points for pasty and semiformed stools that did not adhere to the anus, and 4 points for liquid stools that did adhere to the anus. For bleeding, 0 was assigned for no blood, as determined using hemoccult (Beckman Coulter), 1 point for positive hemoccult, and 4 points for gross bleeding.
Histologic analysis
Colons were fixed in 10% neutral-buffered formalin. Paraffin sections were stained with hematoxylin and eosin (H&E). Histological scoring was performed by determining combined scores for inflammatory cell infiltration (score, 0 to 3) and tissue damage (score, 0 to 3) (40). The presence of occasional inflammatory cells in the lamina propria was scored as 0, increased numbers of inflammatory cells in the lamina propria was scored as 1, confluence of inflammatory cells extending into the submucosa was scored as 2, and transmural extension of the infiltrate was scored as 3. For tissue damage, no mucosal damage was scored as 0, lymphoepithelial lesions were scored as 1, surface mucosal erosion or focal ulceration was scored as 2, and extensive mucosal damage and extension into deeper structures of the bowel wall were scored as 3.
Cockroach allergen–induced mouse asthma model
Eight-week-old female mice were sensitized by intratracheal inhalation of 20 μg of CRE (GREER Laboratories) per mouse on days 1, 2, 3, and 14 and challenged on days 20 and 22 with the same amount of CRE. Control mice received PBS during the sensitization and challenge phases. On day 23, mice were sacrificed and lung tissues were dissected and analyzed for inflammation. BALF was harvested.
Analysis of lung inflammation
Mouse lungs were fixed in 10% neutral-buffered formalin. Sections (5 μm) were stained for H&E and immunohistochemistry (IHC). Antibodies against F4/80 (1: 250; Cell Signaling Technology) and CD206 (1:100; Abcam) were used for IHC analysis. For analysis of BALF, cells were stained for eosinophils, macrophages, neutrophils, and lymphocytes using flow cytometry. Eosinophils were defined as SSChigh SiglecF+ Mac-3− cells, alveolar macrophages were identified as SSChigh SiglecF+ Mac-3+ cells, granulocytes were recognized as SSChigh Gr-1+ cells, and lymphocytes were identified as FSClow/SSClow cells expressing CD3 or CD19.
Human study subjects
Children with allergic asthma and age-matched healthy controls were recruited from the Children’s Hospital of Fudan University after informed consent was obtained from all patients. Human PBMCs were isolated using Ficoll-Paque density gradient solution (density = 1.077 g/ml; GE Healthcare). Peripheral blood was mixed with PBS and overlaid on top of Ficoll. Following centrifugation at 400g for 0.5 hours, PBMCs were aspirated from the Ficoll-plasma interface. The cell pellet was washed twice with PBS and resuspended in TRIzol. The study was approved by the Research Ethics Board of the Children’s Hospital of Fudan University.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7.0 software. Three independent experiments were performed to confirm the reproducibility of each experiment in vitro. Bars represent the means ± SEM. The two-tailed Student’s t tests and one-way analysis of variance were used to evaluate the data. The relationship between PTPRE-AS1 and PTPRE, or CD206, was tested using Pearson’s correlation and linear regression. P values of <0.05 were deemed statistically significant.
Supplementary Material
Acknowledgments
We thank Y. Wang for flow cytometry analysis. Funding: This work was supported by the National Key R&D Program of China (2016YFC1305102 to Y.Z.); the National Natural Science Foundation of China (81671561 and 81974248 to Y.Z.; 81900751 to X.H.); 1000 Young Talents Plan Program of China; Initial Funding for New PI; Fudan Children’s Hospital and Fudan University; Program for Outstanding Medical Academic Leader (2019LJ19 to Y.Z.); the International Joint Laboratory Program of National Children’s Medical Center (EK1125180109 to Y.Z.); Shanghai Municipal Planning Commission of Science and Research Fund (201740065 to Y.Z. and 20174Y0079 to X.H.); Shanghai Pujiang Program 16PJ1401600 (to J.F.); Shanghai Committee of Science and Technology (no. 19ZR1406400 to J.F.); National Health Research Institutes, Taiwan (EOPP10-014 and EOSP07-014 to S.-K.H.); Kaohsiung Medical University “The Talent Plan” (106KMUOR04 to S.-K.H.); and Taiwan and Shenzhen Science and Technology Peacock Team Project (KQTD20170331145453160 to S.-K.H.). Author contributions: X.H., S.H., P.X., L.L., C.Z., L.Y., L.X., L.S., and J.F. designed and performed experiments, and analyzed data. S.-K.H. provided intellectual input and aided in the experimental design. X.H. and Y.Z. wrote the manuscript. Y.Z. planned, designed, supervised, and coordinated the overall research efforts. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Microarray data have been deposited in the GEO under accession number GSE122150. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data are available from authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaax9230/DC1
Fig. S1. Differentially expressed lncRNAs in LPS- and IL-4–stimulated BMDMs.
Fig. S2. PTPRE-AS1 mediates PTPRE expression.
Fig. S3. Neither PTPRE-AS1 nor PTPRE influences IL-4–induced STAT6 phosphorylation levels.
Fig. S4. Generation of PTPRE-AS1 KO mice.
Fig. S5. PTPRE-AS1 and PTPRE are reduced in both DSS-induced colitis and CRE-induced allergic asthma.
Fig. S6. PTPRE-AS1 inhibits the expression of M2 macrophage–associated genes in THP1 macrophages.
Fig. S7. PTPRE-AS1 deficiency promotes the proliferation and expansion of lung resident macrophages stimulated by IL-4.
Table S1. Demographic and respiratory health characteristics of patients with allergic asthmatic and healthy controls.
Table S2. The primer sequences.
Table S3. The sequences of PTPRE-AS1 ASO, shRNA, PTPRE siRNA, and WDR5 siRNA.
REFERENCES AND NOTES
- 1.Murray P. J., Wynn T. A., Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galli S. J., Borregaard N., Wynn T. A., Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 12, 1035–1044 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mosser D. M., Edwards J. P., Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon S., Martinez F. O., Alternative activation of macrophages: Mechanism and functions. Immunity 32, 593–604 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Martinez F. O., Helming L., Gordon S., Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Moreira A. P., Cavassani K. A., Hullinger R., Rosada R. S., Fong D. J., Murray L., Hesson D. P., Hogaboam C. M., Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J. Allergy Clin. Immunol. 126, 712–721.e7 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Mercer T. R., Dinger M. E., Mattick J. S., Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 10, 155–159 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Wilusz J. E., Sunwoo H., Spector D. L., Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 23, 1494–1504 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Z., Chao T.-C., Chang K.-Y., Lin N., Patil V. S., Shimizu C., Head S. R., Burns J. C., Rana T. M., The long noncoding RNA THRIL regulates TNFα expression through its interaction with hnRNPL. Proc. Natl. Acad. Sci. U.S.A. 111, 1002–1007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atianand M. K., Hu W., Satpathy A. T., Shen Y., Ricci E. P., Alvarez-Dominguez J. R., Bhatta A., Schattgen S. A., McGowan J. D., Blin J., Braun J. E., Gandhi P., Moore M. J., Chang H. Y., Lodish H. F., Caffrey D. R., Fitzgerald K. A., A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell 165, 1672–1685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wabakken T., Hauge H., Finne E. F., Wiedlocha A., Aasheim H.-C., Expression of human protein tyrosine phosphatase epsilon in leucocytes: A potential ERK pathway-regulating phosphatase. Scand. J. Immunol. 56, 195–203 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Tanuma N., Nakamura K., Shima H., Kikuchi K., Protein-tyrosine phosphatase PTPεC inhibits Jak-STAT signaling and differentiation induced by Il-6 and LIF in M1 leukemia cells. J. Biol. Chem. 275, 28216–28221 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Wang K. C., Yang Y. W., Liu B., Sanyal A., Corces-Zimmerman R., Chen Y., Lajoie B. R., Protacio A., Flynn R. A., Gupta R. A., Wysocka J., Lei M., Dekker J., Helms J. A., Chang H. Y., A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guarnaccia A. D., Tansey W. P., Moonlighting with WDR5: A cellular multitasker. J. Clin. Med. 7, E21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Y., Do D. C., Ishmael F. T., Squadrito M. L., Tang H. M., Tang H. L., Hsu M.-H., Qiu L., Li C., Zhang Y., Becker K. G., Wan M., Huang S.-K., Gao P., Mannose receptor modulates macrophage polarization and allergic inflammation through miR-511-3p. J. Allergy Clin. Immunol. 141, 350–364.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carpenter S., Aiello D., Atianand M. K., Ricci E. P., Gandhi P., Hall L. L., Byron M., Monks B., Henry-Bezy M., Lawrence J. B., O'Neill L. A. J., Moore M. J., Caffrey D. R., Fitzgerald K. A., A long noncoding RNA mediates both activation and repression of immune response genes. Science 341, 789–792 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilott N. E., Heward J. A., Roux B., Tsitsiou E., Fenwick P. S., Lenzi L., Goodhead I., Hertz-Fowler C., Heger A., Hall N., Donnelly L. E., Sims D., Lindsay M. A., Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat. Commun. 5, 3979 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rinn J. L., Kertesz M., Wang J. K., Squazzo S. L., Xu X., Brugmann S. A., Goodnough L. H., Helms J. A., Farnham P. J., Segal E., Chang H. Y., Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez J. A., Wapinski O. L., Yang Y. W., Bureau J.-F., Gopinath S., Monack D. M., Chang H. Y., Brahic M., Kirkegaard K., The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell 152, 743–754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sully V., Pownall S., Vincan E., Bassal S., Borowski A. H., Hart P. H., Rockman S. P., Phillips W. A., Functional abnormalities in protein tyrosine phosphatase ε-deficient macrophages. Biochem. Biophys. Res. Commun. 286, 184–188 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Gour N., Wills-Karp M., IL-4 and IL-13 signaling in allergic airway disease. Cytokine 75, 68–78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calvo K. R., Dabir B., Kovach A., Devor C., Bandle R., Bond A., Shih J. H., Jaffe E. S., IL-4 protein expression and basal activation of Erk in vivo in follicular lymphoma. Blood 112, 3818–3826 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanuma N., Shima H., Nakamura K., Kikuchi K., Protein tyrosine phosphatase εC selectively inhibits interleukin-6–and interleukin-10–induced JAK-STAT signaling. Blood 98, 3030–3034 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Jenkins S. J., Ruckerl D., Cook P. C., Jones L. H., Finkelman F. D., van Rooijen N., MacDonald A. S., Allen J. E., Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332, 1284–1288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gundra U. M., Girgis N. M., Ruckerl D., Jenkins S., Ward L. N., Kurtz Z. D., Wiens K. E., Tang M. S., Basu-Roy U., Mansukhani A., Allen J. E., Loke P., Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood 123, e110–e122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Y., Pollard J. W., Casadevall A., Fcγ receptor cross-linking stimulates cell proliferation of macrophages via the ERK pathway. J. Biol. Chem. 285, 4232–4242 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khalil A. M., Guttman M., Huarte M., Garber M., Raj A., Rivea Morales D., Thomas K., Presser A., Bernstein B. E., van Oudenaarden A., Regev A., Lander E. S., Rinn J. L., Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu P., Chen X., Xie R., Han J., Xie W., Wang B., Dong W., Chen C., Yang M., Jiang J., Chen Z., Huang J., Lin T., lncRNA HOXD-AS1 regulates proliferation and chemo-resistance of castration-resistant prostate cancer via recruiting WDR5. Mol. Ther. 25, 1959–1973 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun T.-T., He J., Liang Q., Ren L.-L., Yan T.-T., Yu T.-C., Tang J.-Y., Bao Y.-J., Hu Y., Lin Y., Sun D., Chen Y.-X., Hong J., Chen H., Zou W., Fang J.-Y., LncRNA GClnc1 promotes gastric carcinogenesis and may act as a modular scaffold of WDR5 and KAT2A complexes to specify the histone modification pattern. Cancer Discov. 6, 784–801 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Zhang E., Han L., Yin D., He X., Hong L., Si X., Qiu M., Xu T., De W., Xu L., Shu Y., Chen J., H3K27 acetylation activated-long non-coding RNA CCAT1 affects cell proliferation and migration by regulating SPRY4 and HOXB13 expression in esophageal squamous cell carcinoma. Nucleic Acids Res. 45, 3086–3101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wahlestedt C., Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov. 12, 433–446 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Zhu W., Yu J., Nie Y., Shi X., Liu Y., Li F., Zhang X.-l., Disequilibrium of M1 and M2 macrophages correlates with the development of experimental inflammatory bowel diseases. Immunol. Invest. 43, 638–652 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Hunter M. M., Wang A., Hirota C. L., McKay D. M., Neutralizing anti-IL-10 antibody blocks the protective effect of tapeworm infection in a murine model of chemically induced colitis. J. Immunol. 174, 7368–7375 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Hunter M. M., Wang A., Parhar K. S., Johnston M. J., Van Rooijen N., Beck P. L., McKay D. M., In vitro-derived alternatively activated macrophages reduce colonic inflammation in mice. Gastroenterology 138, 1395–1405 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Chupp G. L., Lee C. G., Jarjour N., Shim Y. M., Holm C. T., He S., Dziura J. D., Reed J., Coyle A. J., Kiener P., Cullen M., Grandsaigne M., Dombret M. C., Aubier M., Pretolani M., Elias J. A., A chitinase-like protein in the lung and circulation of patients with severe asthma. N. Engl. J. Med. 357, 2016–2027 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Melgert B. N., ten Hacken N. H., Rutgers B., Timens W., Postma D. S., Hylkema M. N., More alternative activation of macrophages in lungs of asthmatic patients. J. Allergy Clin. Immunol. 127, 831–833 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Draijer C., Robbe P., Boorsma C. E., Hylkema M. N., Melgert B. N., Characterization of macrophage phenotypes in three murine models of house-dust-mite-induced asthma. Mediators Inflamm. 2013, 632049 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ford A. Q., Dasgupta P., Mikhailenko I., Smith E. M. P., Noben-Trauth N., Keegan A. D., Adoptive transfer of IL-4Rα+ macrophages is sufficient to enhance eosinophilic inflammation in a mouse model of allergic lung inflammation. BMC Immunol. 13, 6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murk W., Walsh K., Hsu L. I., Zhao L., Bracken M. B., DeWan A. T., Attempted replication of 50 reported asthma risk genes identifies a SNP in RAD50 as associated with childhood atopic asthma. Hum. Hered. 71, 97–105 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Siegmund B., Lehr H.-A., Fantuzzi G., Dinarello C. A., IL-1β-converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. U.S.A. 98, 13249–13254 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaax9230/DC1
Fig. S1. Differentially expressed lncRNAs in LPS- and IL-4–stimulated BMDMs.
Fig. S2. PTPRE-AS1 mediates PTPRE expression.
Fig. S3. Neither PTPRE-AS1 nor PTPRE influences IL-4–induced STAT6 phosphorylation levels.
Fig. S4. Generation of PTPRE-AS1 KO mice.
Fig. S5. PTPRE-AS1 and PTPRE are reduced in both DSS-induced colitis and CRE-induced allergic asthma.
Fig. S6. PTPRE-AS1 inhibits the expression of M2 macrophage–associated genes in THP1 macrophages.
Fig. S7. PTPRE-AS1 deficiency promotes the proliferation and expansion of lung resident macrophages stimulated by IL-4.
Table S1. Demographic and respiratory health characteristics of patients with allergic asthmatic and healthy controls.
Table S2. The primer sequences.
Table S3. The sequences of PTPRE-AS1 ASO, shRNA, PTPRE siRNA, and WDR5 siRNA.