

Abstract
RNA polymerase III (Pol III) is an essential 17-subunit complex responsible for the transcription of small housekeeping RNAs such as transfer RNAs and 5S ribosomal RNA. Biallelic variants in four genes (POLR3A, POLR3B, and POLR1C and POLR3K) encoding Pol III subunits have previously been found in individuals with (neuro-) developmental disorders. In this report, we describe three individuals with biallelic variants in POLR3GL, a gene encoding a Pol III subunit that has not been associated with disease before. Using whole exome sequencing in a monozygotic twin and an unrelated individual, we detected homozygous and compound heterozygous POLR3GL splice acceptor site variants. RNA sequencing confirmed the loss of full-length POLR3GL RNA transcripts in blood samples of the individuals. The phenotypes of the described individuals are mainly characterized by axial endosteal hyperostosis, oligodontia, short stature, and mild facial dysmorphisms. These features largely fit within the spectrum of phenotypes caused by previously described biallelic variants in POLR3A, POLR3B, POLR1C, and POLR3K. These findings further expand the spectrum of POLR3-related disorders and implicate that POLR3GL should be included in genetic testing if such disorders are suspected.
Subject terms: Genetic testing, Clinical genetics
Introduction
RNA polymerase III (Pol III) is one of the three polymerase complexes involved in eukaryotic RNA synthesis. RNAs synthesized by Pol III include the small transfer RNAs (tRNA), 5S ribosomal RNA (rRNA), U6 small nuclear RNA, U5 rRNA, and several other non-coding RNAs [1, 2]. Pol III is a highly conserved essential enzyme complex consisting of 17 subunits (Fig. 1a). Twelve of these subunits are shared with RNA polymerase I and/or RNA polymerase II [3]. Germline variants in four genes encoding for Pol III subunits POLR3A, POLR3B, POLR1C, and POLR3K have been shown to cause a spectrum of phenotypes termed POLR3-related disorders, which were previously appointed as tremor-ataxia with central hypomyelinisation (TACH), leukodystrophy with oligodontia (LO, MIM: 607694), 4H (hypomyelination, hypodontia, hypogonadotropic hypogonadism) syndrome (MIM: 612440) or isolated hypogonadotropic hypogonadism [4–21]. Frequently occurring phenotypic features in individuals with POLR3-related disorders are white matter abnormalities, cerebellar signs, motor delay and/or regression, dental abnormalities, myopia, short stature, and hypogonadotropic hypogonadism [21]. Specific combinations of biallelic splicing and truncating variants in POLR3A can also lead to the distinct neonatal progeroid (Wiedemann-Rautenstrauch) syndrome (MIM: 264090) [22–24].
Fig. 1.
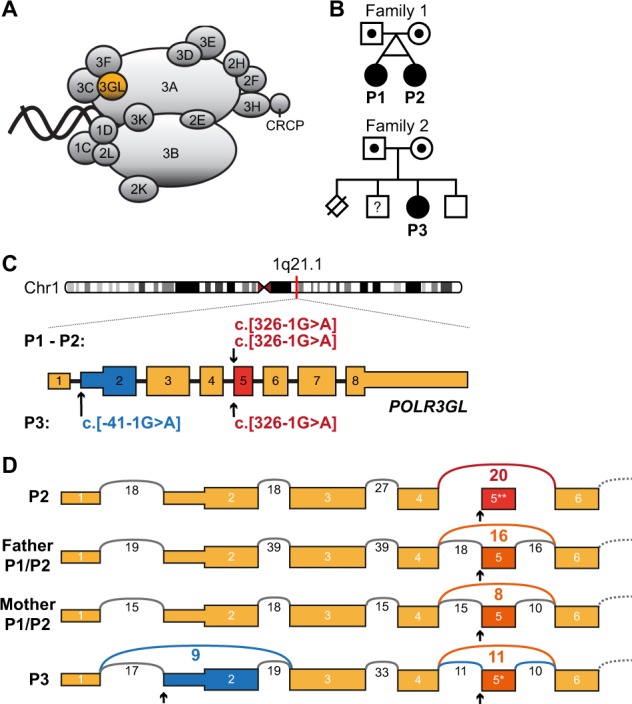
Biallelic POLR3GL splice site variants in three individuals with endosteal hyperostosis and oligodontia. a Schematic representation of Pol-III protein complex with the highlighted subunit POLR3GL which forms a trimer subcomplex with POLR3C and POLR3F. Adapted from Flores et al. [38]. b Pedigrees of the two families included in this study. Filled shapes denote affected individuals and dotted shapes denote heterozygous carriers. The affected monozygotic twin of family 1 inherited the POLR3GL c.326-1G > A homozygous variant from their healthy, distantly related carrier parents. Individual P3 from family 2 inherited a heterozygous c.-41-1G > A variant from her mother and a c.326-1G > A variant from her father. She has one healthy brother and one possibly affected brother who was not available for genetic counseling (square with question mark). One perinatal death of a sib due to a neural tube defect was reported. Nomenclature of variants is according to HGVS nomenclature. c Schematic representation of the POLR3GL coding sequence (ENSG00000121851, mRNA NM_032305.1, GRCh37/hg19) showing the positions of the homozygous splice site variants in individuals P1 and P2 and the compound heterozygous variants in individual P3. Variant c.326-1G > A (red) is predicted to cause an skip of exon 5 (ENSE00003501352) in the transcript and variant c.-41-1G > A (blue) may cause aberrant splicing of exon 2 (ENSE00003603498) containing the POLR3GL translation initiation site. d Schematic sashimi plot showing the number of RNA-seq reads crossing the junctions of POLR3GL exons 1 to 6 in blood cells of individual P2, father and mother of individuals P1/P2 and individual P3. The reads that skip either exon 2 or exon 5 are displayed in color. The arrows denote the location of the splice site variants. *p < 0.05, **p < 0.01 Benjamini-Hochberg adjusted p-values for differential exon usage of the exons of all transcripts of POLR3GL calculated with the R-package DEXseq
The vertebrate Pol III complex contains either the widely expressed POLR3GL (also known as RPC7L or RPC32β) or its isoform POLR3G (also known as RPC32α) [25, 26]. The two paralogous genes likely originated from a gene duplication event in a common ancestor of vertebrates and their protein sequences share 46% amino acid identity [26]. POLR3GL is part of a detachable Pol III subcomplex important for transcription initiation together with POLR3C and POLR3F [3]. Here we report biallelic variants in the POLR3GL gene in three individuals with syndromic forms of endosteal hyperostosis in combination with oligodontia.
Materials and methods
Whole exome sequencing
Written informed consent was obtained from all included individuals and all procedures were performed in accordance with the guidelines of the Medical Ethics Committee (METC) of the University Medical Center Utrecht. Research has been performed in accordance with the Declaration of Helsinki. After referral for routine diagnostic whole exome sequencing (WES), exomes of individuals P1 and P3 and their parents were enriched using the Agilent SureSelect XT Human All Exon kit V5 and sequenced on the HiSeq2500 sequencing system (Illumina, San Diego, CA, USA) in rapid 2 × 100 bp run mode with a mean target depth of 100×. Reads were aligned to hg19 using BWA (BWA-MEM v0.7.5a) and variants were called using GATK haplotype caller (V2.7-2).
Variant filtering and reporting
Detected variants were annotated, filtered and prioritized using the Bench NGS Lab platform (Agilent-Cartagenia, Santa Clara, CA, USA). Only variants that fitted a de novo or recessive inheritance model were analyzed. Variants dominantly inherited from one of the parents were excluded from the analysis. Reporting of de novo variants in candidate genes (genes of uncertain clinical significance) was restricted to putative protein changing variants in genes that are intolerant to missense and loss-of-function variants [27]. For the recessive inheritance hypothesis, homozygous and compound heterozygous putative protein changing variants were filtered using a population allele frequency cutoff of 0.5% (ExAC database [27]). Variants in candidate recessive genes were only reported if at least one allele carried a putative loss-of-function variant. Larger deletions/duplications, missense, synonymous, and intronic variants affecting protein function of other genes cannot be excluded. No putative protein changing de novo variants were identified in individuals P1 and P3. The only variants that fulfilled the stringent diagnostic filtering and reporting criteria were homozygous and compound heterozygous variants in the POLR3GL gene in P1 and P3. No putative causative variants were identified in the other POLR3-related genes. The presence of the POLR3GL variants was confirmed in all three individuals and their parents by Sanger sequencing on an ABI 3730 analyzer with BigDye chemistry V.3.1. Monozygosity between individuals P1 and P2 was confirmed by Short Tandem Repeat (STR) analysis. POLR3GL variants are annotated according to reference NC_000001.10 (NM_032305.1). POLR3GL exons are numbered according to ENST00000369314.1 (GRCh37.p13, exon 2 is named ENSE00003603498 and exon 5 is named ENSE00003501352).
RNA sequencing
Peripheral blood mononuclear cells (PBMCs) were isolated from fresh blood samples using Ficoll-Paque PLUS (GE Healthcare Life Sciences, Cleveland, Ohio) and SepMate tubes (STEMCELL Technologies, Köln, Germany) according to the manufacturer’s protocols. Total RNA was isolated from the PBMCs using the QIAsymphony (Qiagen, Venlo, The Netherlands) or the RNeasy Kit (Qiagen). RNA-sequencing (RNA-seq) libraries were prepared using TruSeq Stranded Total RNA Library Prep Kit (Illumina) according to the manufacturer’s protocol. RNA-seq libraries were pooled and sequenced on a NextSeq500 (Illumina) in 2 × 75 bp paired-end mode. RNA sequencing data analysis was performed using a custom in-house pipeline (https://github.com/UMCUGenetics/RNASeq). Briefly, the reads were mapped against the human reference genome (CRCh37/hg19) using STAR [28]. Mapped reads were quantified using HTseq-count [29] and read counts were normalized using the R-package DESeq2 [30]. DESeq2 was also used to perform differential gene expression analysis. Differential POLR3GL (ENST00000369314.1, CRCh37) exon expression analysis of individual P2, father of individuals P1/P2, mother of individuals P1/P2, individual P3 and 20 control subjects from an in-house database was performed using the R-package DEXSeq [31, 32].
PCR validation of exon skipping
cDNA was synthesized with OligoDT and SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, California) following manufacturer’s protocol. PCR reactions were prepared in 20 µl volumes containing 2× Phusion High-Fidelity master mix (New England Biolabs, Ipswich, MA, USA), 3% DMSO, 0.5 µM forward primer, 0.5 µM reverse primer and 10% template cDNA and were performed using the following PCR conditions: one step of 98 °C for 30 s; 16 thermal cycles (including touchdown steps of −0.5 °C each annealing step) of: 98 °C for 10 s, 61 °C (−0.5 °C per cycle) for 30 s and 72 °C for 30 s; followed by 20 thermal cycles of: 98 °C for 10 s, 58 °C for 30 s, and 72 °C for 30 s; followed by one step of 72 °C for 10 min. PCR products were analyzed on 1% agarose gel with 1:15000 SYBR safe DNA gel stain (ThermoFisher Scientific, Waltham, MA, USA). The following primer sequences have been used (Fig. S5B): Pr1: GCCCAGTACATTTCAAGTTGG, Pr2: GCAGCAGGTTTATTCACTGG, Pr5: TGTAATCCGTTCCTTCTGTAGC, Pr6: CCTTATCTTCTGTGGTCTTAGGG.
Results
Detection of biallelic POLR3GL variants by WES
Diagnostic trio-based WES identified biallelic POLR3GL variants in a female of 19 years old (individual P1) and one unrelated female of 36 years old (individual P3). Individual P1 has a monozygotic twin sister (individual P2, Fig. 1b) and the presence of the same POLR3GL variants in her sister was validated by Sanger sequencing. The siblings and the unrelated individual show syndromic forms of endosteal hyperostosis in combination with oligodontia, but they were only matched after the identification of the POLR3GL variants. Individuals P1 and P2 have a homozygous c.326-1G>A p.? splice acceptor site variant upstream of exon 5 of POLR3GL. They inherited these variants from their healthy parents who are heterozygous carriers of this variant (Fig. 1b, c). Genealogical analysis indicates that the father and mother have a shared ancestor seven generations ago and are therefore distantly related (Fig. 1b). Individual P3 carries compound heterozygous variants (c.[-41-1G > A];[326-1G > A] p.[?]; [?]) in the splice acceptor sites of POLR3GL exons 2 and 5 (Fig. 1c). The variant c.326-1G > A p.?, which was also found in P1 and P2, was inherited from her father and the variant upstream of exon 2 c.-41-1G > A p.? (rs782661984) was inherited from her mother (Fig. 1b, c). The genealogical study did not find indications for a relationship between the two families. Both POLR3GL variants are reported in the GnomAD database (v2.1), but only in a heterozygous state at very low allele frequencies (c.-41-1G > A: 0.00002475 and c.326-1G > A: 0.000007955) [27]. Variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Individuals with biallelic POLR3GL variants show endosteal hyperostosis and oligodontia
The three affected individuals with POLR3GL variants show overlapping phenotypes (Table 1, Supplementary Case Reports). Individuals P1 and P2 are monozygotic twin sisters who were born by vacuum extraction because of umbilical cord prolapse at 36 weeks. Individual P1 showed a delayed speech and motor development with hypotonia. She has a disharmonic IQ profile with an average verbal IQ and a low performance IQ (40 points below her verbal IQ). She is diagnosed with a pervasive developmental disorder and currently attends a school for physically handicapped children. Ophthalmological examination at the age of 18 years showed mild hypermetropia with good vision. She developed normal secondary sexual characteristics, but puberty was delayed and periods started at age 17 years. She is frequently using a wheelchair because of pains in her back and feet since the age of 16 years. Neurological investigations showed no cerebellar signs like ataxia or intention tremor, but there was hypotonia in arms and legs, with relatively low reflexes and proximal weakness of leg muscles. In addition, she has neurogenic bladder dysfunction with recurrent cystitis and an MRI scan of the lumbar spine showed bulging discs of L2-L3 and L3-L4 affecting the cauda equina.
Table 1.
Clinical features of individuals with biallelic POLR3GL variants
| P1 | P2 | P3 | |
|---|---|---|---|
| Age at follow-up | 19 years | 19 years | 36 years |
| Gender | Female | Female | Female |
| POLR3GL variants | c.[326-1G > A]; [326-1G > A] | c.[326-1G > A]; [326-1G > A] | c.[-41-1G > A]; [326-1G > A] |
| Neurological features | |||
| Intellectual disability | −, Mean verbal IQ, low performance IQ, PDD-NOS | −, Mean verbal IQ, low performance IQ | −, Mild learning problems |
| Motor retardation | + | + | + |
| Cerebellar signs (ataxia, dysmetria) | – | – | – |
| Microcephaly | – | + | – |
| Upper motor signs (pyramidal) | – | +, non-progressive spastic paraparesis | – |
| Seizures | – | – | – |
| Dysarthria | – | +, mild | – |
| Ophthalmological | |||
| Myopia | – | −, astigmatism, cerebral visual impairment | – |
| Hearing | |||
| Hearing loss | – | – | +, mainly conductive |
| Dental | |||
| Oligodontia | + | + | + |
| Orthopedic | |||
| Club feet | + | + | +, very mild |
| Growth impairment (<p5) | + | + | + |
| Endocrine | |||
| Growth hormone deficiency | – | – | – |
| Delayed puberty | + | – | + |
| Radiological | |||
| Endosteal sclerosis | + | + | + |
| MRI | |||
| Cerebellar hypoplasia | U | – | U |
| Diffuse hypomyelination | U | − (at age 3.5 years) | U |
| Hypoplasia of corpus callosum | U | +, localized | U |
| Other | |||
| Motor regression | +, frequent wheelchair use because of back pain | stable, wheelchair dependent, spasticity, hip problems | +, due to coxarthrosis |
| Dysmorphic facial features | + | + | + |
Her sister, individual P2, has a non-progressive congenital spastic diplegia. She has severe motor problems and has never walked independently. Periventricular localized white matter abnormalities including a focal thinning of the corpus callosum were detected on an MRI scan of the brain at the age of 3.5 years (Fig. S1). These abnormalities were consistent with brain lesions caused by perinatal asphyxia and there were no signs of a diffuse hypomyelination typically observed in individuals with POLR3-related leukodystrophy [10, 21, 33]. Like her sister, she has a mean verbal IQ and a significant lower performance IQ. She attends a special school. Ophthalmological examination at the age of 18 years showed cerebral visual impairment in combination with mild hypermetropia. Puberty began at the age of 13 years and her periods started spontaneously at 16 years. Neurological examination at the age of 18 years revealed mild pseudobulbar dysarthria, bilateral spastic paresis on the right side more pronounced than on the left side and bilateral Babinski reflexes. No ataxia or intention tremor was noted.
Both sisters were born with club feet and mild syndactyly of the second and third toe (Fig. S2a–d). In addition they have oligodontia with only 8 (P1) and 14 teeth (P2) in their permanent dentition (Fig. 2b, c, e, f). Radiological examination showed that both sisters have a mainly axial localized form of endosteal sclerosis (Fig. 3a–d and Fig. S3a–c). They also have a short stature (the height of P1 was 140 cm (−4,7 standard deviations (SD)) at age 18 years and the height of P2 was 136 cm (−4,59SD) at age 15 years), but endocrinological analyses of IGF-1, IGFBP3, free T4 and TSH levels at age of 18 years did not indicate growth hormone deficiency (Supplementary Case Reports). In addition, normal levels of bone metabolism markers were detected at the age of 18 years (Supplementary Case Reports). The twins have mild facial dysmorphisms including upslanting palpebral fissures, thin lips with downturned corners of the mouth, a flat philtrum and a beaked nose with relatively long columella (Fig. 2a, d).
Fig. 2.

Facial appearances and dental abnormalities of the three individuals carrying biallelic POLR3GL variants. a Photograph showing the facial characteristics of individual P1 at the age of 3 years including upslanting palpebral fissures, thin lips, a flat philtrum and relatively long columella. b Intra-oral photograph of dental abnormalities of individual P1 showing an anterior open bite extending to the buccal teeth whereby only the most distal molars occlude. The central lower deciduous teeth are worn because of prolonged use. c Orthopantomographic radiograph of the full dentition of individual P1. The Orthopantomograph shows that the permanent dentition consists of the first and second molars, all other teeth are congenitally absent. In addition, it shows that the upper- and lower molar pulp chamber have a taurodontic shape. d Photograph of the face of individual P2 at the age of 3 years showing upslanting palpebral fissures, a flat philtrum and thin lips with relatively long columella. e Intraoral photograph of dental abnormalities of individual P2. Only the distal molars occlude. There is spacing in between the teeth due to the growth of the jaws and absence of permanent teeth. f Radiograph of deciduous upper frontal teeth of individual P2 showing short erratic shape of the teeth and obliterated root canals. The original shape of the crowns and the covering with composite can be seen. g Photograph showing the facial characteristics of individual P3 at the age of 8 years which include a high nasal bridge with a relatively long columella and thin lips. h Orthopantomographic radiograph of the full dentition of individual P3. Upper medial incisors and 5 molars are present in the permanent dentition with taurodontic shape of the upper- and lower molars
Fig. 3.
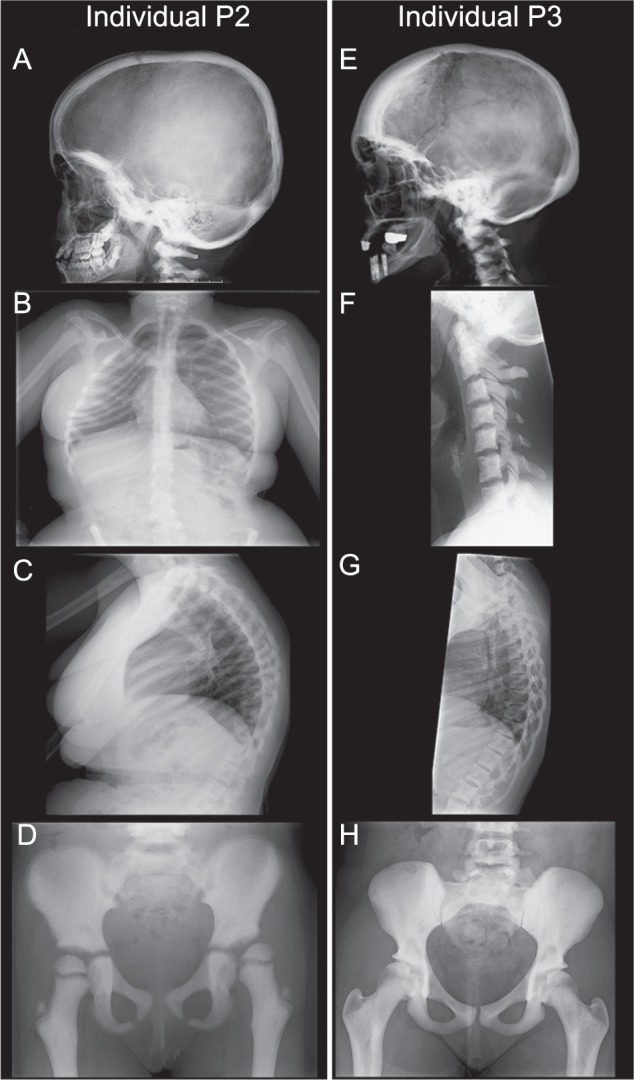
Skeletal X-ray scans showing endosteal hyperostosis in individuals P2 and P3. a–d X-rays of individual P2 at different ages. a X-ray at the age of 9 years showing sclerotic thickening of the neurocranium and the cranial base with sclerosis of C1 and C2. b Sclerotic ribs with scoliosis at the age of 18 years. c Sclerotic margins of the vertebrae with increased kyphosis of the thoracic spine at the age of 18 years. d X-ray of the pelvis at the age of 6 years showing diffuse hyperostosis, coxa valga and lateral displacement of the left femoral head. e–h X-rays of individual P3 at the age of 26 years. e Adult lateral skull showing sclerotic thickening, especially in the frontal and occipital area of the neurocranium. f Diffuse hyperostosis of the vertebral bodies and arches of the cervical spine. g Sclerotic margins of the vertebral bodies and arches of the thoracic spine. h Diffuse hyperostosis of the iliac and public bones and to a lesser extent of the proximal femora
Individual P3 is a 36-year old female who was born after an uneventful pregnancy. She had a growth retardation in her childhood and she has a short stature (142.8 cm, −4.5SD at adulthood), but growth hormone levels measured at ages 7, 18, and 34 were normal (Supplementary Case Reports). She had a normal speech development and a slightly delayed motor development and attended regular primary and secondary school with some additional support. She had amblyopia of the left eye for which she was treated with a patch. Ophthalmological examination at the age of 34 years revealed suboptimal vision with mild subcapsular posterior cataract and a bilateral refraction abnormality (Supplementary Case Reports). She had a delayed puberty with breast development starting around 15 years and a menarche at 17 years and 9 months, but she developed normal secondary sexual characteristics. She has oligodontia with only seven teeth in her permanent dentition (Fig. 2h). Like the twins, she has several dysmorphic features including mild proptosis, a high nasal bridge with a relatively long columella, a broad nasal tip, downturned corners of the mouth and retrognathia (Fig. 2g). She has syndactyly of the second and third toe and brachydactyly with short and stubby toes (Fig. S2e, f). Skeletal surveys showed that she has a mainly axial localized form of endosteal hyperostosis like the other two individuals (Fig. 3e–h, Fig. S3d–i and Fig. S4). Bone metabolism marker levels were normal at the age of 34 years (Supplementary Case Reports). She has moderate, mainly conductive and progressive hearing loss since the age of 18 years possibly due to tympanosclerosis. She is having increasing pain in the hips due to bilateral coxarthrosis since the age of 33 years and she recently underwent arthroplasty of the right hip at the age of 34 years. Neurological examination at the age of 34 years showed bilateral hypotonia and proximal muscle weakness (MRC4) in the arms, but normal distal muscle strength. No signs of cerebellar involvement like ataxia, nystagmus or coordination difficulties were present at that age.
Thus, the three individuals with biallelic POLR3GL variants show overlapping phenotypic features, including axial endosteal hyperostosis, oligodontia, short stature and mild facial dysmorphisms.
Splice site variants in POLR3GL cause exon skipping
The detected biallelic POLR3GL variants are predicted to disrupt splice acceptor sites which could cause aberrant splicing of POLR3GL exon 5 and/or exon 2. We performed RNA-seq on blood samples of individual P2, the parents of individuals P1/P2 and individual P3 to determine the effects of the variants on splicing of POLR3GL RNA transcripts. Exon 5 is skipped in the POLR3GL RNA transcripts of individual P2 containing homozygous c. [326-1G > A]; [326-1G > A] variants and this exon is only partially included in the heterozygous carrier parents (Fig.1d). The compound heterozygous POLR3GL splice variants c. [-41-1G > A] and c. [326-1G > A] in individual P3 are predicted to cause either skipping of exon 2 or exon 5. Indeed, RNA-seq shows that both exons are only partially included in the POLR3GL transcripts of this individual (Fig. 1d). PCR analysis confirms that this individual has no expression of full-length POLR3GL transcripts containing both exon 2 and exon 5 (Fig. S5). The abundance of POLR3GL transcripts is not significantly altered in the individuals and their parents compared to the expression levels in unaffected controls (Fig. S6). There is no indication for partially compensating cryptic POLR3GL splice acceptor sites in the RNA-seq data. Overall, these results confirm that the splice acceptor variants disrupt POLR3GL RNA transcripts in the affected individuals.
Discussion
Here we describe the presence of biallelic POLR3GL splice site variants in three individuals with axial endosteal hyperostosis, oligodontia, short stature, and mild facial dysmorphisms. Biallelic variants in POLR3A, POLR3B, POLR3K and POLR1C, which encode other Pol III subunits, have previously been associated with a spectrum of phenotypes (Table 2, Table S1). Patients with a severe POLR3-related disorder show a progressive disease with leukodystrophy, motor regression, oligodontia, hypogonadotropic hypogonadism, myopia, and intellectual disability. In contrast, some individuals at the mild end of the spectrum present only with learning difficulties and a delay in motor development [21] or with isolated hypogonadotropic hypogonadism [17]. The oligodontia, short stature and delayed puberty in the individuals described here are present in more than half of the individuals with POLR3-related disorders (Table 2). Most, but not all [12, 17], described individuals with biallelic variants in Pol III genes have hypomyelination and cerebellar signs [21]. It is uncertain whether the individuals described here have developed white matter abnormalities during follow-up, because MRI scans of the brain have not been performed at later ages due to ethical and practical considerations. The three individuals do not suffer from a progressive neurological disorder with cerebellar, pyramidal or extrapyramidal signs. In addition, they do not have progressive myopia, which has been described for the majority of individuals with POLR3-related disorders. The endosteal hyperostosis in the individuals with POLR3GL variants is remarkable and has only been reported in two individuals with POLR3B variants [16, 19]. However, skeletal X-rays are not always performed and the presence of endosteal hyperostosis in other individuals with POLR3-related disorders might therefore be underestimated [16]. We therefore recommend to perform skeletal surveys in all individuals with POLR3-related disorders. Taken together, the extraneurologic phenotypic features of the three individuals with POLR3GL variants fit in the spectrum of POLR3-related disorders, but the absence of progressive cerebellar, pyramidal or extrapyramidal features is remarkable.
Table 2.
Summary of the main clinical features in individuals with POLR3-related disorders
| POLR3A+POLR3B [5, 10, 12, 17, 21] | POLR1C [18] | POLR3K [20] | POLR3GL | |
|---|---|---|---|---|
| Number of individuals | n = 147 | n = 8 | n = 2 | n = 3 |
| Intellectual disability | 13/42 | 6/8 | 2/2 | 0/3 |
| Motor delay | 58/116 | 7/8 | 2/2 | 3/3 |
| Cerebellar signs (ataxia, dysmetria) | 142/147 | 8/8 | 8/8 | 0/3 |
| Endosteal hyperostosis | U | U | U | 3/3 |
| Myopia | 87/121 | 3/8 | 1/1 | 0/3 |
| Oligo-/hypodontia | 86/131 | 3/8 | 1/2 | 3/3 |
| Hypogonadotropic hypogonadism/Delayed puberty | 53/69 | 0/2 | 1/2 | 2/3 |
| Short stature | 54/126 | U | 2/2 | 3/3 |
| Diffuse white matter abnormalities/Hypomyelination | 100/126 | 8/8 | 2/2 | U |
U unknown
Not all phenotypic characteristics have been explicitly specified or examined for each of the individuals described in literature and this may lead to an under- or over appreciation of some phenotypic characteristics in the spectrum of POLR3-related disorders. The clinical presentation of cohorts of individuals with POLR3A and POLR3B variants are frequently discussed together in literature and therefore the data for these two genes are merged in the table. More detailed information can be found in Table S1
It remains unclear how the newly identified POLR3GL variants lead to the observed phenotypes. The POLR3GL transcripts of the individuals described here lack exon 2, which contains the translation initiation site, or exon 5, which is part of the core domain essential for interacting with other Pol III subunits [34]. Nevertheless, it is likely that these variants do not lead to a full loss-of-function of POLR3GL due to the essential functions of the Pol III complex. In addition, biallelic nonsense or full loss of function variants of the other Pol III subunits have not been reported. The skip of exon 5 does not cause a frameshift in the POLR3GL RNA sequence and likely results in disrupted POLR3GL protein missing 19 of the 218 amino acids (p.(Asp109_Arg128delinsGly)). In contrast, loss of exon 2 causes the loss of the canonical translation initiation site and the next potential downstream translation initiation site is in exon 3 located at p.Met71. Therefore, the POLR3GL allele missing exon 2 is either not functional or misses the first 70 amino acids (p.(Met1_Ala70del)). In theory, the isoform POLR3G could compensate for the reduced POLR3GL function. It seems that POLR3G or POLR3GL largely bind to the same target genes, but that their activity is controlled by different mechanisms [26]. However, it has been shown that, at least in HeLa and Huh7 human cancer cell lines, POLR3G cannot fully compensate for the loss of POLR3GL [25]. Homozygous knockout variants of Polr3c and Polr3f, which form a subcomplex of Pol III together with POLR3GL, as well as homozygous knockout variants of Polr3a and Polr3b are lethal in mice [35]. POLR3GL knockout animal models have not been described to our knowledge. These observations suggest that the variants are hypomorphic and that the affected POLR3GL retains some form of essential functioning.
The phenotypic overlap between individuals carrying variants affecting the functions of Pol III subunits suggests that a common function of this complex is affected. Deficiency of tRNAs normally generated by Pol III could be an important cause of the phenotypes [2]. However, it is unknown why such a deficiency would lead to the observed cell-type specific phenotypes. Neuronal tissues seem to be highly sensitive to pathogenic variants in tRNA genes such as n-Tr20 in mice or genes coding for tRNA processing enzymes such as CLP1 [36]. Deficiencies of other Pol III transcribed RNAs such as U5 rRNA cannot be excluded, although ribosomopathies mainly lead to other phenotypes [37]. The teeth and bone phenotypes in the individuals with biallelic variants in POLR3GL and other Pol III-subunit genes suggest that osteoblasts or osteoclasts are affected [21], but it is unclear why these specific cell types would be sensitive to reduced Pol III activity.
In conclusion, biallelic splice site variants in POLR3GL can cause endosteal hyperostosis, oligodontia and short stature. These phenotypes fit within the spectrum of phenotypes observed in individuals carrying variants in other Pol III subunits. These findings show that it is important to include POLR3GL in genetic testing if a POLR3-related disorder is suspected, especially if endosteal hyperostosis and oligodontia are observed.
Supplementary information
Acknowledgements
We thank the individuals and their parents for their participation in this study. We thank Utrecht Sequencing Facility (USEQ) for providing RNA-sequencing service and data. USEQ is subsidized by the University Medical Center Utrecht, Hubrecht Institute and Utrecht University. We thank Jacques Giltay for his contribution to the inclusion of one of the families. We thank F.A.M. Hennekam for performing the genealogical studies in the families. This work was financially supported by a Vici grant (865.13.004) from the Netherlands Science Foundation (NWO) to Edwin Cuppen.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Paulien A. Terhal, Judith M. Vlaar, Sjors Middelkamp
Supplementary information
The online version of this article (10.1038/s41431-019-0427-0) contains supplementary material, which is available to authorized users.
References
- 1.White RJ. Transcription by RNA polymerase III: more complex than we thought. Nat Rev Genet. 2011;12:459–63. doi: 10.1038/nrg3001. [DOI] [PubMed] [Google Scholar]
- 2.Arimbasseri AG, Maraia RJ. RNA polymerase III advances: structural and tRNA functional views. Trends Biochem Sci. 2016;41:546–59. doi: 10.1016/j.tibs.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vannini A, Cramer P. Conservation between the RNA polymerase I, II, and III transcription initiation machineries. Mol Cell. 2012;45:439–46. doi: 10.1016/j.molcel.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 4.Bernard G, Chouery E, Putorti ML, Tétreault M, Takanohashi A, Carosso G, et al. Mutations of POLR3A encoding a catalytic subunit of RNA Polymerase Pol III cause a recessive hypomyelinating leukodystrophy p415. Am J Hum Genet. 2012;91:972. doi: 10.1016/j.ajhg.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saitsu H, Osaka H, Sasaki M, Takanashi JI, Hamada K, Yamashita A, et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet. 2011;89:644–51. doi: 10.1016/j.ajhg.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potic A, Brais B, Choquet K, Schiffmann R, Bernard G. 4H syndrome with late-onset growth hormone deficiency caused by POLR3A mutations. Arch Neurol. 2012;69:920–3. doi: 10.1001/archneurol.2011.1963. [DOI] [PubMed] [Google Scholar]
- 7.Terao Y, Saitsu H, Segawa M, Kondo Y, Sakamoto K, Matsumoto N, et al. Diffuse central hypomyelination presenting as 4H syndrome caused by compound heterozygous mutations in POLR3A encoding the catalytic subunit of polymerase III. J Neurol Sci. 2012;320:102–5. doi: 10.1016/j.jns.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Daoud H, Tétreault M, Gibson W, Guerrero K, Cohen A, Gburek-Augustat J, et al. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J Med Genet. 2013;50:194–7. doi: 10.1136/jmedgenet-2012-101357. [DOI] [PubMed] [Google Scholar]
- 9.Takanashi JI, Osaka H, Saitsu H, Sasaki M, Mori H, Shibayama H, et al. Different patterns of cerebellar abnormality and hypomyelination between POLR3A and POLR3B mutations. Brain Dev. 2014;36:259–63. doi: 10.1016/j.braindev.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 10.La Piana R, Cayami FK, Tran LT, Guerrero K, van Spaendonk R, Õunap K, et al. Diffuse hypomyelination is not obligate for POLR3-related disorders. Neurology. 2016;86:1622–6. doi: 10.1212/WNL.0000000000002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimojima K, Shimada S, Tamasaki A, Akaboshi S, Komoike Y, Saito A, et al. Novel compound heterozygous mutations of POLR3A revealed by whole-exome sequencing in a patient with hypomyelination. Brain Dev. 2014;36:315–21. doi: 10.1016/j.braindev.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 12.Minnerop M, Kurzwelly D, Wagner H, Soehn AS, Reichbauer J, Tao F, et al. Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain. 2017;140:1561–78. doi: 10.1093/brain/awx095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tétreault M, Choquet K, Orcesi S, Tonduti D, Balottin U, Teichmann M, et al. Recessive mutations in POLR3B, encoding the second largest subunit of pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:652–5. doi: 10.1016/j.ajhg.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutierrez M, Thiffault I, Guerrero K, Martos-Moreno GAacute, Tran LT, Benko W, et al. Large exonic deletions in POLR3B gene cause POLR3-related leukodystrophy. Orphanet J Rare Dis. 2015;10:69. doi: 10.1186/s13023-015-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cayami FK, La Piana R, van Spaendonk RML, Nickel M, Bley A, Guerrero K, et al. POLR3A and POLR3B mutations in unclassified hypomyelination. Neuropediatrics. 2015;46:221–8. doi: 10.1055/s-0035-1550148. [DOI] [PubMed] [Google Scholar]
- 16.Ghoumid J, Petit F, Boute-Benejean O, Frenois F, Cartigny M, Vanlerberghe C, et al. Cerebellar hypoplasia with endosteal sclerosis is a POLR3-related disorder. Eur J Hum Genet. 2017;25:1011–4. doi: 10.1038/ejhg.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards MR, Plummer L, Chan YM, Lippincott MF, Quinton R, Kumanov P, et al. Phenotypic spectrum of POLR3 Bmutations: isolated hypogonadotropic hypogonadism without neurological or dental anomalies. J Med Genet. 2016;54:19–25. doi: 10.1136/jmedgenet-2016-104064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiffault I, Wolf NI, Forget D, Guerrero K, Tran LT, Choquet K, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun. 2015;6. 10.1038/ncomms8623. [DOI] [PMC free article] [PubMed]
- 19.Ozgen HM, Overweg-Plandsoen WCG, Blees-Pelk J, Besselaar PP, Hennekam RCM. Cerebellar hypoplasia-endosteal sclerosis: a long term follow-up. Am J Med Genet A. 2005;134A:215–9. doi: 10.1002/ajmg.a.30589. [DOI] [PubMed] [Google Scholar]
- 20.Dorboz I, Dumay-Odelot H, Boussaid K, Bouyacoub Y, Barreau P, Samaan S, et al. Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurology. Genetics. 2018;4:e289. doi: 10.1212/NXG.0000000000000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolf NI, Vanderver A, van Spaendonk RML, Schiffmann R, Brais B, Bugiani M, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology. 2014;83:1898–905. doi: 10.1212/WNL.0000000000001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jay AM, Conway RL, Thiffault I, Saunders C, Farrow E, Adams J, et al. Neonatal progeriod syndrome associated with biallelic truncating variants in POLR3A. Am J Med Genet A. 2016;170:3343–6. doi: 10.1002/ajmg.a.37960. [DOI] [PubMed] [Google Scholar]
- 23.Paolacci S, Li Y, Agolini E, Bellacchio E, Arboleda-Bustos CE, Carrero D, et al. Specific combinations of biallelic POLR3A variants cause Wiedemann-Rautenstrauch syndrome. J Med Genet. 2018;55:837–46. doi: 10.1136/jmedgenet-2018-105528. [DOI] [PubMed] [Google Scholar]
- 24.Wambach JA, Wegner DJ, Patni N, Kircher M, Willing MC, Baldridge D, et al. Bi-allelic POLR3A loss-of-function variants cause autosomal-recessive Wiedemann-Rautenstrauch syndrome. Am J Hum Genet. 2018;103:968–75. doi: 10.1016/j.ajhg.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haurie V, Durrieu-Gaillard S, Dumay-Odelot H, Da Silva D, Rey C, Prochazkova M, et al. Two isoforms of human RNA polymerase III with specific functions in cell growth and transformation. Proc Natl Acad Sci USA. 2010;107:4176–81. doi: 10.1073/pnas.0914980107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renaud M, Praz V, Vieu E, Florens L, Washburn MP, l’Hôte P, et al. Gene duplication and neofunctionalization: POLR3G and POLR3GL. Genome Res. 2014;24:37–51. doi: 10.1101/gr.161570.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. Bioinformatics. STAR: ultrafast universal RNA-seq aligner; 2013. pp. 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–17. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reyes A, Anders S, Weatheritt RJ, Gibson TJ, Steinmetz LM, Huber W. Drift and conservation of differential exon usage across tissues in primate species. Proc Natl Acad Sci USA. 2013;110:15377–82. doi: 10.1073/pnas.1307202110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.La Piana R, Tonduti D, Gordish Dressman H, Schmidt JL, Murnick J, Brais B, et al. Brain magnetic resonance imaging (MRI) pattern recognition in Pol III-related leukodystrophies. J Child Neurol. 2014;29:214–20. doi: 10.1177/0883073813503902. [DOI] [PubMed] [Google Scholar]
- 34.Boissier F, Dumay-Odelot H, Teichmann M, Fribourg S. Structural analysis of human RPC32β-RPC62 complex. J Struct Biol. 2015;192:313–9. doi: 10.1016/j.jsb.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Koscielny G, Yaikhom G, Iyer V, Meehan TF, Morgan H, Atienza-Herrero J, et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2013;42(D1):D802–9. doi: 10.1093/nar/gkt977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirchner S, Ignatova Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet. 2015;16:98–112. doi: 10.1038/nrg3861. [DOI] [PubMed] [Google Scholar]
- 37.Yelick PC, Trainor PA. Ribosomopathies: global process, tissue specific defects. Rare Dis. 2015;3:e1025185. doi: 10.1080/21675511.2015.1025185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flores A, Briand JF, Gadal O, Andrau JC, Rubbi L, Van Mullem V, et al. A protein-protein interaction map of yeast RNA polymerase III. Proc Natl Acad Sci USA. 1999;96:7815–20. doi: 10.1073/pnas.96.14.7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.