

Abstract
Whether wind pollination in trees can offset the negative genetic consequences of anthropogenic forest fragmentation is not clearly established. To answer this question, we examined the demographic genetics of Quercus bambusifolia over a 70-year recovery period in highly fragmented forests in Hong Kong. We sampled 1138 individuals from 37 locations, and genetically analysed the chronosequence through the classification of tree diameters from the same populations using 13 microsatellite markers. Our study reveals that severe fragmentation caused a significant genetic bottleneck with very few remaining but genetically diverse individuals. We observed an enhanced genetic diversity during demographic recovery. We found full-sibs within populations and half-sibs across the study range. This reflects a limited seed dispersal and extensive pollen flow. Despite reduced genetic structure both among and within populations, overall a strong persisting genetic differentiation (F′ST = 0.240, P < 0.01) and significant small-scale spatial genetic structure (F(1) = 0.13, Sp = 0.024, P < 0.01) were observed. Existing bottlenecks and low effective population sizes within the temporal chronosequence suggest that the long-term effect of severe fragmentation cannot be entirely eliminated by wind pollination with demographic recovery in the absence of effective seed dispersal. Our results lead to recommendations for forest management.
Subject terms: Genetic variation, Ecological genetics, Plant ecology
Introduction
Tree species have higher levels of genetic diversity and gene flow compared to other life forms in the plant kingdom (Petit and Hampe 2006), which makes them especially resistant to genetic erosion resulting from fragmentation (Hamrick 2004). This is mainly attributed to life history traits such as their longevity, that allows trees to persist in remnant populations by coexistence of multiple generations, and their large body size with abundant pollen and seed production, which guarantees efficient reproduction and rejuvenation. Especially wind-pollinated trees have no need to rely on biotic dispersal vectors by having small, light-weight pollen (Mulcahy 1979), which potentially can travel great distances. It has been hypothesised that these traits offer the greatest advantage in offsetting negative genetic effects of forest fragmentation (Hamrick 2004; Shohami and Nathan 2014).
Nonetheless, there are contrasting reports regarding the genetic recovery of fragmented populations for wind-pollinated trees. On one hand, there is increasing evidence that wind pollination is an efficient mechanism that sustained long-distance gene flow between isolated populations from both primary and fragmented forests (Schuster and Mitton 2000; Wang et al. 2011). On the other hand, it was reported that small populations and disrupted gene flow led to a high risk of genetic drift in severely fragmented forests (Robledo‐Arnuncio et al. 2004; Sebbenn et al. 2011). The efficiency of wind pollination drops sharply for small populations (Jump and Peñuelas 2006; Knapp et al. 2001) or for large distances between fragmented populations (Austerlitz et al. 2004; Bacles and Ennos 2008). In addition, fragmentation affects the seed flow of wind-pollinated species (Parejo-Farnés et al. 2017). Localised seed shadow, i.e. clumped seed distribution close to the mother tree, caused by reduced numbers of dispersal agents after fragmentation can lead to elevated inbreeding and bottleneck effects (Sebbenn et al. 2011). These effects are particularly important for tree species with a prodigious fecundity, where a few maternal individuals are able to produce large numbers of seeds and will contribute the majority of offspring. Thus, the ability of wind pollination to counteract genetic erosion or inbreeding caused by fragmentation is largely influenced by seed dispersal, severe level of isolation, and reduced size of fragmented populations.
The cumulated population demographic changes over time can modulate the genetic effects of habitat fragmentation, as different tree generations do not respond to fragmentation in the same way. Old generations often precede habitat fragmentation, and thus decreased genetic variation is more easily detected in the progeny because of reduced population size, limited pollen dispersal, localised seed shadow or increased inbreeding after fragmentation (Vranckx et al. 2012). New recruits after fragmentation represent a smaller subsample of maternal sources, which can create genetic bottlenecks (Grivet et al. 2009).
The genetic loss in future generations could be avoided if reductions of population sizes do not cause a loss of genetic variation over time (Ezard and Travis 2006). The continuous decline of effective population size causes genetic drift (Nei et al. 1975). Thus, demographic recovery after short-time fragmentation events can contribute to overcoming genetic erosion. Once the driving forces of fragmentation are alleviated, fragmented populations may recover by demographic expansion through increased population size and reduced isolation, leading to increased genetic connectivity and diversity (Born et al. 2008; Oostermeijer et al. 2003).
According to traditional expectations, demographic expansion is accompanied by a range expansion as the result of migration or recolonisation (Jones and Hubbell 2006; Pannell and Dorken 2006). Most tree species are dispersed by animals, both in tropical and temperate forests (Howe and Smallwood 1982). Consequently, without seed-dispersing animals in the highly fragmented landscapes of these regions, demographic expansion is expected to be very local, consisting in an increase of population size without significant expansion in distribution range where new populations are established. Particularly in forests disturbed on a landscape scale, without old-growth remnants acting as refuge for wildlife, young cohorts can remain clustered around mother trees for centuries (Voigt et al. 2009).
In population genetics, the effect of demographic expansion based on range expansion has been studied in theory and simulation models (Excoffier et al. 2009; Pannell and Dorken 2006), mostly with animals and herbs (Helsen et al. 2015; Zenger et al. 2003), and in few tree species (but see Echt et al. 1998; Jones and Hubbell 2006). Moreover, only one simulation study explored the genetic consequences of simultaneous habitat fragmentation and range expansion (Mona et al. 2014). In contrast, the scenario of recovery with no range expansion has received no consideration by previous studies.
In the last few decades, Asia has been among the regions with the most extensive forest fragmentation, because of human-induced land use change (FAO 2015). It is still unanswered to which extent the genetic recovery of tree species is hampered by such drastic population declines, and how limited gene pools influence future population expansions and survival. An extreme case is Hong Kong, where deforestation and fragmentation started hundreds of years ago and had a low point of almost zero forest cover after the Second World War (WWII) as seen on historic photos (Abbas et al. 2016; Corlett 1999). Following WWII, increasing efforts have been invested in the conservation of natural forests leading to expansion of forest cover over the last 70 years. Forest cover increased mainly by natural regeneration from remnant trees in steep valleys, ravines, and small patches of village forests. Despite forest expansion, many animal species went extinct during the fragmentation phase, causing late-successional trees with limited dispersal to occur with an inconsistent and clustered distribution (Nichol et al. 2017). Wind pollination was reported to facilitate the recovery of gene pools after severe fragmentation (Hamrick 2004). Our study attempts to address the question: How does the genetic makeup of populations respond to the demographic recovery after severe fragmentation? Our hypotheses are that: (1) Fragmentation significantly reduces the population size of species, causing genetic drift of wind-pollinated tree species, and (2) limited seed dispersal reduces range expansions and counteracts the positive effects of wind pollination during the demographic recovery after fragmentation.
Quercus bambusifolia, a wind-pollinated tree species with large acorns, is commonly found in primary forests in South China (Xu et al. 2015). It has a restricted and scattered distribution in Hong Kong’s forests. Using this species as a model, we checked the demographic genetics by comparing the genetic diversity, genetic structure, and effective population size of different age classes in the populations after fragmentation. With the comparison of genetic properties of different age classes, we analysed the effects of fragmentation and demographic recovery on the genetic makeup of populations.
Materials and methods
Study species
Q. bambusifolia Hance also known in the literature as Quercus neglecta (Schott.) Koidz. or Cyclobalanopsis neglecta Schottky (Fagaceae) is an evergreen oak tree species native to South China and Vietnam. It can grow to more than 20 m in height and 80 cm in diameter at breast height (DBH). It is found mainly on ridge tops, upper or dry slopes in mature or primary forests (Kuznetsov and Guigue 2000; Xu et al. 2015). Although no specific study about the mating system of the species exists, we expect it to be a monoecious, wind-pollinated, and self-incompatible species, as most oaks (Ducousso et al. 1993). The acorns are the food source for scatter-hoarding rodents, like squirrels and forest-dwelling rats (Corlett 2017). In our study region, their habitat has been severely disturbed. Although there are few potential seed dispersal rodents, like introduced squirrel (Callosciurus erythraeus) and some hillside rats (Rattus andamanensis, Niviventer fulvescens), they are observed to directly eat the seeds without hoarding behaviour (Shek 2006). Some crows are known as scatter-hoarding birds for oaks. Only one scatter-hoarding crow (Garrulus glandarius) is resident to the north Asian tropics where Hong Kong is located, but it was not found to act as a seed dispersal agent in the region (Corlett 2017). Some omnivorous carnivores in Hong Kong, such as the civet (Paguma larvata), could be potential seed dispersal agents as they can ingest large seeds and disperse them in their droppings (Shek 2006). In general, we expect that Q. bambusifolia has limited secondary seed dispersal and that most acorns are dispersed by gravity alone.
Study area and populations
Hong Kong is located at the southern coast of China. The landscape of Hong Kong is fairly hilly to mountainous with steep slopes (Fig. 1). Lowland forests are dominated by tropical vegetation gradually shifting into subtropical vegetation with increasing altitude (Nichol et al. 2017). Most of the landscape is now covered in a rather species-poor secondary vegetation dominated by a mixture of woodland, scrubland, and grassland (Nichol et al. 2017).
Fig. 1.
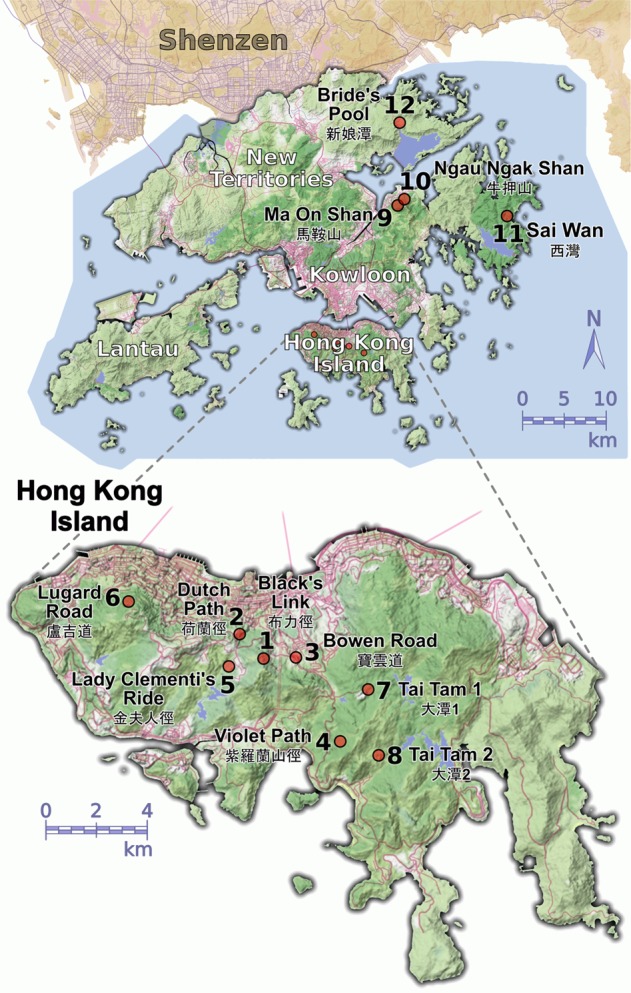
Location of the sampling sites in Hong Kong. Red dots indicate the 12 pooled populations of Quercus bambusifolia. The relief of the terrain is shown in different shades of green, major roads are in magenta, while reservoirs, lakes, and wetlands are in deep blue. The bottom panel presents an enlarged representation of Hong Kong Island, which contains eight populations. Map tiles were rendered by Stamen Design (http://maps.stamen.com, under CC BY 3.0.) using data from OpenStreetMap (http://openstreetmap.org, under ODbL)
Q. bambusifolia is one of the canopy species in Hong Kong with a clustered distribution pattern in forests. We sampled a total of 1138 individuals from 37 locations in six protected areas (country parks), three in the New Territories, and three on Hong Kong Island in 2014 and 2015. We grouped all the sampling locations into 12 populations based on the proximity of locations (Fig. 1). The habitat and environmental conditions within populations were uniform. The individuals in different sampling locations were not continuously distributed. The distance between the locations was usually >100 m, and we only included a few distances shorter than 100 m. We sampled randomly around 30 individuals in each location, except when fewer individuals were available, and for one population (Bowen Road) where we sampled along the entire distribution. We kept the distance between sampled individuals >4 m.
Since trees can live for centuries, it is difficult in practice to monitor population demographic changes over time. Alternatively, past demographic changes of populations can be assessed by a chronosequence of extant tree diameters although mortality caused by environmental filtering and inter-/intraspecific competition may inflict a bias in age class distributions (Lusk and Smith 1998). However, as the numbers of old Q. bambusifolia trees remaining after fragmentation in general have been low and sparsely distributed, it is very unlikely that inter/intraspecific competition and environmental filtering during the early stage of forest recovery caused a high rate of mortality of old and well-established individuals. Such factors mainly dictate the spatial distribution patterns of young trees by stem exclusion during secondary succession (Ashton et al. 2001), whereas old and much larger individuals remain largely unaffected. The long life expectancy of oaks of several hundreds of years, very hard wood, comparatively slow growth rate, the capability of coppicing and improved legal protection of old trees by Hong Kong Government further reduces the possibility of high mortality rates.
Nonetheless, some random mortality has to be expected over the last 70 years, even if the likelihood is not very high. Therefore, we sampled all individuals larger than the average diameter to include as many large individuals as possible. This allows tracking back the gene pool of founding generations with a greater accuracy, and also counteracts bias effects from the lower sampling rate of old individuals.
The DBH was measured for each sampled individual and their geographic coordinates were recorded (Table 1). Q. bambusifolia can grow <1 cm in diameter annually for young individuals depending on habitat conditions, with reduced growth rate <0.5 cm in diameter for mature individuals (personal observation in Kadoorie Farm and Botanic Garden). Thus, we assume that individuals with >40 cm of diameter already existed or coppiced during the last severe fragmentation during and after WWII around 70 years ago.
Table 1.
Population sampling information and genetic variation of Quercus bambusifolia
| Country park | Population | Location ID | Ind. no. | Ho | He | No. of alleles | Ar | Fi | Ar5% | Private alleles | DBH_max (cm) | DBH_ave (cm) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aberdeen | Black’s link | 1 | 18 | 0.56 | 0.68 | 6.54 | 5.94 | 0.064a | 24 | 2 | 41.70 | 16.32 |
| Black’s link | 2 | 30 | 0.59 | 0.60 | 7.08 | 5.30 | 0.007 | 47 | 2 | 30.88 | 17.35 | |
| Black’s link | 3 | 30 | 0.57 | 0.67 | 8.46 | 6.50 | 0.019a | 48 | 0 | 26.10 | 12.87 | |
| Black’s link | 4 | 31 | 0.56 | 0.63 | 7.92 | 5.83 | 0.015 | 57 | 2 | 29.28 | 14.43 | |
| Black’s link | 5 | 31 | 0.55 | 0.65 | 7.08 | 5.61 | 0.020a | 40 | 0 | 27.69 | 16.74 | |
| Dutch path | 6 | 31 | 0.56 | 0.62 | 6.23 | 5.21 | 0.054a | 30 | 2 | 41.70 | 22.08 | |
| Dutch path | 7 | 31 | 0.52 | 0.57 | 6.54 | 4.91 | 0.015 | 43 | 1 | 30.88 | 17.59 | |
| Bowen road | 8 | 73 | 0.62 | 0.69 | 10.31 | 6.77 | 0.013 | 75 | 3 | 33.10 | 16.60 | |
| Lady Clementi’s ride | 11 | 27 | 0.50 | 0.62 | 6.15 | 5.12 | 0.027a | 30 | 0 | 57.61 | 13.46 | |
| Lady Clementi’s ide | 12 | 32 | 0.58 | 0.65 | 6.92 | 5.31 | 0.013 | 43 | 0 | 34.06 | 11.96 | |
| Lady Clementi’s ride | 13 | 31 | 0.62 | 0.61 | 5.69 | 4.83 | 0.007 | 25 | 0 | 56.34 | 21.53 | |
| Pok Fu Lam | Lugard road | 14 | 18 | 0.56 | 0.64 | 4.69 | 4.49 | 0.012a | 6 | 0 | 46.15 | 21.33 |
| Lugard road | 15 | 30 | 0.58 | 0.60 | 7.31 | 5.66 | 0.008 | 38 | 1 | 37.24 | 16.28 | |
| Lugard road | 16 | 33 | 0.62 | 0.61 | 6.85 | 4.97 | 0.005 | 46 | 2 | 25.46 | 16.47 | |
| Lugard road | 17 | 30 | 0.64 | 0.66 | 6.85 | 5.46 | 0.013 | 35 | 0 | 28.97 | 17.61 | |
| Tai Tam | Violet path | 9 | 31 | 0.51 | 0.63 | 7.69 | 5.93 | 0.102 | 47 | 2 | 27.69 | 13.24 |
| Violet path | 10 | 31 | 0.57 | 0.62 | 5.23 | 4.43 | 0.014 | 23 | 1 | 40.11 | 16.13 | |
| Tai Tam1 | 18 | 31 | 0.69 | 0.67 | 7.15 | 5.72 | 0.014 | 42 | 1 | 36.29 | 16.25 | |
| Tai Tam2 | 19 | 31 | 0.58 | 0.62 | 7.15 | 5.68 | 0.010 | 43 | 0 | 25.46 | 13.37 | |
| Tai Tam2 | 20 | 31 | 0.58 | 0.60 | 5.31 | 4.39 | 0.016 | 26 | 0 | 40.74 | 15.86 | |
| Tai Tam2 | 21 | 31 | 0.58 | 0.57 | 5.46 | 4.58 | 0.007 | 25 | 0 | 53.48 | 15.37 | |
| Ma On Shan | Ma On Shan | 22 | 32 | 0.59 | 0.62 | 6.62 | 5.25 | 0.013 | 35 | 0 | 34.06 | 14.94 |
| Ma On Shan | 23 | 31 | 0.56 | 0.60 | 6.38 | 5.24 | 0.030a | 33 | 2 | 24.51 | 13.36 | |
| Ngau Ngak Shan | 25 | 31 | 0.55 | 0.61 | 5.15 | 4.39 | 0.021a | 22 | 1 | 26.10 | 19.08 | |
| Ngau Ngak Shan | 28 | 32 | 0.52 | 0.60 | 6.15 | 4.92 | 0.010a | 34 | 0 | 36.29 | 19.54 | |
| Ngau Ngak Shan | 29 | 32 | 0.57 | 0.67 | 7.38 | 5.95 | 0.014 | 40 | 1 | 35.33 | 17.09 | |
| Ngau Ngak Shan | 30 | 33 | 0.56 | 0.66 | 7.08 | 5.66 | 0.009a | 39 | 0 | 20.37 | 11.03 | |
| Ngau Ngak Shan | 31 | 32 | 0.51 | 0.60 | 6.31 | 5.04 | 0.011 | 31 | 0 | 21.33 | 14.18 | |
| Ngau Ngak Shan | 32 | 13 | 0.59 | 0.68 | 5.38 | 5.38 | 0.020 | 18 | 1 | 25.15 | 13.17 | |
| Sai Kung East | Sai Wan | 24 | 32 | 0.52 | 0.64 | 7.77 | 5.93 | 0.063 | 49 | 1 | 25.46 | 9.52 |
| Sai Wan | 26 | 32 | 0.56 | 0.67 | 8.15 | 6.40 | 0.028a | 54 | 0 | 27.06 | 10.97 | |
| Sai Wan | 27 | 30 | 0.55 | 0.66 | 7.00 | 5.71 | 0.037a | 30 | 0 | 29.28 | 9.01 | |
| Sai Wan | 33 | 31 | 0.58 | 0.65 | 7.38 | 5.93 | 0.016a | 43 | 0 | 16.87 | 8.43 | |
| Sai Wan | 35 | 16 | 0.66 | 0.70 | 6.23 | 5.91 | 0.010a | 20 | 1 | 21.96 | 9.89 | |
| Plover Cove | Bride’s pool | 34 | 33 | 0.49 | 0.61 | 6.92 | 5.39 | 0.026 | 40 | 0 | 47.75 | 16.33 |
| Bride’s pool | 36 | 33 | 0.57 | 0.63 | 6.69 | 5.49 | 0.031 | 33 | 1 | 30.88 | 15.10 | |
| Bride’s pool | 37 | 33 | 0.55 | 0.63 | 6.62 | 5.45 | 0.011 | 27 | 1 | 28.97 | 17.14 |
Ho observed heterozygosity, He expected heterozygosity, Ar allelic richness based on 13 individuals, Ar5% number of rare alleles, Fi mean inbreeding coefficient corrected for null alleles in INEst, DBH_max maximum DBH, DBH_ave average DBH
aSuggests the inbreeding model with lower DIC value outperforms the model without inbreeding, which indicates a presence of inbreeding
Nuclear microsatellite genotyping and genetic diversity
Genomic DNA of leaf samples was extracted using the DNeasy Plant mini-kit (Qiagen). In total, 35 microsatellite loci from three related species were tested and modified for Q. bambusifolia (Isagi and Suhandono 1997; Lee et al. 2010; Tong et al. 2012). We finally selected 13 highly polymorphic microsatellite loci for genotyping (Supplementary Table S1). Polymerase chain reactions (PCR) were performed using the Multiplex PCR Kit (Qiagen) in a reaction volume of 25 μl, including 12.5 μl Multiplex Mix, 10 μl water, 0.5 μl of 10 μM dye-labelled front primer, 1 μl of 10 μM rear primer, and 1 μl (>10 ng) DNA. The PCR conditions included 2 min at 95 °C, 40 cycles of 15 s at 95 °C, 15 s at annealing temperature (Supplementary Table S1), 30 s at 72 °C, 5 min at 72 °C, and cool-down at 16 °C. Fragment analysis was performed on 3730xl DNA Analyzer (Applied Biosystems) with GeneScan500 LIZ dye size standard (Applied Biosystems). Genotypes were analysed with the software GeneMarker v.2.4.0 (SoftGenetics, LLC, PA, USA). PCRs with primers were repeated until a high quality of microsatellite sequences was achieved.
We calculated the population genetic characters with Fstat v.2.9.3.2 (Goudet 2001). Genetic diversity within locations was characterised by observed heterozygosity (Ho), expected heterozygosity (He), number of alleles, allelic richness (Ar, i.e. estimated number of alleles based on the minimum sample size of 13 individuals), number of rare alleles (total abundance < 5%, Ar5%), and private alleles. Genetic differentiation among all locations was estimated as FST (Weir and Cockerham 1984), and F′ST, the standardised measure of genetic differentiation for highly polymorphic markers (Hedrick 2005). The presence of null alleles by locus in each location was checked with the Van Oosterhout null allele estimator using Micro-Checker 2.2.3 (Van Oosterhout et al. 2004). The inbreeding coefficients (Fi) corrected for null alleles were estimated with the programme INEst 2.2 (Chybicki and Burczyk 2009). We performed all analyses twice to evaluate the influence of null alleles, either including all loci or including only loci presenting no null alleles.
Genetic structure among and within population
The isolation-by-distance (IBD) patterns among locations were checked by correlating the pairwise log geographical and genetic distances (F′ST/(1 − F′ST)). The significance of the relationships was tested by a Mantel test with 1000 randomisations. Spatial genetic structure within populations was assessed by pairwise kinship coefficients (Fij) (Loiselle et al. 1995) within defined geographic distance classes with the programme SPAGeDi 1.2 (Hardy and Vekemans 2002). We set up the distance classes of 5, 10, 50, 100, 200, 500, 800, and 1000 m to ensure a sufficient number of individual pairs in each distance class. The significance of mean Fij per class and 95% confidence intervals (CI) were assessed with 1000 permutations of multi-locus genotypes.
We pooled all individuals and performed population structuring with clustering analysis to assign individual plants to distinct genetic clusters with the discriminant analysis of principal components (DAPC) approach (Jombart et al. 2010). The DAPC analysis provides a visual assignment of individuals based on allelic information, which can present the quantity and the identity of genetic diversity of populations. It was conducted using the R package ADEGENET (Jombart 2008) in R v.3.2.5 (R Development Core Team 2016) (Method S1 in Supporting Information).
Demographic genetics with tree diameters
Analysing genetic diversity and genetic structure of multiple age classes with different diameters from the same populations can express the genetic dynamics from different demographic stages at a temporal scale. The demographic genetics was checked with the genetic compositions in different diameter classes of the species. We assigned all the individuals into seven classes with diameter intervals of 0–10, 10–15, 15–20, 20–25, 25–30, 30–40, and >40 cm. Demographic genetic diversity was represented by the cluster assignment based on the above DAPC analysis of all samples, He and allele numbers along the diameter classes. To avoid any change in genetic diversity because of different habitat conditions, we presented the cluster assignment, He and allele numbers mentioned above by each population, which have consistent environment conditions.
The demographic genetic structure within populations was described through the spatial genetic structure at different tree diameters with spatial autocorrelation method in SPAGeDi (Hardy and Vekemans 2002). We assigned all the individuals into three diameter groups (0–10, 10–20 and >20 cm). We monitored the distance classes used in kinship analysis from 10 to 1000 m, and the mean Fij per class from different diameters was compared.
Stronger gene flow in young generations could reduce the genetic differentiation among locations, which would lead to different isolation-by-distance patterns in young generations compared to old generations. Demographic genetic structure among locations was analysed using FST among locations with individuals having different DBH. We assigned the individuals from each location into two groups with an equal individual number, one group of young generation with smaller diameters and another group of old generation with bigger diameters. Genetic distances F′ST/(1 − F′ST) between locations were calculated separately for the old and young generations. We identified the difference between the old and young generations with the pairwise Wilcoxon test. The isolation-by-distance patterns among locations, i.e. the slope of the relationship between geographic distance and genetic differentiation, were calculated and the Mantel tests were performed for both old and young generations. We tested for a difference in the slope of the pairwise genetic differentiation–geographic distance relationship between old and young individuals by using a linear regression approach.
Bottleneck test and effective population size
The extent of recent demographic history can be characterised by genetic bottleneck methods (Garza and Williamson 2001; Piry et al. 1999). To infer the effects of old bottlenecks of extant populations of Q. bambusifolia, the M-ratio test was chosen and conducted in six protected areas using the software M_P_VAL (Garza and Williamson 2001). The value of the test (M) is the mean ratio of the number of alleles to the range in allele size. A lower M-ratio indicates a more severe bottleneck (Garza and Williamson 2001). We ran the test with all pooled individuals from the same protected area using the two-phase model with 10,000 replications with the conservative assumption for the M-ratio test (pg = 0.1, Δg = 3.5, θ = 10) (Garza and Williamson 2001).
The population development can be present by the population stages, where founding individuals are now present with a large diameter, and population expansion proceeded with continuous add-in of smaller individuals. We divided population stages with individuals DBH > 30, >25, >20, >15, >10, and >0 cm, respectively. To check whether the add-in individuals mitigate the bottleneck of populations, we estimated effective population size (Ne) of the aforementioned population stages in each of the six protected areas using the linkage disequilibrium method by the programme NeEstimator 2.1 (Do et al. 2014). Alleles with frequencies < 0.05 were excluded to avoid the influence of rare alleles, and 95% CIs for each estimate were assessed using the Jackknife procedure (Waples and Do 2008).
Sibship structure
Analysing the genealogical relationships between individuals can further reveal the efficiency of gene flow. We expected the neighbouring sibling and parent–offspring relationships because of clustered seed shadows. The sibship structure was analysed using the full-pedigree likelihood method of the software COLONY 2.0 (Jones and Wang 2010). We regarded individuals with a DBH < 20 cm as offspring (886 individuals) and those with a DBH ≥ 20 cm as parents (252 individuals). We performed a total of five runs using 13 loci, with the locus-specific allelic dropout rate specified by the Micro-Checker programme and a value of 0.5% for other types of error. The best estimates of sib relationship with the maximum likelihood were obtained in COLONY. We regarded the relationships with associated probabilities ≥80% as conservative estimates. In addition, we grouped the best estimates of full-sibs, half-sibs, and parents based on the distance between the paired individuals. The mean distance between individuals was calculated for each group.
Results
Population demography
The diameter at breast height (DBH) ranged between 5 and 20 cm for the majority of individuals, whereas individual numbers in smaller or larger diameter classes were rather scarce (Supplementary Fig. S1a). The largest DBH was 57 cm, which is much smaller than the maximum DBH (80 cm) detected from undisturbed natural populations in other parts of the species distribution range (Kuznetsov and Guigue 2000; Xu et al. 2015). Only 11 out of 1138 individuals had a DBH above 40 cm. This distribution confirms that only a few remnant individuals survived extensive disturbances during and after WWII in Hong Kong. The remnant old individuals, having similar sizes as the 11 individuals of our samples, assumed as seed sources, led to a gradual increase in each population over time.
We compared our population structure with the population structure of Q. bambusifolia in a 50 ha forest dynamics plot in an undisturbed primary forest in a nature reserve in Hainan province of China (Supplementary Fig. S1) (Xu et al. 2015). Our populations have a high proportion of comparatively young individuals (10–20 cm of DBH) and reduced numbers of individuals with >20 cm DBH. In contrast, the undisturbed populations have mostly equalised cohorts of middle- and old-aged individuals (20–60 cm of DBH). In addition our populations lack mature generations with >60 cm DBH, which are an important component in undisturbed populations (Supplementary Fig. S1). On the basis of this comparison, we can conclude that our populations are expanding as a consequence of past fragmentation.
We found similar diameter distributions for most locations, supporting a synchronal starting point for the demographic expansion. The recruitment of the species around remnant patches of putative mother trees formed clustered cohorts of young generations and led to an increase in population densities. However, no newly established population or any indication of range expansions could be detected.
An exception is the population in Sai Wan, with smaller diameter classes from some locations, suggesting the possibility of a later demographic expansion (Table 1, Fig. 2). On the basis of our observation, however, the smaller diameter classes in Sai Wan could be largely attributed to the harsh habitat condition rather than a later expansion. This population is located on rocky and very windy slopes with thin soil layer, limiting the possible diameter growth.
Fig. 2.

Genetic composition of the pooled populations along the diameter classes. The clustering analysis was performed using the discriminant analysis of principal components (DAPC) approach that revealed an optimal number of clusters (K = 14). Each genetic cluster is indicated by different colour. Trees of increasing diameters are represented by diameter classes ranging from 1 to 7, with diameter at breast height (DBH) intervals from 0 to 10 cm, 10 to 15 cm, 15 to 20 cm, 20 to 25 cm, 25 to 30 cm, 30 to 40 cm, and >40 cm, respectively
Genetic diversity
Out of 1138 samples and 13 microsatellite markers, we detected 220 alleles in total with 5 to 32 alleles per markers (Supplementary Table S1). The populations had a high gene diversity (He) ranging from 0.57 to 0.70 with a mean of 0.63 (SD = 0.03) (Table 1). Allelic richness (Ar) ranged from 4.39 to 6.77 with a mean of 5.42 (SD = 0.58). The numbers of private alleles within populations were rather low. Six loci showed significant positive fixation indices (FIS) (Supplementary Table S1). Null alleles were detected at these loci, with significant null allele frequencies ranging from 0.06 to 0.37 within populations. After the correction for null alleles, the presence of inbreeding is observed in 14 out of 37 locations, but with rather small mean inbreeding coefficients (Fi) (≤0.064, Table 1). All analyses show similar pattern when including or excluding the loci with null alleles. Consequently, we use results including all loci for the whole study.
Genetic structure among and within populations
Overall, we found that all populations were significantly differentiated from each other (FST = 0.088, F′ST = 0.240, P < 0.01). Up to 8 km the isolation-by-distance pattern indicates a gene flow-drift equilibrium (Fig. 3). Genetic differentiation between locations was randomly changed at scales larger than 8 km, for example across the gap between Hong Kong Island and New Territories or between the country parks in the New Territories (inset in Fig. 3).
Fig. 3.
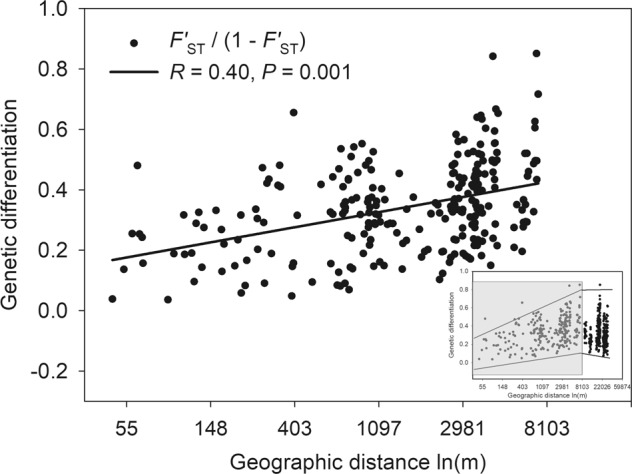
Correlation between genetic differentiation and log geographical distance for Q. bambusifolia. The correlation is shown for sampling locations within an 8 km radius (main figure) and across the whole study region (22 km, right bottom inset). In the inset, solid lines suggest the range of genetic differentiation and the geographical radius of 8 km is shown by the shaded area
On the basis of our DAPC of the entire dataset, the optimal cluster number was determined to be K = 14. Old generations had different clusters in different populations, representing the diverse gene pools of founding remnants. Individuals from the same population were assigned to multiple clusters with one or two dominant clusters (Fig. 2).
The spatial autocorrelation analysis in SPAGeDi indicated the presence of significant spatial genetic structure within sampled populations (Sp = 0.024, P < 0.01) with a high kinship coefficient from the first distance class (F(1) = 0.13) (Fig. 4a). The genetic similarity was much higher among neighbouring individuals, especially in the first four geographic distance classes (≤100 m), than among more distant individuals.
Fig. 4.
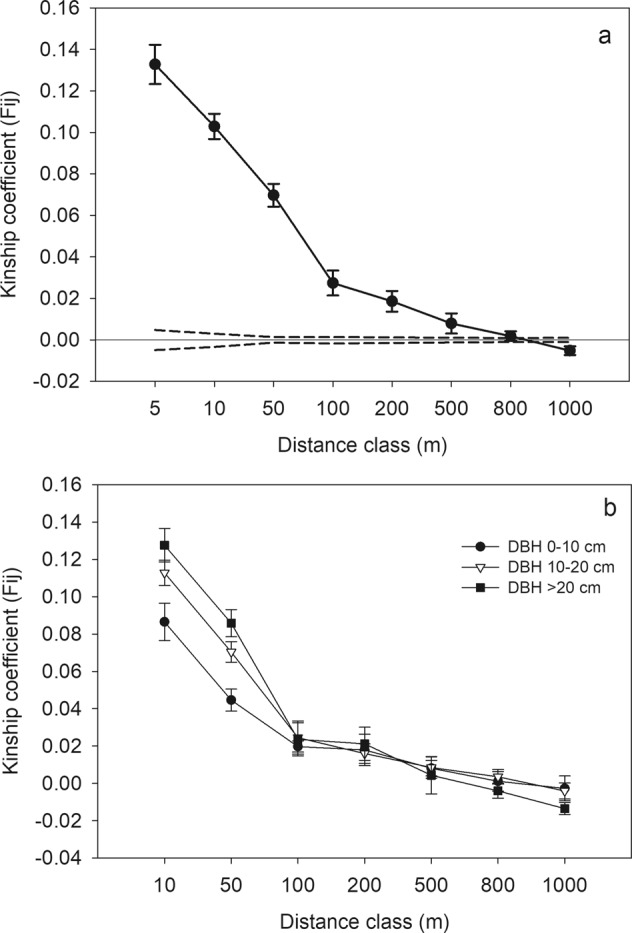
Correlogram of kinship coefficient (Fij) along geographic distances. The genetic spatial autocorrelation is depicted for Q. bambusifolia a with all individuals and b by grouping tree individuals according to their DBH. The dashed lines in a represent 95% Jackknife confidence intervals (CI)
Demographic genetics among diameter classes
Regarding genetic diversity, we found that the amount of diversity—defined by the cluster number and the allele number—increased in younger generations (smaller DBH) for all sampled populations (Figs. 2, 5a, details by locus and by population, see in Supplementary Fig. S2). High correlations were detected between genetic diversity and number of individuals from each diameter class of sampled populations (R = 0.97, P < 0.001 for allele number and number of individuals—log transformed; R = 0.94, P < 0.001 for cluster number and number of individuals). He increased at first, and became stable later on with reduced diameter classes from old to young generations (Fig. 5b). With all individuals pooled, new genetic clusters were formed during the development of young generations until the maximum value was achieved in the fifth diameter class (Fig. 5c). It was consistent with the pattern observed with allele numbers, which increased during demographic expansion and stabilised in the fourth diameter class (Fig. 5c).
Fig. 5.
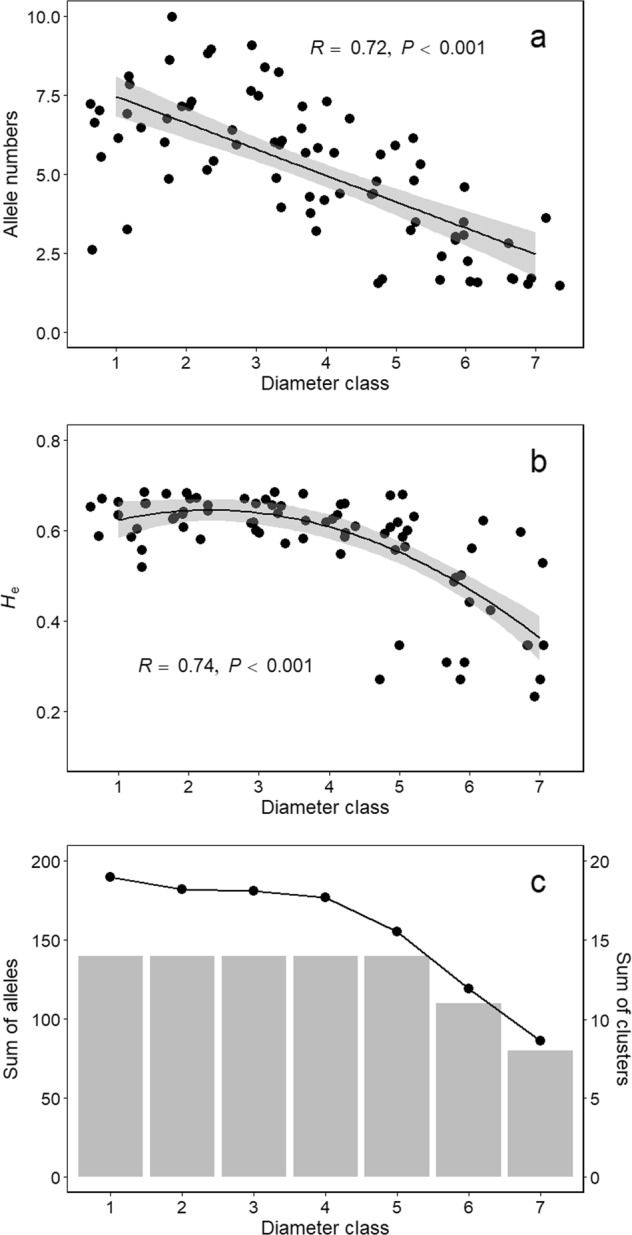
Genetic composition depicted for different diameter classes. We show the genetic composition a by average allele numbers per locus of each pooled population, b by expected heterozygosity (He) of each pooled population, and c by the sum of allele numbers (line) and clusters (bar chart). The shade in a and b represents the 95% confidence interval (CI)
Regarding the genetic structure of Q. bambusifolia, the analysis of kinship coefficients (Fij) reveals that small-scale spatial genetic structure reduced following demographic expansion. Kinship coefficients increase significantly for larger tree diameters in the first two distance classes (10 m, 50 m) when individuals are separated into different diameter groups (Fig. 4b). In most populations, we also detected stronger spatial genetic structure in old generations compared to young generations, with higher Fij in the first or second distance classes for old generations (8 out of 12 populations, Supplementary Fig. S3). Among populations, old generations presented higher genetic differentiation (F′ST/(1 − F′ST)) compared to young generations (pairwise Wilcoxon test P < 0.02). However, no significant interaction between generation and geographic distance was detected by the linear regression model, suggesting no significant difference in the isolation-by-distance pattern between old and young generations (Supplementary Fig. S4).
Bottleneck effect with demographic expansion
In Aberdeen, Ma On Shan, Tai Tam, and Sai Kung East, the increased effective population size (Ne) was detected with the increased young individuals, strongly indicating a reduced bottleneck during population expansion (Table 2). It is consistent with the results that no significant bottlenecks were detected in these protected areas. In Pok Fu Lam and Plover Cove, Ne did not change greatly with the increased young generations (Table 2), which would be explained by the significant bottlenecks (M = 0.724, P = 0.02 for Pok Fu Lam; M = 0.0.696, P = 0.004 for Plover Cove).
Table 2.
Results of estimated effective population size by linkage disequilibrium (Ne) method for different DBH ranges
| Country park | DBH range (cm) | Ind. no. | Ne | 95% CI |
|---|---|---|---|---|
| Aberdeen | >30 | 19 | 14.6 | 5.9–60.1 |
| >25 | 54 | 41.6 | 24.0–89.2 | |
| >20 | 99 | 56.1 | 36.7–93.9 | |
| >15 | 193 | 81.9 | 59.2–117.5 | |
| >10 | 298 | 106.4 | 65.8–184.4 | |
| All | 365 | 100.8 | 73.8–140.0 | |
| Pok Fu Lam | >25 | 13 | 15.0 | 5.4–180.9 |
| >20 | 36 | 22.6 | 14.6–38.3 | |
| >15 | 66 | 22.1 | 15.8–31.5 | |
| >10 | 97 | 18.7 | 14.3–24.2 | |
| All | 111 | 20.3 | 15.5–26.4 | |
| Tai Tam | >25 | 19 | 16.7 | 6.1–140.6 |
| >20 | 40 | 27.7 | 15.3–61.8 | |
| >15 | 80 | 27.8 | 17.0–47.9 | |
| >10 | 137 | 31.9 | 22.8–44.7 | |
| All | 186 | 40.5 | 30.2–54.4 | |
| Ma On Shan | >25 | 14 | 22.4 | 8.3–∞ |
| >20 | 49 | 28.7 | 15.6–63.3 | |
| >15 | 113 | 35.9 | 24.4–54.2 | |
| >10 | 195 | 46.6 | 36.7–59.5 | |
| All | 236 | 53.9 | 37.2–78.8 | |
| Sai Kung East | >15 | 17 | 16.6 | 4.4–∞ |
| >10 | 50 | 117.4 | 40.4–∞ | |
| All | 141 | 74.3 | 46.1–134.3 | |
| Plover Cove | >25 | 11 | 25.3 | 9.9–∞ |
| >20 | 22 | 37.7 | 19.6–139.6 | |
| >15 | 53 | 24.3 | 14.9–42.4 | |
| >10 | 81 | 34.2 | 23.6–51.9 | |
| All | 99 | 33.1 | 21.1–54.0 |
95% Jackknife confidence intervals (95% CI) for the effective population size (Ne) in NeEstimator 2.1 are presented. The lowest allele frequency employed for the linkage disequilibrium method was 0.05
Sibship reconstruction
In the full-pedigree likelihood analysis of COLONY, all the young individuals (886) were assigned as half-sibs, and 210 individuals were assigned as full-sibs (Table 3). Among them, 60% of half-sibs and 82% of full-sibs had probabilities ≥80% for the assignment, suggesting a high correlation among individuals (Table 3). Almost all the full-sib pairs were detected within locations, with the mean distance of 15 m between individuals (Table 3). For half-sib pairs, the highest portion was detected within locations (44% of the conservative estimates). Percentages obtained for the conservative pairs were as follows: 14% (within same populations), 12% (<8 km), and 30% (>8 km) (Table 3).
Table 3.
Sibship distribution with the maximum likelihood obtained in COLONY 2.0
| Groups | Full-sib pairs | Half-sib pairs | Parent pairs | |||
|---|---|---|---|---|---|---|
| Prob. ≥ 80% | Total | Prob. ≥ 80% | Total | Full-sib | Half-sib | |
| Pairs within locations | 136 | 183 (15 m) | 200 | 1583 (21 m) | 75 (19 m) | 252 (19 m) |
| Pairs within populations | 2 | 2 (125 m) | 65 | 561 (172 m) | 2 (90 m) | 26 (131 m) |
| Pairs < 8 km | 2 | 3 (2999 m) | 53 | 461 (2495 m) | 0 | 32 (2542 m) |
| Pairs > 8 km | 1 | 4 (18,246 m) | 133 | 1125 (20,633 m) | 2 (12,105 m) | 48 (22,540 m) |
| Total number of pairs | 141 | 192 | 451 | 3730 | 79 | 358 |
| Number of individuals | 173 | 210 | 532 | 886 | 29 | 59 |
We considered individuals with DBH < 20 cm as offspring and those with DBH ≥ 20 cm as candidate parents. We report the number of pairs (or individuals) according to the maximum likelihood obtained in COLONY 2.0, as well as assigned with ≥80% relationship probability for full-sibs and half-sibs. The average distances between individuals for the specific pair groups are denoted in parenthesis
For the parentage assignment, 29 estimated parents were assigned to full-sibs with 95% of parent–offspring pairs detected within locations, and 59 estimated parents were assigned to half-sibs with 70% of pairs from the same locations, 7% within the same populations, 9% within 8 km, and 13% outside 8 km (Table 3). The high number of sibships within locations and the half-sibs crossing the whole study region represents the contribution of gene flow, originating from seed dispersal and pollen flow, respectively.
Discussion
Q. bambusifolia has shown a strong regeneration capability in its recovery, with a rapid population expansion after the severe fragmentation that occurred in Hong Kong. The recovered populations increased in density rather than in distribution range, because of the absence of seed dispersers. In the following sections, we discuss and interpret the genetic pattern that has formed for this species during the recovery, both among and within populations.
Increased connectivity
Despite the highly localised seed pool within populations of Q. bambusifolia, our study finds an increase in genetic connectivity of fragments. This is concomitant with an increased genetic diversity, i.e. increased heterozygosity, genetic clusters and allele numbers, and a reduced genetic differentiation (FST) in younger generations. These results can be attributed to three main aspects of pollen flow of the species, i.e. the range, quantity, and quality of wind pollination.
First, wind pollination has an advantage over other strategies of pollen flow in increasing the genetic connectivity of remote populations. In our study, pollen flow is most probably the only medium contributing to >50% of the reconstructed half-sib pairs and 30% of the parent–offspring (for half-sib) pairs across long distances, from >100 m among sampling locations and up to 22 km across the whole study region. An earlier study has shown that extensive long-distance pollen flow of oak species can efficiently maintain the genetic diversity of populations on small, isolated islands within a few kilometres (Wang et al. 2011). This is comparable to the range of isolation-by-distance detected in our study (8 km). Other studies on oak species found that a concentrated pollen cloud could extend up to 30 km based on simulation modelling (Schueler and Schlünzen 2006), and efficient pollen immigration was detected across more than 80 km (Buschbom et al. 2011). Both cases cover the range of family relationships within our study area (22 km).
Second, the highly positive correlation between the size of the gene pool (cluster number and allele number) and the number of individuals in our study indicates that the recovery of genetic diversity largely benefits from the increasing population size. Population expansion leads to both a continuous accumulation of genes by the coexistence of multiple generations, and to a higher quantity of pollen donors, which increases the chances of successful pollination (Bacles and Jump 2011).
Third, population genetic recovery is influenced by the quality of pollen sources, i.e. the genetic diversity of incoming pollens from the founding generations. A high risk of genetic variation loss has been attributed to the homogeneous incoming genes rather than limited gene dispersal (Sork and Smouse 2006). In our study, old individuals in scattered populations have a diverse genetic composition, demonstrating that Q. bambusifolia was able to maintain a diverse gene pool even after severe fragmentation. Owing to the leptokurtic pattern of pollen dispersal (Austerlitz et al. 2004), small and isolated remnant populations could only receive the very far tails of realised pollen dispersal kernels. However, these far-dispersed pollens generally sample a very diverse gene pool (Bacles and Ennos 2008). As suggested in another study for young oak cohorts (Buschbom et al. 2011), genetically diverse founders served as pollen pool, facilitating the exchange of genes through long-distance wind pollination between fragmented populations. Q. bambusifolia populations thereby were able to gain genetic diversity over time. Besides, following the general pattern of oaks (Buschbom et al. 2011), not all Q. bambusifolia individuals flower synchronously and every year, which also increases the probability of remote outcrossing.
Limitations in pollen flow
We detected an existing genetic structure among our populations, with a differentiation (F′ST = 0.240) that is rather high compared to other oak studies employing microsatellites (mean F′ST = 0.145, range from 0.029 to 0.202, summarised in Shi et al. 2011). Moreover, we observed a lack of whole regional equilibrium of an isolation-by-distance pattern, which existed only within the range of 8 km with no reduced isolation in the young generations. Following Hutchison and Templeton (1999) (Case IV in Fig. 1), the pattern represents strong genetic drift at greater distances, whereas effective gene flow exists among populations of Q. bambusifolia at shorter distances (<8 km). The scattered distribution of Q. bambusifolia populations observed in Hong Kong’s forests suggests a strong historical fragmentation and the loss of many populations.
The success of pollination largely drops after a short distance because of the randomised wind dispersal (Bacles and Ennos 2008). Most long-distance pollen dispersal cannot actually contribute to the gene flow without continuous recipients. Topographic features such as mountain ranges and valleys, composing most land of Hong Kong, can also prevent gene flow (Ashley et al. 2015). Without continuous populations, it is uncertain whether a regional equilibrium between gene flow and genetic drift could eventually be achieved (Browne and Karubian 2018).
Reduced genetic clustering
In Q. bambusifolia, the spatial genetic structure was reduced within populations during recovery (Fig. 4b, Supplementary Fig. S3). In tree species, spatial clumping patterns can be either decayed (Jones and Hubbell 2006; Ueno et al. 2002) or strengthened in young generations (Sebbenn et al. 2011; Troupin et al. 2006). The reduced spatial genetic structure of Q. bambusifolia can be attributed to the overlapping seed shadows and pollen flow of young generations.
First, we observed wide variations in diameters among the old generations. High spatial genetic structure in old generations can be caused by extinction of the majority of genotypes during the disturbance (Troupin et al. 2006). The small-scale spatial genetic structure is increased when the individuals that remain after disturbance are originating from overlapping generations (Batista Leite et al. 2014; Latouche-Halle et al. 2003), especially when old generations suffer from a bottleneck (Jones and Hubbell 2006; Latouche-Halle et al. 2003) as is the case for Q. bambusifolia populations in Hong Kong.
Second, in contrast to overlapping generations, overlapping seed shadows were reported to reduce small-scale spatial genetic structure (Ueno et al. 2002; Yoon Chung et al. 2007). The key for the opposite effects could lie in the size and relatedness of the cohorts. The overlap in generations is not possible in young cohorts composed by mostly immature saplings (DBH < 15 cm). With an increasing single generation, increasing overlaps of seed shadow lead to reduced relatedness in young cohorts of Q. bambusifolia.
Finally, extensive pollen flow acts as a homogenising factor and blurs the clusters induced by limited seed dispersal (Berg and Hamrick 1995; Streiff et al. 1998), especially for the species with outcrossing breeding system (Ueno et al. 2000).
Long-term negative effects of overlapping seed shadow
We obtained values for kinship coefficient from the first distance class (F(1) = 0.13) and spatial genetic structure (Sp = 0.024) that demonstrate a strong small-scale spatial genetic structure for Q. bambusifolia. These values are higher compared to studies on other oaks (0.011 ≤ F(1) ≤ 0.031; 0.00298 ≤ Sp ≤ 0.0113) (Streiff et al. 1998; Wang et al. 2011), and compared to the average level for tree species (mean Sp = 0.0102) and wind-pollinated species (mean Sp = 0.0064) (Vekemans and Hardy 2004).
Most oak species are regarded as outcrossing species. In our study, inbreeding is detected for some populations, but with a very low inbreeding coefficient. The genetic structure within populations of Q. bambusifolia is most probably caused by clumping seed shadows rather than inbreeding. The high percentages of sibship within populations confirm localised seed shadows.
Although the efficient pollen flow and outcrossing of Q. bambusifolia are highly probable within the current populations, it needs to be emphasised that the 70-year recovery is still a short period compared to the long life span of a late-successional tree species, which would last centuries. Continuously clustered populations with overlapping seed shadows will lead to overlapping generations and a highly clumped distribution of genetically close pollen donors. Chances of future inbreeding increase as remote outcrossing cannot outcompete pollination by dense and local pollen clouds from siblings or related generations (Jump and Peñuelas 2006).
Thus, there is still a risk in current populations of Q. bambusifolia that restricted seed and pollen flow contribute to increased genetic structure because of overlapping generations of cumulated seeds, elevated inbreeding, and reduced genetic connectivity with ineffective long-distance pollen flow (Bacilieri et al. 1994).
Demographic recovery from bottlenecks
The pattern of increased effective population sizes found in our study populations illustrates the dynamics where demographic expansion with gene flow continuously erases the “genetic signature” left by historical fragmentation. Despite a partial recovery, existing bottlenecks and low effective population sizes in Pok Fu Lam and Plover Cove give a striking impression of the long-term effects of a severe historical fragmentation.
Estimating effective population sizes in multiple populations and monitoring the changes over time can provide a convincing proof of genetic dynamics (Wang et al. 2016). To our knowledge, the current study is the first to track the effective population size on a temporal chronosequence of populations. The consistent results of bottlenecks and effective population sizes from our studies confirm the sustained prediction of the genetic consequences of population demographic change, rather than interrupted results caused by occasional events or unrepresentative sampling (Garza and Williamson 2001; Wang et al. 2016).
We tracked the population demographic change with a chronosequence of individual groups with different diameters in a one-time census. A caveat of this method is that it may lead to a biased distribution of old individuals caused by high mortality rates after the fragmentation. The expected mortality depends on the degree of fragmentation and successional pathways. As outlined previously the expected mortality in our case was low but it is recommended before applying the same sampling method elsewhere to do a thorough context analysis of past disturbance factors and successional pathways to avoid any sampling bias.
Implications for sustainable forest management
Understanding the interactions between population demography and genetic makeup of tree species at the temporal scale helps us to better evaluate the consequences of past disturbances and to predict future impacts of forest fragmentation. It therefore facilitates decision-making for forest restoration and species management.
We demonstrated that wind pollination combined with demographic expansion, despite the absence of range expansion, accelerated the process of genetic recovery of Q. bambusifolia from diverse founders that suffered from severe fragmentation. However, our results show that a sustainable mitigation of strong genetic structure, within and among populations at both local and landscape scales, can hardly be achieved by pollen flow alone. To overcome these limitations, we recommend planting different genotypes within populations to reduce future inbreeding between genetically related neighbours. Among fragmented populations, a range expansion by planting new populations in suitable habitats would compensate for the missing dispersal agents and enhance the connectivity of populations at the landscape scale. As the seed-dispersing animals rely on trees for the provision of food and habitat, it is essential to first actively restore populations of key tree species demographically and genetically at the landscape scale. Second, extinct seed dispersers need to be reintroduced to guarantee seed dispersal services. This will thereby ensure the recovery and persistence of a healthy and resilient natural forest.
Data archiving
Sampling locations, DBH data, and microsatellite genotypes available from the Dryad Digital Repository 10.5061/dryad.6g97722.
Supplementary information
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xueqin Zeng, Phone: +852 24837130, Email: xqzeng@kfbg.org.
Gunter A. Fischer, Email: gfischer@kfbg.org
Supplementary information
The online version of this article (10.1038/s41437-019-0258-3) contains supplementary material, which is available to authorized users.
References
- Abbas S, Nichol JE, Fischer GA. A 70-year perspective on tropical forest regeneration. Sci Total Environ. 2016;544:544–552. doi: 10.1016/j.scitotenv.2015.11.171. [DOI] [PubMed] [Google Scholar]
- Ashley MV, Abraham ST, Backs JR, Koenig WD. Landscape genetics and population structure in Valley Oak (Quercus lobata Née) Am J Bot. 2015;102:2124–2131. doi: 10.3732/ajb.1500182. [DOI] [PubMed] [Google Scholar]
- Ashton MS, Gunatilleke CVS, Singhakumara BMP, Gunatilleke IAUN. Restoration pathways for rain forest in southwest Sri Lanka: a review of concepts and models. Ecol Manag. 2001;154:409–430. [Google Scholar]
- Austerlitz F, Dick CW, Dutech C, Klein EK, Oddou-Muratorio S, Smouse PE, et al. Using genetic markers to estimate the pollen dispersal curve. Mol Ecol. 2004;13:937–954. doi: 10.1111/j.1365-294x.2004.02100.x. [DOI] [PubMed] [Google Scholar]
- Bacilieri R, Labbe T, Kremer A. Intraspecific genetic structure in a mixed population of Quercus petraea (Matt.) Leibl and Q. robur L. Heredity. 1994;73:130. [Google Scholar]
- Bacles CFE, Ennos RA. Paternity analysis of pollen-mediated gene flow for Fraxinus excelsior L. in a chronically fragmented landscape. Heredity. 2008;101:368–380. doi: 10.1038/hdy.2008.66. [DOI] [PubMed] [Google Scholar]
- Bacles CFE, Jump AS. Taking a tree’s perspective on forest fragmentation genetics. Trends Plant Sci. 2011;16:13–18. doi: 10.1016/j.tplants.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Batista Leite F, Brandão R, Buzatti RO, de Lemos-Filho J, Lovato M. Fine-scale genetic structure of the threatened rosewood Dalbergia nigra from the Atlantic Forest: comparing saplings versus adults and small fragment versus continuous forest. Tree Genet Genomes. 2014;10:307–316. [Google Scholar]
- Berg EE, Hamrick JL. Fine-scale genetic structure of a Turkey oak forest. Evolution. 1995;49:110–120. doi: 10.1111/j.1558-5646.1995.tb05963.x. [DOI] [PubMed] [Google Scholar]
- Born C, Hardy OJ, Chevallier MH, Ossari S, Atteke C, Wickings J, et al. Small-scale spatial genetic structure in the Central African rainforest tree species Aucoumea klaineana: a stepwise approach to infer the impact of limited gene dispersal, population history and habitat fragmentation. Mol Ecol. 2008;17:2041–2050. doi: 10.1111/j.1365-294X.2007.03685.x. [DOI] [PubMed] [Google Scholar]
- Browne L, Karubian J. Habitat loss and fragmentation reduce effective gene flow by disrupting seed dispersal in a Neotropical palm. Mol Ecol. 2018;15:1–15. doi: 10.1111/mec.14765. [DOI] [PubMed] [Google Scholar]
- Buschbom J, Yanbaev Y, Degen B. Efficient long-distance gene flow into an isolated relict oak stand. J Hered. 2011;102:464–472. doi: 10.1093/jhered/esr023. [DOI] [PubMed] [Google Scholar]
- Chung MY, Nason JD, Chung MG. Effects of population succession on demographic and genetic processes: predictions and tests in the daylily Hemerocallis thunbergii (Liliaceae) Mol Ecol. 2007;16:2816–2829. doi: 10.1111/j.1365-294X.2007.03361.x. [DOI] [PubMed] [Google Scholar]
- Chybicki IJ, Burczyk J. Simultaneous estimation of null alleles and inbreeding coefficients. J Hered. 2009;100:106–113. doi: 10.1093/jhered/esn088. [DOI] [PubMed] [Google Scholar]
- Corlett RT. Environmental forestry in Hong Kong: 1871–1997. Ecol Manag. 1999;116:93–105. [Google Scholar]
- Corlett RT. Frugivory and seed dispersal by vertebrates in tropical and subtropical Asia: an update. Glob Ecol Conserv. 2017;11:1–22. [Google Scholar]
- Do C, Waples RS, Peel D, Macbeth G, Tillett BJ, Ovenden JR. NeEstimator v2: re‐implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour. 2014;14:209–214. doi: 10.1111/1755-0998.12157. [DOI] [PubMed] [Google Scholar]
- Ducousso A, Michaud H, Lumaret R. Reproduction and gene flow in the genus Quercus L. Ann Sci. 1993;50:91s–106s. [Google Scholar]
- Echt CS, DeVerno L, Anzidei M, Vendramin G. Chloroplast microsatellites reveal population genetic diversity in red pine, Pinus resinosa Ait. Mol Ecol. 1998;7:307–316. [Google Scholar]
- Excoffier L, Foll M, Petit RJ. Genetic consequences of range expansions. Annu Rev Ecol Evol Syst. 2009;40:481–501. [Google Scholar]
- Ezard THG, Travis JMJ. The impact of habitat loss and fragmentation on genetic drift and fixation time. Oikos. 2006;114:367–375. [Google Scholar]
- FAO . Global forest resurces assessment 2015. Rome: The Food and Agricultural Organization of the United Nations (FAO); 2015. [Google Scholar]
- Garza JC, Williamson EG. Detection of reduction in population size using data from microsatellite loci. Mol Ecol. 2001;10:305–318. doi: 10.1046/j.1365-294x.2001.01190.x. [DOI] [PubMed] [Google Scholar]
- Goudet J (2001) FSTAT, A program to estimate and test gene diversities and fixation indices (version 2.9.3). http://www2.unil.ch/popgen/softwares/fstat.htm
- Grivet D, Robledo-Arnuncio JJ, Smouse PE, Sork VL. Relative contribution of contemporary pollen and seed dispersal to the effective parental size of seedling population of California valley oak (Quercus lobata, Nee) Mol Ecol. 2009;18:3967–3979. doi: 10.1111/j.1365-294X.2009.04326.x. [DOI] [PubMed] [Google Scholar]
- Hamrick JL. Response of forest trees to global environmental changes. Ecol Manag. 2004;197:323–335. [Google Scholar]
- Hardy OJ, Vekemans X. SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes. 2002;2:618–620. [Google Scholar]
- Hedrick PW. A standardized genetic differentiation measure. Evolution. 2005;59:1633–1638. [PubMed] [Google Scholar]
- Helsen K, Jacquemyn H, Honnay O. Hidden founder effects: small-scale spatial genetic structure in recently established populations of the grassland specialist plant Anthyllis vulneraria. Mol Ecol. 2015;24:2715–2728. doi: 10.1111/mec.13207. [DOI] [PubMed] [Google Scholar]
- Howe HF, Smallwood J. Ecology of seed dispersal. Annu Rev Ecol Syst. 1982;13:201–228. [Google Scholar]
- Hutchison DW, Templeton AR. Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution. 1999;53:1898–1914. doi: 10.1111/j.1558-5646.1999.tb04571.x. [DOI] [PubMed] [Google Scholar]
- Isagi Y, Suhandono S. PCR primers amplifying microsatellite loci of Quercus myrsinifolia Blume and their conservation between oak species. Mol Ecol. 1997;6:897–899. doi: 10.1111/j.1365-294x.1997.tb00147.x. [DOI] [PubMed] [Google Scholar]
- Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones FA, Hubbell SP. Demographic spatial genetic structure of the Neotropical tree, Jacaranda copaia. Mol Ecol. 2006;15:3205–3217. doi: 10.1111/j.1365-294X.2006.03023.x. [DOI] [PubMed] [Google Scholar]
- Jones OR, Wang J. COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour. 2010;10:551–555. doi: 10.1111/j.1755-0998.2009.02787.x. [DOI] [PubMed] [Google Scholar]
- Jump AS, Peñuelas J. Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc Natl Acad Sci USA. 2006;103:8096–8100. doi: 10.1073/pnas.0510127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp EE, Goedde MA, Rice KJ. Pollen-limited reproduction in blue oak: implications for wind pollination in fragmented populations. Oecologia. 2001;128:48–55. doi: 10.1007/s004420000623. [DOI] [PubMed] [Google Scholar]
- Kuznetsov A, Guigue AM. The forests of Vu Quang Nature Reserve. Ha Tinh Province, Vietnam: Vu Quang Nature Reserve Conservation Project; 2000. [Google Scholar]
- Latouche-Halle C, Ramboer A, Bandou E, Caron H, Kremer A. Nuclear and chloroplast genetic structure indicate fine-scale spatial dynamics in a neotropical tree population. Heredity. 2003;91:181–190. doi: 10.1038/sj.hdy.6800296. [DOI] [PubMed] [Google Scholar]
- Lee J-H, Park M-H, Min G-S, Choi B-H. Isolation and Characterization of 13 Microsatellite Loci from Korean Quercus acuta (Fagaceae) J Plant Biol. 2010;53:201–204. [Google Scholar]
- Loiselle BA, Sork VL, Nason J, Graham C. Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae) Am J Bot. 1995;82:1420–1425. [Google Scholar]
- Lusk CH, Smith B. Life history differences and tree species coexistence in an old-growth New Zealand rain forest. Ecology. 1998;79:795–806. [Google Scholar]
- Mona S, Ray N, Arenas M, Excoffier L. Genetic consequences of habitat fragmentation during a range expansion. Heredity. 2014;112:291. doi: 10.1038/hdy.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy DL. The rise of the angiosperms: a genecological factor. Science. 1979;206:20–23. doi: 10.1126/science.206.4414.20. [DOI] [PubMed] [Google Scholar]
- Nei M, Maruyama T, Chakraborty R. The bottleneck effect and genetic variability in populations. Evolution. 1975;29:1–10. doi: 10.1111/j.1558-5646.1975.tb00807.x. [DOI] [PubMed] [Google Scholar]
- Nichol JE, Abbas S, Fischer GA. Spatial patterns of degraded tropical forest and biodiversity restoration over 70-years of succession. Glob Ecol Conserv. 2017;11:134–145. [Google Scholar]
- Oostermeijer JGB, Luijten SH, den Nijs JCM. Integrating demographic and genetic approaches in plant conservation. Biol Conserv. 2003;113:389–398. [Google Scholar]
- Pannell JR, Dorken ME. Colonisation as a common denominator in plant metapopulations and range expansions: effects on genetic diversity and sexual systems. Landsc Ecol. 2006;21:837–848. [Google Scholar]
- Parejo-Farnés C, Robledo-Arnuncio JJ, Albaladejo RG, Rubio-Pérez E, Aparicio A. Effects of habitat fragmentation on parental correlations in the seed rain of a bird-dispersed species. Tree Genet Genomes. 2017;13:17. [Google Scholar]
- Petit RJ, Hampe A. Some evolutionary consequences of being a tree. Annu Rev Ecol Evol Syst. 2006;37:187–214. [Google Scholar]
- Piry S, Luikart G, Cornuet J-M. BOTTLENECK: a program for detecting recent effective population size reductions from allele data frequencies. J Hered. 1999;90:502–503. [Google Scholar]
- R Development Core Team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2016. [Google Scholar]
- Robledo‐Arnuncio JJ, Alia R, Gil L. Increased selfing and correlated paternity in a small population of a predominantly outcrossing conifer, Pinus sylvestris. Mol Ecol. 2004;13:2567–2577. doi: 10.1111/j.1365-294X.2004.02251.x. [DOI] [PubMed] [Google Scholar]
- Schueler S, Schlünzen KH. Modeling of oak pollen dispersal on the landscape level with a mesoscale atmospheric model. Environ Model Assess. 2006;11:179. [Google Scholar]
- Schuster WSF, Mitton JB. Paternity and gene dispersal in limber pine (Pinus flexilis James) Heredity. 2000;84:348–361. doi: 10.1046/j.1365-2540.2000.00684.x. [DOI] [PubMed] [Google Scholar]
- Sebbenn AM, Carvalho ACM, Freitas MLM, Moraes SMB, Gaino A, da Silva JM, et al. Low levels of realized seed and pollen gene flow and strong spatial genetic structure in a small, isolated and fragmented population of the tropical tree Copaifera langsdorffii Desf. Heredity. 2011;106:134–145. doi: 10.1038/hdy.2010.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shek CT. A Field Guide to the Terrestrial Mammals of Hong Kong. Hong Kong: Agriculture, Fisheries and Conservation Department, Friends of the Country Parks and Cosmos Books Ltd.; 2006. [Google Scholar]
- Shi M-M, Michalski SG, Chen X-Y, Durka W. Isolation by elevation: genetic structure at neutral and putatively non-neutral loci in a dominant tree of subtropical forests, Castanopsis eyrei. PLoS ONE. 2011;6:e21302. doi: 10.1371/journal.pone.0021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohami D, Nathan R. Fire‐induced population reduction and landscape opening increases gene flow via pollen dispersal in Pinus halepensis. Mol Ecol. 2014;23:70–81. doi: 10.1111/mec.12506. [DOI] [PubMed] [Google Scholar]
- Sork VL, Smouse PE. Genetic analysis of landscape connectivity in tree populations. Landsc Ecol. 2006;21:821–836. [Google Scholar]
- Streiff R, Labbe T, Bacilieri R, Steinkellner H, Kremer A. Within-population genetic structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol Ecol. 1998;7:317–328. [Google Scholar]
- Tong X, Xu N-N, Li L, Chen X-Y. Development and characterization of polymorphic microsatellite markers in Cyclobalanopsis glauca (Fagaceae) Am J Bot. 2012;99:e120–e122. doi: 10.3732/ajb.1100448. [DOI] [PubMed] [Google Scholar]
- Troupin D, Nathan R, Vendramin GG. Analysis of spatial genetic structure in an expanding Pinus halepensis population reveals development of fine-scale genetic clustering over time. Mol Ecol. 2006;15:3617–3630. doi: 10.1111/j.1365-294X.2006.03047.x. [DOI] [PubMed] [Google Scholar]
- Ueno S, Tomaru N, Yoshimaru H, Manabe T, Yamamoto S. Genetic structure of Camellia japonica L. in an old‐growth evergreen forest, Tsushima, Japan. Mol Ecol. 2000;9:647–656. doi: 10.1046/j.1365-294x.2000.00891.x. [DOI] [PubMed] [Google Scholar]
- Ueno S, Tomaru N, Yoshimaru H, Manabe T, Yamamoto S. Size-class differences in genetic structure and individual distribution of Camellia japonica L. in a Japanese old-growth evergreen forest. Heredity. 2002;89:120–126. doi: 10.1038/sj.hdy.6800111. [DOI] [PubMed] [Google Scholar]
- Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P. MICRO‐CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Resour. 2004;4:535–538. [Google Scholar]
- Vekemans X, Hardy OJ. New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol. 2004;13:921–935. doi: 10.1046/j.1365-294x.2004.02076.x. [DOI] [PubMed] [Google Scholar]
- Voigt FA, Arafeh R, Farwig N, Griebeler EM, Bohning-Gaese K. Linking seed dispersal and genetic structure of trees: a biogeographical approach. J Biogeogr. 2009;36:242–254. [Google Scholar]
- Vranckx G, Jacquemyn H, Muys B, Honnay O. Meta‐analysis of susceptibility of woody plants to loss of genetic diversity through habitat fragmentation. Conserv Biol. 2012;26:228–237. doi: 10.1111/j.1523-1739.2011.01778.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Santiago E, Caballero A. Prediction and estimation of effective population size. Heredity. 2016;117:193. doi: 10.1038/hdy.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Compton SG, Chen X-Y. Fragmentation can increase spatial genetic structure without decreasing pollen‐mediated gene flow in a wind‐pollinated tree. Mol Ecol. 2011;20:4421–4432. doi: 10.1111/j.1365-294X.2011.05293.x. [DOI] [PubMed] [Google Scholar]
- Waples RS, Do C. LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour. 2008;8:753–756. doi: 10.1111/j.1755-0998.2007.02061.x. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Xu H, Li Y-D, Luo T-S. Jianfengling tropical mountain rain forest dynamic plot: community characteristics, tree species and their distribution patterns. Beijing: China Forestry Publishing House; 2015. [Google Scholar]
- Zenger KR, Richardson BJ, Vachot-Griffin AM. A rapid population expansion retains genetic diversity within European rabbits in Australia. Mol Ecol. 2003;12:789–794. doi: 10.1046/j.1365-294x.2003.01759.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sampling locations, DBH data, and microsatellite genotypes available from the Dryad Digital Repository 10.5061/dryad.6g97722.