

Summary
Increased sensitivity of cancer cells to viruses is a prerequisite for the success of oncolytic virotherapy. One of the major causes of such a phenotype is the disruption of innate antiviral defenses associated with dysfunction of type 1 interferons (IFNs) that permits unlimited replication of viruses in cancer cells. Defects in IFN pathways help cancer progression by providing additional advantages to tumor cells. However, while these defects promote the survival and accelerated proliferation of malignant cells, they facilitate viral replication and thus enhance the efficiency of viral oncolysis. This review describes a broad spectrum of defects in genes that participate in IFN induction and IFN response pathways. Expression levels and/or functional activities of these genes are frequently low or absent in cancer cells, making them sensitive to virus infection. Therefore, certain specific defects in IFN signaling cascades might serve as potential biomarkers to help in identifying individual cancer patients who are likely to benefit from oncolytic virotherapy.
Keywords: biomarkers, defects in IFN pathways, defects in IFN induction, defects in IFN response, IFN defects in cancer cell, malfunction of IFN signaling cascade, oncolytic viruses, oncolytic virotherapy, viral oncolysis
Abbreviations
- cGAS
cyclic GMP‐AMP synthase
- ERK
extracellular signal‐regulated kinase (also known as mitogen‐activated protein kinase 1 [MAPK1] or p42MAPK)
- GBP‐1
guanylate binding protein 1
- HSV
herpes simplex virus
- IRF
interferon regulatory factor
- ISG
interferon stimulated gene
- JAK
Janus kinase
- LGP2
laboratory of genetics and physiology 2 (also known as RIG‐I‐like receptor 3 [RLR‐3] or RIG‐I‐like receptor LGP2)
- MDA5
melanoma differentiation‐associated protein 5
- MEK
mitogen‐activated protein kinase (also known as MAP kinase)
- MV
measles virus
- Mx
myxovirus resistance proteins
- NDV
Newcastle disease virus
- OAS
oligoadenylate synthetase
- PAMP
pathogen‐associated molecular pattern
- PKR
protein kinase R
- PRR
pattern recognition receptor
- PTEN
phosphatase and tensin homolog tumor suppressor gene
- RAF
serine/threonine‐specific protein kinase
- RIG‐I
retinoic acid‐inducible gene‐1
- RLR
RIG‐I‐like receptor
- RNase L
ribonuclease L
- RSV
respiratory syncytial virus
- STAT
signal transducer and activator of transcription protein
- STING
stimulator of IFN genes (also known as transmembrane protein [TMEM173])
- TLR
toll‐like receptor
- TYK
tyrosine kinase
- VSV
vesicular stomatitis virus
1. INTRODUCTION
Oncolytic viruses represent a promising new addition to existing approaches to cancer therapy. However, despite impressive examples of occasional long‐term remissions or even cures, clinical trials with oncolytic virotherapy still demonstrate rather modest results: A significant percentage of patients may not show any response at all. The difference in response to viruses highlights the tremendous variability of cancer cells in different patients. Analysis of the underlying roots of the difference forms an active area of research that can potentially lead to identification of reliable biomarkers, which would identify patients, who are most likely to respond to virotherapy.
Normal cells have a variety of mechanisms that protect them from pathogens. Key players in cellular antiviral defenses are type 1 interferons (IFNs). In response to initial virus intrusion, IFNs signal surrounding cells and stimulate them to enter an antiviral state that includes restricted proliferation, downregulated metabolism, and other specific changes that restrict viral replication.1, 2 Interferons also play an important role in immune surveillance, which reduces the probability of malignant transformation.3 A malignant cell with dysfunctional IFN induction or response has numerous survival and growth advantages. Thus, accumulation of genetic defects in IFN signaling cascades and cancerogenesis go hand in hand.4
Half of the silencing epigenetic changes associated with immortalization of cells belong to genes involved in IFN pathways.5 The deletion of such genes is often observed in gliomas,6 leukemias,7 and bladder cancer.8 Low expression of IFN receptors is characteristic of hepatocellular,9, 10 pancreatic,11 gastric,12 colon rectal,13 and many other cancers.4 Moreover, immune cells of cancer patients often have impaired IFN signaling.14
Thus, along with the importance of IFN pathways for cellular defense against viruses, defects in these pathways promote viral oncolysis. The purpose of this review is to describe the broad range of these defects along with their role in promotion of malignant cell sensitivity to oncolytic viruses.
2. DEFECTIVE INDUCTION OF TYPE 1 IFNs IN CANCER CELLS
In normal cells, virus infection triggers an antiviral mechanism which consists of 2 phases. First, as a result of recognition of viral components, expression and secretion of type 1 IFNs are initiated. Interferons then interact with specific receptors and stimulate a second phase, the IFN response. During the second phase, the secretion of IFNs is additionally stimulated by a positive regulatory feedback loop. The induction of IFN production is triggered by a set of molecular events that includes the interaction of viral components with specific cellular receptors and their activation. The activated receptors form a complex with adaptor proteins, and this complex promotes phosphorylation of transcription factors. Finally, the phosphorylated transcription factors move to the nucleus and initiate the transcription of IFN mRNAs, leading to production and secretion of IFNs. In cancer cells, this chain of molecular events might be broken at different levels and by a variety of mechanisms.
The genes of the IFN induction pathway whose defects have been shown to be associated with sensitivity to viruses are described below and listed in Table 1.
Table 1.
Biomarkers related to the IFN induction pathway
| Category | Gene/Protein Name | Sensor of Viral | Type of Defect | Sensitivity to | Ref | |
|---|---|---|---|---|---|---|
| Membrane‐associated pattern recognition receptors (PRRs) | Toll‐like receptors (TLRs) | TLR2 | DNA | Absent | Adenovirus | 15 |
| TLR9 | Dislocated | |||||
| TLR3 | RNA | Low expression | Sendai virus | 16 | ||
| TLR7 | ||||||
| Cytosolic pattern recognition receptors (PRRs) | Cyclic GMP‐AMP synthase | cGAS | DNA | Epigenetic silencing | HSV1 vaccinia virus | 17 |
| RIG‐I like receptors (RLRs) | RIG‐I | RNA | Downstream signaling is blocked | Reovirus | 18 | |
| Low expression | Measles virus | 19 | ||||
| MDA5 | Experimental knockdown or low natural expression | Sindbis virus | 20 | |||
| Protein kinase | PKR | Deletion | NDV | 21 | ||
| Experimental inhibition | VSV | 22 | ||||
| RNase | RNase L | Experimental inhibition | VSV | 22 | ||
| OAS gene family | OAS2 | Low expression | VSV | 23 | ||
| Adaptor protein | STING | Loss of function | HSV1 | 24 | ||
| Epigenetic silencing | HSV1, vaccinia virus | 17 | ||||
| Interferon regulating factors | IRF1 | MEK activation suppresses IRF1 binding to RIG‐I promoter | VSV | 25, 26, 27 | ||
| Reovirus | 18 | |||||
| IRF3 | Aberrant splicing | VSV | 28 | |||
| IRF3/7 | Low basal expression | NDV | 29 | |||
| IRF5/7 | Hyper‐methylated promoters | VSV | 30 | |||
| IFN‐regulatory factor 9 (P48) | Expression is low or absent | VSV | 31 | |||
| Tumor suppressor, promoter of IRF3 nuclear import | PTEN | Deletion | VSV | 32 | ||
3. PATTERN RECOGNITION RECEPTORS FOR VIRAL NUCLEIC ACIDS
Pattern recognition receptors (PRRs; also called innate immune receptors) are proteins that initiate induction pathways for IFNs and proinflammatory cytokines. Pattern recognition receptors are activated by pathogen‐associated molecular patterns (PAMPs), molecular features of pathogens that are absent in host organisms. Viral nucleic acids are among the PAMPs, and their sensing by PRRs forms the first line of host defense against viral infection. Viruses are highly diverse pathogens, and correspondingly, cells evolved multiple types of PRRs with different functions and cellular locations. Some of these PRRs recognize only viral DNA and some only viral RNA. Some PRRs recognize viral nucleic acids at the cell surface, even before the virus enters the cell, while others recognize viral components inside the cell.33
3.1. Membrane‐associated PRRs
Toll‐like receptors (TLRs) are membrane‐bound PRRs that initiate pathways for induction of IFNs and proinflammatory cytokines (Figure 1). Toll‐like receptors become activated after interaction with PAMPs, which might be viral nucleic acids or other pathogen‐derived molecules.34 Particular types of PAMPs activate particular types of TLRs that in turn recruit TLR type‐specific adaptors for further signal transduction.34 Thus, some TLRs (TLR2 and TLR9) recognize viral DNA, some (TLR7 and TLR8) recognize single‐stranded viral RNA, and some (TLR3) recognize double‐stranded viral RNA.34 Toll‐like receptor 2/7/8 and TLR9 use the MYD88 protein as adaptor, while TLR3 instead uses TRIF protein.34 Toll‐like receptors have various cellular locations: TLR2 is present at the cell surface, while TLR3, TLR7/8, and TLR9 are associated with endosomal membranes.34, 35
Figure 1.
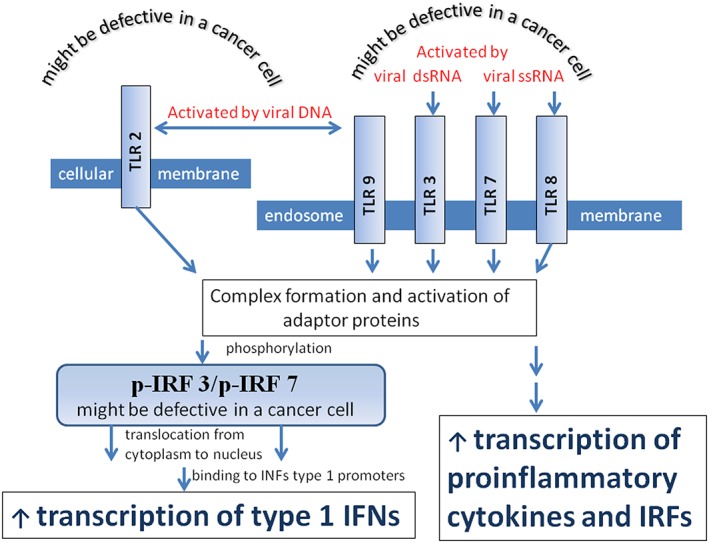
Toll‐like receptors (TLRs) and their downstream signaling. Toll‐like receptors are membrane attached pattern recognition receptors (PRRs) that initiate pathways that produce IFN and proinflammatory cytokines. TLR 2 and TLR9 are activated by viral DNA, TLR3 by viral double stranded RNA, and TLR7/8 by single‐stranded RNA. Toll‐like receptor 2 is located on the outer membrane of the cell, while TLR 3, TLR7/8, and TLR9 are associated with an endosome membrane. After activation by viral nucleic acids, TLRs engage adapter proteins and activate them. The complexes of TLRs with activated adaptor proteins phosphorylate interferon regulatory factors 3 and/or 7 (IRF3 and/or IRF7). Phosphorylated IRF3 and IRF7 relocate from the cytoplasm to the nucleus, where they trigger type 1 IFN production. Through an alternative pathway, the TLR/adaptor protein complexes trigger proinflammatory cytokine production. Malfunction of TLR receptors or IRFs disrupts downstream pathways, making cancer cells vulnerable to oncolytic virus infection
3.2. Cytosolic PRRs
Cytosolic PRRs are represented by viral RNA and DNA sensing proteins. The RNA sensing proteins include retinoic acid‐inducible gene (RIG)‐I‐like receptors (RLRs), protein kinase R (PKR), ribonuclease L (RNase L), and the oligoadenylate synthetase (OAS) enzyme family. RIG‐I‐like receptors include 3 proteins, RIG‐I, LGP2 (also known as RLR‐3), and melanoma differentiation‐associated protein 5 (MDA5). The DNA sensors include GMP‐AMP synthase (cGAS), gamma‐interferon‐inducible protein (IFI16), HIN200, DAI, AIM‐2, LRRFIP1, RNA polymerase III, and some other proteins.36, 37, 38, 39 It is interesting that cytosolic viral RNA can also serve as cGAS activating ligand by forming DNA: RNA hybrid.40 In general, it should be noted that some viral RNA intermediately transcribed from the cytoplasmic genome of a DNA virus can be also be recognized by cytosolic RNA sensors.41, 42, 43 Moreover, DNA reverse transcribed from genomic RNA of retroviruses can be recognized by cytosolic DNA sensors.44
3.3. A type 1 interferon autocrine‐paracrine signal amplification loop
Receptors that recognize viral nucleic acids initiate signaling cascades that lead to the induction of IFNs and some other proinflammatory cytokines. In particular, some TLRs34 and RLRs,45 along with PKR46 and cGAS,47 trigger signaling events that lead to increased transcription of type 1 IFNs. In turn, type 1 IFNs stimulate production of virus‐sensing proteins. For example, IFN‐beta promotes transcription of RIG‐I,48 MDA5,49 and PKR50, 51 by increasing binding of interferon regulatory factor (IRF) 1 to the promoters of these genes. Increased transcription from these genes results in increased production of the corresponding proteins (Figure 2). Thus, RIG‐I, MDA5, and PKR are functionally activated by viral RNA, but their transcription is activated by IFN‐beta via a positive feedback loop.48, 52 This autocrine or paracrine signal amplification cycle is crucial for the antiviral protection of cells, but it can be disrupted in cancer cells by various mechanisms.
Figure 2.
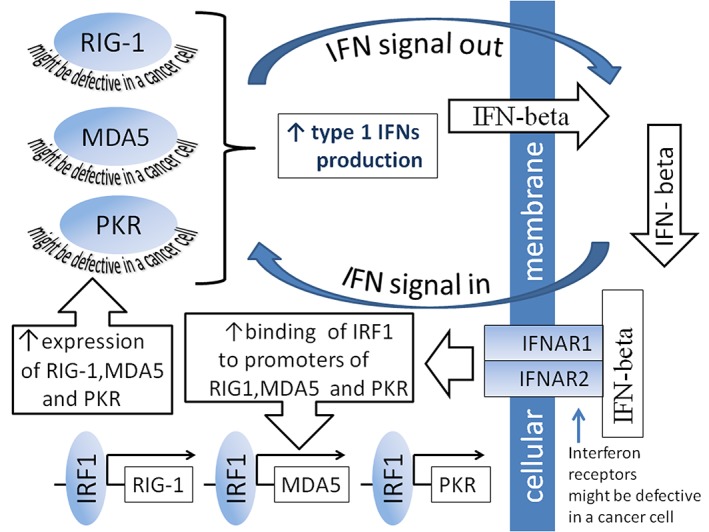
Interferon (IFN) signal amplification loop. RIG‐I, MDA5, and PKR are functionally activated by viral RNAs. After activation, they trigger signaling cascades that result in increased production and secretion of type 1 IFNs. In turn, secreted IFN‐beta interacts with intramembrane receptors of the cell in which it was produced or of another cell. Such interaction initiates autocrine or paracrine downstream signaling pathways, resulting in increased binding of IRF1 to promoters of RIG‐I, MDA5, and PKR. Such binding stimulates transcription of these genes and ultimately increases production of the relevant proteins
3.4. Sensors of viral DNA
Among PRRs that sense viral DNA, TLR2 is located in the surface membranes of cells, TLR9 is located in endosomal membranes,34, 35 and cGAS is a cytosolic PRR.47 Any serious defect in the TLR receptor circuit disrupts the IFN induction pathway and increases susceptibility to viral infection. For example, in some breast cancer stem cells, the TLR2 receptor is not detectable, whereas TLR9 is associated with endoplasmic reticulum (ER) and Golgi‐like structures instead of endosomes. Consequently, in such cells, the virus is unable to induce expression of IFN‐alpha, IFN‐beta, or signal transducer and activator of transcription protein 1 (STAT1). Deficiency of these components promotes sensitivity of cells to adenovirus infection.15
Type 1 IFN response is triggered by cGAS though activation of secondary messengers (Figure 3).47 Epigenetic silencing of the cGAS gene was found in some melanoma cell lines. These cell lines were highly susceptible to herpes simplex virus 1 (HSV1) and vaccinia virus.17
Figure 3.
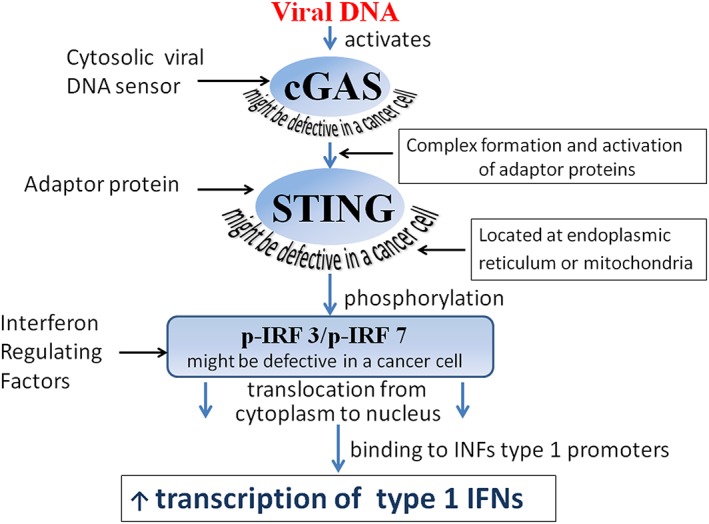
cGAS viral DNA sensor and its downstream signaling. GMP‐AMP synthase (cGAS) is a cytosolic sensor of viral DNA that, after activation by foreign DNA, triggers a type 1 IFN response. cGAS forms a complex with adaptor protein STING, which promotes phosphorylation of interferon regulatory factors 3 and/or 7 (IRF3 and/or IRF7). Phosphorylated IRF3 and IRF7 relocate from the cytoplasm to the nucleus, where they trigger type 1 IFN production. Malfunction of cGAS, STING, IRF3, or IRF7 disrupts downstream IFN production pathways, making cancer cells vulnerable to oncolytic virus infection
3.5. Sensors of viral RNA
Among PRRs that sense viral RNA are TLR3, TLR7/8,34 RLRs,53, 54, 55 PKR,33 RNase L,56 and representatives of the 2′‐5′‐OAS enzyme family.41 All of these proteins are functionally activated by viral RNAs and initiate signaling cascades (Figure 4) that trigger type 1 IFN production and limit virus spread.
Figure 4.
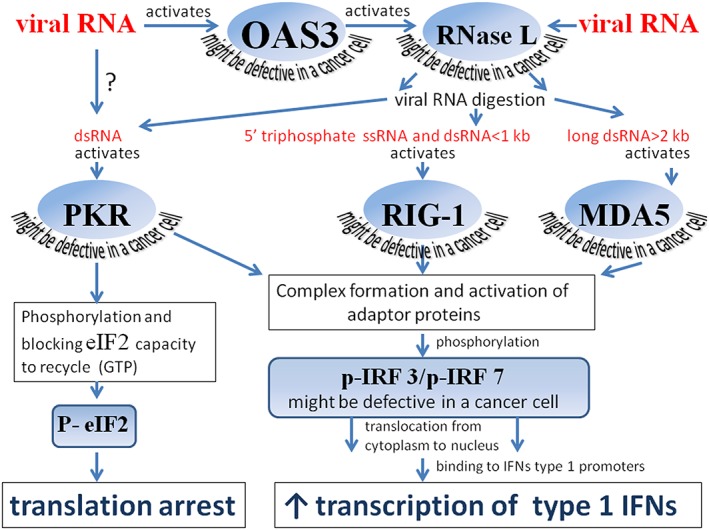
Viral RNA sensing proteins and their downstream signaling. Viral RNA activates the protein product of the OAS3 gene, which in turn activates RNase L that cleaves cytosolic viral RNA. RNA digestion products activate protein kinase PKR and the 2 helicases RIG1 and MDA5. After activation, PKR undergoes autophosphorylation and phosphorylates the translation initiation factor eIF2α. The phosphorylation of the initiation factor leads to translational arrests of both cellular and viral mRNAs, and these arrests promote apoptosis. Protein kinase R, RIG1, and MDA5 also participate in a signal transduction pathway that triggers IFN‐beta transcription and limits viral spread. Protein kinase R, RIG1, and MDA5 form complexes with adaptor proteins that trigger phosphorylation of interferon regulatory factors 3 and/or 7 (IRF3 and/or IRF7). After phosphorylation, IRF3 and IRF7 relocate from the cytoplasm to the nucleus, where they trigger type 1 IFN transcription. Malfunction of OAS3, RNase L, PKR, RIG‐I, MDA5, IRF3, or IRF7 disrupts downstream IFN induction pathways, making cancer cells vulnerable to oncolytic virus infection
Toll‐like receptors that recognize viral RNAs might be located in cell surface membranes (TLR3) or in endosomal membranes (TLR7/8). Sensitivity of primary prostatic adenocarcinoma cell lines to Sendai virus inversely correlates with expression levels of TLR3 and TLR7 mRNA. Cell lines with lower expression levels of these TLRs were more sensitive to virus infection.16
Retinoic acid‐inducible gene‐I‐like receptors include protein products of RIG‐I and MDA5 genes. These proteins are RNA helicases that are ubiquitously expressed in most tissues.53, 54, 55
Retinoic acid‐inducible gene‐I is a cytosolic viral RNA‐sensing protein that is functionally activated after its interaction with ssRNAs containing 5′‐triphosphate or with dsRNAs shorter than 1 kb57, 58 (Figure 4). Several studies suggest a contribution of RIG‐I deficiency to virus‐mediated oncolysis. In multiple myeloma, glioblastoma, and astrocytoma derived cells, the ability of Newcastle disease virus (NDV) to induce the expression of RIG‐I is critical for resistance to viral infection.59 Similar results were obtained in another study, in which normal cells were compared with sarcoma, breast adenocarcinoma, and macrophage‐derived tumor cells. A negative correlation was found between the efficiency of viral oncolysis and constitutive or NDV‐stimulated expression of RIG‐I.29, 60 Retinoic acid‐inducible gene‐I downstream signaling is blocked in some RAS transformed cells. The block causes susceptibility to reovirus infection, whereas overexpression of RIG‐I restores virus resistance.18 It has also been demonstrated that resistance to measles virus positively correlates with viral ability to initiate the expression of RIG‐I in sarcoma cell lines. This expression was strong in resistant and weak in susceptible cell lines.19
Melanoma differentiation‐associated protein 5 is a cytosolic viral RNA sensing protein that is activated after interaction with dsRNAs larger than 1 kb55 (Figure 4). Impaired expression of the MDA5 gene leads to inhibition of type 1 IFN transcription that contributes to viral oncolysis. Although in normal cells treatment with an artificial analog of dsRNAs, poly (I:C), stimulates IFN‐beta production, in hepatocellular carcinoma and some other forms of cancer in which there is weak, if any, expression of the MDA5 gene, similar treatment does not affect IFN‐beta expression. Such lack of IFN‐beta induction renders these cells sensitive to Sindbis virus. However, forced expression of MDA5 restores both cellular ability to produce IFN‐beta and resistance to the virus.20
Protein kinase R is a cytosolic RNA‐sensing protein that is activated by viral dsRNAs of variable length. During the activation process, PKR undergoes autophosphorylation and then phosphorylates the translation initiation factor eIF2α (Figure 4), leading to arrested translation of both cellular and viral mRNAs and even to apoptosis.42, 43 Protein kinase R also participates in a signal transduction pathway that further upregulates IFN‐beta transcription46 (Figure 4). By these mechanisms, the activated PKR restrains virus replication. Similar to RIG‐I and MDA5 genes, the transcription of PKR mRNA is activated by type 1 IFNs, forming a self‐activation regulatory loop.50, 51
Malignant progression often leads to impaired PKR function.61 Truncations of the PKR gene have been observed in many leukemia cell lines.62 Moreover, in most of the leukocyte samples from patients with chronic B‐cell leukemia, PKR enzymatic activity was undetectable, despite the presence of a full‐length PKR gene.62 Decreased levels of PKR expression were found in samples obtained from patients with chronic lymphocytic leukemia63 and nonsmall cell lung cancer.64, 65, 66 Low levels of PKR expression were also associated with higher incidence of disease relapse and reduced overall survival of patients with rectal cancers.67 In breast cancer samples, substantial decrease in PKR functional activity was also observed, despite an increase in its expression.68 In response to viral infection, cancer cells with low or absent PKR activity could not induce eIF2α phosphorylation and IFN‐beta transcription, resulting in increased NDV replication.21
Ribonuclease L is another cytosolic sensor of viral RNAs.56 After activation by interaction with viral dsRNA, it cleaves both viral and cellular RNAs. The RNA digestion products activate other viral RNA sensors, such as RIG‐I, MDA5, and PKR (Figure 4), which in turn trigger IFN production53 and/or promote apoptosis.69 Some evidence suggests a relationship between tumor progression and RNase L dysfunction. Disabling mutations or deletions of RNAse L have been found in prostate cancer samples70, 71, 72 and are also associated with a predisposition to prostate cancer.73 A decrease in RNase L activity was also observed in lung cancer.74 There is some evidence that impaired RNase L function affects the sensitivity of cancer cells to oncolytic viruses. It was shown that the chemical compound sunitinib promotes infection with vesicular stomatitis virus (VSV) and negatively affects the functions of PKR and RNase L. In ovarian, prostate, and renal cell carcinoma cell lines, sunitinib inhibited RNase L enzymatic activity. In a mouse model, simultaneous sunitinib and VSV treatments led to complete elimination of prostate, breast, and kidney tumors, followed by the animals' recovery.22
The family of OAS enzymes is represented by 3 genes: OAS1, OAS2, and OAS3. All members of the family are cytosolic viral RNA sensors that are functionally activated by viral dsRNAs41 and transcriptionally activated by type 1 IFNs.50, 51 During viral infection, OAS3 activates latent RNase L (Figure 4), whereas OAS 1 and OAS2 have other antiviral functions.41 Without OAS3 activation, RNase L does not sense viral RNAs.41 Oligoadenylate synthetase dysfunction is connected with carcinogenesis: Polymorphism of OAS1 is associated with prostate cancer75; low or absent OAS2 expression characterizes some pancreatic cancers23; and polymorphism of OAS3, which decreases its gene expression level, is associated with risk of chronic lymphocytic leukemia.76 It has been shown that defective expression of members of the OAS family causes cellular susceptibility to viral oncolysis. Thus, pancreatic malignant cells with low or absent levels of OAS2 are often sensitive to VSV. In contrast, similar cells with constitutive expression of OAS2 are virus resistant.23
4. ADAPTOR PROTEINS FOR PRRs
After activation with viral nucleic acid, PRRs recruit adapter proteins and form PRR‐adaptor protein pairs that further facilitate downstream antiviral signaling (Figure 4). The PRR‐adapter protein pairs initiate phosphorylation of transcription factors IRF3 and IRF7 via a mediator protein kinase. Phosphorylated IRFs activate transcription of type I IFN genes77, 78 (Figure 4).
The pairing adaptor for cGAS is the stimulator of IFN genes (STING) protein, also known as transmembrane protein TMEM173 or MITA (Figure 3). Stimulator of IFN genes is located on the ER and/or the mitochondria‐associated ER membranes.79 In some tumors, immune adaptor proteins' expression and/or activity is deregulated. Thus, STING function is disabled in numerous colorectal adenocarcinomas. This loss of function is highly predictive of HSV1‐mediated oncolytic activity.24 Melanoma cells often lose STING signaling through the epigenetic silencing of either STING or cGAS genes. Because of this loss, these cells are unable to produce type 1 IFNs in response to DNA viruses and are highly susceptible to HSV1 and vaccinia virus.17
5. ANTIVIRAL IRFs
Antiviral IRFs belong to a family of transcription factors that control many cellular processes, including the induction of antiviral cytokines and type I IFNs. Viral nucleic acid sensors such as TLR3/7/8/9,34 RLRs,55 PKR,46 and cGAS,47 by interacting with adaptor proteins, initiate phosphorylation of IRF3 and/or IRF7 (Figures 3 and 4).80 This phosphorylation promotes IRF3 and/or IRF7 translocation from cytoplasm to nucleus. In the nucleus, these phosphorylated IRFs trigger transcription of type 1 IFNs, which initiate autocrine and paracrine loops of signal amplification. Interferon‐beta stimulates transcription of viral RNA sensors such as RIG‐I,48 MDA5,49 and PKR51 by increasing the binding of IRF1 to promoter regions of these genes. Thus, on the one hand, IRFs could be activated by virus‐sensing proteins; on the other hand, IRFs promote the signal amplification process, leading to the activation of transcription from the genes encoding the virus‐sensing proteins (Figure 2). As documented below, aberrant expression of IRFs, which characterizes many cancers,81 contributes to viral oncolysis.
Interferon regulatory factor 1 is a transcription factor that activates transcription of the genes containing IFN‐stimulated response elements. Viral nucleic acid‐sensing proteins such as RIG‐I, MDA5, and PKR are among these genes. Interferon regulatory factor 1 also participates in the upregulation of many genes that restrain cell proliferation and exert antitumor effects.82 In cancer cells, the transcription activation function of IRF1 is often disabled. So, in acute myeloid leukemias83 and gastric cancers,84 the IRF1 gene is frequently deleted or silent; in many breast85, 86 and invasive hepatocellular carcinomas,87 its expression is low or absent.
In cancer cells, IRF1 function can be suppressed by an activated Ras‐Raf‐MEK‐ERK pathway. Specifically, activated MEK inhibits IRF1 binding to target promoter binding sites and suppresses transcription of viral RNA sensing genes. This inhibition often occurs in malignant cells, which on the one hand eliminates the antiproliferative constraints of IRF1‐controlled genes, and on the other makes the cells vulnerable to viral infection. MEK inhibitors restore transcription of IRF1‐controlled genes.82 It was also shown that a permanently activated Ras‐Raf‐MEK‐ERK pathway not only suppresses IRF1 transcriptional activity but also inhibits the function of IRF1 as such, whereas MEK inhibitors restore it.82 Dysfunctional activation of MEK increases susceptibility of Ras‐transformed malignant cells to VSV infection. In contrast, inhibition of MEK restores their VSV resistance.25, 26, 27 , 88 Similar observations were made on another Ras‐transformed cell line, which has defects in the MEK/ERK pathway and is vulnerable to reovirus infection. In these cells, activation of MEK suppressed the binding of IRF1 to RIG‐I promoter and thus blocked RIG‐I transcription and downstream signaling. Overexpression of RIG‐I restored downstream signaling and resistance to reovirus.18 However, malignant cells displaying an activated Ras‐Raf‐MEK‐ERK pathway are not necessarily susceptible to virus infection. For instance, fibrosarcoma cells with constitutively activated RAS may develop reovirus resistance associated with suppression of some factors required for effective viral entry into cells.89
In normal cells, viruses induce activation of IRF3, IRF5, and IRF7 and thereby trigger production of type 1 IFNs. However, in cancer cells, activation may be impaired, leading to virus sensitivity. For example, in normal hepatocytes, VSV infection induces IEF3 expression accompanied by type 1 IFN production and the acquisition of virus resistance. In contrast, in hepatocellular carcinoma cells, VSV does not activate IRF3 expression due to IRF3 mRNA's aberrant splicing. This defective splicing, which causes low IRF3 expression levels, prevents activation of IFN‐beta transcription and leads to virus sensitivity.28
The comparison of normal and malignant macrophage derived cells demonstrated that basal and NDV‐induced expression levels of IRF3 and IRF7 were reduced in cancer cells. Because the IRF3‐IRF7 complex triggers transcription of type 1 IFNs, reduced levels of the complex are associated with low levels of type 1 IFNs, which are insufficient for protection against viruses. In cancer cells, a negative correlation was found between NDV sensitivity and the basal expression levels of IRF3, IRF7, and IFN‐beta.29 In lung cancer cell lines, IRF7 and/or IRF5 epigenetic silencing were associated with high VSV sensitivity. In cells that were the most sensitive to VSV, promoters of both IRF5 and IRF7 were hypermethylated. Moreover, experimental knockdown of IRF5 and IRF7 by siRNAs increased cell susceptibility to viral infection. In contrast, IRF5 and IRF7 overexpression reduced this susceptibility.30
The phosphatase and tensin homolog (PTEN) acts as a tumor suppressor gene and participates in IRF3 import into the nucleus, which is required for the transcription activation of IFN genes.41 Therefore, it plays a critical role in antiviral innate immunity.90 Phosphatase and tensin homolog tumor suppressor gene is commonly inactivated by mutations or deletions in human cancers,91 resulting in VSV sensitivity.32
6. DEFECTIVE IFN RESPONSE IN MALIGNANT CELLS
The type 1 IFN response pathway, schematically shown in Figure 5, is triggered by interaction of either IFN‐alpha or IFN‐beta with a receptor on the cell surface. The receptor consists of a complex of 2 transmembrane subunits, the products of the IFNAR1 and IFNAR2 genes.92 After the interaction, the complex activates receptor‐associated Janus kinase (JAK) 1 and tyrosine kinase 2. The kinases phosphorylate the STAT1 and STAT2 proteins, which form a dimer and interact with IFN‐regulatory factor 9 (also known as p48) to form a trimolecular complex called IFN‐stimulated gene factor 3. IFN‐stimulated gene factor 3 moves to the nucleus and activates the transcription of interferon‐stimulated genes92 (Figure 5). Interferon‐stimulated genes encode a family of proteins that inhibit multiple stages of a viral infection, including virus entry, translation, replication, assembly, and dissemination.93 Other types of IFNs employ different receptor molecules, although their signal transduction pathways utilize similar JAK kinase and STAT proteins. Janus kinase 1 participates in type 1 and type 3 IFN signaling, while JAK2 participates only in the type 2 IFN signaling cascade.94, 95
Figure 5.
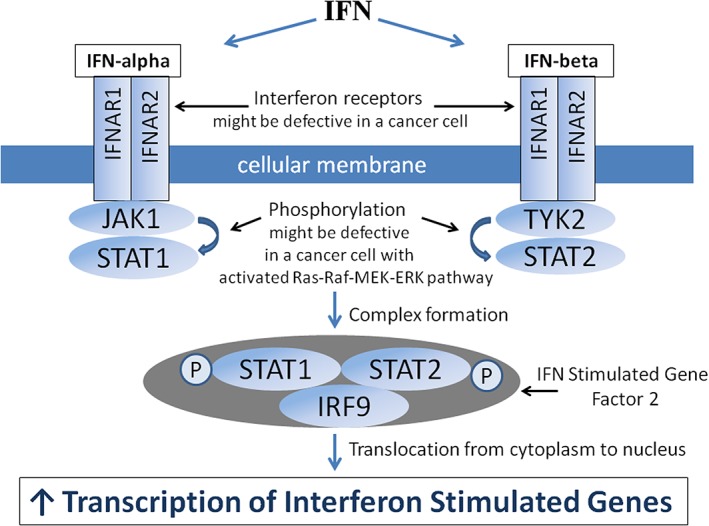
IFN‐alpha and IFN‐beta response pathways. Type 1 IFN response is triggered by a cell surface receptor represented by a complex of 2 transmembrane subunits IFNAR1 and IFNAR2. After interaction with IFN‐alpha or IFN‐beta, the complex activates receptor‐associated Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK 2). The kinases phosphorylate the signal transducer and activator of transcription 1 (STAT1) and STAT2 proteins. These phosphorylated proteins (STAT1 and STAT2), in the form of a dimer, interact with IFN‐regulatory factor 9 (IRF9, also known as p48) and form a trimolecular complex called IFN‐stimulated gene factor 3. Interferon‐stimulated gene factor 3 relocates from the cytoplasm to the nucleus, where it activates transcription of interferon stimulated genes. Interferon stimulated genes encode a family of proteins that inhibit multiple stages of viral infection, including virus entry, translation, replication, assembly, and spread. Malfunction of IFNAR1, IFNAR2, JAK1, STAT1, STAT2, and/or IRF9 disrupts transcription of IFN‐stimulated genes, making cancer cells vulnerable to oncolytic virus infection
An impaired IFN response pathway is a common defect in cancer cells. The genes that belong to the pathway whose defects were shown to be associated with sensitivity to viral oncolysis are described below and listed in Table 2.
Table 2.
Biomarkers related to the IFN response pathway
| Category | Gene/Protein Name | Type of Defect | Sensitivity to | Reference | |
|---|---|---|---|---|---|
| Interferon receptors | IFNAR1 | Deletion | NDV | 60 | |
| IFNAR1/2 | Expression is low or absent | VSV | 31 | ||
| IFNAR2 | VSV | 96 | |||
| Genes of JAK/STAT pathway | JAK1 | Expression is low or absent | VSV | 31 | |
| JAK1/2 | Experimental inhibition | VSV | 23, 97 | ||
| Tyrosine kinase 2 | Expression is low or absent | VSV | 31 | ||
| STAT1 | Transcription is absent | Respiratory syncytial virus | 98 | ||
| STAT1 | Experimental knockdown | Sindbis virus | 20 | ||
| STAT1 | Delayed and low phosphorylation | Measles virus (MV) | 19 | ||
| STAT1, STAT2 | Low expression, absent phosphorylation | NDV | 99 | ||
| STAT1, STAT2 | Expression is low or absent | VSV | 31 | ||
| STAT1, STAT2 | Low phosphorylation | VSV | 100 | ||
| STAT2 | Expression is low | 100 | |||
| Genes of Ras, Raf, MEK, and ERK pathway | MEK2 | Dysfunctionally activated | VSV | 88 | |
| B‐RAF | 101 | ||||
| Interferon stimulated genes (ISGs) | GTPase | GBP‐1 | Expression is absent | VSV | 102 |
| Apoptosis inducer | XAF1 | ||||
| Anti‐apoptotic protein | EPSTI1 | ||||
| Proteins with tetratricopeptide repeats | IFN‐induced protein with tetratricopeptide repeats 1 | Expression is low or absent | MV | 19 | |
| GTPase | MX1/MxA | Low expression | VSV | 23 | |
| Experimental knockdown | 102 | ||||
| Low expression | Adenovirus 5 and adeno‐associated viruses 5 and 6 | 103 | |||
7. IFN RECEPTORS
Low expression levels of IFNAR1 and/or IFNAR2 protein and/or mRNA are characteristic of different malignancies, including melanomas,104 mesotheliomas,31 and carcinomas of hepatocellular,9, 10 pancreatic,11 and gastric12 origin. IFNAR1 expression was also missing in approximately 25% of 48 mesothelioma tumor biopsies.31 In addition, low IFNAR protein levels are common in various bladder cancer cell lines and in clinical samples of bladder tumors. IFNAR expression level correlated with tumor grade: The content of IFNAR was relatively high in more differentiated tumors and relatively low in less differentiated tumors.96
IFNAR expression levels also correlated with cell sensitivity to viral infection. It was shown that in primary macrophages, deletion of the IFNAR1 gene was associated with NDV susceptibility.60 In mesothelioma cell lines, which were sensitive to VSV, IFNAR1 and/or IFNAR2 expression were significantly downregulated or undetectable.31 In bladder carcinoma cell lines, an inverse correlation was found between the inhibition of cellular proliferation induced by type 1 IFN treatment and VSV susceptibility. Cells that most actively proliferated despite IFN treatment were most sensitive to the virus. It has been suggested that VSV sensitivity can be caused by decreased IFNAR expression. This assumption was confirmed by the experimental knockdown of IFNAR either by siRNA or by neutralizing antibodies, both of which led to sensitization of the bladder carcinoma cells to VSV.96
IFNAR1 protein expression in clinical tumor samples can be conveniently and inexpensively evaluated by immunochemistry. Therefore, such easily measurable proteins as IFNAR1 can be particularly useful markers for the selection of cancer patients who are most likely to benefit from oncolytic virotherapy.
8. JAK/STAT PATHWAY
Alterations in the JAK/STAT pathway were found in a number of malignancies. In some lymphomas,105, 106 myeloid leukemias,107 and prostate cancers,98 there was no expression or functional activation of STAT1. In chronic lymphocytic leukemia, STAT1 and STAT3 phosphorylation was impaired.108 In some prostate cancers, JAK1 expression was not detected,109 while in some leukemias and lymphomas, disabling mutations or chromosomal rearrangements of JAK 1 and/or JAK2 have been observed.110, 111, 112, 113
There is a relation between JAK/STAT pathway dysfunction and virus sensitivity. Janus kinase 1/2 inhibitors make cancer cells sensitive to VSV infection. Treatment of virus‐resistant head and neck cancer cells with these inhibitors before or simultaneously with administration of VSV enhanced the spread of infection and increased the virus progeny yield by few orders of magnitude.97 Similarly, treatment of VSV‐resistant pancreatic ductal adenocarcinoma cells with JAK1/2 inhibitor promoted VSV sensitivity.23
In fibrosarcoma cells, a defect in the IFN response pathway at the level of STAT protein promotes the cells' NDV sensitivity. Phosphorylation of STAT1 and STAT2 that occurs in normal fibroblasts in response to IFN treatment was not detected in these cells. Moreover, pretreatment of these cells with IFN‐beta induced a much smaller number of IFN‐stimulated genes and did not inhibit NDV spread.99
Similar observations were made in prostate cancer cells with respect to their susceptibility to respiratory syncytial virus. Unlike normal prostate cells, prostate cancer cells could not activate STAT1 transcription, despite their ability to induce IFN production in response to respiratory syncytial virus. It was concluded that a defect in the IFN response pathway at the level of STAT1 protein led to VSV sensitivity.98 Similarly, STAT1 experimental knockdown rendered hepatocellular carcinoma cells susceptible to Sindbis virus.20
More examples of the relationship between a deficient JAK/STAT pathway and viral oncolysis were found in studies with mesothelioma and sarcoma cells. Vesicular stomatitis virus‐sensitive mesothelioma cells, in addition to reduced expression levels of IFNAR1 and/or IFNAR2 mRNA, also displayed downregulation of one or several transcripts of STAT1, STAT2, JAK1, tyrosine kinase 2, PKR, and IFN‐regulatory factor 9 (p48).31 It also has been shown that measles virus‐sensitive sarcoma cells exhibit weak, delayed, or only transient phosphorylation of STAT1 and weak or undetectable expression of IFN‐induced protein with tetratricopeptide repeats 1.19
In another study, a decrease in IFN‐induced phosphorylation of STAT1 and STAT2 proteins was observed when the Ras‐Raf‐MEK‐ERK pathway was activated. Moreover, in cells with the activated pathway, the total amount of STAT2 was reduced. However, STAT2 overexpression partially restored the transcription of IFN‐induced genes along with VSV resistance.100
It is sometimes difficult to distinguish whether cancer cell sensitivity to viruses is caused by defects in the IFN induction or IFN‐response pathways. For example, it was found that differences in the sensitivity of melanoma cell lines to VSV were associated with a mutation in the B‐RAF gene, a representative of the RAF serine/threonine specific protein kinase family and a participant in the Ras‐Raf‐MEK‐ERK pathway.101 The mechanism that underlies this sensitivity is not yet known, and it is not clear if the mutation is associated with downregulation of genes involved in type 1 IFN induction or response pathways.
9. IFN STIMULATED GENES
The products of the MX genes are proteins with a protective function against both RNA and DNA viruses. Most mammals have two MX genes, MX1 and MX2, whose protein products are called MxA and MxB.114 Mx proteins bind GTP and act as GTPases.115 MX genes belong to a family of IFN stimulated genes; upregulation of their expression in response to viruses depends on the production of type 1 or type 3 IFNs.
Constitutive expression of Mx1 in normal and malignant pancreatic cells correlates with VSV resistance. Cell lines with particularly low or absent Mx1 and OAS gene expression are often sensitive to VSV.23 Moreover, shRNA‐mediated knockdown of Mx1 promotes VSV replication in virus‐resistant cells.102 Low expression of Mx1 mRNA and MxA protein in primary or established cell lines of pancreatic adenocarcinoma is associated with sensitivity to adenovirus 5 and adeno‐associated viruses 5 and 6.103
In the pleural mesothelioma cell lines that were most VSV sensitive, neither basal nor IFN‐beta stimulated expression of MxA, PKR, and OAS was detected. However, in cell lines that were less sensitive to VSV, IFN‐beta treatment stimulated, to various degrees, transcription from these genes.31 Thus, IFN‐beta treatment response, in the form of induction of MxA, PKR, and OAS transcription, correlates with VSV sensitivity.
A relationship was also found between the expression of GBP1, XAF1, and/or EPSTI1 genes and viral oncolysis in pancreatic ductal adenocarcinoma cells. These genes were constitutively expressed in VSV resistant cells and were not expressed at all in VSV‐sensitive cells.102
10. DIRECTIONS FOR FUTURE RESEARCH
There are genes that are not involved in the IFN signaling pathway, whose expression is necessary for a productive viral infection of tumor cells (Figure 6). The virus replication cycle is a multistep process that involves the virus initial entry into the host cell, synthesis of viral proteins and nucleic acids, the assembly of progeny virions, and their release from the cell. Host cells must provide all the necessary conditions for this process. Viruses require that host cells express on their surface virus receptors, which control the efficiency of virus entry into the cell. Some viruses additionally require cells to express processing enzymes that perform modifications or cleavage of viral proteins necessary for the formation of mature infectious virions.
Figure 6.
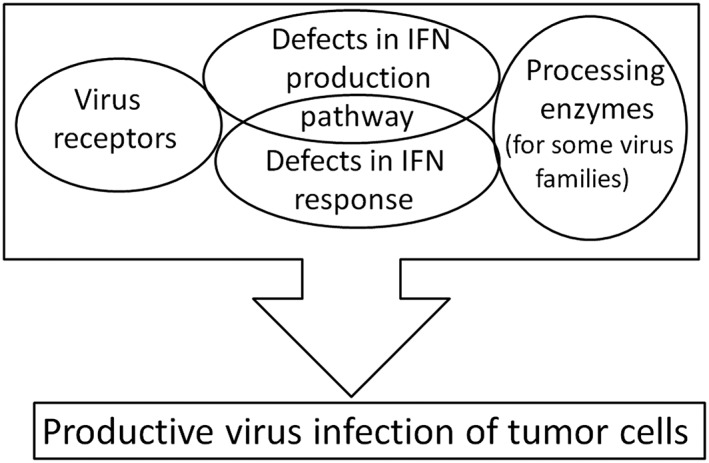
Host requirements for viral infection of tumor cells. For productive infection, viruses require cell to express virus receptors and to have a malfunctioning IFN pathway. Some virus families also require cells to express processing enzymes, without which infective virions cannot be formed
Expression levels of the viral receptors, processing enzymes and other genes needed for productive viral infection can vary considerably in different cancer cells. This variation along with variations in expression and functional activity of the genes involved in the IFN signaling pathways cause differences in cancer cell sensitivity to a particular virus. Therefore, all these genes could potentially serve as predictive biomarkers for identifying individual cancer patients who can most likely benefit from oncolytic virotherapy. Until now, relationships between the efficacy of viral oncolysis and the expression levels or functional abilities of these genes have been poorly understood, but future research should fill this gap.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this article.
ACKNOWLEDGEMENT
The study was supported by the grant from the Center of Strategic Planning, The Ministry of Healthcare of the Russian Federation (project code 1.1096) and by SATOR Therapeutics BioEnterprise.
Matveeva OV, Chumakov PM. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev Med Virol. 2018;28:e2008 10.1002/rmv.2008
[Correction added on 12 December 2018, after first online publication: SATOR Therapeutics BioEnterprise was added in the Acknowledgment and Funding Information.]
The copyright line for this article was change on 8 January 2019 after original online publication.
REFERENCES
- 1. Stetson DB, Medzhitov R. Antiviral defense: interferons and beyond. J Exp Med. 2006;203(8):1837‐1841. 10.1084/jem.20061377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Negishi H, Taniguchi T, Yanai H. The interferon (IFN) class of cytokines and the IFN regulatory factor (IRF) transcription factor family. Cold Spring Harb Perspect Biol. 2017. 10.1101/cshperspect.a028423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121(1):1‐14. 10.1111/j.1365-2567.2007.02587.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Katsoulidis E, Kaur S, Platanias LC. Deregulation of interferon signaling in malignant cells. Pharmaceuticals. 2010;3(2):406‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kulaeva OI, Draghici S, Tang L, Kraniak JM, Land SJ, Tainsky MA. Epigenetic silencing of multiple interferon pathway genes after cellular immortalization. Oncogene. 2003;22(26):4118‐4127. 10.1038/sj.onc.1206594 [DOI] [PubMed] [Google Scholar]
- 6. Olopade OI, Jenkins RB, Ransom DT, et al. Molecular analysis of deletions of the short arm of chromosome 9 in human gliomas. Cancer Res. 1992;52(9):2523‐2529. [PubMed] [Google Scholar]
- 7. Diaz MO, Ziemin S, Le Beau MM, et al. Homozygous deletion of the alpha‐ and beta 1‐interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci U S A. 1988;85(14):5259‐5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cairns P, Tokino K, Eby Y, Sidransky D. Homozygous deletions of 9p21 in primary human bladder tumors detected by comparative multiplex polymerase chain reaction. Cancer Res. 1994;54(6):1422‐1424. [PubMed] [Google Scholar]
- 9. Kondo M, Nagano H, Sakon M, et al. Expression of interferon alpha/beta receptor in human hepatocellular carcinoma. Int J Oncol. 2000;17(1):83‐88. [PubMed] [Google Scholar]
- 10. Damdinsuren B, Nagano H, Wada H, et al. Interferon alpha receptors are important for antiproliferative effect of interferon‐alpha against human hepatocellular carcinoma cells. Hepatol Res. 2007;37(1):77‐83. 10.1111/j.1872-034X.2007.00007.x [DOI] [PubMed] [Google Scholar]
- 11. Saidi RF, Williams F, Silberberg B, Mittal VK, ReMine SG, Jacobs MJ. Expression of interferon receptors in pancreatic cancer: identification of a novel prognostic factor. Surgery. 2006;139(6):743‐748. 10.1016/j.surg.2005.11.010 [DOI] [PubMed] [Google Scholar]
- 12. Chen HM, Tanaka N, Mitani Y, et al. Critical role for constitutive type I interferon signaling in the prevention of cellular transformation. Cancer Sci. 2009;100(3):449‐456. 10.1111/j.1349-7006.2008.01051.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slattery ML, Lundgreen A, Bondurant KL, Wolff RK. Interferon‐signaling pathway: associations with colon and rectal cancer risk and subsequent survival. Carcinogenesis. 2011;32(11):1660‐1667. 10.1093/carcin/bgr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Critchley‐Thorne RJ, Simons DL, Yan N, et al. Impaired interferon signaling is a common immune defect in human cancer. Proc Natl Acad Sci U S A. 2009;106(22):9010‐9015. 10.1073/pnas.0901329106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahtiainen L, Mirantes C, Jahkola T, et al. Defects in innate immunity render breast cancer initiating cells permissive to oncolytic adenovirus. PLoS One. 2010;5(11):e13859 10.1371/journal.pone.0013859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Belova AA, Sosnovtseva AO, Lipatova AV, et al. Biomarkers of prostate cancer sensitivity to the Sendai virus. Mol Biol (Mosk). 2017;51(1):94‐103. 10.7868/S0026898417010049 [DOI] [PubMed] [Google Scholar]
- 17. Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76(22):6747‐6759. 10.1158/0008-5472.CAN-16-1404 [DOI] [PubMed] [Google Scholar]
- 18. Shmulevitz M, Pan LZ, Garant K, Pan D, Lee PW. Oncogenic Ras promotes reovirus spread by suppressing IFN‐beta production through negative regulation of RIG‐I signaling. Cancer Res. 2010;70(12):4912‐4921. [DOI] [PubMed] [Google Scholar]
- 19. Berchtold S, Lampe J, Weiland T, et al. Innate immune defense defines susceptibility of sarcoma cells to measles vaccine virus‐based oncolysis. J Virol. 2013;87(6):3484‐3501. 10.1128/JVI.02106-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang PY, Guo JH, Hwang LH. Oncolytic Sindbis virus targets tumors defective in the interferon response and induces significant bystander antitumor immunity in vivo. Mol Ther. 2012;20(2):298‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang S, Sun Y, Chen H, et al. Activation of the PKR/eIF2alpha signaling cascade inhibits replication of Newcastle disease virus. Virol J. 2014;11(1):62 10.1186/1743-422X-11-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jha BK, Dong B, Nguyen CT, Polyakova I, Silverman RH. Suppression of antiviral innate immunity by sunitinib enhances oncolytic virotherapy. Mol Ther. 2013;21(9):1749‐1757. 10.1038/mt.2013.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moerdyk‐Schauwecker M, Shah NR, Murphy AM, Hastie E, Mukherjee P, Grdzelishvili VZ. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: role of type I interferon signaling. Virology. 2013;436(1):221‐234. 10.1016/j.virol.2012.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14(2):282‐297. 10.1016/j.celrep.2015.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noser JA, Mael AA, Sakuma R, et al. The RAS/Raf1/MEK/ERK signaling pathway facilitates VSV‐mediated oncolysis: implication for the defective interferon response in cancer cells. Mol Ther. 2007;15(8):1531‐1536. 10.1038/sj.mt.6300193 [DOI] [PubMed] [Google Scholar]
- 26. Christian SL, Zu D, Licursi M, et al. Suppression of IFN‐induced transcription underlies IFN defects generated by activated Ras/MEK in human cancer cells. PLoS One. 2012;7(9):e44267 10.1371/journal.pone.0044267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Komatsu Y, Christian SL, Ho N, Pongnopparat T, Licursi M, Hirasawa K. Oncogenic Ras inhibits IRF1 to promote viral oncolysis. Oncogene. 2015;34(30):3985‐3993. 10.1038/onc.2014.331 [DOI] [PubMed] [Google Scholar]
- 28. Marozin S, Altomonte J, Stadler F, Thasler WE, Schmid RM, Ebert O. Inhibition of the IFN‐beta response in hepatocellular carcinoma by alternative spliced isoform of IFN regulatory Factor‐3. Mol Ther. 2008;16(11):1789‐1797. 10.1038/mt.2008.201 [DOI] [PubMed] [Google Scholar]
- 29. Wilden H, Fournier P, Zawatzky R, Schirrmacher V. Expression of RIG‐I, IRF3, IFN‐beta and IRF7 determines resistance or susceptibility of cells to infection by Newcastle disease virus. Int J Oncol. 2009;34(4):971‐982. [DOI] [PubMed] [Google Scholar]
- 30. Li Q, Tainsky MA. Epigenetic silencing of IRF7 and/or IRF5 in lung cancer cells leads to increased sensitivity to oncolytic viruses. PLoS One. 2011;6(12):e28683 10.1371/journal.pone.0028683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saloura V, Wang LC, Fridlender ZG, et al. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon‐beta for use in malignant pleural mesothelioma: heterogeneity in interferon responsiveness defines potential efficacy. Hum Gene Ther. 2010;21(1):51‐64. 10.1089/hum.2009.088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu N, Puckett S, Antinozzi PA, Cramer SD, Lyles DS. Changes in susceptibility to oncolytic vesicular stomatitis virus during progression of prostate cancer. J Virol. 2015;89(10):5250‐5263. 10.1128/JVI.00257-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240‐273, Table of Contents. 10.1128/CMR.00046-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown J, Wang H, Hajishengallis GN, Martin M. TLR‐signaling networks: an integration of adaptor molecules, kinases, and cross‐talk. J Dent Res. 2011;90(4):417‐427. 10.1177/0022034510381264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tatematsu M, Seya T, Matsumoto M. Beyond dsRNA: toll‐like receptor 3 signalling in RNA‐induced immune responses. Biochem J. 2014;458(2):195‐201. 10.1042/BJ20131492 [DOI] [PubMed] [Google Scholar]
- 36. Wang L, Ning S. "Toll‐free" pathways for production of type I interferons. AIMS Allergy Immunol. 2017;1(3):143‐163. 10.3934/Allergy.2017.3.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Diner BA, Lum KK, Cristea IM. The emerging role of nuclear viral DNA sensors. J Biol Chem. 2015;290(44):26412‐26421. 10.1074/jbc.R115.652289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38(5):870‐880. 10.1016/j.immuni.2013.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang X, Brann TW, Zhou M, et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J Immunol. 2011;186(8):4541‐4545. 10.4049/jimmunol.1003389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fung S‐Y, Yuen C‐K, Zhu W, et al. Activation of innate sensing cGAS‐STING pathway by Sindbis virus. J Immunol. 2017;198. [Google Scholar]
- 41. Li Y, Banerjee S, Wang Y, et al. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci U S A. 2016;113(8):2241‐2246. 10.1073/pnas.1519657113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garcia MA, Meurs EF, Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89(6‐7):799‐811. 10.1016/j.biochi.2007.03.001 [DOI] [PubMed] [Google Scholar]
- 43. Husain B, Hesler S, Cole JL. Regulation of PKR by RNA: formation of active and inactive dimers. Biochemistry. 2015;54(44):6663‐6672. 10.1021/acs.biochem.5b01046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang J, Kang L, Song D, et al. Ku70 senses HTLV‐1 DNA and modulates HTLV‐1 replication. J Immunol. 2017;199(7):2475‐2482. 10.4049/jimmunol.1700111 [DOI] [PubMed] [Google Scholar]
- 45. Oshiumi H, Mifsud EJ, Daito T. Links between recognition and degradation of cytoplasmic viral RNA in innate immune response. Rev Med Virol. 2016;26(2):90‐101. 10.1002/rmv.1865 [DOI] [PubMed] [Google Scholar]
- 46. McAllister CS, Taghavi N, Samuel CE. Protein kinase PKR amplification of interferon beta induction occurs through initiation factor eIF‐2alpha‐mediated translational control. J Biol Chem. 2012;287(43):36384‐36392. 10.1074/jbc.M112.390039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786‐791. 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Su ZZ, Sarkar D, Emdad L, Barral PM, Fisher PB. Central role of interferon regulatory factor‐1 (IRF‐1) in controlling retinoic acid inducible gene‐I (RIG‐I) expression. J Cell Physiol. 2007;213(2):502‐510. 10.1002/jcp.21128 [DOI] [PubMed] [Google Scholar]
- 49. Kang DC, Gopalkrishnan RV, Lin L, et al. Expression analysis and genomic characterization of human melanoma differentiation associated gene‐5, mda‐5: a novel type I interferon‐responsive apoptosis‐inducing gene. Oncogene. 2004;23(9):1789‐1800. 10.1038/sj.onc.1207300 [DOI] [PubMed] [Google Scholar]
- 50. Hovanessian AG. Interferon‐induced and double‐stranded RNA‐activated enzymes: a specific protein kinase and 2',5'‐oligoadenylate synthetases. J Interferon Res. 1991;11(4):199‐205. [DOI] [PubMed] [Google Scholar]
- 51. Hovanessian AG. On the discovery of interferon‐inducible, double‐stranded RNA activated enzymes: the 2'‐5'oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev. 2007;18(5‐6):351‐361. 10.1016/j.cytogfr.2007.06.003 [DOI] [PubMed] [Google Scholar]
- 52. Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda‐5: an interferon‐inducible putative RNA helicase with double‐stranded RNA‐dependent ATPase activity and melanoma growth‐suppressive properties. Proc Natl Acad Sci U S A. 2002;99(2):637‐642. 10.1073/pnas.022637199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86(6):2900‐2910. 10.1128/JVI.05738-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berke IC, Li Y, Modis Y. Structural basis of innate immune recognition of viral RNA. Cell Microbiol. 2013;15(3):386‐394. 10.1111/cmi.12061 [DOI] [PubMed] [Google Scholar]
- 55. Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG‐I and MDA5. Front Immunol. 2014;5:342 10.3389/fimmu.2014.00342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liang SL, Quirk D, Zhou A. RNase L: its biological roles and regulation. IUBMB Life. 2006;58(9):508‐514. 10.1080/15216540600838232 [DOI] [PubMed] [Google Scholar]
- 57. Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol. 2004;5(7):730‐737. 10.1038/ni1087 [DOI] [PubMed] [Google Scholar]
- 58. Hornung V, Ellegast J, Kim S, et al. 5'‐Triphosphate RNA is the ligand for RIG‐I. Science. 2006;314(5801):994‐997. 10.1126/science.1132505 [DOI] [PubMed] [Google Scholar]
- 59. Biswas M, Kumar SR, Allen A, et al. Cell‐type‐specific innate immune response to oncolytic Newcastle disease virus. Viral Immunol. 2012;25(4):268‐276. 10.1089/vim.2012.0020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fournier P, Bian H, Szeberenyi J, Schirrmacher V. Analysis of three properties of Newcastle disease virus for fighting cancer: tumor‐selective replication, antitumor cytotoxicity, And Immunostimulation. Methods Mol Biol. 2012;797:177‐204. [DOI] [PubMed] [Google Scholar]
- 61. Balachandran S, Barber GN. PKR in innate immunity, cancer, and viral oncolysis. Methods Mol Biol. 2007;383:277‐301. [DOI] [PubMed] [Google Scholar]
- 62. Hii SI, Hardy L, Crough T, et al. Loss of PKR activity in chronic lymphocytic leukemia. Int J Cancer. 2004;109(3):329‐335. 10.1002/ijc.11714 [DOI] [PubMed] [Google Scholar]
- 63. Basu S, Panayiotidis P, Hart SM, et al. Role of double‐stranded RNA‐activated protein kinase in human hematological malignancies. Cancer Res. 1997;57(5):943‐947. [PubMed] [Google Scholar]
- 64. Pataer A, Raso MG, Correa AM, et al. Prognostic significance of RNA‐dependent protein kinase on non‐small cell lung cancer patients. Clin Cancer Res. 2010;16(22):5522‐5528. 10.1158/1078-0432.CCR-10-0753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. He Y, Correa AM, Raso MG, et al. The role of PKR/eIF2alpha signaling pathway in prognosis of non‐small cell lung cancer. PLoS One. 2011;6(11):e24855 10.1371/journal.pone.0024855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Guo C, Shao R, Correa AM, et al. Prognostic significance of combinations of RNA‐dependent protein kinase and EphA2 biomarkers for NSCLC. J Thorac Oncol. 2013;8(3):301‐308. 10.1097/JTO.0b013e318282def7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kwon HC, Moon CH, Kim SH, et al. Expression of double‐stranded RNA‐activated protein kinase (PKR) and its prognostic significance in lymph node negative rectal cancer. Jpn J Clin Oncol. 2005;35(9):545‐550. 10.1093/jjco/hyi146 [DOI] [PubMed] [Google Scholar]
- 68. Savinova O, Joshi B, Jagus R. Abnormal levels and minimal activity of the dsRNA‐activated protein kinase, PKR, in breast carcinoma cells. Int J Biochem Cell Biol. 1999;31(1):175‐189. [DOI] [PubMed] [Google Scholar]
- 69. Brennan‐Laun SE, Ezelle HJ, Li XL, Hassel BA. RNase‐L control of cellular mRNAs: roles in biologic functions and mechanisms of substrate targeting. J Interferon Cytokine Res. 2014;34(4):275‐288. 10.1089/jir.2013.0147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rokman A, Ikonen T, Seppala EH, et al. Germline alterations of the RNASEL gene, a candidate HPC1 gene at 1q25, in patients and families with prostate cancer. Am J Hum Genet. 2002;70(5):1299‐1304. 10.1086/340450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Casey G, Neville PJ, Plummer SJ, et al. RNASEL Arg462Gln variant is implicated in up to 13% of prostate cancer cases. Nat Genet. 2002;32(4):581‐583. 10.1038/ng1021 [DOI] [PubMed] [Google Scholar]
- 72. Rennert H, Bercovich D, Hubert A, et al. A novel founder mutation in the RNASEL gene, 471delAAAG, is associated with prostate cancer in Ashkenazi Jews. Am J Hum Genet. 2002;71(4):981‐984. 10.1086/342775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Xiang Y, Wang Z, Murakami J, et al. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2',5'‐oligoadenylates. Cancer Res. 2003;63(20):6795‐6801. [PubMed] [Google Scholar]
- 74. Yin H, Zhou A, Dai Y. The association of elevated 2',5'‐oligoadenylate‐dependent RNase L with lung cancer correlated with deficient enzymatic activity and decreased capacity of RNase L dimerization. Lung Cancer. 2012;78(1):30‐38. 10.1016/j.lungcan.2012.07.010 [DOI] [PubMed] [Google Scholar]
- 75. Mandal S, Abebe F, Chaudhary J. 2'‐5' oligoadenylate synthetase 1 polymorphism is associated with prostate cancer. Cancer. 2011;117(24):5509‐5518. 10.1002/cncr.26219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sava GP, Speedy HE, Di Bernardo MC, et al. Common variation at 12q24.13 (OAS3) influences chronic lymphocytic leukemia risk. Leukemia. 2015;29(3):748‐751. 10.1038/leu.2014.311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347(6227):aaa2630 10.1126/science.aaa2630 [DOI] [PubMed] [Google Scholar]
- 78. Zevini A, Olagnier D, Hiscott J. Crosstalk between cytoplasmic RIG‐I and STING sensing pathways. Trends Immunol. 2017;38(3):194‐205. 10.1016/j.it.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674‐678. 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol. 2013;78(0):105‐116. 10.1101/sqb.2013.78.020321 [DOI] [PubMed] [Google Scholar]
- 81. Yanai H, Negishi H, Taniguchi T. The IRF family of transcription factors: inception, impact and implications in oncogenesis. Oncoimmunology. 2012;1(8):1376‐1386. 10.4161/onci.22475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. AbuSara N, Razavi S, Derwish L, Komatsu Y, Licursi M, Hirasawa K. Restoration of IRF1‐dependent anticancer effects by MEK inhibition in human cancer cells. Cancer Lett. 2015;357(2):575‐581. 10.1016/j.canlet.2014.12.017 [DOI] [PubMed] [Google Scholar]
- 83. Green WB, Slovak ML, Chen IM, Pallavicini M, Hecht JL, Willman CL. Lack of IRF‐1 expression in acute promyelocytic leukemia and in a subset of acute myeloid leukemias with del(5)(q31). Leukemia. 1999;13(12):1960‐1971. [DOI] [PubMed] [Google Scholar]
- 84. Nozawa H, Oda E, Ueda S, et al. Functionally inactivating point mutation in the tumor‐suppressor IRF‐1 gene identified in human gastric cancer. Int J Cancer. 1998;77(4):522‐527. [DOI] [PubMed] [Google Scholar]
- 85. Doherty GM, Boucher L, Sorenson K, Lowney J. Interferon regulatory factor expression in human breast cancer. Ann Surg. 2001;233(5):623‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cavalli LR, Riggins RB, Wang A, Clarke R, Haddad BR. Frequent loss of heterozygosity at the interferon regulatory factor‐1 gene locus in breast cancer. Breast Cancer Res Treat. 2010;121(1):227‐231. 10.1007/s10549-009-0509-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yi Y, Wu H, Gao Q, et al. Interferon regulatory factor (IRF)‐1 and IRF‐2 are associated with prognosis and tumor invasion in HCC. Ann Surg Oncol. 2013;20(1):267‐276. 10.1245/s10434-012-2487-z [DOI] [PubMed] [Google Scholar]
- 88. Battcock SM, Collier TW, Zu D, Hirasawa K. Negative regulation of the alpha interferon‐induced antiviral response by the Ras/Raf/MEK pathway. J Virol. 2006;80(9):4422‐4430. 10.1128/JVI.80.9.4422-4430.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kim M, Egan C, Alain T, et al. Acquired resistance to reoviral oncolysis in Ras‐transformed fibrosarcoma cells. Oncogene. 2007;26(28):4124‐4134. 10.1038/sj.onc.1210189 [DOI] [PubMed] [Google Scholar]
- 90. Li S, Zhu M, Pan R, et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat Immunol. 2016;17(3):241‐249. 10.1038/ni.3311 [DOI] [PubMed] [Google Scholar]
- 91. Chalhoub N, Baker SJ. PTEN and the PI3‐kinase pathway in cancer. Annu Rev Pathol. 2009;4(1):127‐150. 10.1146/annurev.pathol.4.110807.092311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36‐49. 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Diamond MS. IFIT1: a dual sensor and effector molecule that detects non‐2'‐O methylated viral RNA and inhibits its translation. Cytokine Growth Factor Rev. 2014;25(5):543‐550. 10.1016/j.cytogfr.2014.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. de Weerd NA, Nguyen T. The interferons and their receptors—distribution and regulation. Immunol Cell Biol. 2012;90(5):483‐491. 10.1038/icb.2012.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Stark GR, Darnell JE Jr. The JAK‐STAT pathway at twenty. Immunity. 2012;36(4):503‐514. 10.1016/j.immuni.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang KX, Matsui Y, Hadaschik BA, et al. Down‐regulation of type I interferon receptor sensitizes bladder cancer cells to vesicular stomatitis virus‐induced cell death. Int J Cancer. 2010;127:830‐838. 10.1002/ijc.25088 [DOI] [PubMed] [Google Scholar]
- 97. Escobar‐Zarate D, Liu YP, Suksanpaisan L, Russell SJ, Peng KW. Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther. 2013;20(10):582‐589. 10.1038/cgt.2013.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Echchgadda I, Chang TH, Sabbah A, et al. Oncolytic targeting of androgen‐sensitive prostate tumor by the respiratory syncytial virus (RSV): consequences of deficient interferon‐dependent antiviral defense. BMC Cancer. 2011;11(1):43 10.1186/1471-2407-11-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Krishnamurthy S, Takimoto T, Scroggs RA, Portner A. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J Virol. 2006;80(11):5145‐5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Christian SL, Collier TW, Zu D, Licursi M, Hough CM, Hirasawa K. Activated Ras/MEK inhibits the antiviral response of alpha interferon by reducing STAT2 levels. J Virol. 2009;83(13):6717‐6726. 10.1128/JVI.02213-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wollmann G, Davis JN, Bosenberg MW, van den Pol AN. Vesicular stomatitis virus variants selectively infect and kill human melanomas but not normal melanocytes. J Virol. 2013;87(12):6644‐6659. 10.1128/JVI.03311-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hastie E, Cataldi M, Moerdyk‐Schauwecker MJ, Felt SA, Steuerwald N, Grdzelishvili VZ. Novel biomarkers of resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus. Oncotarget. 2016;7(38):61601‐61618. 10.18632/oncotarget.11202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Monsurro V, Beghelli S, Wang R, et al. Anti‐viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: potential relevance for adenoviral gene therapy. J Transl Med. 2010;8(1):10 10.1186/1479-5876-8-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Katlinskaya YV, Katlinski KV, Yu Q, et al. Suppression of type I interferon signaling overcomes oncogene‐induced senescence and mediates melanoma development and progression. Cell Rep. 2016;15(1):171‐180. 10.1016/j.celrep.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sun WH, Pabon C, Alsayed Y, et al. Interferon‐alpha resistance in a cutaneous T‐cell lymphoma cell line is associated with lack of STAT1 expression. Blood. 1998;91(2):570‐576. [PubMed] [Google Scholar]
- 106. Dummer R, Dobbeling U, Geertsen R, Willers J, Burg G, Pavlovic J. Interferon resistance of cutaneous T‐cell lymphoma‐derived clonal T‐helper 2 cells allows selective viral replication. Blood. 2001;97(2):523‐527. [DOI] [PubMed] [Google Scholar]
- 107. Landolfo S, Guarini A, Riera L, et al. Chronic myeloid leukemia cells resistant to interferon‐alpha lack STAT1 expression. Hematol J. 2000;1(1):7‐14. 10.1038/sj/thj/6200004 [DOI] [PubMed] [Google Scholar]
- 108. Frank DA, Mahajan S, Ritz J. B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J Clin Invest. 1997;100(12):3140‐3148. 10.1172/JCI119869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Dunn GP, Sheehan KC, Old LJ, Schreiber RD. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 2005;65(8):3447‐3453. 10.1158/0008-5472.CAN-04-4316 [DOI] [PubMed] [Google Scholar]
- 110. Smith CA, Fan G. The saga of JAK2 mutations and translocations in hematologic disorders: pathogenesis, diagnostic and therapeutic prospects, and revised World Health Organization diagnostic criteria for myeloproliferative neoplasms. Hum Pathol. 2008;39(6):795‐810. 10.1016/j.humpath.2008.02.004 [DOI] [PubMed] [Google Scholar]
- 111. Salmoiraghi S, Montalvo ML, D'Agostini E, et al. Mutations and chromosomal rearrangements of JAK2: not only a myeloid issue. Expert Rev Hematol. 2013;6(4):429‐439. 10.1586/17474086.2013.826910 [DOI] [PubMed] [Google Scholar]
- 112. Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high‐risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009;106(23):9414‐9418. 10.1073/pnas.0811761106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol. 2008;19(4):385‐393. 10.1016/j.semcdb.2008.07.002 [DOI] [PubMed] [Google Scholar]
- 114. Verhelst J, Hulpiau P, Saelens X. Mx proteins: antiviral gatekeepers that restrain the uninvited. Microbiol Mol Biol Rev. 2013;77(4):551‐566. 10.1128/MMBR.00024-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Richter MF, Schwemmle M, Herrmann C, Wittinghofer A, Staeheli P. Interferon‐induced MxA protein. GTP Binding and GTP Hydrolysis Properties. J Biol Chem. 1995;270(22):13512‐13517. [PubMed] [Google Scholar]