

Dexamethasone is used to manage cerebral oedema in patients with glioblastoma, despite significant drawbacks. Herting et al. show that dexamethasone reduces oedema via inhibition of interleukin-1 signalling. They highlight a likely interaction between dexamethasone and immunotherapy, and propose that specific interleukin-1 inhibition may be preferable to dexamethasone for managing oedema.
Keywords: glioblastoma, cerebral oedema, microglia, macrophage, interleukin-1
Abstract
Glioblastoma is the most common and uncompromising primary brain tumour and is characterized by a dismal prognosis despite aggressive treatment regimens. At the cellular level, these tumours are composed of a mixture of neoplastic cells and non-neoplastic cells, including tumour-associated macrophages and endothelial cells. Cerebral oedema is a near-universal occurrence in patients afflicted with glioblastoma and it is almost exclusively managed with the corticosteroid dexamethasone despite significant drawbacks associated with its use. Here, we demonstrate that dexamethasone blocks interleukin-1 production in both bone marrow-derived and brain resident macrophage populations following stimulation with lipopolysaccharide and interferon gamma. Additionally, dexamethasone is shown to inhibit downstream effectors of interleukin-1 signalling in both macrophage populations. Co-culture of bone marrow-derived macrophages with organotypic tumour slices results in an upregulation of interleukin-1 cytokines, an effect that is absent in co-cultured microglia. Genetic ablation of interleukin-1 ligands or receptor in mice bearing RCAS/tv-a-induced platelet-derived growth factor B-overexpressing glioblastoma results in reduced oedema and partial restoration of the integrity of the blood–brain barrier, respectively; similar to results obtained with vascular endothelial growth factor neutralization. We establish that tumours from dexamethasone-treated mice exhibit reduced infiltration of cells of the myeloid and lymphoid compartments, an effect that should be considered during clinical trials for immunotherapy in glioblastoma patients. Additionally, we emphasize that caution should be used when immune profiling and single-cell RNA sequencing data are interpreted from fresh glioblastoma patient samples, as nearly all patients receive dexamethasone after diagnosis. Collectively, this evidence suggests that interleukin-1 signalling inhibition and dexamethasone treatment share therapeutic efficacies and establishes interleukin-1 signalling as an attractive and specific therapeutic target for the management of glioblastoma-associated cerebral oedema.
Introduction
Glioblastoma is the most common primary brain tumour and is characterized by a dismal median survival of ∼15 months despite an aggressive treatment regimen consisting of maximal surgical resection followed by radiation and concomitant and maintenance temozolomide (Stupp et al., 2005). During tumour progression, the development of glioblastoma-associated cerebral oedema is ubiquitous, and it is nearly exclusively managed with the corticosteroid dexamethasone (Raslan and Bhardwaj, 2007). Dexamethasone has been the gold standard for management of cerebral oedema since its introduction for this purpose in the 1960s (Galicich et al., 1961), to the extent that it has even been referred to as ‘one of the greatest contributions in the history of neurosurgery’ (Kostaras et al., 2014). Although it is exceptionally efficacious for peri-surgical management of inflammation, historical hurdles and recent discoveries highlight significant drawbacks associated with long-term dexamethasone-based management of oedema in patients with glioblastoma. The notion that an alternative therapy is necessary has received significant attention recently (Wong and Swanson, 2019).
Since the introduction of glucocorticoid therapy, side effects including abnormal fat deposition, myopathy, mental excitement, and hyperglycaemia have been well-documented (Kofman et al., 1957). Initial trials of dexamethasone therapy for the management of cerebral oedema additionally encountered difficulties in weaning patients off the drug due to suppressed production of endogenous glucocorticoids caused by prolonged administration (French, 1966). Moreover, in these initial trials, uncertainty around ideal dosing was identified (Galicich et al., 1961), a problem that still has not been resolved conclusively (Kostaras et al., 2014). Recent work has demonstrated that dexamethasone therapy significantly interferes with anti-neoplastic therapies in glioblastoma and is associated with worse prognosis in both mice and humans (Pitter et al., 2016; Luedi et al., 2017). This interference has been proven to be dose-dependent, with maximal interference occurring at the highest daily doses of dexamethasone (Wong et al., 2015). The immunosuppressive effects of dexamethasone have also been highlighted (Coutinho and Chapman, 2011), although the exact impact on the composition and behaviour of the immune microenvironment in glioblastoma remains undefined. It has nonetheless been suggested that dexamethasone may interfere with novel immunotherapies entering clinical trials (Wong et al., 2015). Considering this evidence, there is significant rationale to support the notion that an alternative to dexamethasone for the management of cerebral oedema will be beneficial for patients with brain tumours and the clinicians who manage their treatment.
Studies suggest that the effects of dexamethasone in management of vasogenic oedema are mediated by decreasing blood–brain barrier permeability (Ostergaard et al., 1999; Kotsarini et al., 2010). The exact molecular and cellular mechanisms of action of dexamethasone for the management of cerebral oedema, however, remain elusive. Specific to brain tumours, it has been suggested that the mechanism of action of dexamethasone stems from suppressed production of vascular endothelial growth factor (VEGF) by the tumour (Heiss et al., 1996; Machein et al., 1999). Analysis of human tumour samples with microarrays, however, illustrated no significant effect between dexamethasone use and VEGF expression in malignant gliomas (Carlson et al., 2007). Regardless, VEGF inhibition with bevacizumab or cediranib has been shown to be effective in managing glioblastoma-associated cerebral oedema (Kamoun et al., 2009; Pitter et al., 2016).
The founding members of the IL-1 family of cytokines, IL-1α and IL-1β, were initially discovered and characterized in the 1980s (March et al., 1985) and are now known to signal through the same receptor, although with different potencies (Gabay et al., 2010). IL-1α is produced by nearly all cell types and is typically secreted upon cell death, while IL-1β is produced primarily by activated myeloid cells (Barksby et al., 2007; Dinarello et al., 2012). This exclusivity results from macrophage-specific expression of the NLRP3 inflammasome complex, which is necessary for cleavage of pro-IL-1β into its active form (Guarda et al., 2011). In glioblastoma, tumour-associated macrophages are the largest non-neoplastic cell type, constituting >30% of the tumour bulk and contributing significantly to tumour progression and treatment resistance (Hambardzumyan et al., 2016). Specifically in models of proneural glioblastoma driven by platelet-derived growth factor B (PDGFB) overexpression, the tumour-associated macrophage population is composed of both brain-resident microglia as well as infiltrating bone marrow-derived macrophages (BMDM) from the circulation (Chen et al., 2017). Considering the prevalence of macrophages in glioblastoma, and their capacity to produce and cleave IL-1β to its active form in the tumour microenvironment, we found IL-1 signalling to be an intriguing avenue of investigation in glioblastoma-associated oedema. Bearing in mind that dexamethasone is known to alter cytokine production in glioblastoma (Gottschall et al., 1992) and that IL-1 signalling has been implicated in both blood–brain barrier disruption (Argaw et al., 2006; Wang et al., 2014) and oedema formation (Clausen et al., 2011) in other neuroinflammatory conditions, we focused on determining the link between dexamethasone and IL-1 signalling as well as the suitability of specific IL-1 inhibition as a strategy for the management of glioblastoma-associated oedema.
Our data demonstrate that dexamethasone inhibits the production of IL-1 cytokines in the context of tumour-associated macrophages in glioblastoma and they further suggest that specific inhibition of IL-1 signalling would provide a therapeutic effect similar to dexamethasone. We found that dexamethasone prevents IL-1 production and dampens IL-1 signalling in primary murine BMDM and microglia. Similar results were obtained in an ex vivo system where organotypic tumour slices and BMDM or microglia were co-cultured. A reduction in Il1a and Il1b mRNA was observed in tumour tissue from dexamethasone-treated tumour-bearing mice. Infiltration of myeloid and lymphoid cells from the blood was impaired in murine tumours following dexamethasone administration. A similar reduction of myeloid cell chemotaxis was also observed in Ntv-a/Il1r1–/– mice with flow cytometry and immunohistochemistry. Ablation of IL-1R1 in vivo additionally resulted in decreased vessel leakage as measured by a Hoechst dye-based assay. Genetic loss of IL-1 ligands resulted in decreased oedema as measured by MRI, serial histology, and a methodology analysing wet/dry tissue weight. The results presented here demonstrate that dexamethasone broadly impairs function of the immune system, a phenomenon that warrants further investigation. The complete impacts of IL-1 inhibition on immune function remain largely undefined outside of myeloid cell chemotaxis and also merit further examination. Finally, by using genetic and pharmacological approaches we demonstrate that targeting IL-1 signalling does not compromise efficacy of radiation therapy in tumour-bearing mice. These results suggest specific IL-1 signalling inhibition may be a preferred alternative to dexamethasone for the management of glioblastoma-associated cerebral oedema.
Materials and methods
Bone marrow-derived macrophage isolation and culture
BMDMs were isolated from 4–12-week-old C57BL/6 mice using a modified established protocol (Weischenfeldt and Porse, 2008). For a detailed description, see the online Supplementary material.
Microglia isolation and culture
Microglia were isolated from postnatal Day 0–3 pups using a modified established protocol (Roy, 2018). For a detailed description, refer to the Supplementary material.
Bone marrow-derived macrophage and microglia stimulation
A 3-h starvation in foetal bovine serum (FBS)-deficient media was carried out prior to stimulation with 100 ng/ml lipopolysaccharide (LPS) (Sigma Aldrich, L5293), 40 ng/ml interferon gamma (IFNγ) (Peprotech, 315-05), 75 pM or 400 pM IL-1α (R&D, 400-ML-005/CF), or 200 pM or 1 nM IL-1β (R&D, 401-ML-005/CF). Six-hour incubations with the indicated stimulants were performed. Dexamethasone (Sigma-Aldrich, D4902) was included as a 2-h pretreatment at a dose of 5 µM where indicated. Gallium nitrate (Sigma-Aldrich, 289892-5G) was included as a 2-h pretreatment at a dose of 500 µM where indicated. Cells were treated with 5 mM ATP (Sigma, A2383) for 30 min following the 6-h simulation to induce cytokine release, as previously described (Wewers and Sarkar, 2009).
Quantitative PCR analysis
RNA was isolated from cell cultures or snap-frozen tumour pieces using a RNeasy® Lipid Tissue Mini Kit (Qiagen, 74804) according to the manufacturer’s instructions. RNA quantity was assessed with a NanoDrop™ 2000 spectrometer, while quality was confirmed by electrophoresis of samples in a 1% bleach gel, as previously described (Aranda et al., 2012). Following RNA validation, cDNA synthesis was carried out with a First Strand SuperScript™ III cDNA synthesis kit (ThermoFisher, 18080051) according to the manufacturer’s instructions and with equal amounts of starting RNA. Quantitative PCR was performed with the validated Bio-Rad PCR primers for murine Il1a, Il1b, Vegfa, Aurka, Cdc20, Plk1, Cenpa, Ccnb1, Kif2c, Ccl2, Ccl5, Ccl7, Ccl8, Ccl12, Aif1, and Actb using a SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad, 1725271). Fold-change in gene expression was determined relative to a defined control group using the 2–ΔΔCt method with Actb/β-actin as a housekeeping gene.
ELISA
Cell lysates for ELISA were collected by sonication of cells in lysis buffer supplemented with protease and phosphatase inhibitors. Tissue lysates were collected via mechanical homogenization in lysis buffer followed by sonication. Protein concentration was determined using a Bradford protein assay (Bio-Rad, 5000001) according to the manufacturer’s instructions. ELISAs were carried out for IL-1β (R&D, DY401-05) on cell lysates and cell supernatants according to the manufacturer’s instructions.
Mice
All procedures related to the care, handling, and usage of animals in this study were reviewed and approved by the IACUC at Emory University. Specific details regarding the mice used can be found in the Supplementary material.
Virus production and tumour generation
Tumours were induced using the RCAS/tv-a system for stereotactic delivery of virus-producing cells expressing RCAS PDGFB-HA or RCAS shp53-RFP, as previously described (Hambardzumyan et al., 2009; Herting et al., 2017). For details regarding viral production and surgical protocols, refer to the Supplementary material.
Organotypic tumour slice culture
At endpoint, Ntv-a/Cdkn2a–/– mice harbouring PDGFB-overexpressing tumours were euthanized by carbon dioxide asphyxiation. Without perfusion, the brain was rapidly extracted and embedded in 4% low-melt agarose in phosphate-buffered saline (PBS). The embedded brain was then mounted on a vibratome (Leica) and submerged in ice-cold Hank’s balanced salt solution (HBSS). The brain was cut into 300-µm thick sections and the slices were transferred to inserts in a 6-well plate. Slices were cultured in NeuroCult™ Basal Medium (STEMCELL, 05700) supplemented with B-27™ supplement (ThermoFisher, 17504044), sodium pyruvate (ThermoFisher, 11360070), and glutamine (ThermoFisher, 35050061). Macrophage-colony stimulating factor (M-CSF) at a concentration of 40 ng/ml, was included in the media during co-culture experiments with BMDM and microglia. Dexamethasone (Sigma-Aldrich, D4902) was included at a dose of 5 µM where administration is indicated.
In vivo dexamethasone administration
Mice were given intraperitoneal injections of either dexamethasone (West-Ward, 462-329-02) at a dose of 10 mg/kg or a corresponding volume of 0.9% saline solution (McKesson, 2718344) for 5 days prior to sacrifice. Treatment was initiated when mice began to show neurological signs of tumour burden. Mice were sacrificed on Day 6 after treatment initiation and tumour tissue was collected for analysis as described.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded sections isolated as previously described (Herting et al., 2017). Detailed methodology is described in the Supplementary material.
Flow cytometry
At endpoint, mice were sacrificed with an overdose of ketamine. Blood was collected via cardiac puncture and decoagulated with EDTA. Mice were then perfused with cold Ringers solution (Sigma-Aldrich, 96724-100TAB) and the brains were extracted and placed in dishes with sterile HBSS. Tumour dissociation was performed with a neural tissue dissociation kit (Milltenyi, 130-092-628) using 1 mg/ml collagenase D (Sigma-Aldrich, 11088882001) in water as a digestive enzyme. The resulting cell suspension was cleaned of debris via Percoll density gradient centrifugation and was blocked with Fc-block (BD Biosciences, 553141), FBS (HyClone, SH30396.03), normal rat serum (ThermoFisher, 10710C), normal mouse serum (ThermoFisher, 10410), and normal rabbit serum (ThermoFisher, 31883) on ice. Staining was performed in the blocking solution with the following antibodies: CD45-APC (BioLegend, 103111), CD11b-PerCP-Cy5.5 (BD Biosciences, 550993), Ly6C-PE-Cy7 (BD Biosciences, 560593), and Ly6G-V450 (BD Biosciences, 560603). Cells were washed and resuspended in a buffer consisting of PBS, bovine serum albumin, EDTA, and sodium azide prior to analysis on an LSRII cytometer (BD).
Tumour tissue isolation
At endpoint, mice were euthanized with an overdose of ketamine and xylazine and perfused with cold Ringer’s solution (Sigma-Aldrich, 96724-100TAB). The brain was extracted and a piece of tumour was immediately snap-frozen in liquid nitrogen for storage at –80°C. The rest of the brain was fixed in 10% formalin for 72 h for histology.
Hoechst dye leakage assay and whole-slice imaging
The Hoechst dye leakage assay was performed as previously described (Herting et al., 2017). A detailed description of this assay can be found in the Supplementary material.
Whole-slice images were obtained using the multi-area time lapse function on the Fluoview FV1000 microscope (Olympus). Four z-planes were acquired at a spacing of 5 µm for each channel analysed. The grid/collection stitching plugin in FIJI was used to combine the individual tiles from the multi-area time lapse into a complete image of the brain slice (Preibisch et al., 2009). Z-projection was used to reduce the separate z-planes into one image. The per cent positive Hoechst area was determined and divided by the total tumour area, as judged by propidium iodide staining, to generate a readout of tumour vessel leakage. All image analysis and quantification was carried out blinded to treatment condition.
B20-4.1.1 treatment
Treatment with B20-4.1.1 (Genentech) commenced 30 days following tumour induction via RCAS injection. Mice were administered B20-4.1.1 at 5 mg/kg via intraperitoneal injection every 4 days until endpoint symptoms developed.
MRI tumour volume reconstruction
All MRI data were acquired as previously described (Herting et al., 2017). Specifics regarding the scanning protocol are described in the Supplementary material. To reconstruct tumour volume, the tumour area in each slice was outlined in the Bruker software and multiplied by the thickness of the slice. The total volume for all slices within each tumour was summed to generate the total tumour volume. All tumours were analysed in a blinded fashion.
Haematoxylin and eosin tumour volume reconstruction
At endpoint, mice were sacrificed with an overdose of ketamine and xylazine and perfused with 4% paraformaldehyde in PBS. The brain was carefully extracted and incubated in 4% paraformaldehyde in PBS for 24 h following a 72-h incubation in 30% sucrose in PBS. The brain was then embedded in O.C.T. compound (VWR, 25608-930) and frozen on dry ice. The entire brain was then serially sectioned on a cryostat (Leica) set to cut 30-µm sections. Every 10th section was collected and mounted on a slide for automated haematoxylin and eosin staining. The slides were scanned at 20× magnification with a whole slide scanner (Hamamatsu). Tumour area in each section was determined in a blinded fashion in NDP.view2 and multiplied by the thickness of 10 slices. The resulting volumes for the slides of each tumour were then summed, producing an estimation of the total volume.
Wet/dry tissue weight oedema measurement
At endpoint, whole brains were extracted without perfusion and immediately weighed. Tissue was then placed in a vacuum oven (Across International) and dried at complete vacuum and 95ºC for 24 h. Dry tissue was then reweighed, and the per cent brain water was calculated as previously described (Tait et al., 2010).
Mouse irradiation and gallium nitrate administration
Irradiation of tumour-bearing mice was performed as previously described (Herting et al., 2017). Gallium nitrate (Sigma-Aldrich, 289892-5G) was administered via intraperitoneal injection in saline at a dose of 50 mg/kg as indicated. Mice were followed until endpoint criteria were met and overall survival was recorded.
Statistical analysis
Statistical analysis and figure generation were performed in GraphPad Prism version 8 and Adobe Illustrator, respectively. Significant effects were considered at P < 0.05. Specific statistical analyses used for each experiment are indicated in the figure legends. All data are presented as mean ± standard error of the mean (SEM).
Data availability
All raw data used for figure generation in this manuscript can be obtained by contacting the corresponding author.
Results
Dexamethasone blocks the production of IL-1 cytokines in primary murine BMDM and microglia in vitro
For in vitro analysis of the effects of dexamethasone on IL-1 production by macrophages, primary murine BMDM and microglia culturing protocols (Fig. 1A and B) were established by modification of existing methods (Weischenfeldt and Porse, 2008; Roy, 2018). To validate the purity of cells isolated with these methodologies, immunofluorescent staining for IBA1 and flow cytometric analysis of CCR2 and CX3CR1 were used (Supplementary Fig. 1).
Figure 1.

Dexamethasone blocks LPS/IFNγ and IL-1-induced IL-1 expression in primary BMDMs and microglia. Primary murine BMDMs (A) and microglia (B) were cultured using modifications of previously established protocols. Il1 expression in BMDMs (n = 4) (C) and microglia (n = 4) (D) following stimulation with LPS and IFNγ. Intracellular (n = 5) and secreted (n = 7) IL-1β protein levels in BMDMs (E) as well as intracellular (n = 6) and secreted (n = 8) IL-1β protein levels in microglia (F) following stimulation with LPS and IFNγ. IL-1α (G) and IL-1β (H) stimulation of BMDMs (n = 4). IL-1α (I) and IL-1β (J) stimulation of microglia (n = 4). One-way ANOVA, ns = not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. MG = microglia.
Macrophages are known to upregulate IL-1 cytokines following stimulation with LPS and IFNγ (Beuscher et al., 1990), and they secrete IL-1β following activation of the P2X7 purinergic receptor by ATP (Wewers and Sarkar, 2009). Therefore, we used a scheme consisting of LPS and INFγ stimulation, followed by ATP treatment, to functionally validate our primary BMDM and microglia cultures (Fig. 1C–F). Dexamethasone pretreatment suppressed LPS and IFNγ-induced Il1a and Il1b RNA expression in both BMDM (Fig. 1C) and microglia (Fig. 1D). Intracellular and secreted IL-1β protein levels were also suppressed by dexamethasone in both cell types (Fig. 1E and F).
IL-1 stimulation has been shown to upregulate IL-1 cytokines in multiple different cell types and we investigated this phenomenon in our primary murine BMDM and microglia (Dinarello et al., 1987; Warner et al., 1987a, b). First, we performed dose-response experiments in BMDM and microglia to determine the optimal doses of IL-1α and IL-1β to use for further experiments (Supplementary Fig. 2). We chose to use doses of 75 pM IL-1α and 200 pM IL-1β in primary BMDMs (Supplementary Fig. 2A and C). Doses of 400 pM IL-1α and 1 nM IL-1β were used in experiments with primary microglia (Supplementary Fig. 2B and D).
We next assessed the effects of dexamethasone on IL-1-induced IL-1 production. In both BMDMs (Fig. 1G and H) and microglia (Fig. 1I and J), dexamethasone was shown to inhibit Il1a and Il1b RNA expression following stimulation with either IL-1 cytokine. These in vitro results confirm that dexamethasone is capable of blocking IL-1α and IL-1β production following multiple different stimulation schemes. A propidium iodide and annexin V-based apoptosis assay confirmed that these results were not due to dexamethasone toxicity in BMDMs or microglia (Supplementary Fig. 3).
Dexamethasone reduces cytokine and chemokine levels in co-cultured myeloid cells and tumour slices
Based on the inhibitory effect of dexamethasone on IL-1 signalling in BMDMs and microglia in vitro, we next designed an ex vivo system to interrogate questions more specific to glioblastoma. Previously by using RNA-sequencing, we showed that BMDMs, and not microglia, primarily express Il1b in glioblastoma and that BMDMs are recruited to the tumour through a monocyte chemoattractant protein (MCP) family chemokine-dependent mechanism (Chen et al., 2017). To evaluate whether BMDMs or microglia upregulate IL-1 family cytokines in the tumour microenvironment, and what effects dexamethasone has on this process, we developed a system for co-culturing BMDMs and microglia with organotypic tumour slices (Fig. 2A).
Figure 2.

Dexamethasone treatment ablates Il1 upregulation by BMDMs exposed to organotypic tumour slices and reduces expression of MCP cytokines by tumour tissue. (A) An experimental outline of the co-culture of BMDMs and microglia (MG) with organotypic tumour slices (TS). (B) Expression of Il1a and Il1b in BMDMs co-cultured with tumour slices (n = 6). (C) Il1a, Il1b, Ccl2, Ccl7, Ccl8, and Ccl12 expression in tumour slices co-cultured with BMDMs (n = 6). (D) Expression of Il1a and Il1b in microglia co-cultured with tumour slices (n = 5). (E) Il1a, Il1b, Ccl2, Ccl7, Ccl8, and Ccl12 expression in tumour slices co-cultured with microglia (n = 6). In all experiments, dexamethasone (DEX) was added at a dose of 5 µM where indicated. One-way ANOVA and two-tailed Student’s t-test. ns = not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. DEX = dexamethasone; VEH = vehicle.
The results showed that BMDMs exposed to organotypic tumour slices upregulate both Il1a and Il1b (Fig. 2B). This effect was shown to be abrogated by dexamethasone treatment. Tumour slices co-cultured with BMDM and treated with dexamethasone were shown to downregulate Il1a and Il1b, as well as Ccl2, Ccl7, and Ccl12, three of the four MCP family members (MCP-1/CCL2, MCP-2/CCL7, MCP-3/CCL8, and MCP-4/CCL12) (Fig. 2C). Contrary to the results in BMDMs, microglia did not upregulate Il1a or Il1b following co-culture with tumour slices (Fig. 2D). In fact, microglia were surprisingly shown to downregulate Il1a and Il1b in this context. Both Il1a and Il1b expression were still downregulated by dexamethasone treatment relative to the control microglia. Tumour slices co-cultured with microglia and treated with dexamethasone were shown to downregulate Il1a and Il1b, as well as Ccl2 and Ccl7, similar to what was seen in tumour slices co-cultured with BMDM (Fig. 2E). The downregulation of MCP cytokine family members suggested that dexamethasone may affect BMDM chemotaxis.
Dexamethasone significantly reduces the amount of tumour-associated macrophages without altering angiogenesis
To assess the effects of dexamethasone on tumour-associated macrophages and angiogenesis in vivo, PDGFB-overexpressing tumours were generated in Ntv-a/Cdkn2a−/− mice as previously described (Herting et al., 2017). The treatment schematic for this experiment (Fig. 3A) involved dosing tumour-bearing mice with vehicle solution (saline) or dexamethasone (10 mg/kg) for 5 days prior to sacrifice, with mice displaying endpoint symptoms. The efficacy of this dexamethasone treatment scheme was verified by assessing the expression levels of Aurka, Cdc20, Plk1, Cenpa, Ccnb1, and Kif2c genes in tumour tissue with quantitative PCR (Supplementary Fig. 4). These genes were previously demonstrated to be downregulated by dexamethasone treatment (Pitter et al., 2016). Quantitative PCR analysis of the isolated tumours indicated a decrease in total Il1a and Il1b post-dexamethasone treatment (Fig. 3B). Contrary to prior reports that dexamethasone alters the expression of Vegfa, we observed no difference in Vegfa expression in tumours isolated from vehicle or dexamethasone-treated mice (Fig. 3B). Aif1, a pan-macrophage marker, was shown to be significantly downregulated by dexamethasone (Fig. 3B).
Figure 3.

Dexamethasone treatment has no effect on tumour angiogenesis, but significantly reduces the amount of tumour-associated macrophages. (A) An experimental outline of the generation and treatment of PDGFB-overexpressing tumours. (B) Quantitative-PCR of tumour samples and interrogation of Il1a, Il1b, Vegfa, and Aif1 expression in tumour tissue samples from vehicle (VEH, n = 13) and dexamethasone (DEX)-treated mice (n = 15). Immunohistochemical analysis of CD31 (C) and IBA1 (D) expression in formalin-fixed, paraffin-embedded tumour slices from vehicle- (n = 9) and dexamethasone-treated mice (n = 11). Scale bars = 100 µm. Two-tailed Student’s t-test, ns = not significant. *P < 0.05, **P < 0.01, ***P < 0.001. RCAS = replication-competent avian sarcoma-leukosis virus long terminal repeat with splice acceptor.
Based on these results, we questioned whether vehicle- and dexamethasone-treated mice would display differences in angiogenesis or tumour-associated macrophage accumulation. Therefore, we stained our tumour specimens for the endothelial cell marker CD31 and the pan-macrophage marker IBA1 (the protein product of the Aif1 gene) (van Mourik et al., 1985; Imai et al., 1996). Quantification of vessel-length density, average vessel size, and vessel calibre based on CD31-positive staining revealed no differences between vehicle or dexamethasone treatment (Fig. 3C). There was a reduction in IBA1-positive area in tumour samples from mice treated with dexamethasone compared to vehicle-treated mice (Fig. 3D). These results suggest that while having no effect on angiogenesis, dexamethasone decreases the amount of tumour-associated macrophages.
Dexamethasone impairs the influx of circulating myeloid and lymphoid cells into glioblastomas in vivo
As IBA1 is a shared marker between BMDMs and microglia it is unable to distinguish between the two cell types by immunohistochemistry. To determine whether dexamethasone decreased tumour-associated macrophages through alterations in BMDMs or microglia, we next used flow cytometry to analyse tumour samples from vehicle- and dexamethasone-treated mice. We previously demonstrated the ability to distinguish BMDMs and microglia based on CD45-protein expression levels by flow cytometry (Chen et al., 2017). PDGFB-overexpressing tumours were generated in Ntv-a mice and treated with vehicle or dexamethasone following the same treatment schematic previously used (Fig. 4A). As expected, we observed a reduction in total myeloid cells (CD45+CD11b+) in tumours of dexamethasone-treated mice (Fig. 4B). When the myeloid cells were divided into BMDMs (CD45high) and microglia (CD45low) by CD45 positivity, the reduction in total myeloid cells could be completely attributed to a reduction in the infiltration of BMDMs from the circulation (Fig. 4B). Additionally, we demonstrated that dexamethasone significantly impairs the ability of lymphoid cells (CD45+CD11b−) to infiltrate the tumours (Fig. 4B).
Figure 4.

Dexamethasone treatment significantly impairs the ability of immune cells to infiltrate the tumour from the circulation. (A) An experimental outline of the treatment of mice bearing PDGFB-overexpressing tumours with vehicle (VEH) or dexamethasone (DEX). (B) Representative flow cytometry plots and quantification of total myeloid cells, BMDMs, microglia (MG), and lymphoid cells in PDGFB-overexpressing tumours from mice treated with vehicle (n = 10) or dexamethasone (n = 10). (C) Representative flow cytometry plots and quantification of circulating myeloid cells, lymphoid cells, inflammatory monocytes, and neutrophils in the blood of tumour-bearing mice treated with vehicle (n = 9) or dexamethasone (n = 9) at endpoint. Two-tailed Student’s t-test, ns = not significant. *P < 0.05, **P < 0.01, ***P < 0.001. RCAS = replication-competent avian sarcoma-leukosis virus long terminal repeat with splice acceptor.
These results indicated that dexamethasone has an inhibitory effect on immune cell infiltration into the tumour. Next, we evaluated whether this decreased infiltration stemmed from effects on immune cell extravasation from the bone marrow or circulation. Therefore, we determined circulating immune cell levels in tumour-bearing mice treated with vehicle or dexamethasone. Our results indicated that dexamethasone has different effects on cells of the myeloid and lymphoid compartments. No significant effect was observed on the circulating levels of total myeloid cells (CD45+CD11b+) or neutrophils (CD45+CD11b+Ly6GhighLy6Cmid) (Fig. 4C). We did, however, observe an increase in circulating inflammatory monocytes (CD45+CD11b+Ly6GlowLy6Chigh) (Fig. 4C). Circulating lymphoid cell (CD45+CD11b−) levels were strongly depressed by dexamethasone administration (Fig. 4C). These effects were confirmed by administration of dexamethasone to tumour-naïve C57BL/6 mice. In fact, in this experiment, significant increases in circulating total myeloid cells (CD45+CD11b+), inflammatory monocytes (CD45+CD11b+Ly6GlowLy6Chigh), and neutrophils (CD45+CD11b+Ly6GhighLy6Cmid) were observed (Supplementary Fig. 5). The significant reduction in circulating lymphoid cells (CD45+CD11b−) observed in tumour-bearing mice was also recapitulated in naïve mice (Supplementary Fig. 5). These experiments illustrated that dexamethasone likely impacts lymphoid cell release from the bone marrow and myeloid cell extravasation from the circulation into inflammatory sites.
IL-1R1 ablation has no effect on angiogenesis but reduces the influx of BMDMs in PDGFB-overexpressing tumours
As dexamethasone was shown to exert such a profound effect on IL-1 signalling in vitro (Fig. 1), ex vivo (Fig. 2), and in vivo (Fig. 3), we questioned if we could recapitulate phenotypes observed following dexamethasone administration by specifically ablating IL-1 signalling. To investigate this hypothesis, PDGFB-overexpressing tumours were generated in Ntv-a and Ntv-a/Il1r1−/− mice. Ablation of IL-1R1 activity was confirmed by stimulating Il1r1−/− BMDMs with IL-1α and IL-1β in vitro (Supplementary Fig. 6). Immunohistochemical analysis of CD31 (Fig. 5A) and IBA1 (Fig. 5B) demonstrated no significant effect on angiogenesis, but a significant reduction in tumour-associated macrophages in tumours from Ntv-a/Il1r1−/− mice compared to tumours from Ntv-a mice. These results closely mimicked those seen following dexamethasone administration (Fig. 3C and D).
Figure 5.

IL-1R1 ablation reduces the influx of circulating myeloid cells in the tumours of mice bearing PDGFB-overexpressing glioblastoma. Immunohistochemical analysis of CD31 (A) in Ntv-a (n = 5) and Ntv-a/Il1r1−/− (n = 9) mice. Immunohistochemical analysis of IBA1 (B) in Ntv-a (n = 14) and Ntv-a/Il1r1−/− (n = 11) mice. (C) Representative flow cytometry plots and quantification of total myeloid cells, BMDMs, microglia (MG), and lymphoid cells in tumours of Ntv-a (n = 5) and Ntv-a/Il1r1−/− (n = 6) mice bearing PDGFB-overexpressing glioblastoma. Two-tailed Student’s t-test, ns = not significant. ***P < 0.001, ****P < 0.0001.
To establish whether the reduction in IBA1-positivity could be attributed to BMDMs, microglia, or both cell types, we used flow cytometry. Our results indicated no significant reduction in total myeloid cells (CD45+CD11b+) in tumours from Ntv-a/Il1r1−/− mice (Fig. 5C). Division of the total myeloid cells into BMDMs (CD45high) and microglia (CD45low) indicated a strong reduction in the influx of BMDMs (Fig. 5C). No decrease in the amount of total lymphoid cells (CD45+CD11b−) was apparent (Fig. 5C). When assessing the circulating immune cell profiles of these mice, no differences were observed in the levels of circulating myeloid cells (CD45+CD11b+), lymphoid cells (CD45+CD11b−), inflammatory monocytes (CD45+CD11b+Ly6GlowLy6Chigh), or neutrophils (CD45+CD11b+Ly6GhighLy6Cmid) (Supplementary Fig. 7).
IL-1R1 ablation reduces blood–brain barrier permeability in PDGFB-overexpressing tumours
Considering the similarities between dexamethasone treatment and IL-1R1 ablation with respect to BMDM infiltration, we next evaluated the ability of IL-1R1 ablation to reduce blood–brain barrier permeability in PDGFB-overexpressing tumours. Additionally, we assessed whether IL-1R1 ablation in conjunction with VEGF neutralization would have a synergistic effect on blood–brain barrier disruption. To do so, we modified a previously established Hoechst dye-based assay (Herting et al., 2017). Tumours were generated in Ntv-a and Ntv-a/Il1r1−/− mice that were treated with vehicle or the VEGF-neutralizing antibody B20-4.1.1 every 4 days at a dose of 5 mg/kg following tumour initiation and up to endpoint (Fig. 6A). Comparison of per cent Hoechst positivity in the tumours of Ntv-a and Ntv-a/Il1r1−/− mice treated with vehicle indicated a significant reduction in blood–brain barrier permeability in Ntv-a/Il1r1−/− mice (Fig. 6B). No significant difference was observed between the two genotypes when treated with the VEGF-neutralizing antibody B20-4.1.1 (Fig. 6C). The leakage in both VEGF-neutralized groups was similar to IL-1R1 ablation alone. These results suggest that IL-1 signalling regulates blood–brain barrier permeability.
Figure 6.

Ablation of IL-1 signalling reduces blood–brain barrier permeability in mice bearing PDGFB-overexpressing tumours. (A) A schematic outlining tumour generation, treatment, and tissue processing. (B) Comparison of Hoechst dye leakage in tumours from Ntv-a (n = 10) and Ntv-a/Il1r1−/− (n = 11) mice treated with vehicle solution. (C) Comparison of Hoechst dye leakage in tumours from Ntv-a (n = 8) and Ntv-a/Il1r1−/− (n = 5) mice treated with the VEGF-neutralizing antibody B20-4.1.1. Two-tailed Student’s t-test, ns = not significant. *P < 0.05.
IL-1 ligand ablation reduces oedema formation in tumour-bearing mice at endpoint
As our results with IL-1R1 ablation indicated a reduction in blood–brain barrier permeability, and both IL-1α and IL-1β signal through the same receptor, we next assessed whether depletion of IL-1β, or IL-1α and IL-1β, would drive a reduction in the formation of cerebral oedema. We used a previously established MRI-based assessment of oedema formation in mice based upon endpoint tumour volume determination, where an increase in endpoint tumour volume is attributed to a decrease in oedema (Pitter et al., 2016). We confirmed the MRI results in this experiment with ex vivo tumour volume reconstruction and a wet/dry tissue weight assay.
The schematic of this experiment used tumour induction in Ntv-a, Ntv-a/Il1b−/−, and Ntv-a/Il1a/b−/− mice and submitted them to T2-weighted MRI at endpoint to determine tumour volume (Fig. 7A). Tumours were then extracted and serially sectioned prior to haematoxylin and eosin staining and ex vivo tumour volume reconstruction to confirm the MRI results. This experiment indicated a significant increase in endpoint tumour volume in the Ntv-a/Il1b−/− and Ntv-a/Il1a/b−/− mice relative to the Ntv-a mice with T2-weighted MRI (Fig. 7B). No significant increase in endpoint tumour volume in the Ntv-a/Il1b−/− group and a significant increase in the Ntv-a/Il1a/b−/− group relative to the Ntv-a group were observed with serial histology (Fig. 7C). Utilizing a wet/dry tissue weight-based measurement of oedema (Fig. 7D) we first validated the assay and the efficacy of dexamethasone in reducing oedema (Supplementary Fig. 8). We then performed the assay in Ntv-a and Ntv-a/Il1a/b−/− mice and confirmed the results obtained with MRI and serial histology (Fig. 7E). These data suggest a decrease in oedema following IL-1 signalling ablation and imply that the effect may be IL-1β-driven.
Figure 7.

IL-1 ligand ablation reduces oedema formation in tumour-bearing mice. (A) A schematic illustration of the MRI and serial histology experimental workflow. (B) Comparison of endpoint tumour volumes between Ntv-a (n = 8), Ntv-a/Il1b−/− (n = 8), and Ntv-a/Il1a/b−/− (n = 6) mice with T2-weighted MRI. (C) Comparison of endpoint tumour volumes between Ntv-a (n = 7), Ntv-a/Il1b−/− (n = 9), and Ntv-a/Il1a/b−/− (n = 7) mice with serial sectioning followed by haematoxylin and eosin staining. (D) A schematic illustration of the wet/dry assay for oedema measurement. (E) Comparison of per cent brain water in naïve Ntv-a (n = 5), naïve Ntv-a/Il1a/b−/− (n = 5), tumour-bearing Ntv-a (n = 7), and tumour-bearing Ntv-a/Il1a/b−/− (n = 8) mice. One-way ANOVA, ns = not significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Inhibition of IL-1 signalling does not compromise radiotherapy efficacy in vivo
The interference of dexamethasone treatment with the response to radiation in mice and humans has been previously documented (Pitter et al., 2016). We next evaluated whether IL-1 inhibition either by genetic or pharmacological approaches compromises the efficacy of radiation therapy in tumour-bearing mice. We first chose to assess the response to radiation in Ntv-a and Ntv-a/Il1a/b−/− tumour-bearing mice (Fig. 8A) and demonstrated no reduction in median survival time following radiation in Ntv-a/Il1a/b−/− compared to Ntv-a mice. We then validated the ability of gallium nitrate to inhibit IL-1 release in bone marrow-derived macrophages (Supplementary Fig. 9) and assessed its impacts on the efficacy of radiation therapy. We used the same treatment schedule as dexamethasone in the experiment demonstrating its interference with radiation (Pitter et al., 2016). Similar to dexamethasone, gallium nitrate alone had no impact on the survival of tumour-bearing mice when compared to the vehicle-treated group. In contrast to dexamethasone, there was no significant difference between gallium nitrate plus radiotherapy-treated animals compared to radiotherapy alone, demonstrating that gallium nitrate treatment does not interfere with radiotherapy efficacy (Fig. 8B).
Figure 8.
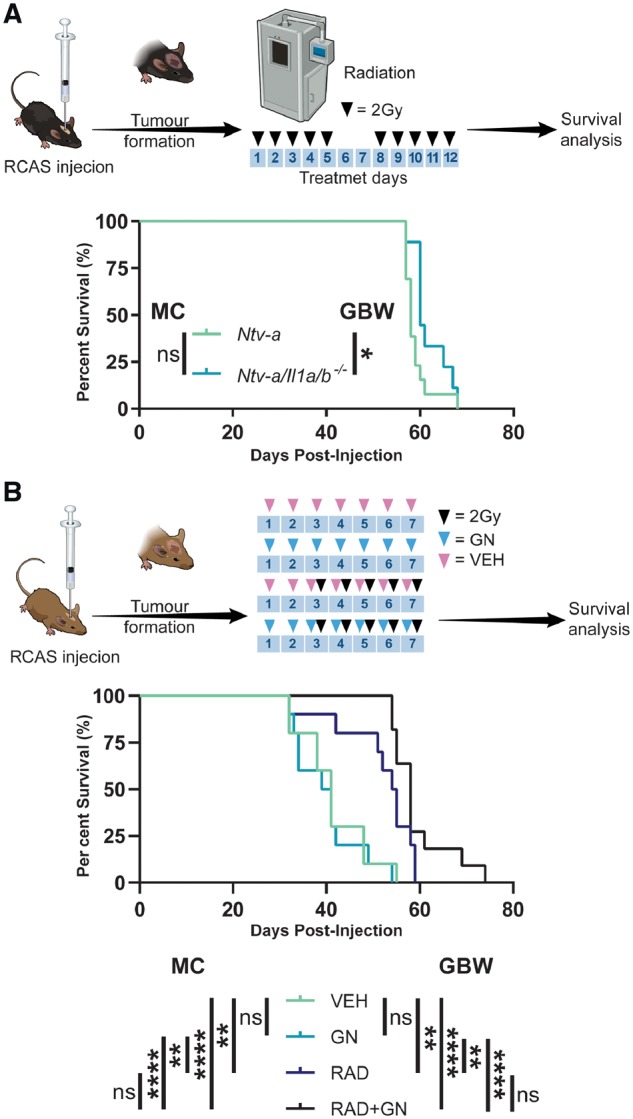
Genetic and pharmacological inhibition of IL-1 signalling have no impact on radiotherapy efficacy. (A) A schematic illustration of the irradiation of tumour-bearing Ntv-a (n = 13) and Ntv-a/Il1a/b−/− (n = 9) mice with the associated survival curves. (B) A schematic illustration of the treatment of tumour-bearing mice with vehicle (n = 10), gallium nitrate (n = 10), vehicle plus radiation (n = 10), or gallium nitrate plus radiation (n = 11) with the associated survival curves. Mantel-Cox (MC) and Gehan-Breslow-Wilcoxon (GBW) tests, ns = not significant. *P < 0.05, **P < 0.01, ****P < 0.0001. RCAS = replication-competent avian sarcoma-leukosis virus long terminal repeat with splice acceptor.
Discussion
Following its introduction, dexamethasone has long been recognized as the gold standard for management of glioblastoma-associated cerebral oedema (Galicich et al., 1961; McClelland and Long, 2008). Recent attention, however, has highlighted the need to find an alternative to dexamethasone for the long-term management of glioblastoma-associated cerebral oedema because of the complications associated with prolonged dexamethasone therapy, as well as the interference of dexamethasone with anti-neoplastic therapies used to combat the tumour (Kostaras et al., 2014; Pitter et al., 2016; Wong and Swanson, 2019).
In the current study we used a murine model of glioblastoma where BMDMs comprise the majority of the tumour-associated macrophage population. RNA sequencing results on these cells suggested that they upregulate the inflammatory mediator Il1b significantly compared to naïve BMDMs or tumour-associated microglia (Chen et al., 2017). In vitro, we demonstrated that BMDMs and microglia both upregulate IL-1 family cytokines when stimulated with either LPS and IFNγ or IL-1; effects that were inhibited by dexamethasone treatment. Our ex vivo experiments demonstrated that BMDMs upregulate Il1a and Il1b when exposed to organotypic tumour slices, and that this effect can be abrogated by dexamethasone treatment. Microglia were surprisingly shown to downregulate IL-1 cytokines when co-cultured with tumour slices. In vivo, we demonstrated a decrease in total tumour Il1a and Il1b levels following dexamethasone treatment.
Our in vivo results further illustrated no effect on the expression levels of Vegfa, or vessel parameters in the tumours of dexamethasone-treated mice. These results differ from previous work that demonstrated a significant reduction in Vegfa produced by glioblastoma tumour cells in response to dexamethasone treatment (Heiss et al., 1996; Machein et al., 1999). This difference in results can be attributed to the genetic make-up of the tumours in these studies. Our data are consistent with results illustrating no effect of dexamethasone therapy on VEGF expression in human glioma samples (Carlson et al., 2007). Our work suggests that dexamethasone impacts the permeability of the blood–brain barrier in an angiogenesis-independent fashion. Experiments in naïve mice, with no cerebral injury or aberrant angiogenesis, have demonstrated that dexamethasone reduces the permeability of the blood–brain barrier to macromolecules (Hedley-Whyte and Hsu, 1986). Moreover, following cerebral injury, dexamethasone has been demonstrated to upregulate tight junction molecules, such as tight junction protein-1, in endothelial cells within the blood–brain barrier (Raslan and Bhardwaj, 2007; Hue et al., 2015). Although cerebral injury and brain tumours are distinct entities, it is plausible that the mechanism of action of drugs used to treat oedema induced by each trauma may be similar.
Although no effect was observed on angiogenesis following dexamethasone treatment, a significant reduction in the amount of tumour-associated macrophages was observed with immunohistochemistry. Further analysis with flow cytometry elucidated that this effect was attributable to a reduction in the influx of BMDM from the circulation. Previous work in cancer and other inflammatory conditions has demonstrated impaired monocyte chemotaxis following treatment with dexamethasone (Badie et al., 2000; Kim et al., 2017). The precise mechanism of this effect has not been determined; however, dexamethasone has been shown to suppress circulating cytokine levels in humans (Schuld et al., 2001; Remmelts et al., 2012). It has been suggested that dexamethasone does so through global suppression of nuclear factor-kappa B, which is a known mediator of IL-1 signalling (Auphan et al., 1995; Albrecht et al., 2007). As macrophage chemotaxis is primarily controlled by the expression of MCP family chemokines, it is plausible that the effects of dexamethasone on macrophage chemotaxis are mediated by its effects on chemokine levels (Jones, 2000; Xuan et al., 2015). This hypothesis is strengthened by our data showing that tumours induced in Ntv-a//Il1r1−/− mice have reduced BMDM infiltration compared to those induced in Ntv-a mice. Evidence also suggests that IL-1 may act as a regulator of MCP family member expression, specifically in the tumour microenvironment, but this hypothesis has not been extensively examined in the context of glioblastoma (Yoshimura, 2017). Our ex vivo data clearly illustrated the ability of dexamethasone to reduce MCP chemokine levels in organotypic tumour slices co-cultured with BMDMs or microglia, lending further support to the hypothesis that this effect is chemokine-dependent.
In addition to the differences observed in myeloid cell infiltration, mice treated with dexamethasone displayed a significant reduction in the influx of lymphoid cells to their tumours. These results are in line with previous studies that demonstrated that dexamethasone decreases circulating lymphocyte levels in patients with brain tumours (Hughes et al., 2005). Direct studies on the effects of dexamethasone on circulating T lymphocytes in a bovine model established that dexamethasone is capable of reducing circulating CD3+ cell levels to 30% of normal, on par with the reduction we observed in mice (Burton and Kehrli, 1996). The effects of dexamethasone on lymphoid cells may not be responsible for its ability to manage oedema; however, they are almost certain to alter the efficacy of T cell-based immunotherapies in glioblastoma. Previous work on ipilimumab in melanoma brain metastases demonstrated that dexamethasone may interfere with its efficacy (Margolin et al., 2012). Additionally, recent work in glioblastoma investigated mechanisms of dexamethasone-induced immunosuppression and their effects on brain tumour immunotherapy (Giles et al., 2018). These investigations must continue if dexamethasone and immunotherapy are to co-exist in the therapeutic repertoire of clinicians in neuro-oncology. Furthermore, if IL-1 inhibition is to be considered as an alternative to dexamethasone for the management of oedema, its precise effects on the action of the immune microenvironment in glioblastoma must be established.
In this work, we used a Hoechst dye-based assay to interrogate blood–brain barrier permeability, as well as an MRI-based assay and wet/dry tissue weight assay for determination of oedema at endpoint as a function of IL-1 signalling. We demonstrated a reduction in blood–brain barrier integrity and oedema formation, respectively, when either IL-1R1, IL-1β, or both IL-1α and IL-1β were homozygous lost compared to wild-type tumour-bearing mice. Considering the collection of clinically-approved drugs for specific IL-1 signalling inhibition (Dinarello et al., 2012), and the demonstration that IL-1β inhibition reduces oedema formation in traumatic brain injury (Clausen et al., 2011), IL-1 signalling inhibition warrants additional investigation in the context of management of glioblastoma-associated cerebral oedema.
Supplementary Material
Acknowledgements
We would like to acknowledge the Emory Flow Cytometry Core, Integrated Cellular Imaging Core, and Pathology Core Lab for their services as well as David R. Schumick for generating illustrations. Thank you to Genentech for graciously providing the B20-4.1.1 for our experiment. C.H. would like to extend significant thanks to the Molecular and Systems Pharmacology program.
Funding
This work was supported by NIH/NINDS R01 NS100864 and from the Aflac Cancer and Blood Disorders Center start-up funds for D.H. PSTP Training Grant 4T32GM008602-20 and NIH/NINDS 1F31NS106887 provided funding for C.H.
Competing interests
The authors have no relevant competing interests to disclose.
Glossary
Abbreviations
- BMDM =
bone marrow-derived macrophage
- LPS =
lipopolysaccharide
- MCP =
monocyte chemoattractant protein
- VEGF =
vascular endothelial growth factor
References
- Albrecht U, Yang X, Asselta R, Keitel V, Tenchini ML, Ludwig S, et al. Activation of NF-kappaB by IL-1beta blocks IL-6-induced sustained STAT3 activation and STAT3-dependent gene expression of the human gamma-fibrinogen gene. Cell Signal 2007; 19(9): 1866–78. [DOI] [PubMed] [Google Scholar]
- Aranda PS, LaJoie DM, Jorcyk CL. Bleach gel: a simple agarose gel for analyzing RNA quality. Electrophoresis 2012; 33: 366–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, et al. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol 2006; 177: 5574–84. [DOI] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 1995; 270: 286–90. [DOI] [PubMed] [Google Scholar]
- Badie B, Schartner JM, Paul J, Bartley BA, Vorpahl J, Preston JK. Dexamethasone-induced abolition of the inflammatory response in an experimental glioma model: a flow cytometry study. J Neurosurg 2000; 93: 634–9. [DOI] [PubMed] [Google Scholar]
- Barksby HE, Lea SR, Preshaw PM, Taylor JJ. The expanding family of interleukin-1 cytokines and their role in destructive inflammatory disorders. Clin Exp Immunol 2007; 149: 217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuscher HU, Gunther C, Rollinghoff M. IL-1 beta is secreted by activated murine macrophages as biologically inactive precursor. J Immunol 1990; 144: 2179–83. [PubMed] [Google Scholar]
- Burton JL, Kehrli ME Jr.. Effects of dexamethasone on bovine circulating T lymphocyte populations. J Leukoc Biol 1996; 59: 90–9. [DOI] [PubMed] [Google Scholar]
- Carlson MR, Pope WB, Horvath S, Braunstein JG, Nghiemphu P, Tso CL, et al. Relationship between survival and edema in malignant gliomas: role of vascular endothelial growth factor and neuronal pentraxin 2. Clin Cancer Res 2007; 13: 2592–8. [DOI] [PubMed] [Google Scholar]
- Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res 2017; 77: 2266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen F, Hanell A, Israelsson C, Hedin J, Ebendal T, Mir AK, et al. Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur J Neurosci 2011; 34: 110–23. [DOI] [PubMed] [Google Scholar]
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol 2011; 335: 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG, et al. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol 1987; 139: 1902–10. [PubMed] [Google Scholar]
- Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 2012; 11: 633–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French LA. The use of steroids in the treatment of cerebral edema. Bull N Y Acad Med 1966; 42: 301–11. [PMC free article] [PubMed] [Google Scholar]
- Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol 2010; 6: 232–41. [DOI] [PubMed] [Google Scholar]
- Galicich JH, French LA, Melby JC. Use of dexamethasone in treatment of cerebral edema associated with brain tumors. J Lancet 1961; 81: 46–53. [PubMed] [Google Scholar]
- Giles AJ, Hutchinson MND, Sonnemann HM, Jung J, Fecci PE, Ratnam NM, et al. Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J Immunother Cancer 2018; 6: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschall PE, Tatsuno I, Arimura A. Increased sensitivity of glioblastoma cells to interleukin 1 after long-term incubation with dexamethasone. Mol Cell Neurosci 1992; 3: 49–55. [DOI] [PubMed] [Google Scholar]
- Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, et al. Differential expression of NLRP3 among hematopoietic cells. J Immunol 2011; 186: 2529–34. [DOI] [PubMed] [Google Scholar]
- Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol 2009; 2: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 2016; 19: 20–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedley-Whyte ET, Hsu DW. Effect of dexamethasone on blood-brain barrier in the normal mouse. Ann Neurol 1986; 19: 373–7. [DOI] [PubMed] [Google Scholar]
- Heiss JD, Papavassiliou E, Merrill MJ, Nieman L, Knightly JJ, Walbridge S, et al. Mechanism of dexamethasone suppression of brain tumor-associated vascular permeability in rats. Involvement of the glucocorticoid receptor and vascular permeability factor. J Clin Invest 1996; 98: 1400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herting CJ, Chen Z, Pitter KL, Szulzewsky F, Kaffes I, Kaluzova M, et al. Genetic driver mutations define the expression signature and microenvironmental composition of high-grade gliomas. Glia 2017; 65: 1914–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue CD, Cho FS, Cao S, Dale Bass CR, Meaney DF, Morrison B 3rd. Dexamethasone potentiates in vitro blood-brain barrier recovery after primary blast injury by glucocorticoid receptor-mediated upregulation of ZO-1 tight junction protein. J Cereb Blood Flow Metab 2015; 35: 1191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MA, Parisi M, Grossman S, Kleinberg L. Primary brain tumors treated with steroids and radiotherapy: low CD4 counts and risk of infection. Int J Radiat Oncol Biol Phys 2005; 62: 1423–6. [DOI] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun 1996; 224: 855–62. [DOI] [PubMed] [Google Scholar]
- Jones GE. Cellular signaling in macrophage migration and chemotaxis. J Leukoc Biol 2000; 68: 593–602. [PubMed] [Google Scholar]
- Kamoun WS, Ley CD, Farrar CT, Duyverman AM, Lahdenranta J, Lacorre DA, et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol 2009; 27: 2542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BY, Son Y, Lee J, Choi J, Kim CD, Bae SS, et al. Dexamethasone inhibits activation of monocytes/macrophages in a milieu rich in 27-oxygenated cholesterol. PLoS One 2017; 12: e0189643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofman S,, Garvin JS, Nagamani D, Taylor SG 3rd. Treatment of cerebral metastases from breast carcinoma with prednisolone. J Am Med Assoc 1957; 163: 1473–6. [DOI] [PubMed] [Google Scholar]
- Kostaras X, Cusano F, Kline GA, Roa W, Easaw J. Use of dexamethasone in patients with high-grade glioma: a clinical practice guideline. Curr Oncol 2014; 21: e493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsarini C, Griffiths PD, Wilkinson ID, Hoggard N. A systematic review of the literature on the effects of dexamethasone on the brain from in vivo human-based studies: implications for physiological brain imaging of patients with intracranial tumors. Neurosurgery 2010; 67: 1799–815; discussion 815. [DOI] [PubMed] [Google Scholar]
- Luedi MM, Singh SK, Mosley JC, Hatami M, Gumin J, Sulman EP, et al. A dexamethasone-regulated gene signature is prognostic for poor survival in glioblastoma patients. J Neurosurg Anesthesiol 2017; 29: 46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machein MR, Kullmer J, Ronicke V, Machein U, Krieg M, Damert A, et al. Differential downregulation of vascular endothelial growth factor by dexamethasone in normoxic and hypoxic rat glioma cells. Neuropathol Appl Neurobiol 1999; 25: 104–12. [DOI] [PubMed] [Google Scholar]
- March CJ, Mosley B, Larsen A, Cerretti DP, Braedt G, Price V, et al. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature 1985; 315: 641–7. [DOI] [PubMed] [Google Scholar]
- Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol 2012; 13: 459–65. [DOI] [PubMed] [Google Scholar]
- McClelland S 3rd, Long DM. Genesis of the use of corticosteroids in the treatment and prevention of brain edema. Neurosurgery 2008; 62: 965–7; discussion 7–8. [DOI] [PubMed] [Google Scholar]
- Ostergaard L, Hochberg FH, Rabinov JD, Sorensen AG, Lev M, Kim L, et al. Early changes measured by magnetic resonance imaging in cerebral blood flow, blood volume, and blood-brain barrier permeability following dexamethasone treatment in patients with brain tumors. J Neurosurg 1999; 90: 300–5. [DOI] [PubMed] [Google Scholar]
- Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, et al. Corticosteroids compromise survival in glioblastoma. Brain 2016; 139 (Pt 5): 1458–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 2009; 25: 1463–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raslan A, Bhardwaj A. Medical management of cerebral edema. Neurosurg Focus 2007; 22: E12. [DOI] [PubMed] [Google Scholar]
- Remmelts HH, Meijvis SC, Biesma DH, van Velzen-Blad H, Voorn GP, Grutters JC, et al. Dexamethasone downregulates the systemic cytokine response in patients with community-acquired pneumonia. Clin Vaccine Immunol 2012; 19: 1532–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy J. Primary microglia isolation from mixed cell cultures of neonatal mouse brain tissue. Brain Res 2018; 1689: 21–9. [DOI] [PubMed] [Google Scholar]
- Schuld A, Kraus T, Haack M, Hinze-Selch D, Zobel AW, Holsboer F, et al. Effects of dexamethasone on cytokine plasma levels and white blood cell counts in depressed patients. Psychoneuroendocrinology 2001; 26: 65–76. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–96. [DOI] [PubMed] [Google Scholar]
- Tait MJ, Saadoun S, Bell BA, Verkman AS, Papadopoulos MC. Increased brain edema in aqp4-null mice in an experimental model of subarachnoid hemorrhage. Neuroscience 2010; 167: 60–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mourik JA, Leeksma OC, Reinders JH, de Groot PG, Zandbergen-Spaargaren J. Vascular endothelial cells synthesize a plasma membrane protein indistinguishable from the platelet membrane glycoprotein IIa. J Biol Chem 1985; 260: 11300–6. [PubMed] [Google Scholar]
- Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, et al. Interleukin-1beta induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS One 2014; 9: e110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner SJ, Auger KR, Libby P. Human interleukin 1 induces interleukin 1 gene expression in human vascular smooth muscle cells. J Exp Med 1987a; 165: 1316–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner SJ, Auger KR, Libby P. Interleukin 1 induces interleukin 1. II. Recombinant human interleukin 1 induces interleukin 1 production by adult human vascular endothelial cells. J Immunol 1987b; 139: 1911–7. [PubMed] [Google Scholar]
- Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc 2008; 2008: pdb prot5080. [DOI] [PubMed] [Google Scholar]
- Wewers MD, Sarkar A. P2X(7) receptor and macrophage function. Purinergic Signal 2009; 5(2): 189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ET, Lok E, Gautam S, Swanson KD. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br J Cancer 2015; 113: 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ET, Swanson KD. Dexamethasone-friend or foe for patients with glioblastoma? JAMA Neurol 2019; 76: 247–8. [DOI] [PubMed] [Google Scholar]
- Xuan W, Qu Q, Zheng B, Xiong S, Fan GH. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J Leukoc Biol 2015; 97: 61–9. [DOI] [PubMed] [Google Scholar]
- Yoshimura T. The production of monocyte chemoattractant protein-1 (MCP-1)/CCL2 in tumor microenvironments. Cytokine 2017; 98: 71–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data used for figure generation in this manuscript can be obtained by contacting the corresponding author.