

Graphical abstract

Keywords: Coevolution, Mutual information, Protein-protein interaction, Protein network, Evolution
Abstract
Here, we investigate the contributions of coevolutive, evolutive and stochastic information in determining protein-protein interactions (PPIs) based on primary sequences of two interacting protein families A and B. Specifically, under the assumption that coevolutive information is imprinted on the interacting amino acids of two proteins in contrast to other (evolutive and stochastic) sources spread over their sequences, we dissect those contributions in terms of compensatory mutations at physically-coupled and uncoupled amino acids of A and B. We find that physically-coupled amino-acids at short range distances store the largest per-contact mutual information content, with a significant fraction of that content resulting from coevolutive sources alone. The information stored in coupled amino acids is shown further to discriminate multi-sequence alignments (MSAs) with the largest expectation fraction of PPI matches – a conclusion that holds against various definitions of intermolecular contacts and binding modes. When compared to the informational content resulting from evolution at long-range interactions, the mutual information in physically-coupled amino-acids is the strongest signal to distinguish PPIs derived from cospeciation and likely, the unique indication in case of molecular coevolution in independent genomes as the evolutive information must vanish for uncorrelated proteins.
1. Introduction
While being selected to be thermodynamically stable and kinetically accessible in a particular fold [1], [2], interacting proteins A and B coevolve to maintain their bound free-energy stability against a vast repertoire of non-specific partners and interaction modes. Protein coevolution, in the form of a time-dependent molecular process, then translates itself into a series of primary-sequence variants of A and B encoding coordinated compensatory mutations [3] and, therefore, specific protein-protein interactions (PPIs) derived from this stability-driven process [4]. As a ubiquitous process in molecular biology, coevolution thus apply to protein interologs, either paralogous or orthologous, under cospeciation or in independent genomes.
Thanks to extensive investigations in the past following ingenious approaches based on the correlation of phylogenetic trees [5], [6], [7] and profiles [8], gene colocalization [9] and fusions [10], maximum coevolutionary interdependencies [11] and correlated mutations [12], [13], the problem of predicting PPIs based on multi-sequence alignments (MSAs) appears to date resolvable, at least for small sets of paralogous sequences – recent improvements [14], [15], [16], [17], [18] resulting from PPI prediction allied to modern coevolutionary approaches [19], [20], [21], [22], [23] to identify interacting amino acids across protein interfaces. In these previous studies, however, the information was taken into account as a whole, and it was not clarified, as discussed in recent reviews [4], [24], the isolated contributions of coevolutive, evolutive and stochastic information in resolving the problem. Differentiating functional coevolution from stochastic and phylogenetic sources remains looked for in the research field and may help introducing models capable of accurately detecting protein-protein interactions and interfaces, especially when the number of sequences or the amount of biological information are limited [25].
Here, by benefiting from much larger data sets made available in the sequence- and structure-rich era, we revisit the field by quantifying the amount of information that protein A stores about protein B stemming from each of these sources and, more importantly, their effective contributions in discriminating PPIs based on MSAs (Scheme 1). Specifically, under the assumption that the coevolutive information is imprinted on the interacting amino acids of protein interologs in contrast to other (evolutive and stochastic) sources spread over their sequences, we want the information to be dissected in terms of compensatory mutations at physically-coupled and uncoupled amino acids of A and B. Given a known set of protein three-dimensional amino-acid contacts and their underlying primary sequences we seek therefore differentiating functional coevolution from stochastic and phylogenetic signals for subsequent evaluation of their contributions in PPI recognition from primary sequences. It is worth emphasizing our study is not aimed at providing a method for prediction of protein-protein interactions nor protein-protein interfaces, hence it differs from previous studies in which sequence covariance is used to predict three-dimensional amino-acid contacts across interfaces and assemble models of protein complexes [26] or protein docking [27]. Anticipating our findings, we show that physically-coupled amino-acids store the largest per-contact mutual information (MI) content to discriminate concatenated MSAs with the largest expectation fraction of PPI matches – a conclusion that holds against various definitions of intermolecular protein contacts and binding modes, including native and non-native decoy structures. A significant fraction of that information results from coevolutive sources alone. Although, our analysis involved protein interologs under cospeciation that is, proteins evolving in the same genome, the derived conclusions are likely general to cases of non-cospeciating interologs given that the underlying thermodynamical principles must be the same for all cases.
Scheme 1.

Structural contacts mapped into M-long multi-sequence alignment (MSA) of protein interologs A and B. A set of pairwise protein-protein interactions is defined by associating each sequence l in MSA B to a sequence k in MSA A in one unique arrangement, {l(k)|z}M, determined by the coevolution process z to which these protein families were subjected. Shown is a “scrambled” concatenated MSA of A and B associated to a given process z (red dashes). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2. Theory and methods
2.1. Decomposition of mutual information
In detail, consider two proteins A and B that interact via formation of i = 1,…,N amino-acid contacts at the molecular level. Proteins A and B are assumed to coevolve throughout M! distinct processes z described by the stochastic variable Z with an uniform probability mass function ρ(z), ∀z ∈ {1,…,M!}. Given any specific process z, their interacting amino-acid sequences are respectively described by two N-length blocks of discrete stochastic variables XN ≡ (X1,…,XN) and YN ≡ (Y1,…,YN) with probability mass functions {ρ(xN),ρ(yN),ρ(xN,yN|z)} such that,
| (1) |
and
| (2) |
for every joint sequence defined in the alphabet χ of size |χ|. Under these considerations, the amount of information that protein A stores about protein B is given by the mutual information I(XN; YN|z) between XN and YN conditional to process z [28]. As made explicit in Eq. (1), we are particularly interested in quantifying I(XN; YN|z) for the situation in which marginals of the N-block variables {ρ(xN), ρ(yN)} are assumed to be independent of process z meaning that, for a fixed sequence composition of proteins A and B only their joint distribution depends on the process. Furthermore, by assuming N-independent contacts, we want that information to be quantified for the least-constrained model ρ*(xN, yN|z) that maximizes the conditional joint entropy between A and B – that condition ensures the mutual information to be written exactly, in terms of the individual contributions of contacts i.
For the least-constrained distribution {ρ*(xN, yN|z)}, the conditional mutual information
| (3) |
writes in terms of the Shannon’s information entropies
| (4) |
associated with the conditional joint distribution {ρ*(xN, yN|z)} and the derived marginals {ρ*(xN), ρ*(yN)} of the N-block variables. From its entropy-maximization property, the critical distribution {ρ*(xN, yN|z)} factorizes into the conditional two-site marginal of every contact i
| (5) |
then allowing Eq. (4) to be written extensively, in terms of the individual entropic contributions
| (6) |
such that,
| (7) |
(cf. SI for details). In Eq. (7), the conditional mutual information achieves its lower bound of zero if XN and YN are conditionally independent given z i.e., ρ*(xN, yN |z) = ρ*(xN) × ρ*(yN). For the case of perfectly correlated variables ρ*(xN, yN |z) = ρ*(xN) = ρ*(yN), the conditional mutual information is bound to a maximum which cannot exceed the entropy of either block variables H(XN) and H(YN).
Given a known set of protein amino-acid contacts and their underlying primary sequence distributions defining the stochastic variables XN and YN, Eq. (7) thus establishes the formal dependence of their mutual information with any given process z. Because “contacts” can be defined for a variety of cutoff distances rc, Eq. (7) is particularly useful to dissect mutual information in terms of physically-coupled and uncoupled protein amino acids. In the following, we explore Eq. (7) in that purpose by obtaining the two-site probabilities in Eq. (5)
| (8) |
from the observed frequencies in the multiple-sequence alignment
where the N-length amino-acid block l of protein B is joint to block k of protein A in one unique arrangement {l(k)|z}M for 1 ≤ k ≤ M (cf. Scheme 1 and Computational Methods).
2.2. Computational methods
Table 1 details the interacting protein systems considered in the study. For each system under investigation, amino-acid contacts defining the discrete stochastic variables XN and YN including physically coupled amino acids at short-range cut-off distances (rc ≤ 8.0 Å) and physically uncoupled amino-acids at long-range cut-off distances (rc > 8.0 Å) were identified from the x-ray crystal structure of the bound state of proteins A and B. The reference (native) multi-sequence alignment {xNk, yNl|z*}M of the joint amino-acid blocks associated to XN and YN was reconstructed from annotated primary-sequence alignments published by Baker and coworkers [22], containing M paired sequences with known protein-protein interactions and defined in the alphabet of 20 amino acids plus the gap symbol (|χ|=21). “Scrambled” MSA models were generated by randomizing the pattern {l(k)|z*}M in which block l is joint to block k in the reference alignment.
Table 1.
Protein system A and B considered in the study.
| Complex description | PDB ID | Protein A | Protein B | M | MSA length | |
|---|---|---|---|---|---|---|
| Obligate Dimers | Carbamoyl Phosphate Synthetase | 1BXR | Chain A: Carbamoyl-Phosphate Synthetase large subunit | Chain B: Carbamoyl-Phosphate Synthetase small subunit | 1004 | 1452 |
| Lactococcus Lactis Dihydroorotate Dehydrogenase B. | 1EP3 | Chain A: Dihydroorotate Dehydrogenase B (PYRD Subunit) | Chain B: Dihydroorotate Dehydrogenase B (pyrk Subunit) | 552 | 572 | |
| Polysulfide reductase native structure | 2VPZ | Chain A: Thiosulfate Reductase | Chain B: NRFC Protein | 676 | 927 | |
| heterohexameric TusBCD proteins | 2D1P | Chain B: Hypothetical UPF0116 protein yheM | Chain C: Hypothetical protein yheL | 216 | 214 | |
| 3-oxoadipate coA-transferase | 3RRL | Chain A: Succinyl-CoA:3-ketoacid-coenzyme A transferase subunit A | Chain B: Succinyl-CoA:3-ketoacid-coenzyme A transferase subunit B | 1330 | 437 | |
| Bovine heart cytochrome c oxidase | 2Y69 | Chain A: Cytochrome C Oxidase Subunit 1 | Chain B: Cytochrome C Oxidase Subunit 2 | 1484 | 740 | |
| Non-Obligate Dimer | Toxin-antitoxin complex RelBE2 from Mycobacterium tuberculosis | 3G5O | ChainA: Protein Rv2865 | ChainB: Protein Rv2866 | 904 | 173 |
For any given MSA model, two-site probabilities were defined from the observable frequencies regularized by a pseudocount effective fraction λ* in case of insufficient data availability as devised by Morcos and coauthors [19]. More specifically, two-site frequencies were calculated according to
| (9) |
where, nmz = |{m′|1 ≤ m′ ≤ M, Hamming Disatnce(m,m′) ≥ δh}| is the number of similar sequences m′ within a certain Hamming distance δh of sequence m and is the effective number of distinguishable primary sequences at that distance threshold – the Kronecker delta ensures counting of (xi,yi) occurrences only. In Eq. (9), two-site frequencies converge to raw occurrences in the sequence alignment for λ* = 0 or approach the uniform distribution for λ* = 1; Eq. (9) is identical to the equation devised by Morcos and coauthors [19] by rewriting λ* = λ/(λ + Meffz). Here, two-site probabilities were computed from Eq. (9) after unbiasing the reference MSA by weighting down primary sequences with amino-acid identity equal to 100%. An effective number of primary sequences Meffz = M (cf. Table S1) was retained for analysis and a pseudocount fraction of λ* = 0.001 was used to regularize data without largely impacting observable frequencies. Single-site probabilities {ρ(xN), ρ(yN)} were derived from ρ*(xi, yi|z) by marginalization via Eq. (1).
The conditional mutual information in Eq. (7) was computed from single- and joint-entropies according to Eq. (3). Given the fact that the maximum value of I(Xi; Yi|z) is bound to the conditional joint entropy, Eq. (7) was computed in practice as a per-contact entropy-weighted conditional mutual information [29], H(Xi; Yi|z)−1 I(Xi; Yi|z), to avoid that contributions of H(Xi, Yi|z) contacts between highly variable sites are overestimated. Because and I(Xi, Yi|z) have units of nats, Eq. (7) is dimensionless in the present form.
3. Results and discussion
Details of all protein systems under investigation are presented in Table 1. Each system involves two families of protein interologs A and B with known PPIs derived from cospeciation in the same genome [26]. We denote by {xNk, yNl|z*}M their reference concatenated MSA associated to the native process z*. For convenience, in the following, we present and discuss results obtained for a representative system A and B – the protein complex TusBCD (chains B and C of 2DIP) which is crucial for tRNA modification in Escherichia coli. Similar results and conclusions hold for all other systems in Table 1 as presented in supplementary Figs. S1 through S4 (cf. SI).
3.1. Decomposition of mutual information
Fig. 1A shows the three-dimensional representation of stochastic variables embodying every possible amino-acid pairs along proteins A and B and their decomposition in terms of physically coupled amino acids at short-range cutoff distances (rc ≤ 8.0 Å) and physically uncoupled amino-acids at long-range cutoff distances (rc > 8.0 Å). In Fig. 1B, the total mutual information (coupled + uncoupled) across every possible amino-acid pairs of A and B amounts to 987.88 in the reference (native) MSA. As estimated from a generated ensemble of “scrambled” MSA models, expectation values for the mutual information <I(XN; YN|z)>M−n decreases significantly as decorrelation or the number of mismatched proteins in the reference MSA increases. The result also holds at the level of individual protein contacts i as the mutual information I(Xi; Yi|z*) for the reference alignment is systematically larger than the mutual information expectation value for “scrambled” MSA models full of sequence mismatches that is, with a total number M of mismatched sequences (Fig. S1).
Fig. 1.
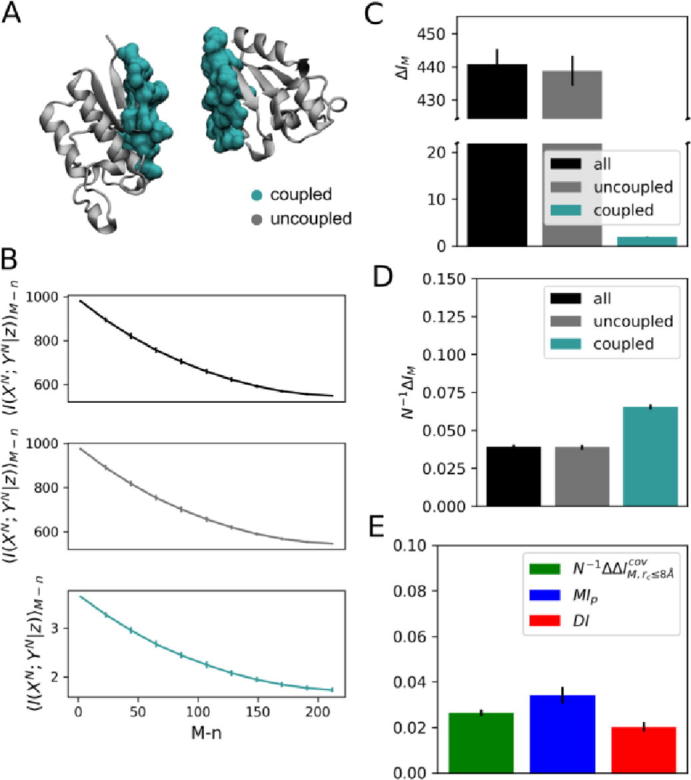
Informational analysis of protein complex TusBCD, chains B and C. (A) Three-dimensional representation of stochastic variables XN and YN as defined from physically coupled amino acids at short-range cutoff distances rc ≤ 8.0 Å (turquoise) and physically uncoupled amino-acids at long-range cutoff distances rc > 8.0 Å (gray). Calculation of rc involved Cβ-Cβ atomic separation distances. (B) Conditional mutual information <I(XN; YN|z)>M−n as a function of the number M − n of randomly paired proteins in the reference (native) MSA, for 0 ≤ n ≤ M. < I(XN; YN|z)>M−n are expectation values estimated from a generated ensemble of 500 MSA models. Mutual information of fully “scrambled” models featuring M unpaired sequences is similar to that calculated from randomized sequence alignments generated by aleatory swapping of lines within columns. (C) Mutual information gap ΔIM between reference and 100 fully “scrambled” models featuring M unpaired sequences. (D) Per-contact mutual information gap N−1ΔIM,rc. (E) Mutual information decomposition according to Eq. (11) and comparison with functional mutual information (MIp,rc≤8Å) and direct information (DIrc≤8Å). In B, C, D and E error bars correspond to standard deviations.
As a measure of correlation, it is not surprising that mutual information in the reference MSA is larger than that of scrambled alignments. Not expected however, is the fact that correlation does not vanish at “scrambled” models meaning that part of the calculated mutual information results at random. Supporting that notion, the mutual information of fully “scrambled” models is found here to be very similar to the same estimate from randomized sequence alignments featuring aleatory swapping of lines within columns. Subtraction of that stochastic source from the native mutual information, as computed in the form of an information gap
| (10) |
between the reference MSA and “scrambled” models full of sequence mismatches, then reveals the isolated nonstochastic contributions to the total correlation between proteins A and B. Here, the information gap amounts to ~440 for every possible amino-acid pairs of A and B.
Fig. 1C shows the individual contributions of physically coupled and uncoupled amino acids to the total mutual information gap, ΔIM = ΔIM,rc≤8.0Å + ΔIM,rc>8.0Å. As a direct consequence of the extensive property of Eq. (7), individual contributions to the total mutual information gap () increase with cutoff distances defining amino-acid contacts (rc) and consequently, with the block length (N) of the corresponding stochastic variables. As such, the information imprinted at physically uncoupled amino acids accounts for most of the total mutual information gap (438.8132 ± 4.5159). When normalized by the block length or the number of amino-acid contacts (Fig. 1D), the mutual-information contribution N−1ΔIM,rc reveals a distinct dependence being larger for physically coupled amino acids than uncoupled ones (0.0653 ± 0.0015 versus 0.039 ± 0.0004). The information-gap profile as a function of amino-acid pair distances shown in Fig. S2 makes sense of the result by showing few larger information-gap values at short distances in contrast to many smaller ones at long distances.
Under the assumption that the coevolutive information is imprinted on the interacting amino acids of interologs in contrast to other (evolutive and stochastic) sources spread over their primary sequences, the difference between short- and long-range contributions provides us with per-contact estimates for the information content resulting from coevolution alone that is,
| (11) |
where, represents the per-contact mutual information resulting from evolution. As shown in Fig. 1E, the information content resulting from coevolution alone amounts to 0.0264 ± 0.0014 which compares well to independent measures of coevolutionary information i.e., functional mutual information () [29] and direct information () [19], 0.0340 ± 0.0037 and 0.0202 ± 0.0019. More specifically, MIp is a metric formulated by Dunn and coworkers [29] in which mutual information is subtracted from structural or functional relationships whereas, DI is based on the direct coupling analysis that removes all kinds of indirect correlations by following a global statistical approach [19]. According to definition in Eq. (11), we then conclude that ~40% of the information content stored in physically coupled amino acids of the protein complex TusBCD results from coevolutive sources alone.
3.2. Degeneracy and error analysis of short and long-range correlations
The present analysis reveals quantitative differences between short- and long-range correlations of proteins A and B. Because the total mutual-information component provides us with an unbiased (intensive) estimate for proper comparison of the information content between coupled and uncoupled amino acids, in the following, we focus our attention on to dissect their effective contributions in determining PPIs based on sequence alignments. Accordingly, let us define the total number ωS of native-like MSA models generated by scrambling of M − n sequence pairs in the reference alignment
| (12) |
in terms of rencontres numbers ωM,n
| (13) |
or permutations of the reference sequence set {l(k)|z*}M with n fixed positions satisfying ∑Mn=0 ωM,n = M! (in combinatorics language). Here, S(rc) denotes the set of fixed positions n
| (14) |
for which the mutual information gap is smaller than a certain resolution δI independently from the corresponding block length N or the number of amino-acid contacts. In simple terms, ωS in Eq. (12) informs us on the degeneracy or the number of “scrambled” MSA models with a similar amount of mutual information of that in the reference (native) alignment.
As shown in Table S1, rencontres numbers ωM,n is an astronomically increasing function of M − n, identical for any definition of the stochastic variables XN and YN derived from the same number M of aligned sequences. For instance, there is 164548102752 alignments for the protein complex TusBCD with M − n = 5 scrambled sequence pairs. In contrast, the total number ωS of native-like MSA models depends on the stochastic variables at various resolutions δI (Fig. 2A). That number is substantially smaller for definitions of XN and YN embodying physically-coupled amino acids in consequence of the smaller number M − n of unpaired sequences required to perturb of a fixed change δI such that ωS accumulates less over MSA models satisfying the condition in Eq. (14) (Fig. 2B).
Fig. 2.
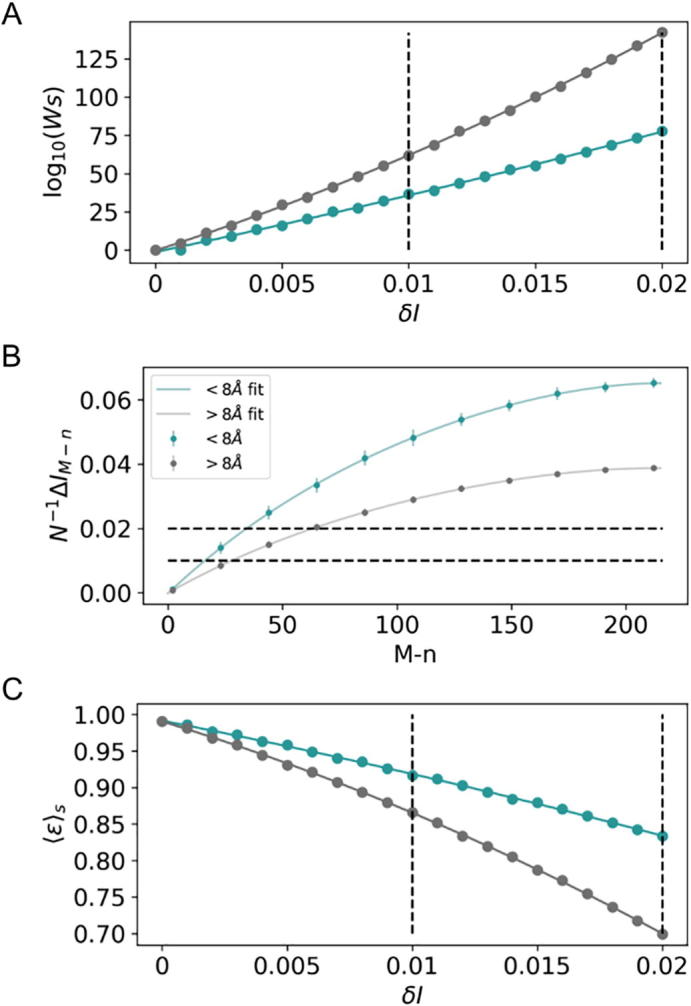
Degeneracy and error analysis for stochastic variables XN and YN involving interacting amino acids at short-range distances rc ≤ 8.0 Å (turquoise) and long-range distances rc > 8.0 Å (gray). (A) Total number ωS of native-like MSA models at various mutual-information resolutions δI. (B) Per-contact gaps of mutual information N−1ΔIM−n,rc as a function of the number M − n of “scrambled” sequence pairs in the reference native alignment. (C) Expectation values <ε>S (Eq. (15)) for the fraction of sequence matches across native-like MSA models at various mutual-information resolutions δI. Dashed lines highlight differences at δI values of 0.01 and 0.02.
The degeneracy of native-like MSA models at a given resolution depends on the cutoff distance defining stochastic variables (Fig. 2A). That condition imposes distinct boundaries for the amount of PPIs amenable of resolution across definitions of the stochastic variables in terms of coupled and uncoupled amino acids. Indeed, the expectation value
| (15) |
for the fraction M−1n of primary sequence matches among native-like MSA models decreases substantially with the degeneracy of such models meaning that <ε>S is systematically larger for physically-coupled amino-acids at various mutual-information resolutions δI (Fig. 2C). For instance, the fraction of matches at δI = 0.02 is ~20% larger for coupled amino-acids than the same estimate for amino acids at long-range distances (0.8333 versus 0.6991). Linear extrapolation in Fig. 2C along increased values of mutual-information resolutions suggests even larger differences in the expectation fraction of PPI matches between short and long-range correlations of A and B.
3.3. Dependence with contact definition and docking decoys
So far, “contact” is actually any given pair of residues “i” in protein A and “j” in protein B within a given distance rc* which can be redefined for a variety of cutoff distances. Specifically, our results were determined by defining physically coupled amino acids at short-range cutoff distances (rc ≤ rc*) and physically uncoupled amino-acids at long-range cutoff distances (rc > rc*) for a typical “contact” geometrical definition involving Cβ-Cβ atomic separation distances of 8.0 Å (that is, Å). In the following, amino-acid “contacts” are loosely redefined for a variety of cutoff distances to study the dependence of the information encoded in short and long-range protein interactions with rc*. Further analysis shows a clear dependence of the per-contact mutual information gap (N−1ΔIM,rc) of coupled amino acids with rc* – which is not the case for uncoupled ones. As shown in Fig. 3A, that distinction is due the coevolutive information stored at short-range distances which reaches a maximum at rc* ≈ 8.0 Å in contrast to evolutive sources uniformly spread over an entire range of rc* values. Particularly interesting, the result strongly support the assumption that coevolutive information is imprinted preferentially on physically-coupled amino acids of interologs in contrast to other (evolutive and stochastic) sources spread over their primary sequences – a conclusion further supported by calculations of the mutual information subtracted from structural-functional relationships (MIP) as a function of rc*.
Fig. 3.

Dependence with contact definition rc* and docking decoys. (A) Per-contact mutual information gap N−1ΔIM,rc and mutual information subtracted from structural-functional relationships MIp,rc at various rc*. (B) Per-contact mutual information gap N−1ΔIM,rc (turquoise), information content resulting from coevolution alone N−1ΔΔICovM,rc (green) and mutual information subtracted from structural or functional relationships MIp,rc (blue) at alternative interfaces generated by docking – only physically coupled amino acids as defined for rc ≤ 8.0 Å were included in the calculations. Black bars represent the root-mean-square deviation (RMSD in Ȧ units) between the native bound structure and docking decoys as generated by GRAMM-X [30]. Docking solutions were selected following a stability binding-energy criterium according to the scoring function of GRAMM – all docking decoys considered in the study are low-energy configurations despite large RMSD values relative to the native structure. (C) Illustration of four docking decoys of chain B in the protein complex TusBCD (chain C is shown in gray). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Still, the information encoded in short and long-range amino-acid interactions was analyzed across the native binding interface between proteins as revealed by x-ray crystallography experiments. The dependence of the per-contact mutual information gap with non-native binding modes or docking decoys of proteins A and B was then analyzed further, at the typical definition of amino-acid contacts ( Å). Shown in Fig. 3B, there is a clear dependence of the information gap with binding modes – the per-contact mutual information gap reaches a maximum at the experimentally-determined native bound configuration of A and B (RMSD = 0.0 Å), meaning that N−1ΔIM,rc embodies coevolutive pressures in the native amino acids contacts beyond their accessibility at the molecular surface of proteins. The conclusion is further supported in Fig. 3B by noticing that the isolated coevolutive content for the bound configuration of A and B or the associated mutual information subtracted from structural-functional relationships are larger than the very same estimates for any docking decoys.
4. Concluding remarks
Overall, molecular coevolution as the maintenance of the binding free-energy of interacting proteins leads their physically coupled amino-acids to store the largest per-contact mutual information at rc* ≈ 8.0 Å, with a significant fraction of the information resulting from coevolutive sources alone. In the present formulation, coupled amino acids are related to the smallest degeneracy of native-like MSA models and, therefore, to the largest expectation fraction of PPI matches across such models. These findings hold against any other definition of protein contacts, either across a variety of limitrophe distances discriminating coupled and uncoupled amino acids or alternative binding interfaces in docking decoys. Although presented for the protein complex TusBCD, results and discussion also extent to other protein systems, including obligate and non-obligate dimers, as shown in supplementary Figs. S1 through S4 (cf. SI).
Advances in PPI prediction [14], [15], [16], [17], [18] are highly welcome in the contexts of paralog matching, host-pathogen PPI network prediction and interacting protein families prediction. Recent studies suggest strategies like maximizing the interfamily coevolutionary signal [14], iterative paralog matching based on sequence “energies” [15] and expectation–maximization [18], which have been capable of accurately matching paralogs for some study cases. Despite these advances, the problem of PPI prediction remains unsolved for sequence ensembles in general, especially for proteins that coevolve in independent genomes though likely resulting from the same free-energy constraints – examples are phage proteins and bacterial receptors, pathogen and host-cell protein, neurotoxins and ion channels, to mention a few. Accordingly, to add efforts in the field, we have addressed the following questions in our study: knowing three-dimensional amino-acid contacts from x-ray crystal structures, what would be the information encoded by them in terms of stochastic, evolutive and coevolutive sources, and what would be the utility of such pieces of information in resolving PPIs from “scrambled” multi-sequence alignments. Since the Direct Information derived from modern coevolutionary approaches [19], [22] already filters out most of the information sources, the decomposition as proposed here does only make sense by considering the Mutual Information embodying unfiltered information. In this regard, it is worth emphasizing that our goals are neither the resolution of pair of residues highly-correlated via direct physical coupling [19], [22] nor to provide with a method for prediction of protein-protein interactions and interfaces [26], [27].
Although our study is not aimed at providing an approach for PPI prediction, the largest amount of non-stochastic information available in primary sequences helpful to differentiate MSA models with the largest expectation fraction of sequence matches as found here, might be of practical relevance in search of more effective heuristics to resolve protein-protein interactions from “scrambled” multi-sequence alignments. When compared to evolutive sources, that information is the strongest signal to characterize protein interactions derived from cospeciation and likely, the unique indication in case of coevolution without cospeciation as the non-stochastic information of uncoupled amino acids must vanish in independent proteins – indeed, low information between amino acid positions of multiple sequence alignments is typically indicative of independently evolved proteins. Developments of more effective heuristics based on that signal would be applied for resolution of the more general problem of PPIs under coevolution in independent genomes, providing us with a highly welcome advance in the field.
We believe the results are of broad interest as the stability principles of protein systems under coevolution must be universal, either under cospeciation or in independent genomes. We therefore anticipate that decomposition of evolutive and coevolutive information imprinted in physically-coupled and uncoupled amino acids and evaluation of their potential utility in resolving MSA models in terms of degeneracy and fraction of PPI matches should guide new developments in the field, aiming at characterizing protein interactions in general.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
Comments of Antônio Francisco P. de Araújo. Fernando Melo, Georgios Pappas and Michael Klein on the manuscript are gratefully acknowledged. The research was supported in part by the Brazilian Agencies CNPq, CAPES and FAPDF under Grants 305008/2015-3, 23038.010052/2013-95 and 193.001.202/2016. WT thanks CAPES for doctoral fellowship to MA and CP.
Author contributions
WT designed research; MA and CP performed research; MA, CP and WT analyzed data; WT wrote original draft; WT, MA and CP reviewed and edited. MA and CP contributed equally to this work.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2019.10.005.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Garcia L.G., Treptow W.L., Pereira de Araújo A.F. Folding simulations of a three-dimensional protein model with a nonspecific hydrophobic energy function. Phys Rev E. 2001;64 doi: 10.1103/PhysRevE.64.011912. [DOI] [PubMed] [Google Scholar]
- 2.Treptow W.L., Barbosa M.A.A., Garcia L.G., de Araújo A.F.P. Non-native interactions, effective contact order, and protein folding: A mutational investigation with the energetically frustrated hydrophobic model. Proteins Struct Funct Bioinforma. 2002;49:167–180. doi: 10.1002/prot.10166. [DOI] [PubMed] [Google Scholar]
- 3.Göbel U., Sander C., Schneider R., Valencia A. Correlated mutations and residue contacts in proteins. Proteins. 1994;18:309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- 4.de Juan D., Pazos F., Valencia A. Emerging methods in protein co-evolution. Nat Rev Genet. 2013;14:249–261. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- 5.Goh C.S., Bogan A.A., Joachimiak M., Walther D., Cohen F.E. Co-evolution of proteins with their interaction partners. J Mol Biol. 2000;299:283–293. doi: 10.1006/jmbi.2000.3732. [DOI] [PubMed] [Google Scholar]
- 6.Pazos F., Valencia A. Similarity of phylogenetic trees as indicator of protein-protein interaction. Protein Eng. 2001;14:609–614. doi: 10.1093/protein/14.9.609. [DOI] [PubMed] [Google Scholar]
- 7.Gertz J. Inferring protein interactions from phylogenetic distance matrices. Bioinforma Oxf Engl. 2003;19:2039–2045. doi: 10.1093/bioinformatics/btg278. [DOI] [PubMed] [Google Scholar]
- 8.Pellegrini M., Marcotte E.M., Thompson M.J., Eisenberg D., Yeates T.O. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc Natl Acad Sci. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dandekar T., Snel B., Huynen M., Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci. 1998;23:324–328. doi: 10.1016/s0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- 10.Marcotte C.J.V., Marcotte E.M. Predicting functional linkages from gene fusions with confidence. Appl Bioinform. 2002;1:93–100. [PubMed] [Google Scholar]
- 11.Tillier E.R.M., Biro L., Li G., Tillo D. Codep: maximizing co-evolutionary interdependencies to discover interacting proteins. Proteins. 2006;63:822–831. doi: 10.1002/prot.20948. [DOI] [PubMed] [Google Scholar]
- 12.Pazos F., Valencia A. In silico two-hybrid system for the selection of physically interacting protein pairs. Proteins. 2002;47:219–227. doi: 10.1002/prot.10074. [DOI] [PubMed] [Google Scholar]
- 13.Burger L., van Nimwegen E. Accurate prediction of protein-protein interactions from sequence alignments using a Bayesian method. Mol Syst Biol. 2008;4:165. doi: 10.1038/msb4100203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gueudré T., Baldassi C., Zamparo M., Weigt M., Pagnani A. Simultaneous identification of specifically interacting paralogs and interprotein contacts by direct coupling analysis. Proc Natl Acad Sci. 2016;113:12186–12191. doi: 10.1073/pnas.1607570113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bitbol A.-F., Dwyer R.S., Colwell L.J., Wingreen N.S. Inferring interaction partners from protein sequences. Proc Natl Acad Sci. 2016;113:12180–12185. doi: 10.1073/pnas.1606762113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Várnai C., Burkoff N.S., Wild D.L. Improving protein-protein interaction prediction using evolutionary information from low-quality MSAs. PLoS ONE. 2017;12 doi: 10.1371/journal.pone.0169356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bitbol A.-F. Inferring interaction partners from protein sequences using mutual information. PLOS Comput Biol. 2018;14 doi: 10.1371/journal.pcbi.1006401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Correa Marrero M, ImminkRGH, de Ridder D, van Dijk ADJ. Improved inference of intermolecular contacts through protein–protein interaction prediction using coevolutionary analysis. Bioinformatics https:/doi.org/10.1093/bioinformatics/bty924 (May 15, 2019). [DOI] [PubMed]
- 19.Morcos F. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci. 2011;108:E1293–E1301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones D.T., Buchan D.W.A., Cozzetto D., Pontil M. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28:184–190. doi: 10.1093/bioinformatics/btr638. [DOI] [PubMed] [Google Scholar]
- 21.Jeong C.-S., Kim D. Reliable and robust detection of coevolving protein residues. Protein Eng Des Sel. 2012;25:705–713. doi: 10.1093/protein/gzs081. [DOI] [PubMed] [Google Scholar]
- 22.Kamisetty H., Ovchinnikov S., Baker D. Assessing the utility of coevolution-based residue–residue contact predictions in a sequence- and structure-rich era. Proc Natl Acad Sci. 2013 doi: 10.1073/pnas.1314045110. 201314045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burger L., van Nimwegen E. Disentangling direct from indirect co-evolution of residues in protein alignments. PLOS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Juan D., Pazos F., Valencia A. Co-evolution and co-adaptation in protein networks. FEBS Lett. 2008;582:1225–1230. doi: 10.1016/j.febslet.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 25.Codoñer FM, Fares MA. Why should we care about molecular coevolution? Evol Bioinform 4, 117693430800400000 (2008). [PMC free article] [PubMed]
- 26.Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. eLife 3, e02030 (2014). [DOI] [PMC free article] [PubMed]
- 27.Nadaradjane A.A., Guerois R., Andreani J. Protein-protein docking using evolutionary information. Methods Mol Biol Clifton NJ. 2018;1764:429–447. doi: 10.1007/978-1-4939-7759-8_28. [DOI] [PubMed] [Google Scholar]
- 28.MacKay DJC, Information theory, inference and learning algorithms, 1st ed., Cambridge University Press; 2003.
- 29.Dunn S.D., Wahl L.M., Gloor G.B. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2008;24:333–340. doi: 10.1093/bioinformatics/btm604. [DOI] [PubMed] [Google Scholar]
- 30.Tovchigrechko A., Vakser I.A. GRAMM-X public web server for protein–protein docking. Nucleic Acids Res. 2006;34:W310–W314. doi: 10.1093/nar/gkl206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.