

Abstract
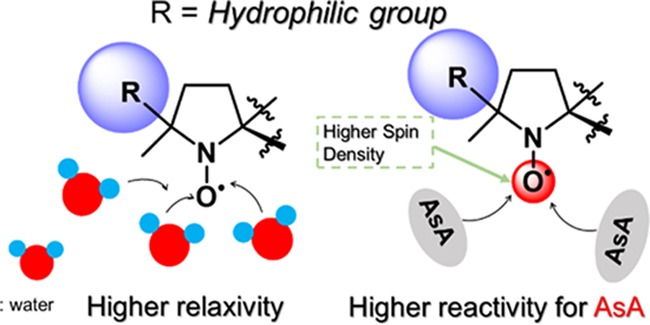
Materials possessing electron spin can shorten the T1 relaxation times in magnetic resonance imaging (MRI). For example, gadolinium (Gd) complexes with seven f-orbital electrons are widely used as contrast agents in clinical applications. However, Gd has severe potential side effects, and thus metal-free alternatives are needed. Toward this end, we synthesized seven NO radicals consisting of a dioxa-azaspiro[4.5]decane framework having various substituents, DAD-X (X = methyl, ethyl, n-propyl, c-propyl, vinyl, phenyl, and 2-pyridyl), that functioned as metal-free MRI contrast agents. The relationship between (i) water–proton relaxivity and log P and (ii) reactivity for ascorbic acid and the spin density of the NO oxygen atom were established, which provided a basis for the rational design of practical metal-free contrast agents.
1. Introduction
As gadolinium (Gd) features the highest possible spin quantum number (S) of 7/2, Gd complexes such as Magnevist and Omniscan are widely used as T1-weighted contrast agents (CAs) for clinical-purpose magnetic resonance imaging (MRI).1 However, these agents can induce renal disorders, accumulate in the brain, etc.,2 which highlights the need for alternative contrast agents.3 Organic radicals possess S = 1/2 and can therefore be employed as metal-free MRI contrast agents; however, the small value of S results in a much lower ability as the contrast agent than that of Gd complexes.4 To address this problem, a dendrimer comprising numerous NO radicals was constructed as a metal-free contrast agent and showed a high ability as the contrast agent and elevated biostability.5
Previously, we reported a series of NO radical-containing self-assembled urea benzene derivatives (UBDs)6−9 as metal-free contrast agents, revealing that these supra-molecules show higher ability as the contrast agents than (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) because of the restricted molecular motion in the NO part (Scheme S1). The ability of Gd complexes is well predicted by the Lipari–Szabo model,10 their abilities depend on the following significant factors; (1) electron relaxation (τιe) of the spin source, (2) rotational correlation time (τR) of the spin source, (3) diffusion rate of water (τm) around the spin source, and (4) the number of water molecules binding to the Gd ion (q). However, for organic radicals, systematical experiments and concepts based on the corresponding results are currently missing. In the case of metal-free contrast agents, water molecules around the NO moiety and their orientation are believed to be of significance for ability enhancement as the contrast agents. Furthermore, resistance toward reductants such as ascorbic acid (AsA) is important for the construction of practical contrast agents. Finally, no systematic experimental assessment of contrast agent reactivity has been performed yet. To understand the fundamental relationship between the presence of water molecules and contrast agent polarity and that between the reactivity for AsA and spin density at NO, we synthesized seven NO radicals with substituents of various hydrophobicity around the NO group (Scheme 1), DAD-X (DAD = dioxa-azaspiro[4.5]decane, X = methyl, ethyl, n-propyl, c-propyl, vinyl, phenyl, 2-pyridyl), and evaluated their abilities as contrast agents. Seven DAD-X are abbreviated as 1(Me), 2(Et), 3(nP), 4(cP), 5(Vin), 6(Ph), and 7(Py) for X = methyl, ethyl, n-propyl, c-propyl, vinyl, phenyl, and 2-pyridyl, respectively. These species were classified into three groups, namely, those bearing substituents without π-orbitals (methyl, ethyl, and n-propyl), with π-orbitals (vinyl, phenyl, and pyridyl), and with a pseudo-π-orbital (c-propyl).10 Despite being strained, the cyclopropyl ring is not labile under ambient conditions because of the presence of Walsh orbitals with symmetric and antisymmetric components.11 Herein, we describe the physical properties of DAD-X, e.g., log P and water–proton relaxivity (r1, a measure of ability as the contrast agent), evaluate the reactivity (k2) of these species for AsA, and establish r1−log P and k2−spin density at NO correlations. In particular, we focused on the log P value as an essential physical parameter. The log P values denote hydrophobicity or hydrophilicity for molecules based on their substituents, therefore, larger and smaller log P values indicate more hydrophobic and more hydrophilic compounds, respectively. If the molecules possess substituents based on smaller log P, a lot of water molecules, which have polarity, might gather around the corresponding hydrophilic molecules. Accordingly, in the case of DAD-X carrying hydrophilic groups, the water molecules gather around the NO group, then the ability as contrast agents are expected to vary. Preparing various substituted-DAD-X, we will herein discuss the abilities as contrast agents based on various log P values.
Scheme 1. Molecular Structures of Seven DAD-X Compounds.

2. Results and Discussion
2.1. Syntheses
DAD-X were prepared as illustrated in Scheme 2. In the presence of an organic acid, 2-bromo-2-nitro-1,3-propanediol and 2,2-dimethoxypropane were condensed to afford the cyclic acetal compound (8), and then bromine was removed via reduction using NaBH4, to afford (9). Next, a Michael addition using the given nitro compound and methyl vinyl ketone in the presence of an organic base was performed to produce (10), and then a reduction using zinc powder was carried out to afford a parent nitron compound (11). Finally, the substituents were introduced into nitrone compounds via similar Grignard reactions. The resulting DAD-X were determined their molecular structures and purities by various spectrometries. Due to the paramagnetic effects of DAD-X, the corresponding hydroxylamine derivatives, which were products by a reduction reaction using ascorbic acid or phenylhydrazine toward DAD-X, were used to conduct NMR spectra (1H and 13C) (Figures S17–S23).
Scheme 2. Syntheses of DAD-X.

(a) (S)-(+)-Camphor-10-sulfonic acid, 2,2-dimethoxypropane, room temperature (rt), 48 h; (b) NaBH4, 75% MeOH solution, rt, 1 h; (c) methyl vinyl ketone, 1,1,3,3-tetramethylguanidine, MeOH, rt, 14 h; (d) Zn, AcOH, EtOH, −10 °C, 20 min; (e) methyl MgI, diethyl ether, 0 °C to rt, 14 h; (f) methyl MgBr, diethyl ether, 0 °C to rt, 1 h; (g) vinyl MgCl, tetrahydrofuran (THF), 0 °C to rt, 4 h; (h) Mg, bromopropane, diethyl ether, 0 °C to rt, 2 h; (i) c-propyl MgBr, THF, 0 °C to rt, 4 h; (j) phenyl MgI, THF, 0 °C to rt, 17 h; (k) 2-bromopyridine, isopropylmagnesium chloride lithium chloride complex, 2-bromopyridine, THF, 0 °C, 6 h, then added dropwise (11), 0 °C to rt, 4 h.
2.2. Physical Properties
The presence of the NO radical moiety was confirmed by UV–visible (vis) (Figure S1), electron spin resonance (ESR, Figure S2), and IR spectroscopies (Figure S3). X-band (9.4 GHz) ESR spectra featured three typical lines with aN = 1.37 mT at g = 2.0006
| 1 |
According to the Kivelson equation (eq 1; Table S1),12 τR values, which are able to represent the molecular motion, were smaller than those of UBD species (5.0–9.5 × 10–11 s), which formed nanoparticles in aqueous solution. Thus, DAD-X compounds were concluded to be present in the monomeric form, which allowed us to exclude self-assembly effects (Scheme S1) for an evaluation of the contrast agents and directly evaluate substituent effects. The stability checks of DAD-X in water solutions were performed by changes of ESR signal intensities. The double integration value of the ESR signal in 1 mM solution was monitored at 23 °C until 82 h. The values were gradually decayed, and after 82 h, the initial values reduced by 85–99%. These results indicated that no positive chemical reaction proceeded even surrounding many water molecules, and the stability of DAD-X was sufficient to perform various examinations such as an MRI, cyclic voltammetry (CV), and a stability check toward an ascorbic acid solution (Figure S4).
For the evaluation of contrast agents, T1-weighted images were acquired at different contrast agent concentrations by a 1.5 T MRI apparatus [Figure 1a for 4(cP) and Figure S5-1 for all of the remaining DAD-X]. With the increasing DAD-X concentration, the image color shifted toward the blue region, i.e., the relaxation time concomitantly decreased. The obtained relaxation rates (inverse of T1) were plotted against the contrast agent concentration [Figure 1b for 4(cP) and Figure S5-2 for all of the remaining DAD-X], and the plots were fitted by a least-squares method. r1 values (Table 1) were estimated from the slope of these plots, with the highest and lowest values obtained for 1(Me) and 6(Ph), respectively.
Figure 1.
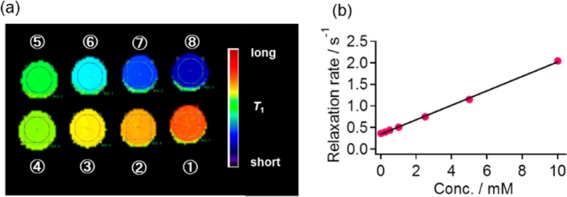
(a) T1-weighted images obtained for various concentrations of 4(cP) and (b) a plot of relaxation rate vs 4(cP) concentration. The numerical numbers in (a) refer to concentrations of (1) 0, (2) 0.156, (3) 0.313, (4) 0.625, (5) 1.25, (6) 2.50, (7) 5.00, and (8) 10.0 mM. The colored bar in (a) represents T1.
Table 1. Selected Physical Parameters of DAD-Xa.
| DAD-X | 1(Me) | 7(Py) | 5(Vin) | 2(Et) | 4(cP) | 3(nP) | 6(Ph) |
|---|---|---|---|---|---|---|---|
| Estimatedbr1 (mM–1 s–1) | |||||||
| r1b | 0.190 | 0.120 | 0.121 | 0.138 | 0.168 | 0.100 | 0.096 |
| (0.001) | (0.001) | (0.001) | (0.001) | (0.001) | (0.001) | (0.001) | |
| Rate Constants (k2) for the Reaction with AsA in PBS (M–1 s–1) | |||||||
| k2b | 3.3 | 6.8 | 7.2 | 2.2 | 2.6 | 1.2 | 3.5 |
| (0.2) | (0.4) | (0.6) | (0.17) | (0.1) | (0.1) | (0.2) | |
| Estimated log Pc | |||||||
| log P | 1.87 | 2.19 | 2.46 | 2.50 | 2.53 | 3.09 | 3.17 |
| Estimated Spin Density of Oxygen and Nitrogen Atoms, and Their Ratioc | |||||||
| O | 0.449 | 0.456 | 0.454 | 0.447 | 0.454 | 0.446 | 0.455 |
| N | 0.503 | 0.494 | 0.491 | 0.503 | 0.496 | 0.504 | 0.496 |
| O/Nd | 0.891 | 0.922 | 0.924 | 0.888 | 0.916 | 0.886 | 0.917 |
| Estimated Volume Percentage (%V) of Filling or Free Space around the Oxygen Atom in NO (%)e | |||||||
| filling (%Vbur) | 44.10 | 47.30 | 44.30 | 46.70 | 48.30 | 45.27 | 47.30 |
| free (%Vfree) | 55.90 | 52.70 | 55.70 | 53.30 | 51.68 | 54.73 | 52.70 |
| Pascal Constant of Individual Substituents (−1.0 × 10–6 cm3 mol–1)f | |||||||
| 14.79 | 47.02 | 15.29 | 26.65 | 26.17 | 38.51 | 53.49 | |
As NO radicals easily react with various reductants such as AsA and glutathione, the potential reactivity for such reductants can be used to directly evaluate the metabolic stabilities of DAD-X.13 To determine this reactivity, we estimated the second-order reaction rate constants k2 by the Ostwald method.14 The decay of the DAD-X (10 μM) ESR signal in the presence of various concentrations of AsA (1–30 mM) was tracked for 2000 s, with decay curves obtained for 5 or 10 mM AsA shown in Figure S6. The curves obtained for individual concentrations in the region of 0–400 s were fitted by a least-squares method using eq 2 to afford the pseudo k1 rate constant, namely, kobs. The values of kobs estimated for various concentrations were plotted vs AsA concentration [Figure 2 for 3(nP) and 4(cP), and Figures S6–S8 for the remaining DAD-X], and the slope of these plots gave k2 (eq 3)
| 2 |
| 3 |
The thus obtained k2 values decreased in the order of 5(Vin) > 7(Py) > 6(Ph) > 1(Me) > 4(cP) > 2(Et) > 3(nP), i.e., compounds with π-orbitals (including pseudo-π) featured obviously larger k2 values than those without π-orbitals (Table 1). It is worth noting that the reactivities of DAD-X were comparable or much lower than one of TEMPO derivatives (Figure S9),15 therefore, the DAD-X might prolong the function as MRI CA for in vivo examination.
Figure 2.
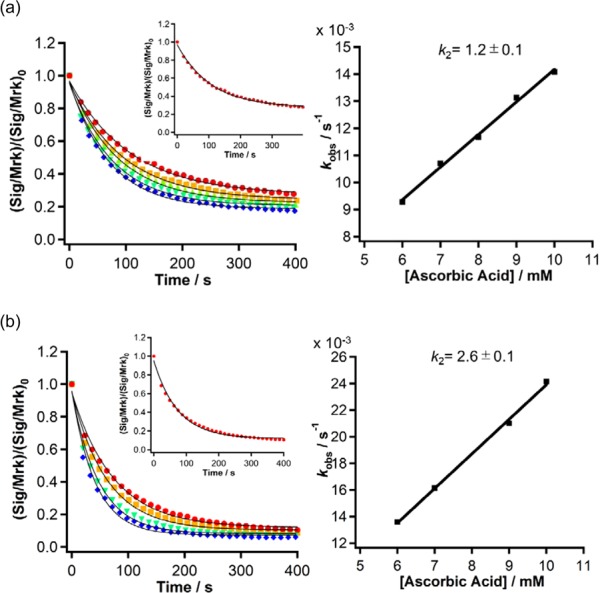
(Left) Time-dependent decay curves obtained from the ESR intensities of (a) 3(nP) and (b) 4(cP) at various concentrations of ascorbic acid (AsA). The solid line indicates curve fitting according to eq 2. Red, orange, green, sky blue, and blue colored circles represent 6, 7, 8, 9, and 10 mM of AsA solutions. The inset indicates the result of 6 mM. (Right) Plots of the resulting kobs values vs concentration of AsA together with k2 values.
One-electron reduction or oxidation of NO radicals affords labile anions (NO–) or cations (+N=O), respectively (Scheme 3). Thus, redox potentials were estimated by cyclic voltammetry (CV) measurements as indices of resistance to reduction and oxidation. The absolute oxidation potentials were similar to each other, whereas the reduction potentials decreased in the order of 1(Me) > 5(Vin) > 4(cP) ≈ −2(Et) > 7(Py) > 3(nP) > 6(Ph), which was different from the order of estimated k2 values (Table S2 and Figure S9).
Scheme 3. Reversible One-Electron Oxidation and Reduction Reactions of NO Radicals.

To find potential correlations of r1, k2, and reduction potential with the substitution pattern of DAD-X, we estimated parameters such as log P, spin density at NO by density functional theory (DFT) calculations.16 In particular, the log P value is predicted to work as an essential parameter among the metal-free contrast agents (vide supra). Due to the formation of emulsion, the calculated log P values were used as the valid one. In addition, free space around NO was estimated as the percent buried volume (%Vbur) and free volume (%Vfree), using the SambVca 2.0 web tool,17 and the values of diamagnetic susceptibility (χD) as Pascal’s constant were taken from a previous report.18 The optimized structures are shown in Figure S10, and the estimated parameters are summarized in Table 1 for estimated volume percentage and Table S3 for solvation Gibbs free energies. DAD-X with longer alkyl chains featured higher log P values [3(nP) > 2(Et) > 1(Me)], and 6(Ph) possessed a higher value than −Py. As the NO radical resonates between the oxygen radical (O•) and nitrogen cation radical (N+•) forms (Scheme 4), the O•/N+• spin density ratio (SDR) depends on the molecule and the environment. Interestingly, the SDRs of DAD-X with π-orbitals exceeded those of species without π-orbitals, i.e., spin density at oxygen was higher in the former case (Table 1). This behavior was explained by the through-space stabilization of O• by π-orbitals.
Scheme 4. Resonance Structures of NO Radical.

The estimated volume of free space (%Vfree) around NO (Table 1 and Figure S11) was independent of alkyl chain length [1(Me), 2(Et), or 3(nP)] and number of carbon atoms [3(nP) vs 4(cP) and 5(Vin) vs 2(Et)]. The r2/r1 ratio was influenced by overall magnetic susceptibility (χmeas),19 which is a sum of a paramagnetic term (χP) and χD. Compared to those without π-orbitals, DAD-X with π-orbitals had a higher absolute χD, except for 5(Vin), which was ascribed to the shielding effect of ring current in phenyl, pyridyl, and c-propyl rings.
2.3. Estimation of Ability as Contrast Agents
The obtained parameters were plotted against r1, as shown in Figure 3. In Figure 3a, r1 was relatively well correlated with log P, especially in the case of alkyl substituents. The above correlation was negative, which implied that the hydrophobic groups kept water molecules away from the NO radicals, i.e., displayed pseudo-dehydrated behavior. However, a deviation was observed for DAD-X with π-orbitals. A good correlation between r1 and χD or SDR was observed for alkyl-substituted compounds (Figure 3b or c), while no such correlation was observed in the case of DAD-X with π-orbitals. Therefore, r1 was plotted vs a new function (function 1 = log P + 0.009χD) accounting for log P and χD, and the resulting correlation was better than those obtained using log P and χD as sole parameters (Figure 3d).
Figure 3.
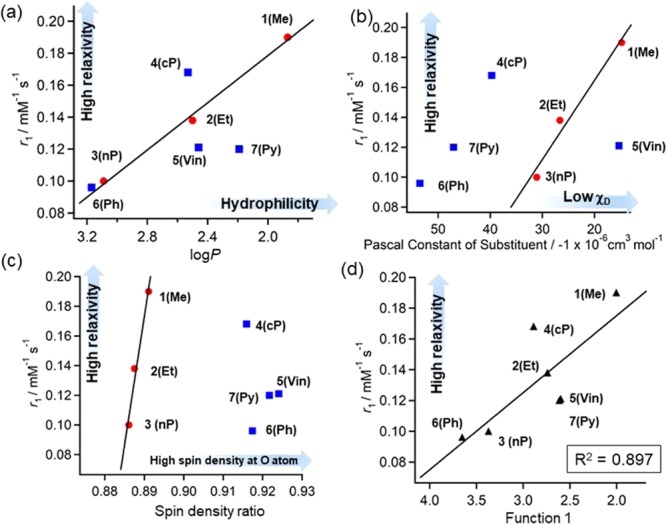
Plots of (a) r1 vs log P, (b) r1 vs Pascal’s constant (χD), (c) r1 vs spin density ratio (SDR), (d) r1 vs function 1. Solid lines indicate least-squares fits obtained using (a–c) alkyl analogue data and (d) all data. In (d), function 1 = log P + 0.009χD (see text). The numerical number “0.009” in function 1 was determined by the smallest R2 value according to the least-squares fitting.
Figure 4 presents the correlations between k2 and various parameters. Figure 4a shows that reasonable negative correlations between k2 and log P were observed for two groups of DAD-X (with and without π-orbitals), which suggested that high hydrophilicity enhanced the ability of DAD-X to interact with the highly polar AsA. To explain the fact that two separate correlations were observed for compounds with and without π-orbitals, we plotted k2 as a function of SDR (Figure 4b), and again, two separate strong positive correlations were observed. To support this behavior, we calculated SDR values for TEMPO(20) and PROXYL(21) as 0.8952 and 0.8266, respectively. These values agreed with the higher reactivity of the former compound for AsA,22 i.e., SDR was confirmed to be positively correlated with reactivity for AsA.
Figure 4.
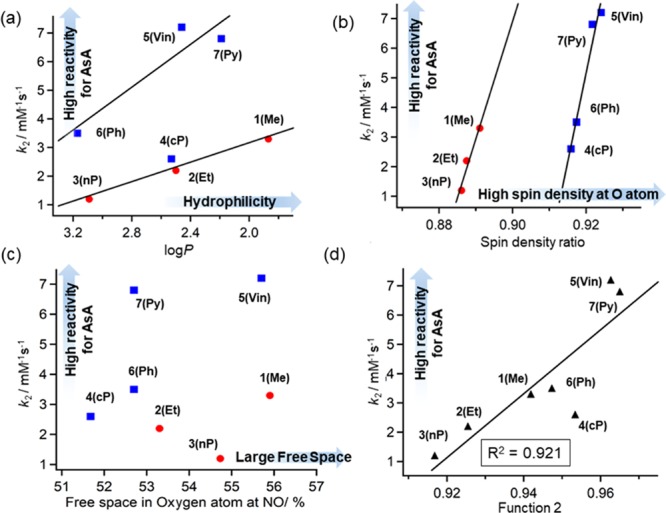
Plots of (a) k2 vs log P, (b) k2 vs SDR, (c) k2 vs FrS, (d) k2 vs function 2. Solid lines indicate least-squares fits obtained for data including and excluding species with π-orbitals [(a) and (b) respectively] and for all data (d). In (d), function 2 is defined as SDR + 0.095(1/log P) (see the text). The numerical number “0.095” in function 2 was determined by the smallest R2 value according to the least-squares fitting.
Moreover, the correlation between k2 and free space (%Vfree) was taken into account (Figure 4c), as %Vfree can be influenced by substituents in DAD-X and was expected to be positively correlated with k2. However, no clear relationship between %Vfree and k2 was revealed. Notably, TEMPO (%Vfree = 53.7) features a higher reactivity for AsA than PROXYL (%Vfree = 56.6) despite featuring a smaller %Vfree (Figure 5). Thus, we concluded that reactivity for AsA is dominantly controlled by SDR and not by %Vfree. Finally, we defined a new function, function 2, as SDR + 0.095 (1/log P) and showed that this function was better correlated with k2 than the individual parameters of SDR and log P (Figure 4d).
Figure 5.

Space-filling models (top panels) and molecular structures with coordinates (down panels) of TEMPO (left) and PROXYL (right). %Vbur indicates the percentage of the buried spaces. The values of free space (%Vfree) were estimated according to 100 – %Vbur.
Surprisingly, the absolute value of reduction potential decreased with increasing hydrophobicity and decreasing SDR, which indicated that the NO radical was mainly present as N+• when reacting with e–. The N+• form may be stabilized by electron-donating (e.g., alkyl) groups because of their inductive effect. However, no correlation between reduction potential and the σp parameter of the Hammett equation23 was observed (Figure S12).
3. Conclusions
Based on the above, we concluded that metal-free contrast agents need to possess high r1 and low reactivity for AsA. As systematic investigations of NO radicals as contrast agents have been missing, we prepared seven DAD-X with various substituents on the NO-fused five-membered ring, revealing the strong negative correlation between r1 and hydrophobicity (represented by log P) (Figure 6). Furthermore, the reactivity for AsA (represented by k2) was strongly positively correlated with the spin density at oxygen (SDR), which was higher for DAD-X with π-orbitals. Interestingly, the correlations observed for reduction potential were opposite to those observed for k2, which suggested that reduction by AsA dominantly occurred at O•, whereas that by e– occurred at N+• (Figure 6). Furthermore, an acetal ring in the structure of DAD-X is capable of deprotecting under acidic conditions, to form a diol analogue, which is a hydrophilic compound and the r1 value increases compared to the one before deprotection (Scheme 5). Thus, under acidic tissue, such as a tumor, the deprotected DAD-X might enhance the contrast imaging, and the tumor recognition using DAD-X might be a valid system. We believe that the obtained insights can be effectively used for the construction of practical metal-free contrast agents in the near future.
Figure 6.
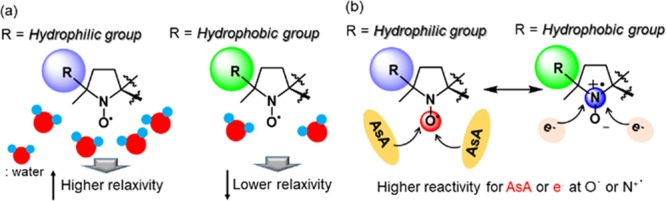
Correlations between (a) log P and r1, and (b) SDR and reactivity for AsA or e–.
Scheme 5. Deprotection of Acetal Rings and Their Physical Property Changes.

4. Experimental Section
4.1. General Information
Infrared spectra were recorded using a JASCO 4600 Fourier transform infrared (FT-IR) spectrometer. UV–vis spectra were recorded using the JASCO V570 and V760 spectrometers or the Agilent 8453 spectrometer. The 1H and 13C NMR spectra were measured by a Bruker Biospin AVANCE III 500 spectrometer using CDCl3 or D2O as solvents and tetramethylsilane as a reference. High-resolution electrospray ionization mass spectra were recorded using a Bruker micrOTOF spectrometer. The melting points were found using the Büchi melting point apparatus M-560 (uncalibrated).
4.2. Cyclic Voltammetry (CV)
Cyclic voltammograms were recorded using a BAS chemical analyzer (model 660C). Individual 1 mM DAD-X solutions in phosphate-buffered saline (PBS, pH = 7.4) were prepared and were bubbled in a N2 atmosphere several times. The potentials of each DAD-X solution were measured using a glass carbon as the working electrode, the Ag/AgCl as the reference electrode, and the Pt as the counter electrode. All of the scanning processes were performed at a scanning rate of 4 mV s–1.
4.3. Electron Spin Resonance (ESR)
ESR spectra were recorded using a JEOL JES-X310 spectrometer. Individual 50 μM DAD-X solutions in a phosphate-buffered saline (PBS, pH = 7.4) were prepared and approximately 40 μL solutions were added into a glass tube (Drummond Microcap 50 μL, ϕinside: 0.80 mm and ϕoutside: 1.09 mm) until the solution height from the bottom of the tube became equal to 8.0 cm. A quartz tube, including the DAD-X solution in the glass tube, was used to perform ESR with the following parameters: microwave power: 1.0 mW, modulation width: 0.1 mT, modulation frequency: 100 kHz, and time constant: 0.03 s. The values of the parameters of the center field, receiver gain, and sweep width were set based on the optimized value of the individual DAD-X solutions.
4.4. Stability Evaluation against the Ascorbic Acid Using ESR
DAD-X solutions (50 μM, 100 μM) and solutions of ascorbic acid (AsA) (1.0–20 mM) were prepared in PBS. The solution which contained a high concentration of AsA was neutralized using NaOH powder. ESR was performed using a 50 μL solution of DAD-X (50 μM), and the spectral intensity was recorded as the reference value for comparisons. A mixture of DAD-X (500 μL, 100 μM) and AsA (500 μL, 1.0–20 mM) solutions was added in a 2.0 mL tube and was stirred using VORTEX for 3 s. After stirring, the resulting solutions were transferred immediately into 50 μL ESR tubes, and ESR was performed continually using the SEQUENTIAL MEASUREMENT mode 40 times. The time spent for the preparation of the mixture of DAD-X and AsA mentioned above was regarded as the second measurement time point. A stopwatch was used to record this time.
The resulting data were used to estimate the intensity ratio between any given spectrum and the Mn(II) peak that was used as an internal standard. DAD-X was used as the reference standard for comparisons at the beginning of the measurements. Its intensity value in relation to Mn(II) was estimated as the ratio of (Sig/Mrk)0. All of the acquired spectra from the 40 continuous measurements were analyzed individually using the same method.
The given data of (Sig/Mrk)/(Sig/Mrk)0 were plotted as a function of reaction times. The plot was fitted with the use of a pseudo-first-order reaction rate equation and yielded a pseudo-first-order reaction rate constant (kobs). The same analyses were conducted for different concentrations of AsA, and the corresponding kobs values were obtained. The given kobs values were plotted as a function of concentrations and yielded the second-order reaction rate constants estimated based on the slopes of the plots. ESR was performed using the following parameters: sweep time: 10.0 s, center field: 333.300 mT, sweep width: 2.5 mT, modulation frequency: 100.00 kHz, microwave power: 10 mW, modulation width: 1.0 × 10–1 mT, amplitude: 7.00 × 10, and time constant: 0.03 s.
4.5. Relaxivity
T1-weighted MRI and relaxivity of the samples were estimated based on MRI acquisitions of CAs with a 1.5 T MRI scanner (Japan REDOX, Fukuoka, Japan), and the use of a volume coil (transmission–reception coil with an internal diameter of 38.5 mm, Japan REDOX, Fukuoka, Japan). Aqueous solutions of the contrast agents were initially placed into 1 mL disposable syringes. These were bundled and positioned in the center of the volume coil. The sample temperature was maintained at 25.0 ± 0.5 °C throughout the experiments with the use of the gradient coil cooling system and air conditioners. Using the MRI scanner, horizontal single-slice, T1-weighted, MR images were acquired with a gradient echo pulse sequence with the following parameters: TR/TE = 80/10 ms, slice thickness = 1.0 mm, acquisition matrix = 256 × 256, field-of-view (FOV) = 50 × 50 mm2, number of averages (NA) = 1, number of slices = 1. For longitudinal relaxation time (T1) and longitudinal relaxivity (r1) calculations, horizontal, single-slice inversion recovery MRI was performed using an inversion recovery fast spin-echo acquisition with the following parameters: TR = 10 s, TE = 12 ms, inversion time = 5, 40, 80, 200, 300, 500, 700, 900, 1200, 1500, 1800, 2100, 2500, 3000, and 4000 ms, acquisition matrix size = 256 × 128, FOV = 60 × 30 mm2, slice thickness = 3.0 mm, RARE factor = 16, and NA = 1.
4.6. log P Calculation
log P values were obtained using the following calculation methods. The modeling studies were performed using Spartan’08 (Wavefunction, Inc.). The conformational analysis of organic radicals, DAD-X [1(Me), 2(Et), 3(nP), 4(cP), 5(Vin), 6(Ph), 7(Py)] was conducted using the conformer distribution method (RM1 semiempirical method). The stable conformations were subsequently optimized in the gas phase using DFT at the UB3LYP-D3(BJ)/6-31G* level with the use of Gaussian 16 (revision A.03). Subsequently, frequencies were analytically computed at the UB3LYP-D3(BJ)/6-311G** level of theory to yield Gibbs free energies (298 K, 1 atm).
The value of log P was estimated from the computed free transfer energy according to eq 4(24)
| 4 |
where R is the molar gas constant, and T is the temperature.
To estimate the log P (n-octanol/water) values, gas-phase DFT-optimized conformers of DAD-X were reoptimized in n-octanol and water, respectively (UB3LYP-D3(BJ)/6-311G**//UB3LYP-D3(BJ)/6-31G*: SMD = n-octanol or water). The results of each optimization were used to evaluate the free energy difference for the two solvents.
4.7. Toxicity
No cytotoxicity assays were conducted in vitro or in vivo for DAD-X.
4.8. Materials
Unless otherwise stated, all of the reagents and solvents were used as received without further purification. Specifically, 2-bromo-2-nitro-1,3-propanediol, (S)-(+)-camphor-10-sulfonic acid, 2,2-dimethoxypropane, triethylamine, sodium borohydride, methyl vinyl ketone, 1,1,3,3-tetramethylguanidine, MgSO4, zinc powder, AcOH, concd HCl, NH4Cl, methylmagnesium iodide (3.0 M diethyl ether solution), ethylmagnesium bromide (1.0 M THF solution), magnesium turning, bromopropane, cyclopropylmagnesium bromide (0.5 M diethyl ether solution), vinylmagnesium bromide (1.0 M diethyl ether solution), phenylmagnesium bromide (3.0 M diethyl ether solution), 2-bromopyridine, and isopropylmagnesium chloride lithium chloride complex THF solution, were purchased from Nacalai Tesque Co. Ltd., Fujifilm Wako Pure Chemical Co. Ltd., Sigma-Aldrich, or from the Tokyo Chemical Industry Co. Ltd. DAD-X analogues were prepared according to the procedure previously reported.25 Silica gel for column chromatography was purchased from Kanto Chemical Ltd. in Japan. Thin-layer chromatography was performed on silica gel plates, using a 60 F254 (Merck).
4.8.1. 5-Bromo-2,2-dimethyl-5-nitro-1,3-dioxane (8)
A mixture of 2-bromo-2-nitro-1,3-propanediol (17.8 g, 89.1 mmol), (S)-(+)-camphor-10-sulfonic acid (1.86 g, 17.8 mmol), and 2,2-dimethoxypropane (131 mL, 1.78 mol) was stirred at rt for 48 h. The reaction mixture was neutralized with triethylamine and was then evaporated. The resulting residue was purified by silica gel column chromatography with the use of a mixture of n-hexane/AcOEt as the eluent to afford (8) as a white solid in 72% yield. FT-IR spectra (KBr pellet) 3000, 2945, 1565, 1433, 1375, 1330, 1259, 1196, 1127, 1080, 1039, 946, 898, 844, and 818 cm–1; 1H NMR (CDCl3, 500 MHz) δ 1.37 (s, 3H), 1.53 (s, 3H), 4.27 (d, J = 13.0 Hz, 2H), and 4.77 (d, J = 13.5 Hz, 2H) ppm; 13C NMR spectra (CDCl3, 126 MHz) δ 18.5, 27.8, 66.1, 81.9, and 99.5 ppm, and a mp of 45 °C.
4.8.2. 2,2-Dimethyl-5-nitro-1,3-dioxane (9)
Sodium borohydride (3.86 g, 102 mmol) was added slowly and in a dropwise manner at 0 °C to a 75% MeOH solution that contained 6.93 g of compound (8) (29.0 mmol) in an ice bath, and was stirred until all of the generated bubbles disappeared. The reaction mixture was stirred for 1 h until the temperature reached rt. The mixture was neutralized using a 10% HCl solution and extracted using CH2Cl2 three consecutive times. The combined organic layer was dried over MgSO4 and was evaporated to afford (9) as a pale yellowish solid (4.48 g, 27.8 mmol) in 96% yield. FT-IR spectra (KBr pellet) 2997, 2978, 1555, 1470, 1450, 1435, 1380, 1362, 1279, 1249, 1199, 1119, 1096, 1065, 947, 875, 849, and 820 cm–1; 1H NMR (CDCl3, 500 MHz) δ 1.42 (s, 3H), 1.46 (s, 3H), 4.25 (dd, J = 13.1 and 4.1 Hz, 2H), 4.36 (quin, J = 4.2 Hz, 1H), and 4.50 (dd, J = 13.1 and 4.1 Hz, 2H) ppm; 13C NMR (CDCl3, 126 MHz) δ 21.0, 25.8, 59.8, 77.1, and 99.1 ppm, and a mp of 39.3 °C.
4.8.3. 4-(2,2-Dimethyl-5-nitro-1,3-dioxane-5-yl)butan-2-one (10)
Methyl vinyl ketone (5.43 mL, 67.0 mmol) and 1,1,3,3-tetramethylguanidine (0.668 mL, 5.30 mmol) were added dropwise to an 80 mL of MeOH solution, which contained 9.81 g of compound (9) (60.9 mmol). The solution was stirred at rt for 14 h. The reaction mixture was neutralized using a 10% HCl solution, and the resulting solution was extracted with CH2Cl2 three consecutive times. The combined organic layer was dried over MgSO4 and was evaporated. The resulting residue was purified by a silica gel column chromatography using a mixture of n-hexane/AcOEt as the eluent to afford (10) as a white solid (13.3 g, 57.6 mmol) in 95% yield. FT-IR spectra (KBr pellet) 2989, 2938, 1716, 1548, 1539, 1446, 1425, 1379, 1353, 1324, 1262, 1201, 1147, 1091, 1041, 850, and 828 cm–1; 1H NMR (CDCl3, 500 MHz) δ 1.37 (s, 3H), 1.44 (s, 3H), 2.08 (t, J = 7.4 Hz, 2H), 2.16 (s, 3H), 2.43 (t, J = 7.4 Hz, 2H), 3.92 (d, J = 12.9 Hz, 2H), and 4.46 (d, J = 12.9 Hz, 2H) ppm; 13C NMR (CDCl3, 126 MHz) δ 20.1, 26.5, 27.1, 30.0, 36.5, 64.0, 85.5, 99.1, and 205.6 ppm, and a mp of 41.6 °C.
4.8.4. 5-(2,2-Dimethyl-1,3-dioxane-5-yl)-2-methylpyrrolidine-N-oxide (11)
Zinc powder (2.86 g, 43.8 mmol) was added to a 30 mL of MeOH solution which contained 3.38 g of compound (10) (14.6 mmol) at a temperature below −10 °C in an ice NaCl bath. AcOH (2.51 mL, 43.8 mmol) was added to the reaction mixture in a dropwise manner at temperatures below −10 °C. The reaction mixture was gradually warmed to rt and was stirred for 20 min. The resulting zinc dust was removed from the solution with the use of a filtrate paper and was sufficiently washed using MeOH. The MeOH in a filtrate was evaporated, and the residue was extracted using CHCl3 three consecutive times. The combined organic layer was dried over MgSO4 and was evaporated. The resulting residue was purified by silica gel column chromatography using a mixture of AcOEt/MeOH as the eluent to afford (11) as a yellowish-white colored solid (1.93 g, 9.68 mmol) in 66% yield. FT-IR spectra (KBr pellet) 2998, 2888, 1597, 1491, 1456, 1402, 1374, 1281, 1263, 1234, 1222, 1203, 1157, 1090, 1037, 974, and 829 cm–1; 1H NMR (CDCl3, 500 MHz) δ 1.41 (s, 3H), 1.57 (s, 3H), 2.04 (s, 3H), 2.34 (t, J = 7.5 Hz, 2H), 2.64 (t, J = 7.3 Hz, 2H); 3.57 (d, J = 11.5 Hz, 2H), and 4.55 (d, J = 11.4 Hz, 2H) ppm; 13C NMR (CDCl3, 126 MHz) δ 12.9, 18.7, 27.6, 28.6, 29.1, 65.2, 71.2, 98.6, and 114.8 ppm, and a mp 121.7 °C.
4.8.5. 2,2,8,8-Tetramethyl-7,9-dioxa-1-azaspiro[4.5]decan1-1-yloxyl (1(Me))
A solid compound (11) (2.35 g, 11.8 mmol) was dissolved using a distilled (distd) THF solution (30 mL) in a N2 atmosphere. To the solution, a 16.5 mL (49.6 mmol) of methylmagnesium iodide (3.0 M solution in diethyl ether) was added slowly and in a dropwise manner, and the reaction mixture was stirred at rt for 17 h in a N2 atmosphere. To the reaction mixture, a saturated NH4Cl solution was added and the resulting solution was extracted with the use of CHCl3 three consecutive times. The combined organic layer was dried over MgSO4 and was evaporated. The residue was purified by a silica gel column chromatography using a mixture of n-hexane/AcOEt to afford compound (1(Me)) as an orange colored solid 1.57 g (7.35 mmol) in 62% yield. FT-IR spectra (KBr pellet) 2998, 2990, 2972, 2937, 2884, 1459, 1383, 1373, 1361, 1270, 1247, 1230, 1208, 1193, 1105, 1091, 835, and 829 cm–1; 1H NMR (D2O + ascorbic acid, 500 MHz) δ 1.24 (s, 6H), 1.41 (s, 6H), 1.81 (t, J = 7.3 Hz, 2H), 1.91 (t, J = 7.3 Hz, 2H), 3.81 (d, J = 12.6 Hz, 2H), and 4.17 (d, J = 12.6 Hz, 2H) ppm; 13C NMR (D2O + ascorbic acid, 126 MHz) δ 22.1, 22.7, 23.1, 29.0, 33.2, 64.7, 72.7, 77.7, 87.4, 99.5, and 105.4 ppm; a mp of 96.1 °C, and HRMS [electrospray ionization-time-of-flight (ESI-TOF) in MeOH]: calcd for C11H20NO3 [M + Na]+: 237.1335, found 237.1345.
4.8.6. 2,8,8-Trimethyl-2-ethyl-7,9-dioxa-1-azaspiro[4.5]decan1-1-yloxyl (2(Et))
This compound (2(Et)) was prepared in a manner similar to (1(Me)) using ethylmagnesium bromide (1.0 M solution in THF) instead of methylmagnesium iodide (3.0 M solution in diethyl ether) to afford (2(Et)) as an orange colored solid in 14% yield. FT-IR spectra (KBr pellet) 2993, 2970, 2936, 2881, 1461, 1453, 1379, 1370, 1345, 1263, 1204, 1189, 1146, 1093, 1035, 1005, 971, 943, 937,877, 823, 791, 769, 729, 637, 578, and 522 cm–1; 1H NMR (D2O + ascorbic acid, 500 MHz) δ 0.89 (t, J = 7.5 Hz, 3H), 1.26 (s, 3H), 1.40 (s, 3H), 1.42 (s, 3H), 1.59–1.64 (m, 1H), 1.74–1.79 (m, 1H), 1.84–1.87 (m, 2H), 1.91–1.96 (m, 2H), 3.88–3.96 (m, 3H), and 4.13–4.25 (m, 2H) ppm; 13C NMR (D2O + ascorbic acid, 126 MHz) δ 8.09, 18.3, 28.5, 31.3, 63.8, 64.5, 72.6, 76.0, 87.4, 99.6, and 105.4 ppm; a mp of 47.5 °C, and HRMS (ESI-TOF in MeOH): calcd for C12H22NO3 [M + Na]+: 251.1492, found 251.1496.
4.8.7. 2,8,8-Trimethyl-2-propyl-7,9-dioxa-1-azaspiro[4.5]decan1-1-yloxyl (3(nP))
THF solution (20 mL distd) which contained bromopropane 1.83 mL (20.1 mmol) was added slowly and in a dropwise manner to magnesium turning (488 mg, 20.1 mmol), and the reaction mixture was stirred at rt until the reaction turned into an orange solution in a N2 atmosphere. A distd THF solution (30 mL)—which contained 1.00 mg (5.02 mmol) of compound (3(nP))—was added to the reaction mixture slowly and in a dropwise manner in a N2 atmosphere, and was stirred at rt for 2 h. The reaction was quenched by a saturated NH4Cl solution and the resulting mixture was extracted with CH2Cl2 three consecutive times. The combined organic layer was dried over MgSO4 and was evaporated. The residue was purified by silica gel column chromatography using a mixture of n-hexane/AcOEt as the eluent to afford compound (3(nP)) as an orange colored solid in 11% yield. FT-IR spectra (KBr pellet) 2994, 2978, 2959, 2874, 1467, 1456, 1408, 1383, 1369, 1302, 1270, 1204, 1190, 1155, 1092, 1038, 938, and 833 cm–1; 1H NMR (D2O + ascorbic acid, 500 MHz) δ 0.87 (t, J = 7.2 Hz, 3H), 1.28–1.43 (m, 11H), 1.57 (td, J = 12.2 and 4.70 Hz, 1H), 1.76 (td, J = 12.3 and 4.8 Hz, 1H), 1.86–1.96 (m, 4H), and 3.95–4.24 (m, 4H) ppm; 13C NMR (D2O + ascorbic acid, 126 MHz) δ 3.7, 17.3, 19.9, 25.3, 28.3, 31.7, 64.0, 72.6, 76.0, 87.3, 99.7, 105.4, and 116.5 ppm, and a mp of 48.3 °C. HRMS (ESI-TOF in MeOH): calcd for C13H24NO3 [M + Na]+: 265.1648, found 265.1662.
4.8.8. 2,8,8-Trimethyl-2-cyclopropyl-7,9-dioxa-1-azaspiro[4.5]decan1-1-yloxyl (4(cP))
Compound (4(cP)) was prepared in a manner similar to compound (1(Me)) using cyclopropylmagnesium bromide (0.5 M solution in diethyl ether) instead of methylmagnesium iodide (3.0 M solution in diethyl ether) to afford compound (4(cP)) as an orange colored solid in 26% yield. FT-IR spectra (KBr pellet) 2989, 2975, 2936, 2885, 1469, 1454, 1383, 1373, 1275, 1265, 1203, 1191, 1150, 1088, 1024, 992, 976, 936, 903, 877, 827, 784, 768, 734, 653, 632, 602, 580, 521, and 511 cm–1; 1H NMR (D2O + ascorbic acid, 500 MHz) δ 0.36–0.60 (m, 4H), 1.12–1.15 (m, 1H), 1.19 (s, 3H), 1.41 (s, 6H), 1.64–1.72 (m, 2H), 1.83–1.98 (m, 2H), 1.89 (d, J = 12.9 Hz, 2H), 4.22 (dd, J = 18.8 and 13.6 Hz, 2H) ppm; 13C NMR (D2O + ascorbic acid, 126 MHz) δ 0.0036, 0.16, 14.2, 17.1, 26.7, 27.6, 60.5, 61.8, 70.8, 74.1, 85.5, 97.7, and 103.6 ppm, and a mp 48.1 °C. HRMS (ESI-TOF in MeOH): calcd for C13H22NO3 [M + Na]+: 263.1492, found 263.1493.
4.8.9. 2,8,8-Trimethyl-2-vinyl-7,9-dioxa-1-azaspiro[4.5]decan1-1-yloxyl (5(Vin))
Compound (5(Vin)) was prepared in a manner similar to compound (1(Me)) using vinylmagnesium bromide (1.0 M solution in diethyl ether) instead of methylmagnesium iodide (3.0 M solution in diethyl ether), to afford compound (5(Vin)) as an orange colored solid in 6% yield. FT-IR spectra (KBr pellet) yielded peaks at 3000, 2991, 2970, 1454, 1381, 1371, 1262, 1200, 1186, 1152, 1082, 1031, 991, 970, 945, 936, 831, 774, 757, 732, 693, 635, 582, 550, and 521 cm–1; 1H NMR (CDCl3 + phenylhydrazine, 500 MHz) δ 1.23 (s, 3H), 1.37 (s, 3H), 1.45 (s, 3H), 1.59–1.64 (m, 1H), 1.80–1.85 (m, 1H), 1.96 (t, J = 7.8 Hz, 2H), 3.48–3.52 (m, 2H), 4.14 (t, J = 25.5 and 11.5 Hz, 2H), 5.06–5.15 (m, 2H), and 6.03 (dd, J = 17.7 and 10.9 Hz, 1H); 13C NMR (CDCl3 + phenylhydrazine, 126 MHz) δ 20.1, 22.3, 27.5, 30.7, 32.2, 61.9, 66.8, 67.1, 67.8, 97.9, 113.1, and 142.9 ppm; a mp of 43.6 °C, and HRMS (ESI-TOF in MeOH): calcd for C12H20NO3 [M + Na]+: 249.1335, found 249.1333.
4.8.10. 2,2,8-Trimethyl-2-phenyl-7,9-dioxa-1-azaspiro[4.5]decan-1-yloxyl (6(Ph))
Compound (6(Ph)) was prepared in a manner similar to compound (1(Me)) using phenylmagnesium bromide (3.0 M solution in diethyl ether) instead of methylmagnesium iodide (3.0 M solution in diethyl ether) to afford (6(Ph)) as a yellowish solid in 68% yield.
FT-IR spectra (KBr pellet) 2988, 2977, 2882, 1496, 1455, 1384, 1369, 1219, 1204, 1185, 1151, 1098, 1042, 937, 881, 850, 832, 766, and 702 cm–1; 1H NMR (CDCl3 + phenylhydrazine, 500 MHz) δ 1.39 (s, 3H), 1.48 (s, 3H), 1.53 (s, 3H), 1.90–2.01 (m, 3H), 2.08–2.14 (m, 1H), 3.37 (dd, J = 11.2 and 1.8 Hz, 1H), 3.75 (dd, J = 11.5 and 1.8 Hz, 1H), 3.93 (d, J = 11.2 Hz, 1H), 4.45 (d, J = 11.5 Hz, 1H), 7.16–7.32 (m, 3H), and 7.49 (d, J = 7.20 Hz, 2H); 13C NMR (CDCl3 + phenylhydrazine, 126 MHz) δ 20.2, 21.8, 27.4, 31.2, 36.2, 62.3, 66.1, 68.1, 69.3, 98.0, 125.8, 126.5, 128.2, and 147.5 ppm; a mp of 51.3 °C, and HRMS (ESI-TOF in MeOH): calcd for C16H22NO3 [M + Na]+: 299.1492, found 299.1499.
4.8.11. 2,2,8-Trimethyl-2-pyridyl-7,9-dioxa-1-azaspiro[4.5]decan-1-yloxyl (7(Py))
Isopropylmagnesium chloride lithium chloride complex THF solution (8.78 mL, 7.53 mmol) was added slowly and in dropwise manner at 0 °C to a 15 mL distd THF solution which contained 2-bromopyridine (0.730 mL, 7.53 mmol) in an ice bath in a N2 atmosphere. The reaction mixture was stirred for 6 h until its temperature increased to rt To the reaction mixture, a 15 mL distd THF solution containing 500 mg of compound (11) (2.51 mmol) was added slowly and in a dropwise manner at 0 °C in an ice bath in a N2 atmosphere. The resulting mixture was stirred at rt for 4 h and was quenched by a saturated NH4Cl solution. The resulting mixture was extracted with CH2Cl2 three consecutive times and the combined organic layer was dried over MgSO4. The organic layer was evaporated and the resulting residue was purified by a silica gel column chromatography using a mixture of n-hexane/AcOEt as the eluent to afford compound (7(Py)) 76.9 mg (0.277 mmol) as a yellowish solid in 11% yield. FT-IR spectra (KBr pellet) 2981, 2931, 2889, 1586, 1572, 1466, 1423, 1373, 1263, 1199, 1154, 1086, 1034, 991, 970, 945, 936, 826, 798, 728, 665, 606, 591, 560, 518, and at 501 cm–1; 1H NMR (CDCl3 + phenylhydrazine, 500 MHz) δ 1.37 (s, 3H), 1.50 (s, 3H), 1.55 (s, 3H), 1.86–1.91 (m, 1H), 1.98–2.04 (m, 1H), 2.09–2.16 (m, 2H), 3.25 (d, J = 11.2 Hz, 1H), 3.83 (d, J = 11.4 Hz, 1H), 4.18 (d, J = 11.2 Hz, 1H), 4.33 (d, J = 11.4 Hz, 1H), 7.15–7.17 (m, 1H), 7.33 (d, J = 8.00 Hz, 1H), 7.67 (t, J = 7.9 Hz, 1H), and 8.16 (d, J = 4.9 Hz, 1H); 13C NMR spectra (CDCl3 + phenylhydrazine, 126 MHz) yielded δ values at 19.3, 24.2, 28.2, 30.9, 33.4, 61.0, 66.9, 67.4, 69.2, 97.8, 120.3, and 121.9 ppm; a mp of 106.7 °C, and HRMS (ESI-TOF in MeOH): calcd for C15H21N2O3 [M + Na]+: 300.1444, found 300.1444.
Acknowledgments
This work was partially supported by the JST PRESTO (grant numbers JPMJPR1337 for K.-i.Y. and JPMJPR14K4 for S.K.) and a Grant-in-Aid for Scientific Research (C) (19K07016 for S.K.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03003.
1H and 13C NMR spectra; UV–vis, IR, and ESR spectra for DAD-X; phantom images and plots of relaxation rate vs concentration of DAD-X solutions; time-dependent decay curves of DAD-X in the presence of AsA; cyclic voltammograms and redox potentials of DAD-X in solutions; optimized structures and Mulliken charges of the oxygen atom in NO with Gibbs free energies and log P values obtained from a DFT calculation; space-filling model with the percentage of the filling space of DAD-X, TEMPO, and PROXYL; Cartesian coordinates of DAD-X (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Caravan P.; Ellison J. J.; McMury T. J.; Lauffer R. B. Gadolin-ium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications. Chem. Rev. 1999, 99, 2293–2352. 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- Kanda T.; Ishii K.; Kawaguchi H.; Kitajima K.; Takenaka D. High Signal Intensity in The Dentate Nucleus and Globus Pallidus on Unenhanced T1-weighted MR Images: Relationship with Increasing Cumulative Dose of A Gadolinium-based Contrast Material. Radiology 2014, 270, 834–841. 10.1148/radiol.13131669. [DOI] [PubMed] [Google Scholar]
- Gale E. M.; Atanasova I. P.; Blasi F.; Ay I.; Caravan P. A Manganese Alternative to Gadolinium for MRI Contrast. J. Am. Chem. Soc. 2015, 137, 15548–15557. 10.1021/jacs.5b10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Lei X.; Lawler R. G.; Murata Y.; Komatsu K.; Turro N. J. Comparison of Nuclear Spin Relaxation of H2O@C60 and H2@C60 and Their Nitroxide Derivatives. J. Phys. Chem. Lett. 2010, 1, 2135–2138. 10.1021/jz100645w. [DOI] [PubMed] [Google Scholar]
- Rajca A.; Wang Y.; Boska M.; Paletta J. T.; Olankitwanit A.; Swanson M. A.; Mitchell D. G.; Eaton S. S.; Eaton G. R.; Rajca S. Organic Radical Contrast Agents for Magnetic Resonance Imaging. J. Am. Chem. Soc. 2012, 134, 15724–15727. 10.1021/ja3079829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T.; Murayama S.; Usui K.; Shimada T.; Aoki I.; Karasawa S. Self-Assembly Behavior of Emissive Urea Benzene Derivatives Enables Heat-Induced Accumulation in Tumor Tissue. Nano Lett. 2017, 17, 2397–2403. 10.1021/acs.nanolett.6b05371. [DOI] [PubMed] [Google Scholar]
- Morishita K.; Murayama S.; Araki T.; Aoki I.; Karasawa S. Thermal- and pH-Dependent Size Variable Radical Nanoparticles and Its Water Proton Relaxivity for Metal-Free MRI Functional Contrast Agents. J. Org. Chem. 2016, 81, 8351–8362. 10.1021/acs.joc.6b01509. [DOI] [PubMed] [Google Scholar]
- Morishita K.; Okamoto Y.; Murayama S.; Usui K.; Ohashi E.; Hirai G.; Aoki I.; Karasawa S. Water-Proton Relaxivities of Radical Nanoparticles Self-Assembled via Hydration or Dehydration Processes. Langmuir 2017, 33, 7810–7817. 10.1021/acs.langmuir.7b01126. [DOI] [PubMed] [Google Scholar]
- Morishita K.; Ueki S.; Fuchi Y.; Murayama S.; Kaneko T.; Narita N.; Kobayashi S.; Hirai G.; Aoki I.; Karasawa S. Self-Assembled Biradical Ureabenzene Nanoparticles for Magnetic Resonance Imaging. ACS Appl. Nano Mater. 2018, 1, 6967–6975. 10.1021/acsanm.8b01774. [DOI] [Google Scholar]
- Walsh A. D. The Structures of Ethylene Oxide, Cyclopropane, and Related Molecules. Trans. Faraday Soc. 1949, 45, 179–190. 10.1039/tf9494500179. [DOI] [Google Scholar]
- Momeni M. R.; Shakib F. A.; Azizi Z. New Generation of Dialkylsilylenes with Stabilities Comparable to Diaminosilylenes: A Theoretical Study. J. Phys. Chem. A 2011, 115, 10550–10555. 10.1021/jp201067r. [DOI] [PubMed] [Google Scholar]
- Atkins P. W.; Kivelson D. ESR Linewidths in Solution. II. Analysis of Spin—Rotational Relaxation Data. J. Chem. Phys. 1966, 44, 169–174. 10.1063/1.1726440. [DOI] [Google Scholar]
- Saphier O.; Silberstein T.; Shames A. I.; Likhtenshtein G. I.; Maimon E.; Mankuta D.; Mazor M.; Katz M.; Meyerstein D.; Meyerstein N. The Reduction of A Nitroxide Spin Label as A Probe of Human Blood Antioxidant Properties. Free Radical Res. 2003, 37, 301–308. 10.1080/1071576021000050410. [DOI] [PubMed] [Google Scholar]
- Brigham E. C.; Meyer G. J. Comparison of Permeation Measurements and Hybrid Density-Functional Calculations on Oxygen Vacancy Transport in Complex Perovskite Oxides. J. Phys. Chem. C 2014, 118, 7886–7893. 10.1021/jp501814r. [DOI] [Google Scholar]
- Paletta J. T.; Pink M.; Foley B.; Rajca S.; Rajca A. Synthesis and Reduction Kinetics of Sterically Shielded Pyrrolidine Nitroxides. Org. Lett. 2012, 14, 5322–5325. 10.1021/ol302506f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16; Gaussian, Inc.: Wallingford, CT, 2016.
- Falivene L.; Credendino R.; Poater A.; Petta A.; Serra L.; Oliva R.; Scarano V.; Cavallo L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. 10.1021/acs.organomet.6b00371. [DOI] [Google Scholar]
- a Bain G. A.; Berry J. F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532–536. 10.1021/ed085p532. [DOI] [Google Scholar]; b Wiberg K. B.Cyclobutane–Physical Properties and Theoretical Studies. In The Chemistry of Cyclobutanes;Rappoport Z., Liebman J., Eds.; Wiley, 2005; Chapter 1, pp 1–15. [Google Scholar]
- Kim D.-H.; Zeng H.; Ng T. C.; Brazel C. S. T1 and T2 Relaxivities of Succimer-coated MFe23+O4 (M = Mn2+, Fe2+ and Co2+) Inverse Spinel Ferrites for Potential Use as Phase-contrast Agents in Medical MRI. J. Magn. Magn. Mater. 2009, 321, 3899–3904. 10.1016/j.jmmm.2009.07.057. [DOI] [Google Scholar]
- Beejapur H. A.; Zhang Q.; Hu K.; Zhu L.; Wang J.; Ye Z. TEMPO in Chemical Transformations: From Homogeneous to Heter-ogeneous. ACS Catal. 2019, 9, 2777–2839. 10.1021/acscatal.8b05001. [DOI] [Google Scholar]
- Yamato M.; Shibata T.; Naganuma T.; Ichikawa K.; tsumi H.; Yamada K. Overhauser-enhanced Magnetic Resonance Imaging Characterization of Mitochondria Functional Changes in The 6-Hydroxydopamine Rat Model. Neurochem. Int. 2011, 59, 804–811. 10.1016/j.neuint.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Lampp L.; Morgenstern U.; Merzweiler K.; Imming P.; Seidel R. W. Synthesis and Characterization of Sterically and Electrostatical-ly Shielded Pyrrolidine Nitroxide Radicals. J. Mol. Struct. 2019, 1182, 87–94. 10.1016/j.molstruc.2019.01.015. [DOI] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. A Survey of Hammett Substitu-ent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Bayat Z.; Movaffagh J. Evaluation of The 1-Octanol/water Partition Coefficient of Nucleoside Analogs via Free Energy Estimated in Quantum Chemical Calculations. Russ. J. Phys. Chem. A 2010, 84, 2293–2299. 10.1134/S0036024410130157. [DOI] [Google Scholar]
- Hirayama T.; Taki M.; Nakamura M.; Arata T.; Yamamoto Y. Synthesis of a New Water Soluble 2,2-Bifunctionalized Spin Label and Its Application to Troponin C. Chem. Lett. 2006, 35, 834–835. 10.1246/cl.2006.834. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.