

Abstract
PURPOSE
Heterogeneity in tumor mutational burden (TMB) quantification across sequencing platforms limits the application and further study of this potential biomarker of response to immune checkpoint inhibitors (ICIs). We hypothesized that harmonization of TMB across platforms would enable integration of distinct clinical data sets to better characterize the association between TMB and ICI response.
METHODS
Cohorts of patients with non–small-cell lung cancer sequenced by 1 of 3 targeted panels or by whole-exome sequencing (WES) were compared (N = 7,297). TMB was calculated uniformly and compared across cohorts. TMB distributions were harmonized by applying a normal transformation followed by standardization to z scores. In subcohorts of patients treated with ICIs (Dana-Farber Cancer Institute n = 272; Memorial Sloan Kettering Cancer Center n = 227), the association between TMB and outcome was assessed. Durable clinical benefit (DCB) was defined as responsive/stable disease lasting ≥ 6 months.
RESULTS
TMB values were higher in the panel cohorts than in the WES cohort. Average mutation rates per gene were highly concordant across cohorts (Pearson’s correlation coefficients, 0.842-0.866). Subsetting the WES cohort by gene panels only partially reproduced the observed differences in TMB. Standardization of TMB into z scores harmonized TMB distributions and enabled integration of the ICI-treated subcohorts. Simulations indicated that cohorts > 900 are necessary for this approach. TMB did not associate with response in never-smokers or patients who harbor targetable driver alterations, although these analyses were underpowered. An increase of TMB thresholds increased DCB rate, but DCB rates within deciles varied. Receiver operating characteristic curves yielded an area under the curve of 0.614, with no natural inflection point.
CONCLUSION
The z score conversion harmonizes TMB values and enables integration of data sets derived from different sequencing panels. Clinical and biologic features may provide context to the clinical application of TMB and warrant additional study.
INTRODUCTION
Immune checkpoint inhibitors (ICIs) have revolutionized the treatment of multiple advanced cancers.1-6 However, only a minority of patients experience clinical benefit, and clinically actionable biomarkers of response are urgently needed.
To date, the only approved biomarkers of ICI response are mismatch repair deficiency and, in non–small-cell lung cancer (NSCLC), programmed cell death-ligand 1 (PD-L1) expression. However, mounting evidence has demonstrated an association between tumor mutational burden (TMB) and response to ICIs,7-17 and there is considerable interest in developing TMB as a clinical biomarker. TMB quantification from targeted next-generation sequencing (NGS) panels has been shown to correlate with whole-exome sequencing (WES)–derived TMB13,18-20 and to associate with ICI response, which makes the clinical assessment of TMB practically feasible.19,21
However, the proliferation of data related to TMB has also generated confusion because there are now multiple commercial and academic NGS panels in routine use, with important differences in gene panel composition, sequencing pipeline, and TMB algorithm.22,23 How these differences affect TMB quantification is unclear, and how to translate one platform’s TMB values to another for translational discovery or clinical use is not known. Furthermore, the studies that have described an association between TMB and response applied different thresholds to define TMB-high versus TMB-low groups. It is not known whether this threshold heterogeneity reflects different TMB quantification that arises from different platforms, variation across patient cohorts, or unknown clinical or biologic effects on the association between TMB and response.
CONTEXT
Key Objective
It is not known how to account for differences in tumor mutational burden (TMB) generated by different sequencing assays. We sought to address assay heterogeneity in TMB quantification by developing a technique to harmonize TMB across assays and applied this technique to a pool of distinct clinical cohorts to better characterize the association between TMB and response to immune checkpoint inhibitors.
Knowledge Generated
TMB differs across sequencing assays because of differences in gene panels and sequencing pipelines. Standardization of TMB into z scores enabled interassay comparison and pooling of distinct clinical cohorts. From this pooled analysis, we observed that TMB may not associate with response in patients who are never-smokers or harbor targetable oncogenes, and TMB thresholds yield significant trade-offs in sensitivity and specificity.
Relevance
Standardization of z scores harmonizes TMB values across assays for pooled analysis. Clinical and biologic features may modulate the association between TMB and response.
Given these questions, we sought to develop a strategy to harmonize TMB across NGS platforms. We applied this method to integrate multiple clinically annotated cohorts and to more fully characterize the relationship between TMB and ICI response to add nuance and context to our current understanding. We focused on NSCLC because of the early interest in applying TMB to clinical practice in this disease subtype24-26 and to avoid confounding of TMB by tumor type.27
METHODS
Study Population
Three cohorts of patients with NSCLC whose tumors had been profiled by a targeted NGS panel were evaluated. These panel cohorts were compared with a fourth WES cohort from The Cancer Genome Atlas (TCGA).
Dana-Farber Cancer Institute cohort.
Patients at the Dana-Farber Cancer Institute (DFCI) whose tumors had undergone OncoPanel NGS were included if they had advanced NSCLC and had consented to institutional review board–approved protocols. The ICI subcohort consisted of patients treated with ICIs who were evaluable for response.
Memorial Sloan Kettering Cancer Center cohort.
Molecular profiling from the Memorial Sloan-Kettering Cancer Center (MSKCC) MSK-IMPACT NGS panel21 was obtained from the cBioPortal for Cancer Genomics28,29 and limited to NSCLC samples. The ICI subcohort consisted of patients treated with ICIs whose tumors had undergone NGS sequencing.13
Foundation Medicine cohort.
Patient-level mutation calls for samples sequenced by Foundation Medicine were obtained (study accession phs001179)30 and filtered to include only NSCLC samples.
TCGA cohort.
Somatic WES data from NSCLCs sequenced by TCGA31 were downloaded from the cBioPortal for Cancer Genomics.
NGS
The DFCI cohort was sequenced as previously described.32,33 In brief, tumor DNA was extracted and used for custom-designed hybrid capture library preparation. NGS (OncoPanel) was performed and somatic alterations identified by custom pipeline. Given the absence of matched normal tissue, common single nucleotide polymorphisms were filtered if present at > 0.1% in Exome Variant Server, NHLBI GO Exome Sequencing Project, or gnomAD; variants present ≥ 2 times in COSMIC were rescued. All variants were reviewed for technical quality.34 Finally, to minimize inadvertent inclusion of germline variants, consistent with previous aggregation efforts,35 an additional germline filter was applied to exclude events present in the Exome Aggregation Consortium with an allele count > 10, after rescuing known somatic events.
The MSKCC, Foundation Medicine, and TCGA cohorts were sequenced as described.13,30,31,36 The MSK-IMPACT and TCGA WES pipelines use matched normal samples to isolate somatic events. Foundation Medicine uses an internal algorithm to filter putative germline events.
TMB
TMB was uniformly calculated for each sample as the number of nonsynonymous mutations per megabase (Mb) of genome covered. DFCI mutation counts were divided by the number of bases covered in each OncoPanel version: v1, 0.753334 Mb; v2, 0.826167 Mb; and v3, 1.315078 Mb. For MSKCC samples, the mutation count was divided by 0.896665, 1.016478, and 1.139322 Mb for the 341-, 410-, and 468-gene panels, respectively. For Foundation Medicine samples, 1.1 Mb was used as the length of genome covered.19 For TCGA samples, 38 Mb was used to approximate exome size, as previously described.19
PD-L1 Testing
A subset of ICI-treated patients had tissue evaluated for PD-L1 expression, which was reported as the percentage of tumor cells with membranous PD-L1 staining. MSKCC specimens were stained as previously described13; DFCI specimens were stained using clone E1L3N (Cell Signaling Technology, Danvers, MA) at 1:200 dilution with pressure cooker antigen retrieval in citrate buffer.
Immunotherapy Outcomes
Patients in the DFCI and MSKCC ICI subcohorts were annotated for treatment response to anti-PD-(L)1 monotherapy or in combination with anti–cytotoxic T-cell lymphocyte-4. Scans were reviewed by thoracic radiologists at each institution, and response was determined using RECIST version 1.1.37 Progression-free survival was assessed from the start of ICI treatment until the date of progression or death; patients without progression were censored at last scan. Consistent with prior studies,8,13 complete response (CR), partial response (PR), or stable disease (SD) > 6 months was defined as durable clinical benefit (DCB); no durable benefit (NDB) was defined as progressive disease (PD) or SD ≤ 6 months. Patients censored before 6 months of follow-up were considered not evaluable.
Statistical Analysis
Cohort-specific gene mutation averages were calculated by summing the number of mutations in each gene within a cohort and dividing the total by the number of patients in the cohort. The means were then transformed to a normal distribution by natural logarithmic transformation. The linear correlation between log average mutations per gene in the panel cohorts versus TCGA was evaluated using Pearson’s correlation coefficient.
Power transformations were used to normalize cohort-specific TMB distributions; Tukey’s ladder of powers38 in the rcompanion package39 was used to identify the optimal transformation coefficient. The normalized distributions were then standardized into z scores by subtracting the transformed distribution mean and dividing by the standard deviation. Overlap between normalized distributions was calculated using the overlapping package.40
TMB comparisons were made using the Mann-Whitney U test. The Fisher’s exact test was used to test for differences in categorical variables. All P values are two-sided, with P < .05 taken as significant. Receiver operating characteristic (ROC) curve analyses were performed using the pROC and OptimalCutpoints packages.41,42 Exploratory cutoffs were selected to maximize the distance to the y = x line (Youden’s index), maximize specificity with sensitivity > 80%, maximize both sensitivity and specificity, maximize the κ-statistic, and maximize the diagnostic odds ratio. All statistical analyses were performed in R 3.4.2 (R Foundation, Vienna, Austria). Human investigations were performed after approval by a local human investigations committee and in accordance with an assurance filed with and approved by the Department of Health and Human Services, where appropriate.
RESULTS
Comparison of TMB Quantification Across Panel and WES Platforms
Patients with NSCLC whose tumors had undergone sequencing through OncoPanel (n = 1,157), MSK-IMPACT (n = 1,520), Foundation Medicine (n = 3,476), or TCGA (n = 1,144) were included (N = 7,297; see Data Supplement for cohort diagram and clinical characteristics). To determine whether TMB differed between platforms, we plotted the distribution of TMB within each cohort (Fig 1A). TMB distributions differed between cohorts, and targeted panels were associated with higher TMB values than WES. Because targeted panels sequence fewer bases with focused inclusion of mutated cancer genes, we hypothesized that the higher TMB measurements associated with NGS panels were attributable to gene selection. We tested this by subsetting the WES data to include only those genes captured by the targeted panels (downsampling; Data Supplement; Fig 1B) and found that downsampled distributions retained greater TMB counts than the unfiltered TCGA distribution, which suggests that gene panel composition contributes to the observed difference in TMB distributions between cohorts. However, the relative differences were less pronounced than in the real-world cohort comparisons, which suggests that assay-specific differences, such as depth of sequencing and the absence of a paired germline sample, might also contribute to inter-test variation.
FIG 1.
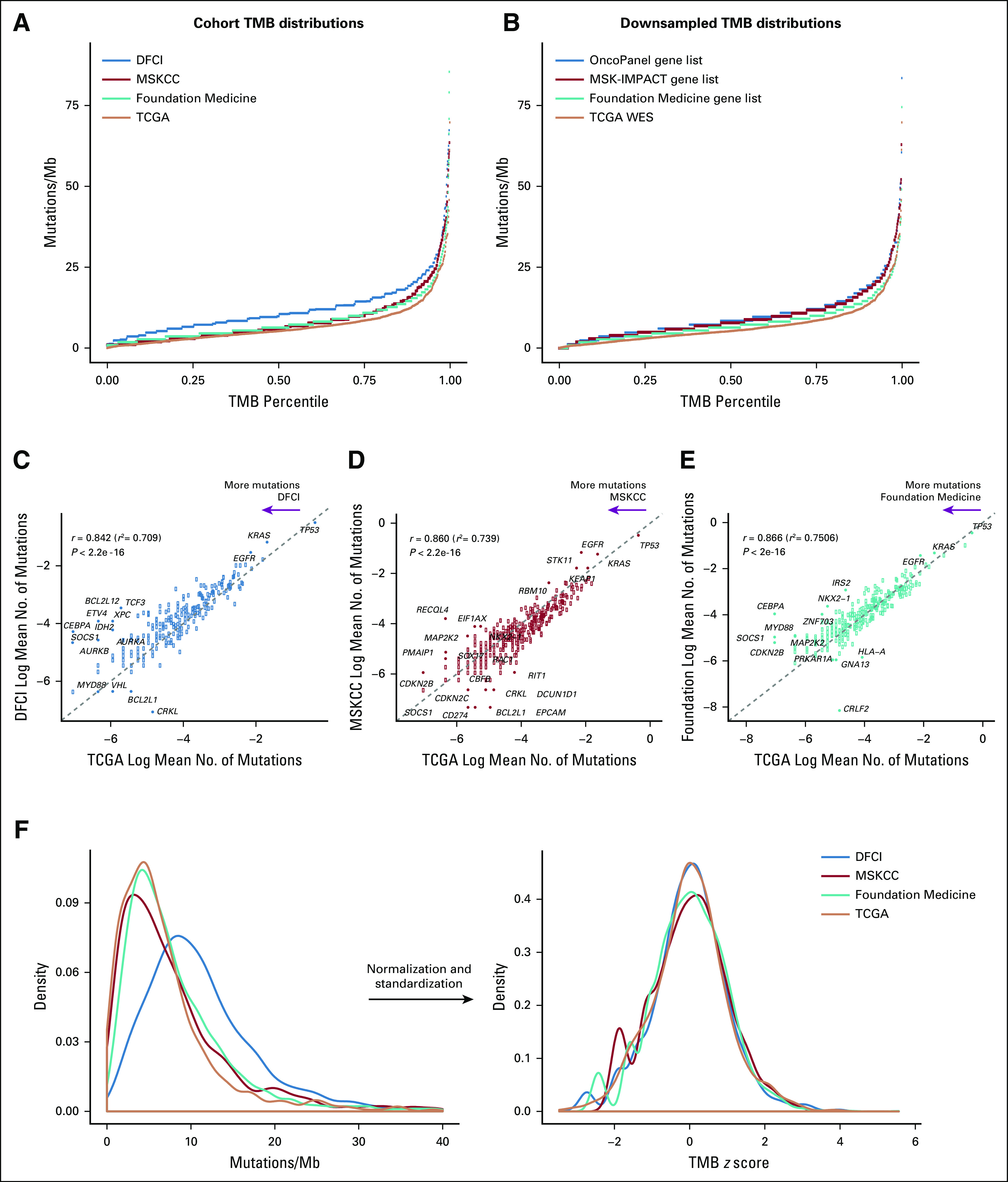
Comparison of tumor mutational burden (TMB) distribution and gene mutation rates across targeted panel and whole-exome sequencing (WES) cohorts. (A and B) TMB distributions in the panel cohorts and obtained by subsetting the WES cohort to the panel gene sets (downsampling) are higher than the WES TMB distribution. The x-axis depicts the percentile of each TMB value; the y-axis depicts TMB in mutations/megabase (Mb). Eight high outliers in panel A are not shown. (C-E) Natural log average mutation rates per gene in each panel cohort are highly correlated with average gene mutation rates in the WES cohort. Each point represents a gene. The dashed line depicts y = x. Pearson’s correlation coefficients are shown. (F) Normalization and standardization of TMB distributions bring the next-generation sequencing and WES cohort distributions into alignment. The left side shows the kernel density plot of unadjusted TMB values in each cohort, and the right side shows the transformed density plot of TMB z scores that demonstrate high overlap. DFCI, Dana-Farber Cancer Institute; MSKCC, Memorial Sloan Kettering Cancer Center; TCGA, The Cancer Genome Atlas.
To further examine assay-specific sources of variation in TMB across panels, we compared the average number of mutations per gene in each cohort against the TCGA averages, surmising that this could reflect differences in assay performance or mutation calling (Figs 1C-E). Concordance between the panel cohorts and TCGA was high (Pearson’s correlation coefficients, 0.842-0.866), and only rarely mutated genes emerged as outliers, which suggests minimal gene-specific variability. Comparison of variant classes demonstrated differential enrichment in the panel versus WES cohorts, which is also consistent with assay-specific differences in mutation filtration (Data Supplement).
Harmonization of TMB Across Platforms Using z Score Standardization
We first attempted to harmonize TMB values across cohorts by linearly mapping panel TMB distributions onto the TCGA TMB distribution (Data Supplement). However, inconsistent variation in TMB across distributions prohibited use of a linear constant. This variability was also diminished but still present when the downsampled TMB values were analyzed (Data Supplement). Consequently, we instead pursued the strategy of transforming unadjusted TMB values into standardized z scores that could be compared across panels. Use of a power transformation converted the right-skewed TMB distributions (Fig 1F) to normal distributions (skewness values ≤ 0.06; Data Supplement), and standardization to z scores brought the TMB distributions into good concordance (Fig 1F) with > 85% overlap (Data Supplement). Cohort size simulation demonstrated that cohorts of > 900 patients are necessary for this approach (Data Supplement).
TMB z Scores Correlate With ICI Outcome and Allow for Cross-Cohort Comparison
We then applied this transformation to TMB in the DFCI and MSKCC ICI-treated subcohorts (n = 272 and 227, respectively; total n = 499; demographic features are listed in Table 1; detailed clinical characteristics are listed in the Data Supplement) and examined whether the derived TMB z scores associated with response.13,24,26 We confirmed that TMB was higher in patients with CR/PR or DCB than in those with PD or NDB in both subcohorts pretransformation (Fig 2A) and that this association held when post-transformation z scores were used (Fig 2B). We noted that while the unadjusted DFCI TMB values were higher than the unadjusted MSKCC TMB values, the z score standardization produced overlapping distributions (Data Supplement), which allowed us to combine the DFCI and MSKCC ICI cohorts. In the merged cohort, TMB z scores remained significantly higher in responders (Fig 2C).
TABLE 1.
Patient Characteristics of Immune Checkpoint Inhibitor–Treated Subcohort
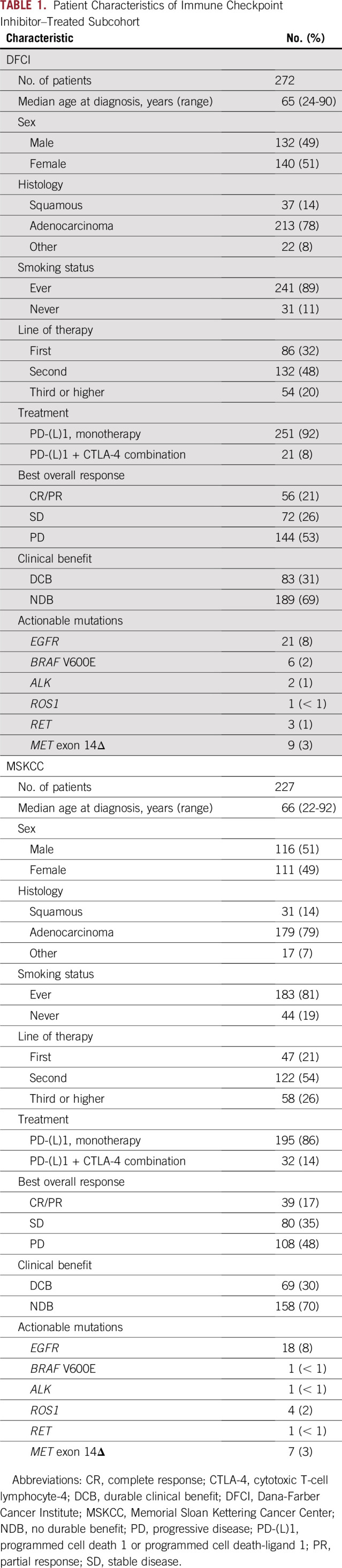
FIG 2.
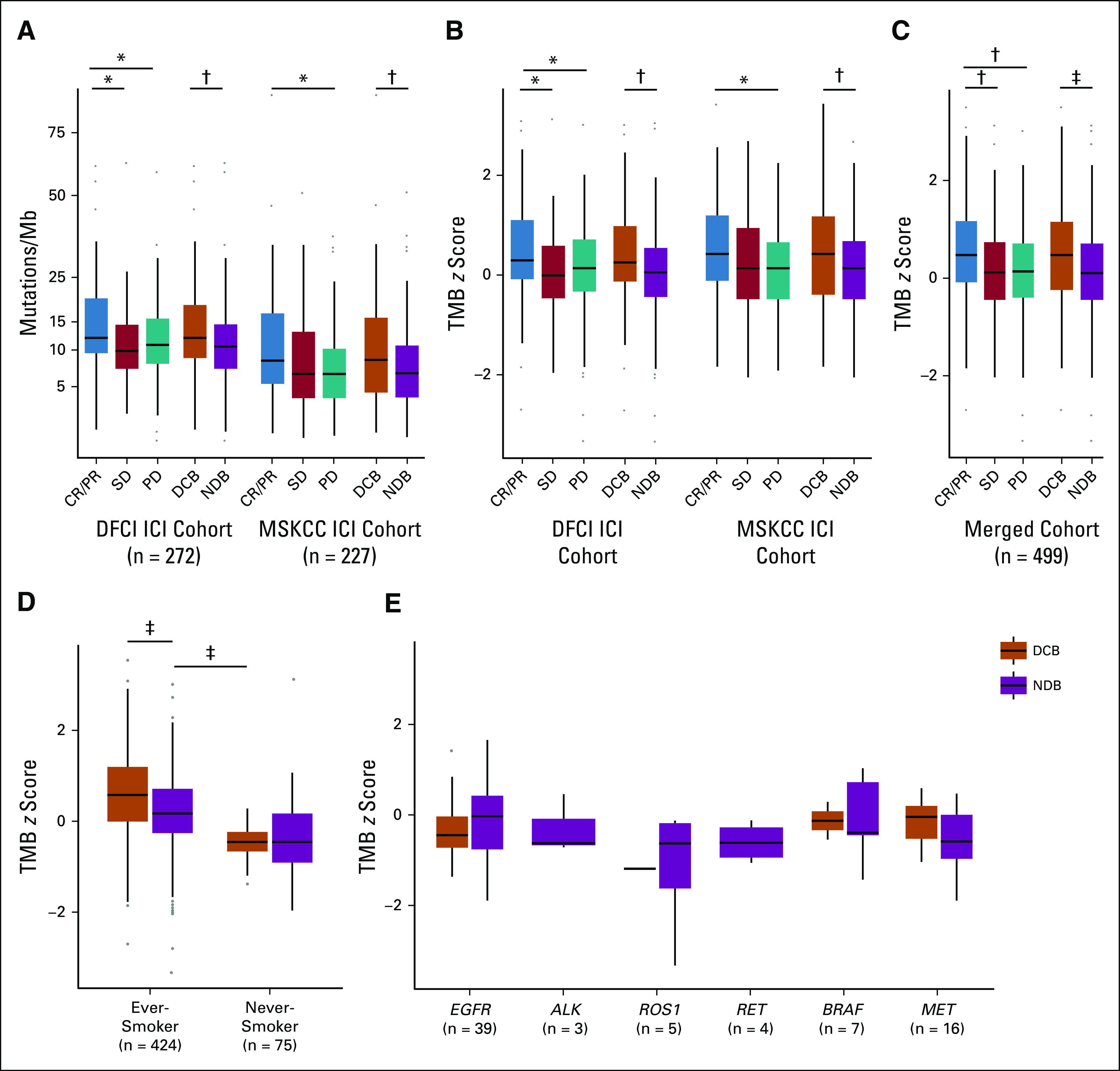
Correlation of tumor mutational burden (TMB) and TMB z scores with outcome. (A) In both the Dana-Farber Cancer Institute (DFCI) and Memorial Sloan Kettering Cancer Center (MSKCC) immune checkpoint inhibitor (ICI) cohorts, TMB is greater in patients with complete response (CR) or partial response (PR) than with progressive disease (PD; DFCI: median, 12.0 v 10.8 mutations/megabase [Mb]; P = .03; MSKCC: median, 8.9 v 6.9 mutations/Mb; P = .02) and in those with durable clinical benefit (DCB) v no durable benefit (NDB; DFCI: median, 12.0 v 10.5 mutations/Mb; P = .003; MSKCC: median, 8.9 v 6.9 mutations/Mb; P = .007). (B) DFCI and MSKCC TMB z score distributions overlap. The z scores are higher in patients with CR/PR v PD and DCB v NDB in each cohort, respectively (DFCI: median CR/PR v PD, 0.30 v 0.14 [P = .03]; median DCB v NDB, 0.30 v 0.10 [P = .003]; MSKCC: median CR/PR v PD, 0.46 v 0.17 [P = .02]; median DCB v NDB, 0.46 v 0.17 [P = .006]). (C) TMB z scores are higher in patients with CR/PR and DCB in the joint cohort (median CR/PR v PD, 0.47 v 0.14 [P = .002]; median CR/PR v stable disease [SD], 0.47 v 0.14 [P = .007]; median DCB v NDB, 0.46 v 0.14 [P = 6.43e-05]). (D) TMB z scores in the joint cohort are higher in ever-smokers with DCB v NDB (median, 0.579 v 0.171; P = 6.046e-05) but were not associated with response in never-smokers (median, −0.456 v −0.456; P = .749). TMB z scores were significantly higher in ever-smokers with NDB than never-smokers with DCB (median, 0.171 v −0.456; P = .00097). (E) TMB z scores in the joint cohort do not associate with response in patients with mutations in targetable oncogenic drivers. Box plots represent medians and interquartile ranges, and vertical lines extend to the 95th percentiles. (*) P < .05, (†) P < .01, (‡) P < .001.
TMB Is Lower in Never-Smokers and May Not Associate With Response
We performed subgroup analyses using the transformed, pooled cohort (DFCI and MSKCC ICI-treated patients, n = 499) to determine whether specific clinical and biologic features affect the association between TMB and response. TMB z scores in ever-smokers were higher than in never-smokers (median, 0.312 v −0.456; P < .0001); notably, TMB in ever-smokers with NDB was higher than that in never-smokers with DCB (median, 0.171 v −0.456; P = .00097; Fig 2D). Among never-smokers, TMB did not differ between DCB and NDB (median, −0.456 v −0.456; P = .749). Sampling simulations (Data Supplement) suggest that this negative finding may be due to decreased power in this subset, although lower TMB values and distinct biology may also contribute (Data Supplement). Similar exploratory analyses of patients who harbor targetable oncogenic drivers did not demonstrate an association between TMB z score and DCB (total n = 74), although power in these small driver subgroups was also limited (Fig 2E). Power simulations suggested that cohort sizes > 300 may be necessary to detect a difference in TMB between patients with DCB and NDB in groups with lower response rates or effect sizes (Data Supplement).
TMB Thresholds and Response
Given the heterogeneity in previously identified thresholds and the percentile cut points used to identify such thresholds, we used our pooled cohort to systematically explore the relationship between TMB and response to ICIs across the TMB distribution. We calculated the rate of DCB and CR/PR with increasing TMB thresholds in the pooled and separate ICI cohorts (Fig 3A; Data Supplement). Table 2 lists the TMB z scores and values associated with each threshold. We observed a gradual increase in the rate of DCB with increasing TMB thresholds. We noted, however, that this could arise from enriching for high TMB outliers and therefore calculated the rate of DCB within each TMB decile (joint cohort shown in Fig 3B; separate cohorts shown in the Data Supplement). In this analysis, we noted high DCB rates in the highest deciles (40.4% in patients with TMB z scores between the 80th and 90th percentiles; 53.1% in patients with TMB z scores ≥ 90th percentile) and low rates in the lowest deciles (16.7% in patients with TMB z scores < 10th percentile). However, the middle deciles exhibited greater heterogeneity in DCB rate. Accordingly, the odds ratio of DCB with increasing TMB thresholds was highest with TMB cutoffs ≥ 80th percentile and more heterogeneous at lower thresholds (Fig 3C). Similar trends were observed in smokers; there was no increase in DCB rates with increasing TMB threshold in never-smokers (Data Supplement). The pattern of association between PD-L1 and DCB was similar to TMB, with increasing rates of DCB with higher PD-L1 thresholds but more variability within PD-L1 score groupings (Data Supplement).
FIG 3.
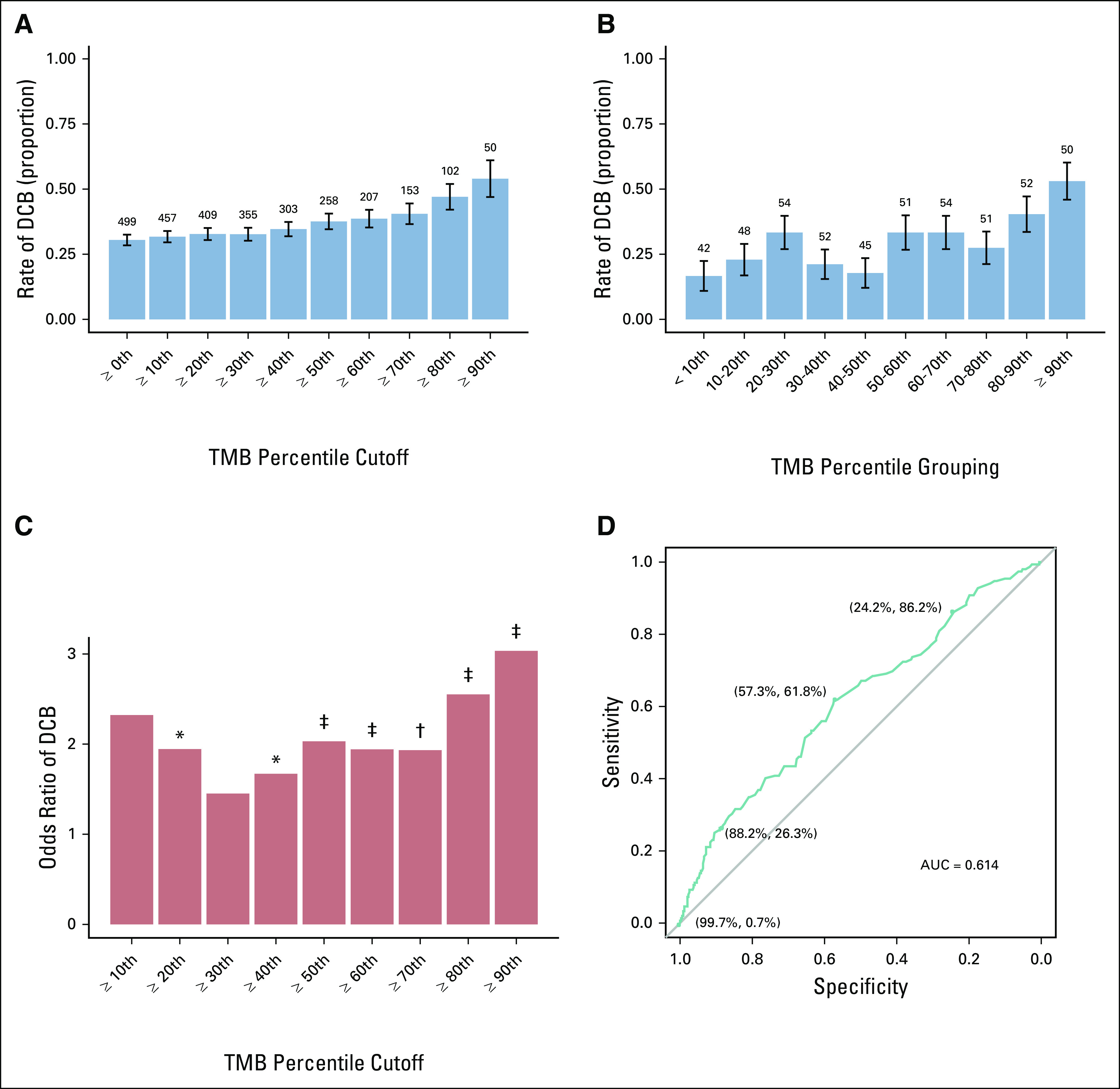
Rate of response to immune checkpoint inhibitors by tumor mutational burden (TMB) threshold. (A) Rate of durable clinical benefit (DCB) increases with increasing TMB cutoff thresholds. TMB z score deciles were selected as cut points, and rate of DCB was calculated for patients in the joint cohorts whose TMB z scores were greater than or equal to the cut point. Bars depict rate of DCB. The leftmost bar depicts rate of DCB in the unselected cohort (TMB ≥ 0th percentile). (B) The rate of DCB within TMB z score deciles is more variable. Bars depict rate of DCB among patients whose TMB z scores are greater than or equal to the lower bound and less than the upper bound. (C) The odds ratio of DCB varies with increasing TMB z score cut points. Bars depict odds ratio; significant P values are indicated above bars. (D) Receiver operating characteristic curve demonstrates a trade-off in sensitivity v specificity of DCB at varying TMB z score values. Exploratory cut points, with their associated specificity and sensitivity, are indicated. Error bars represent standard error. Numbers over bar graphs indicate number of patients in each group. (*) P < .05, (†) P < .01, (‡) P < .001. AUC, area under the curve.
TABLE 2.
TMB z Score Associated With Each Decile Cutoff in the Joint Immune Checkpoint Inhibitor Cohort

Given the heterogeneity of response rates over the mid-TMB distribution, we used ROC curve analysis to formally quantify how well TMB z scores discriminate between DCB and NDB. ROC analysis yielded an area under the curve (AUC) of 0.614. The Youden’s index cutoff was associated with a sensitivity of 61.8% and a specificity of 57.3%, which resulted in undertreatment of 12% of patients and overtreatment of 30% (Fig 3D; Table 3). Application of other thresholds demonstrated better specificity at higher TMB z score thresholds but at the expense of missing patients who would have responded. Cutoffs and their associated sensitivity/specificity were similar in the cohorts considered separately (Data Supplement). Application of the clinically used PD-L1 threshold ≥ 50% was associated with undertreatment of 13% and overtreatment of 19% of patients. TMB z score did not discriminate between DCB and NDB in never-smokers or patients with targetable driver mutations (AUC, 0.493; data not shown). Analysis of TMB thresholds with respect to progression-free survival, rather than response, demonstrated similar results (Data Supplement).
TABLE 3.
Test Characteristics and Performance of TMB Cutoff Values in the Joint Immune Checkpoint Inhibitor Cohort (n = 499)

DISCUSSION
We present a pragmatic comparison of TMB calculated from targeted panels and WES and apply TMB z score conversion to enable harmonized analyses. We demonstrate that this approach can translate TMB values across tests and can be used to integrate distinct data sets for discovery and further analyses. In addition, our use of real-world data sets allows us to incorporate and account for sources of variation not captured by in silico downsampling analyses, such as differences in mutation/indel calling pipelines, depth of coverage, and germline filtration. This approach is distinct from other parallel harmonization efforts,22,43 which focus on standardization of TMB definitions and reporting and eventually aim to generate gold standard cell lines for benchmarking. We anticipate that our approach will be of immediate use to both clinicians and researchers and anticipate that it can be easily applied to other platforms and relevant tumor types.
Although the association between TMB and response to ICIs in NSCLC has been demonstrated, less is understood about how clinical and biologic features affect this association. Here, we found that TMB did not associate with DCB in never-smokers and in patients who harbor targetable oncogenic mutations. Of note, these analyses were underpowered to detect a difference, and our power simulations indicated that larger cohorts are needed and caution against definitive conclusions in these small subgroup analyses. However, we also observed that never-smokers who benefitted from ICIs had markedly lower TMB values than ever-smokers who did not, which suggests that further study to identify TMB-independent predictors of response in never-smokers may be warranted and raises the important possibility that the clinical application of TMB as a biomarker will need to take clinical and biologic features into account.
The importance of context is further emphasized by our analysis of TMB thresholds. Prior analyses have generally focused on identifying a single threshold to define TMB-high and TMB-low subgroups, with variation in selected thresholds across studies.13,15,26,44 Our systematic analysis of TMB thresholds illustrates additional nuances in the relationship between TMB and response. We observed enrichment in DCB with higher TMB thresholds, as expected, but weaker discrimination in the midrange of TMB values without a single, natural biologic inflection point. These findings may account for some of the observed heterogeneity among previously proposed thresholds, as there may be a range of values that discriminate similarly between responders and nonresponders. In addition, our data suggest that the choice of a given threshold must be decided within a goal-specific context that considers the relative efficacy of the alternative treatment; a TMB threshold selected to enrich for response to first-line therapy may be different than a threshold selected for second-line therapy. Of note, TMB is independent of PD-L1 expression,12,13 with similar biomarker performance: Increasing expression is associated with improved efficacy without a natural cut point, there is variability in DCB enrichment within deciles of expression, and distinct thresholds are appropriately applied on the basis of the specific treatment scenario (ie, ≥ 50%, ≥ 1%, none).3,5,45 Ultimately, these data do not answer whether and how TMB should be applied to clinical practice, as this must be examined through prospective clinical trials, but add nuance to our understanding of how TMB associates with response.
One limitation of this study is that our comparison of TMBs assumes that the observed distinctions reflect differences in platforms rather than in patients/samples. We were not able to account for clinical and tumor features because of inconsistent sample annotation but note that our large cohorts help to mitigate sampling bias, and the overall consistencies in shape of distribution are reassuring. Whether TMB distributions should be more narrowly defined by sample features, such as histology or stage, is an open question, and the normalization we describe here can be adjusted as more is learned. At present, however, TMB is compared across patients and biopsy specimens without reference to these sample characteristics, which makes this aggregated approach consistent with current clinical practice.
In conclusion, we provide a practical approach to the challenge of standardizing TMB across platforms, and we apply this approach to integrate distinct data sets to better understand how TMB associates with response. Much remains to be learned about how and why TMB associates with response to ICI and how best to apply TMB in the clinic for precision immunotherapy.
Footnotes
Presented at the International Association for the Study of Lung Cancer Targeted Therapies 2019 World Conference on Lung Cancer, Barcelona, Spain, September 7-10, 2019; 2019 ASCO-SITC Clinical Immuno-Oncology Symposium, San Francisco, CA, February 28-March 2, 2019; and American Association for Cancer Research Annual Meeting 2019, Atlanta, GA, March 29-April 3, 2019.
Supported by the Damon Runyon Foundation (D.L. and E.M.V.A.), National Institutes of Health grants R01CA227388 (E.M.V.A.) and K08CA234458 (DL), the Conquer Cancer Foundation Young Investigator Award (DL), the Society for Immunotherapy of Cancer-Bristol-Myers Squibb Translational Fellowship (DL), Memorial Sloan Kettering Cancer Center (MSKCC) support grant/core grant P30CA008748, and the Druckenmiller Center for Lung Cancer Research at MSKCC. M.D.H. is a Damon Runyon Clinical Investigator supported (in part) by the Damon Runyon Cancer Research Foundation (CI-98-18) and is a member of the Parker Institute for Cancer Immunotherapy. M.N. is supported by National Cancer Institute grants R01CA203636 and U01CA209414.
AUTHOR CONTRIBUTIONS
Conception and design: Natalie I. Vokes, Renato Umeton, Eliezer M. Van Allen, Matthew D. Hellmann, Mark M. Awad
Financial support: Eliezer M. Van Allen
Administrative support: Eliezer M. Van Allen
Provision of study material or patients: Eliezer M. Van Allen, Matthew D. Hellmann
Collection and assembly of data: Natalie I. Vokes, Biagio Ricciuti, Elizabeth Jimenez-Aguilar, Hira Rizvi, Jeffrey Girshman, Anika Adeni, Suzanne Dahlberg, Ahmet Zehir, Mizuki Nishino, Renato Umeton, Lynette M. Sholl, Eliezer M. Van Allen, Matthew D. Hellmann, Mark M. Awad
Data analysis and interpretation: Natalie I. Vokes, David Liu, Felix Dietlein, Meng Xiao He, Claire A. Margolis, Haitham A. Elmarakeby, Francisco Sanchez-Vega, Nikolaus Schultz, Ahmet Zehir, Pasi A. Jänne, Renato Umeton, Lynette M. Sholl, Eliezer M. Van Allen, Matthew D. Hellmann, Mark M. Awad
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Claire A. Margolis
Employment: Verana Health
Suzanne Dahlberg
Consulting or Advisory Role: AstraZeneca
Patents, Royalties, Other Intellectual Property: Patent pending for a statistical model assessing tumor growth (Inst)
Pasi A. Jänne
Stock and Other Ownership Interests: Gatekeeper Pharmaceuticals, Loxo Oncology
Consulting or Advisory Role: Pfizer, Boehringer Ingelheim, AstraZeneca, Merrimack, Chugai Pharma, Roche, Genentech, Loxo Oncology, Mirati Therapeutics, Araxes Pharma, Ignyta, Eli Lilly, Takeda Pharmaceuticals, Novartis, Biocartis, Voronoi, SFJ Pharmaceuticals Group
Research Funding: AstraZeneca, Astellas Pharma, Daiichi Sankyo, Eli Lilly, Boehringer Ingelheim, Puma Biotechnology, Takeda Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Co-inventor on a Dana-Farber Cancer Institute–owned patent on EGFR mutations licensed to Lab Corp; postmarketing royalties received from this invention
Mizuki Nishino
Honoraria: Roche Pharma AG
Consulting or Advisory Role: Daiichi Sankyo, AstraZeneca
Research Funding: Merck (Inst), Toshiba (Inst), AstraZeneca (Inst)
Renato Umeton
Consulting or Advisory Role: Health Catalyst
Patents, Royalties, Other Intellectual Property: Patent on portable medical device and method for quantitative retinal image analysis through a smartphone; patent on Epstein-Barr virus genotypic variants and uses thereof as risk predictors, biomarkers, and therapeutic targets of multiple sclerosis
Lynette M. Sholl
Honoraria: AstraZeneca
Consulting or Advisory Role: Foghorn Therapeutics, Loxo Oncology
Research Funding: Genentech (Inst)
Travel, Accommodations, Expenses: Bristol-Myers Squibb
Eliezer M. Van Allen
Stock and Other Ownership Interests: Syapse, Tango Therapeutics, Genome Medical, Microsoft, Ervaxx
Consulting or Advisory Role: Syapse, Roche, Third Rock Ventures, Takeda Pharmaceuticals, Novartis, Genome Medical, Invitae, Illumina, Tango Therapeutics, Ervaxx
Speakers’ Bureau: Illumina
Research Funding: Bristol-Myers Squibb, Novartis
Patents, Royalties, Other Intellectual Property: Patent on discovery of retained intron as source of cancer neoantigens (Inst), patent on discovery of chromatin regulators as biomarkers of response to cancer immunotherapy (Inst), patent on clinical interpretation algorithms using cancer molecular data (Inst)
Travel, Accommodations, Expenses: Roche, Genentech
Matthew D. Hellmann
Stock and Other Ownership Interests: Shattuck Labs
Honoraria: AstraZeneca, Bristol-Myers Squibb
Consulting or Advisory Role: Bristol-Myers Squibb, Merck, Genentech, AstraZeneca, MedImmune, Novartis, Janssen Pharmaceuticals, Nektar, Syndax, Mirati Therapeutics, Shattuck Labs
Research Funding: Bristol-Myers Squibb (Inst)
Patents, Royalties, Other Intellectual Property: A patent has been filed by Memorial Sloan Kettering (PCT/US2015/062208) for the use of tumor mutation burden for prediction of immunotherapy efficacy and which is licensed to Personal Genome Diagnostics (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Bristol-Myers Squibb
Mark M. Awad
Consulting or Advisory Role: Genentech, Merck, Pfizer, Boehringer Ingelheim, AbbVie, AstraZeneca, MedImmune, Clovis Oncology, Nektar, Bristol-Myers Squibb, ARIAD Pharmaceuticals, Foundation Medicine, Syndax, Novartis, Blueprint Medicines, Maverick Therapeutics
Research Funding: Bristol-Myers Squibb, Genentech (Inst), Roche (Inst), Eli Lilly (Inst), AstraZeneca (Inst), Bristol-Myers Squibb (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 2.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet. 2016;387:1540–1550. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 4.Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–1846. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 5.Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 6.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res. 2016;4:959–967. doi: 10.1158/2326-6066.CIR-16-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carbone DP, Reck M, Paz-Ares L, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376:2415–2426. doi: 10.1056/NEJMoa1613493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rizvi H, Sanchez-Vega F, La K, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36:633–641. doi: 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miao D, Margolis CA, Vokes NI, et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet. 2018;50:1271–1281. doi: 10.1038/s41588-018-0200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gandara DR, Paul SM, Kowanetz M, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24:1441–1448. doi: 10.1038/s41591-018-0134-3. [DOI] [PubMed] [Google Scholar]
- 16.Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362:aar3593. doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garofalo A, Sholl L, Reardon B, et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016;8:79. doi: 10.1186/s13073-016-0333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Szustakowski JD, Green G, Geese WJ, et al: Evaluation of tumor mutation burden as a biomarker for immune checkpoint inhibitor efficacy: A calibration study of whole exome sequencing with FoundationOne. Cancer Res 78:5528, 2018 (suppl; abstr 5528) [Google Scholar]
- 21. doi: 10.1038/nm.4333. Zehir A, Benayed R, Shah RH, et al: Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23:703-713, 2017 [Erratum: Nat Med 23:1004, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. doi: 10.1136/jitc-2019-000147. Fabrizio D, Che S-J, Xie M, et al: In silico assessment of variation in TMB quantification across diagnostic platforms: Phase 1 of the Friends of Cancer Research Harmonization Project. J Immunother Cancer 6, 2018 (suppl 2; abstr O48) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hendriks LE, Rouleau E, Besse B: Clinical utility of tumor mutational burden in patients with non-small cell lung cancer treated with immunotherapy. Transl Lung Cancer Res 7:647-660, 2018. [DOI] [PMC free article] [PubMed]
- 24. Hellmann MD, Nathanson T, Rizvi H, et al: Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33:843-852.e4, 2018. [DOI] [PMC free article] [PubMed]
- 25.Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–2104. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ready N, Hellmann MD, Awad MM, et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol. 2019;37:992–1000. doi: 10.1200/JCO.18.01042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hartmaier RJ, Albacker LA, Chmielecki J, et al. High-throughput genomic profiling of adult solid tumors reveals novel insights into cancer pathogenesis. Cancer Res. 2017;77:2464–2475. doi: 10.1158/0008-5472.CAN-16-2479. [DOI] [PubMed] [Google Scholar]
- 31.Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48:607–616. doi: 10.1038/ng.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia EP, Minkovsky A, Jia Y, et al. Validation of OncoPanel: A targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med. 2017;141:751–758. doi: 10.5858/arpa.2016-0527-OA. [DOI] [PubMed] [Google Scholar]
- 33.Sholl LM, Do K, Shivdasani P, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016;1:e87062. doi: 10.1172/jci.insight.87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23. doi: 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.AACR Project GENIE Consortium AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017;7:818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 38. Tukey JW: Exploratory Data Analysis. Reading, MA, Addison-Wesley, 1977. [Google Scholar]
- 39. Mangiafico SS: Summary and Analysis of Extension Program Evaluation in R, 2016. http://rcompanion.org/handbook.
- 40. Pastore M: Overlapping: A R package for estimating overlapping in empirical distributions. J Open Source Softw 3:1023, 2018. [Google Scholar]
- 41.Robin X, Turck N, Hainard A, et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.López-Ratón M, Rodríguez-Álvarez MX, Cadarso-Suárez C, et al. OptimalCutpoints: An R package for selecting optimal cutpoints in diagnostic tests. J Stat Softw. 2014;1:2014. [Google Scholar]
- 43. Stenzinger A, Allen JD, Maas J, et al: Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer 58:578-255, 2019. [DOI] [PMC free article] [PubMed]
- 44.Heeke S, Hofman P. Tumor mutational burden assessment as a predictive biomarker for immunotherapy in lung cancer patients: Getting ready for prime-time or not? Transl Lung Cancer Res. 2018;7:631–638. doi: 10.21037/tlcr.2018.08.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377:1919–1929. doi: 10.1056/NEJMoa1709937. [DOI] [PubMed] [Google Scholar]