

Abstract
Monitoring the drug susceptibility of Leishmania isolates still largely relies on standard in vitro cell-based susceptibility assays using (patient-isolated) promastigotes for infection. Although this assay is widely used, no fully standardized/harmonized protocol is yet available hence resulting in the application of a wide variety of host cells (primary cells and cell lines), different drug exposure times, detection methods and endpoint criteria. Advocacy for standardization to decrease inter-laboratory variation and improve interpretation of results has already repeatedly been made, unfortunately still with unsatisfactory progress. As a logical next step, it would be useful to reach at least some agreement on the type of host cell and basic experimental design for routine amastigote susceptibility determination. The present laboratory study using different L. infantum strains as a model for visceral leishmaniasis species compared primary cells (mouse peritoneal exudate (PEC), mouse bone marrow derived macrophages and human peripheral blood monocyte derived macrophages) and commercially available cell lines (THP-1, J774, RAW) for either their susceptibility to infection, their role in supporting intracellular amastigote multiplication and overall feasibility/accessibility of experimental assay protocol. The major findings were that primary cells are better than cell lines in supporting infection and intracellular parasite multiplication, with PECs to be preferred for technical reasons. Cell lines require drug exposure of >96h with THP-1 to be preferred but subject to a variable response to PMA stimulation. The fast dividing J774 and RAW cells out-compete parasite-infected cells precluding proper assay read-out. Some findings could possibly also be applicable to cutaneous Leishmania strains, but this still needs cross-checking. Besides inherent limitations in a clinical setting, susceptibility testing of clinical isolates may remain problematic because of the reliance on patient-derived promastigotes which may exhibit variable degrees of metacyclogenesis and infectivity.
Author summary
Leishmaniasis is a neglected tropical disease caused by parasites belonging to the genus of Leishmania and transmitted by the bite of infected female sand flies. Concerns about the effective control of the disease are rising in view of the increasing number of treatment failures that may be related to drug resistance. Monitoring of drug susceptibility in the field should become an essential asset, however, there is still insufficient harmonization in the laboratory assays. This study focused on the standard intracellular amastigote susceptibility assay and compared protocol variables, such as type of macrophage host cell (primary versus cell lines), multiplicity of infection and duration of drug exposure. Primary cells perform best with little difference between cells derived from Swiss mice or BALB/c mice. From a practical point of view, mouse peritoneal exudate cells can be recommended. If mice would not be available, THP-1 cells are the best alternative. For field strains, metacyclic promastigotes should be used at a multiplicity of infection of 10–15 parasites per cell with drug exposure starting at 24h post-infection and continued for 120h. Unfortunately, susceptibility testing of clinical isolates will remain problematic because of the reliance on promastigotes which may exhibit variable degrees of metacyclogenesis and infectivity. Opting for cell-based assays may be complicated by the fact that dedicated laboratory infrastructure may sometimes be lacking in disease-endemic countries.
Introduction
Despite the ongoing search for specific molecular resistance markers [1–3], the approach to evaluate drug resistance in Leishmania still heavily relies on in vitro drug-susceptibility assays. Given the well-known stage-dependent susceptibility differences that can be observed for most drugs [4, 5], intracellular amastigote assays may be regarded as the gold standard [5], although assays on extracellular promastigotes or axenic amastigotes can be informative as well with the advantage that they are much more accessible and less time-consuming. Intracellular assays require a suitable host cell and several types have already been described and compared in literature, covering the THP-1, RAW, J774 and U937 cell lines, and various types of primary macrophages [6–15]. For example, monocyte-derived cell lines require cell stimulation where the different use concentration of phorbol-12-myristate-13-acetate (PMA), retinoic acid or 1,25-dihydroxy-vitamin D3 during variable stimulation periods precludes direct comparison of results [16, 17]. Although most protocols are basically constructed in a similar way, the abundance of marginally different designs (Fig 1) nevertheless complicates drug susceptibility interpretation and inter-laboratory comparison. Despite several pleas for standardization [18–20] and literature on comparative evaluation of several cell types [14, 20, 21], different models are still widely used hence requiring further refinement of the proposed experimental methodology.
Fig 1. Variability in the standard intracellular susceptibility protocol.

Divergences in design related to the application of different host cells, different host and/or cell stimuli and different drug exposure times.
Recognizing the necessity to harmonize Leishmania drug evaluation assays and establish standardized methods to monitor drug resistance, as was done for malaria (WWARN: WorldWide Antimalarial resistance Network; www.wwarn.org) [18–20], the present study aimed to contribute to a pragmatic and straightforward approach to determine the drug susceptibility of (clinical) Leishmania isolates in laboratory conditions, hereby supporting a next step towards a more harmonized operating procedure that could find some application in the clinical setting. Strains of L. infantum were used and specific focus was given to the suitability of different types of primary macrophages and cell lines as permissive host cell, receptivity to infection, capacity to support intracellular multiplication and the susceptibility outcome of the most frequently used antileishmanial reference drugs.
Materials and methods
Media, reagents and chemicals
M199 medium, DMEM-medium, RPMI-1640, MEM non-essential amino acids, L-glutamine, penicillin-streptomycin (P/S) solution, fetal bovine serum (FBS), sodium pyruvate, phosphate buffered saline (PBS) and hemin were purchased from Invitrogen (Ghent, Belgium). Phorbol 12-myristate 13-acetate (PMA), trivalent antimony (SbIII: potassium antimonyl tartrate), EDTA, HEPES, potato starch, Giemsa, adenine, paromomycin (PMM) and Histopaque-1077 were purchased from Sigma Aldrich (Diegem, Belgium). Miltefosine (MIL) was available from Carbosynth (Berkshire, UK), amphotericin B deoxycholate (AmB: Fungizone Bristol-Myers Squibb (Brussels, Belgium), sodium-stibogluconate (SbV; SSG) from Calbiochem (San Diego, US) and 6-biopterine from Schrickx Labs (Bauma, Switzerland).
Preparation of drug stock solutions
Drug stock solutions were prepared as described previously [22]. The SbV stock solution (1 mg SbV eq /ml) was prepared by dissolving 31.9 mg SSG in 10 ml water or PBS by stirring at 37°C for 1 hour until a clear solution was obtained which was divided into small aliquots (1 ml) kept at -20°C for maximally 3 months. To prepare a SbIII stock solution (1 mg SbIII eq /ml), 27.7 mg SbIII tartrate was dissolved in 10 ml water or PBS by stirring until a clear solution was obtained. Similar as for SbV, the suspension was divided into small aliquots and kept at -20°C for maximally 3 months. To obtain the AmB 20 mM stock solution, 41 mg Fungizone powder was dissolved in 1 ml DMSO 100%. Further dilutions were made in water and immediately used because of limited stability. A 20 mM MIL stock solution was prepared by dissolving 81.5 mg MIL in 10 ml water or PBS by stirring until a clear solution was obtained. The MIL stock solution can be kept at 4°C for 3 months. Finally, PMM stock solutions (20 mM) were made by dissolving 142.74 mg PMM-sulfate in 10 ml water or PBS. The stock solution was kept at 4°C for maximally 3 months.
Ethics
The use of laboratory rodents was carried out in strict accordance to all mandatory guidelines (EU directives, including the Revised Directive 2010/63/EU on the Protection of Animals used for Scientific Purposes that came into force on 01/01/2013, and the declaration of Helsinki in its latest version) and was approved by the ethical committee of the University of Antwerp, Belgium (UA-ECD 2016–54 (02-09-2016).
Laboratory animals
Female Swiss and BALB/c mice (body weight 20 g) for the collection of primary peritoneal and bone marrow derived macrophages were purchased from Janvier (Le Genest Saint Isle, France). Food for laboratory rodents (Carfil, Arendonk, Belgium) and drinking water were available ad libitum. Animals were kept in quarantine for at least 5 days before the collection of macrophages.
Leishmania parasites, cultures and infection
All experiments were carried out using the Leishmania infantum laboratory reference strain (MHOM/MA/67/ITMAP263) and three L. infantum clinical isolates with a defined drug susceptibility background to the standard reference drugs antimony (SbV and SbIII), miltefosine (MIL), paromomycin (PMM) and amphotericin B (AmB) (Table 1). LEM3049 (MHOM/FR/95/LEM3049) originated from a French HIV co-infected patient; BH402/60 (MCAN/BR/2002/BH402/60) is a canine isolate from Brazil that was part of a large clinical trial with liposomal meglumine [23] while MHOM/BR/2007/WC (L3015) was obtained from a Brazilian HIV- patient [24]. The French clinical isolate LEM3323 (MHOM/FR/96/LEM3323) with a Sb-resistant background and its experimentally derived MIL-resistant (LEM3323/MIL) and PMM-resistant (LEM3323/PMM) lines [25] were specifically included to assess the discriminating capacities of the different cell types for drug resistant strains. Five recent L. infantum clinical isolates from the recent outbreak in Fuenlabrada (Madrid, Spain) were included in the study to further corroborate the selected assay system (MHOM/ES/2016/LLM-2301, LLM-2309, LLM-2323, LLM-2338, LLM-2346) [26]. Promastigotes were routinely cultured in M199 medium supplemented with HEPES, adenine, hemin, 6-biopterine, L-glutamine, 5% sodium bicarbonate and 10% heat-inactivated (56°C for 30 minutes) FBS (iFBS) [20]. Parasites were sub-cultured twice weekly and the passage number was kept as low as possible to avoid a decrease in virulence due to prolonged cultivation in vitro [27]. Five-day-old promastigote cultures were used for preconditioning, as described earlier [9].
Table 1. Overview of Leishmania infantum strains used.
Origin and drug-susceptibility profile to the current antileishmania drugs (R: resistant; S: susceptible; [R]: resistant after experimental selection).
| L. infantum strain | Code | Origin | Drug susceptibility profile | |||
|---|---|---|---|---|---|---|
| SbV | SbIII | MIL | PMM | |||
| ITMAP263 | MHOM/MA/67/ITMAP263 | Laboratory reference strain | S | S | S | S |
| LEM3049 | MHOM/FR/95/LEM3049 | French field isolate from HIV+ patient | S | S | S | S |
| L3015 | MHOM/BR/2007/WC | Brazilian field isolate from HIV+ patient | S | S | S | S |
| LEM3323 | MHOM/FR/96/LEM3323 | French field isolate from HIV+ patient | R | R | S | S |
| LEM3323/MIL | Experimentally selected MIL-resistant strain | R | R | [R] | S | |
| LEM3323/PMM | Experimentally selected PMM-resistant strain | R | R | S | [R] | |
Preparation of macrophages
A former comparison of mouse-derived primary peritoneal exudate cells (PECs) and bone marrow derived macrophages (BMMφ) already revealed some outcome differences [5, 28]. For in-depth evaluation of the impact of these findings on the susceptibility determination of clinical isolates, BALB/c mouse BMMφ and Swiss mouse PECs were seeded in 96-well plates, as previously described [28]. For the collection of PECs, Swiss mice were stimulated with a 2% starch suspension in PBS 24h prior to macrophage prelevation. PECs were collected via peritoneal lavage with 10mL RPMI-1640 macrophage medium, supplemented with 5% iFBS, 2% PS and 1% L-glutamine. Cells were counted in KOVA counting slides and seeded in 96-well plates at a final concentration of 30,000 cells/well. Bone marrow-derived macrophages (BMMφs) were obtained from the femur and tibia of female BALB/c mice by flushing the bone cavities with cold RPMI-1640 medium. The bone marrow was kept on ice during the isolation, then centrifuged for 20 minutes upon which the red blood cells were lysed with ammonium-chloride-potassium lysis buffer (VWR, Leuven, Belgium). The recovered cells were resuspended in RPMI-1640 medium supplemented with 1% non-essential amino acids, 1% P/S solution, 1% sodium pyruvate, 1% L-glutamine, 10% iFBS and 15% L929 supernatant containing macrophage colony stimulating factor (M-CSF), and incubated in Petri dishes at 37°C and 5% CO2 to obtain macrophage monolayers. After 7 days, the cells were detached from the dishes with dissociation buffer (1% 0.5M EDTA, 2% 1M HEPES in PBS), counted and seeded similarly to the PECs.
Human peripheral blood mononuclear cells (PBMCs) were isolated from the blood of healthy volunteers. A small volume of blood was collected in covered test tubes and allowed to clot 15–30 minutes at room temperature. The clot was separated by centrifugation at 2,000 x g for 10 minutes at 4°C upon which the serum was transferred into a clean tube and inactivated at 56°C for 30 minutes. For the isolation of PBMCs, blood was collected in heparin-coated tubes, layered onto Histopaque-1077 and centrifuged at 400×g for 30 minutes. After gradient separation, the cells were collected and washed three times with RPMI-1640 medium, counted in a KOVA counting chamber and suspended in RPMI-1640 supplemented with 2% P/S and 10% autologous human serum. Cultures were incubated at 37°C and 5% CO2 for 6 days to produce macrophage monolayers.
J774A.1 and RAW264.7 macrophage cell lines were purchased from ATCC and cultured in Dulbecco’s modified minimal essential medium (DMEM) supplemented with 10% iFBS. Cells were grown in T25 culture flasks at 37°C in presence of 5% CO2. Every 2 days, the cells were detached from the culture flask with a cell scraper and sub-cultured. The cells were counted and plated in 96-well plates at a density of 30,000 cells/well 24h before infection.
THP-1 human monocyte cell line was obtained from ATCC and cultured at 37°C and 5% CO2 in RPMI-1640 medium supplemented with 10% iFBS. The cells were sub-cultured every two days with particular attention not to exceed a cell density of 106/mL. Before infection, 30,000 cells/well were plated in 96-well plates and simultaneously stimulated with 100 ng/mL PMA. The PMA stimulus was removed either after 48h by renewing the medium followed by incubation for another 5 days to allow complete macrophage differentiation [17], or after 72h immediately before infection.
Receptivity for infection
To determine the macrophage’s receptivity for infection, the initial rate of phagocytosis was evaluated for each cell type. The infection index was determined microscopically at 2h, 4h and 6h post-infection upon Giemsa staining and was calculated as described previously [29]. In brief, the intracellular parasite burden was quantified microscopically in at least of 50 macrophages (both non-infected and infected), from which the infection index (average number of amastigotes/cell) was calculated.
Parasite and cell replication
Intracellular amastigote replication was evaluated to assess the host cell supportive capacity for infection [25]. Briefly, infection indices of the different types of macrophages were determined microscopically during a 10-day course of experiment. Renewal of the culture medium was omitted to minimize interactions during culture and mainly because no significant decrease in cell viability was observed until up to 168 hpi (S4 Fig). To determine whether continuously dividing cell lines (J774A.1 and RAW264.7) could still be used to determine the IC50, their replication potential was cross-checked to the intracellular replication capacities of the parasites.
Impact of intracellular parasite burden and drug exposure time on drug susceptibility
For each L. infantum isolate, the 50% inhibitory concentration (IC50) against the standard drugs was determined in each of the different cell types. Macrophages were infected at a ratio of 15:1 by pre-conditioned promastigotes of LEM3049 that was selected as drug-susceptible field strain. Macrophages were exposed 24h later to 2-fold drug dilutions of the reference drugs SbV, SbIII, MIL, PMM and AmB. The IC50 value was determined microscopically 96 hours post-infection (hpi) upon methanol fixation and Giemsa-staining.
As several studies in literature used different drug exposure times for susceptibility determination [5, 30–32], the impact of exposure time on the IC50 was evaluated in parallel by determining the IC50 for each drug at 24 hour intervals post-treatment. As previous research also demonstrated that the infection index can impact on cell proliferation or survival [33] and hence may affect the IC50-value [14], a 2-fold dilution of the parasite inoculum combined with a 2-fold dilution of MIL (not done for the other drugs) was included for drug susceptibility determination at different infection indices.
Capacity to discriminate drug resistance
Monitoring drug susceptibility in the field implies the need for a fast and reliable laboratory result. Given the cell-specific susceptibility [21], it is logical to assume that different macrophage types will not equally detect drug resistance against the current reference drugs (breakpoints as used in the present study are listed in Table 2). This was checked by inclusion of LEM3323 having a comparable background and drug susceptibility (except for Sb) to the drug-susceptible LEM3049 and the experimentally resistant selected LEM3323/MIL and LEM3323/PMM lines. The discriminating capacity of each cell type to detect drug resistance was evaluated by calculating the resistance index (RI) for each drug (IC50 resistant strain/IC50 susceptible strain). The higher the RI, the higher the discriminatory capacity of the cell to identify resistance in the field.
Table 2. ‘Breakpoint’ estimates# for categorizing drug-susceptibility and drug resistance for clinical isolates.
[taken from ref 20].
| Drug | Promastigotes (axenic) |
Amastigotes (primary mouse macrophages) |
Susceptibility limits (estimates) |
Cytotoxicity (MRC5) |
|
|---|---|---|---|---|---|
| approx IC50 | Sensitive | Resistant | CC50 | ||
| SbV | >77 | 10–15 | <20 | >70 | >64 |
| SbIII | 40–50 | 5–6 | <15 | >70 | >64 |
| MIL | 2–5 | 3–6 | <10 | >25 | 32 |
| PMM | 15–25 | 40–50 | <60 | >150 | >500 |
| AmB | 0.1–0.3 | 0.01–0.03 | <0.5 | (>2)* | >8 |
# based on results obtained with sensitive reference strains (L. donovani: MHM/ET/67/L82 and L. infantum MHOM/MA/67/ITMAP263)
* AmB-resistant isolates from treated patients are yet not available
CC50: cytotoxic concentration 50%
Comparison of different approaches for the drug susceptibility evaluation of clinical isolates
Based on the obtained results and recommendations on drug exposure time (120h), optimal infection index (≥10:1) and problems with rapid cellular replication, the drug susceptibility of five recent European L. infantum clinical isolates (MHOM/ES/2016/LLM-2301, LLM-2309, LLM-2323, LLM-2338, LLM-2346) was determined and compared upon infection of THP-1 cells, BMMφ and PECs.
Statistical analysis
All statistical analyses were performed using Graphpad Prism version 6.00 software. Statistical differences between the infection ratio of different Leishmania strains at different time points were determined using 2-way ANOVA with Bonferroni post-hoc comparisons. Tests were considered statistically significant if p <0.05.
Results
Receptivity for infection
For each cell type, the receptivity to infection was evaluated by comparing initial phagocytosis rates (S1 Fig). For each Leishmania strain, a same trend of fast uptake by primary cells could be observed: PBMC-derived macrophages demonstrated the quickest uptake, followed by the PECs and BMMφ (Fig 2). The phagocytosis levels in THP-1 cells were slightly higher compared to RAW and J774 cells.
Fig 2. Promastigote phagocytosis by the different cell types upon infection with the laboratory strain ITMAP263.

Results are expressed as the average infection indices at 2h, 4h, 6h and 24hpi ± standard error of the mean (SEM) and are the result of two independent experiments run in duplicate.
Parasite burden and cell replication
The intracellular proliferation of the Leishmania isolates was evaluated in the selected panel of host cells (S2 Fig). As the results between strains were fairly comparable, only the results of the laboratory reference strain ITMAP263 are presented. The higher amastigote survival and replication in BMMφ had already been demonstrated in relation to other primary cell types [28]. Here, PBMC-derived macrophages reach comparable initial infection indices, hereby identifying BMMφ and PBMC-derived macrophages to provide the better support for amastigote survival and replication, followed by PEC. For LEM3049 and BH402/6, intracellular multiplication was better in PEC, demonstrating some strain-dependent effect. Infection indices are systematically lower in the cell lines compared to PEC (Fig 3).
Fig 3. Intracellular amastigote multiplication of the laboratory reference strain ITMAP263.

The number of amastigotes/macrophages (infection index) was determined until 168hpi. The average infection index ± SEM is the result of two independent experiments run in duplicate.
Evaluating the intracellular growth/multiplication rate of parasites in the continuously dividing cell lines proved to be challenging. The rapidly dividing cells tend to outgrow the parasitized cell population within 48h, even for LEM3049 known to have a high multiplication capacity in non-dividing primary cells. The observation that various studies use RPMI-1640 medium to cultivate J774 cells in contrast to the DMEM medium as recommended by ATCC prompted us to compare the multiplication rate of J774 in both media. The J774 multiplication rate was highest in RPMI (Fig 4).
Fig 4. Multiplication rate of J774 and RAW cells in relation to the intracellular growth of ITMAP-263 (not showing significant intracellular multiplication in PECs) and LEM3049 (showing rapid intracellular replication in PECs).

Although both Leishmania strains demonstrate intracellular replication, parasitized cells become outcompeted because of the more rapid expansion of non-infected host cells, resulting in low infection ratios. Multiplication rates are expressed as the average ± SEM of two independent experiments run in duplicate.
Drug susceptibility determination and capacity to discriminate drug resistance
The drug susceptibility of LEM3049 in the various macrophage cell types is shown in Table 3. Variation in drug susceptibility is noted between the different cell types with primary cells showing higher RI values, meaning they may allow easier detection of resistance for the selected panel of reference drugs. The cell types with RI values approximating 1 are less suited for detection of drug resistance. Similar to the IC50-values, the RI is drug-dependent and no particular added benefit can be noted for one particular cell type. In THP-1 cells, the PMA stimulation period and the 5-day period between stimulation and infection did not affect drug susceptibility, except for SbIII where the efficacy significantly increased (p = 0.0006) with longer PMA-stimulation before infection.
Table 3. Drug-susceptibility and resistance indices (RI) of the drug-susceptible LEM3049 isolate for the antileishmanial reference drugs in the different macrophage cell types.
The 50% inhibitory concentration (IC50) of LEM3049 and the RI against pentavalent and trivalent antimonials (SbV and SbIII), miltefosine (MIL), paromomycin (PMM) and amphotericin B (AmB) were determined in primary peritoneal Swiss mouse macrophages (PECs), BALB/c bone-marrow derived macrophages (BMMφ), human blood mononuclear cell-(PBMC) derived macrophages and the commercially available THP-1 and J774 cell lines (not done for RAW cells). Values are expressed as the average IC50 ± SEM and are the result of two independent tests run in duplicate.
| Host cell type | Antileishmanial reference drug: IC50 ± SEM | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SbV (eq.) |
RI | SbIII (eq.) |
RI | MIL (μM) |
RI | PMM (μM) |
RI | AmB (μM) |
RI | |||||||||||
| Primary cells | ||||||||||||||||||||
| PEC | 52.4 | ± | 24.6 | 1.5 | 6.3 | ± | 0.4 | 14 | 0.5 | ± | 0 | 80 | 99.3 | ± | 10.3 | 1.3 | 0.05 | ± | 0.01 | ND |
| BMMφ | >77 | 1 | 11.1 | ± | 1.5 | 7.9 | 0.5 | ± | 0.1 | 70.3 | 25.3 | ± | 7 | 6 | 0.05 | ± | 0.02 | ND | ||
| Human PBMC-derived macrophages | >77 | 2 | 7.9 | ± | 0.1 | 11.1 | 1.3 | ± | 0.2 | 27.4 | 113 | ± | 9.6 | 4.4 | 0.07 | ± | 0.01 | ND | ||
| Cell lines | ||||||||||||||||||||
| THP-1 (48h PMA + 5d rest) | >77 | 1 | 30.2 | ± | 5.8 | 2.1 | 1.8 | ± | 0.1 | 11.2 | 37.2 | ± | 13.9 | 6.1 | 0.16 | ± | 0.02 | ND | ||
| THP-1 (72h PMA) | >77 | 2 | 9.5 | ± | 0.7 | 4.6 | 1.3 | ± | 0.4 | 15.4 | 39.1 | ± | 4 | 5.3 | 0.12 | ± | 0 | ND | ||
| J774 | 5.5 | ± | 0 | ND | >22 | ND | 2.2 | ± | 0.7 | ND | 15.4 | ± | 4.3 | ND | 0.01 | ± | 0 | ND | ||
The impact of drug exposure time and intracellular parasite burden on drug susceptibility
To evaluate the drug exposure time required to reach a stable IC50, the shift in drug IC50 for the various reference drugs over time is shown in Fig 5A. For MIL, SbV and AmB, no significant differences were observed between the IC50 values measured at different time points post-treatment. For SbIII and PMM, a significant decrease was observed with stable IC50 values respectively reached at 120 and 144 hours post-treatment (hpt). A same trend was also observed in BMMφ (Fig 5B). Noteworthy is that the infection ratio can significantly impact on drug susceptibility measured at 120 hpt. For example for MIL, the higher infection burdens obtained after infection with higher infection ratios were associated with lower observed drug susceptibility (Fig 6). Although not investigated here, it is acceptable to assume that the same will apply to the other antileishmanial drugs.
Fig 5. Effect of drug exposure time on parasite drug susceptibility.

The 50% inhibitory concentration (IC50) of the clinical isolate LEM3049 for the reference drugs in Swiss mouse peritoneal macrophages (A) or in BALB/c bone marrow-derived macrophages (B) in function of treatment duration. The cell viability at the IC50 levels at 24h and at 168h remained >95%. Drug susceptibility is expressed as average IC50 values (μM for MIL, PMM and AmB; eq. for SbV and SbIII) ± SEM for two independent tests run in duplicate. (SbV: pentavalent antimony, SbIII: trivalent antimony, MIL: miltefosine, PMM: paromomycin, AmB: amphotericin B).
Fig 6. Effect of the infection ratio on the miltefosine IC50-value of the clinical isolate LEM3049 in Swiss mouse peritoneal macrophages (PECs) at 120hpt.
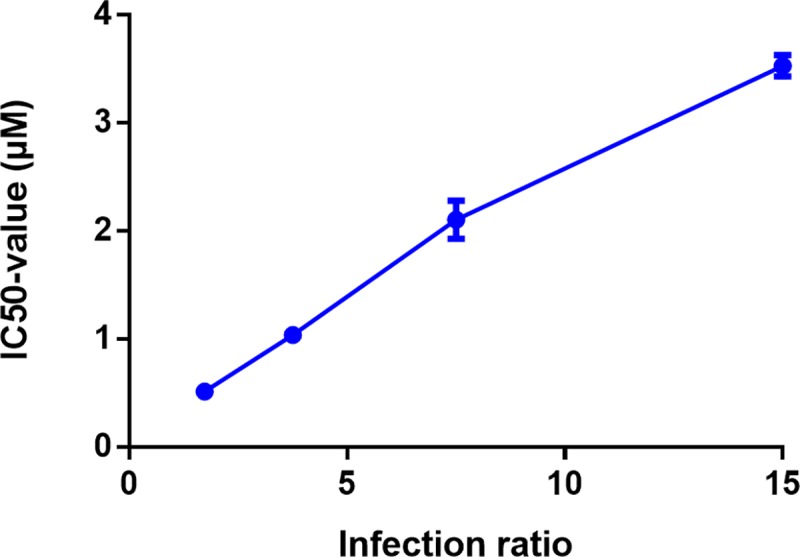
Drug-susceptibility values are expressed as average IC50 value (μM) ± SEM and are the result of two independent tests run in duplicate.
Comparison of different approaches for the drug susceptibility evaluation of clinical isolates
The intracellular drug susceptibility was determined for five recent clinical isolates using the preferred methodology, i.e. primary macrophages or THP-1 cells, infection with promastigotes at 10:1 multiplicity of infection and 120 h of drug exposure (Table 4). Although some variability in drug susceptibility can be noted between the different host cells, the overall susceptibility profile was comparable for the three cell types. Variation in IC50-values was observed only for PMM between the different cells which might obscure the identification of PMM-resistant isolates (resistance cut-off 150 μM) [22]. The higher IC50-values against SbV demonstrated in PEC may also complicate the identification of Sb-resistant strains.
Table 4. In vitro drug susceptibility of a panel of recent clinical L. infantum isolates against antileishmanial reference drugs in THP-1, bone-marrow derived macrophages (BMMφ) and mouse peritoneal exudative cells (PEC).
Intracellular amastigote drug susceptibility against miltefosine (MIL), paromomycin (PMM), pentavalent antimonials (SbV), trivalent antimonials (SbIII) and amphotericin B (AmB). The presented IC50-values are the result of three independent tests run in duplicate.
| Strain | In vitro susceptibility of intracellular amastigotes (IC50 ± SEM) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIL (μM) | PMM (μM) | SbV (μg/ml eq) | SbIII (μg/ml eq) | AmB (μM) | |||||||||||
| mean | ± | SEM | mean | ± | SEM | mean | ± | SEM | mean | ± | SEM | mean | ± | SEM | |
| THP-1 | |||||||||||||||
| LLM-2301 | 0.9 | ± | 0 | 136.2 | ± | 21.6 | 33.6 | ± | 20.8 | 14.4 | ± | 4.6 | 0.12 | ± | 0.02 |
| LLM-2309 | 0.1 | ± | 0 | 29 | ± | 8.4 | 50.8 | ± | 12.5 | 7.6 | ± | 2.6 | 0.04 | ± | 0.01 |
| LLM-2323 | 0.4 | ± | 0.1 | 55 | ± | 8 | 49.8 | ± | 14.9 | 10.6 | ± | 5 | 0.06 | ± | 0.02 |
| LLM-2338 | 0.4 | ± | 0.1 | 324.6 | ± | 104.2 | 43 | ± | 20.1 | 7.8 | ± | 3 | 0.10 | ± | 0.01 |
| LLM-2346 | 0.5 | ± | 0.2 | 87.8 | ± | 26.8 | 18.8 | ± | 3 | 6.8 | ± | 0.1 | 0.10 | ± | 0.01 |
| BMMφ | |||||||||||||||
| LLM-2301 | 0.3 | ± | 0.1 | 157.7 | ± | 45.5 | 54.9 | ± | 10.4 | 12.3 | ± | 1.8 | 0.05 | ± | 0.01 |
| LLM-2309 | ND | ND | ND | ND | ND | ||||||||||
| LLM-2323 | 0.4 | ± | 0.1 | 169.1 | ± | 37.1 | 58.9 | ± | 11.8 | 3.4 | ± | 0.3 | 0.02 | ± | 0.01 |
| LLM-2338 | 0.3 | ± | 0 | 276 | ± | 42.1 | 67.7 | ± | 9.3 | 9.2 | ± | 3.2 | 0.05 | ± | 0.02 |
| LLM-2346 | 3.7 | ± | 1.7 | 365.7 | ± | 97.2 | 65.7 | ± | 11.3 | 6.4 | ± | 0.8 | 0.09 | ± | 0.01 |
| PEC | |||||||||||||||
| LLM-2301 | 0.9 | ± | 0.4 | 354.9 | ± | 60 | 58 | ± | 15.5 | 23.5 | ± | 9.7 | 0.09 | ± | 0.04 |
| LLM-2309 | 0.1 | ± | 0 | 25.7 | ± | 5.7 | 58.8 | ± | 4.4 | 8.4 | ± | 1.4 | 0.04 | ± | 0.01 |
| LLM-2323 | 0.5 | ± | 0.2 | 274.3 | ± | 75.5 | 52 | ± | 12.4 | 24.4 | ± | 14.3 | 0.05 | ± | 0.02 |
| LLM-2338 | 0.6 | ± | 0.2 | 278.1 | ± | 70.3 | 52 | ± | 14.4 | 7.2 | ± | 2.2 | 0.05 | ± | 0.01 |
| LLM-2346 | 3 | ± | 0.5 | 218.1 | ± | 58.2 | >77 | 8.7 | ± | 2.1 | 0.10 | ± | 0.01 | ||
Discussion
Inter-laboratory comparison of drug susceptibility data proves to be very difficult given the large number of laboratory procedures, and although most researchers now agree on the use of amastigote cell-based assays [5], the availability of several host cells, both primary and cell lines, further complicates the progress towards harmonization. Despite earlier recommendations [19, 20], it remains essential to corroborate previous findings and to identify the best suited host cell for rapid and reproducible detection of drug resistance in view of the growing numbers of treatment failures against the current antileishmanial drugs [34–36]. The present laboratory study specifically aimed at comparing the advantages and disadvantages associated with a variety of frequently used primary macrophages and commercially available cell lines next to some important procedure-related differences in an attempt to propose a pragmatic drug susceptibility assay for (clinical) visceral Leishmania isolates. Although it can be assumed that some of the present assay recommendations for VL may also be translatable to the different cutaneous Leishmania species/strains, such cross-check should still be investigated in detail as it fell beyond de scope of the present study.
A first outline for an in vitro assay to determine drug activity against the intracellular L. donovani amastigotes was already published in 1984 [14] demonstrating that the determination of a reproducible drug susceptibililty value is hampered by the macrophage infection ratio (average number of amastigotes/macrophage) which not only influences intracellular amastigote proliferation but also has a direct impact on the measured activity level of the drug. It was also the first description on the use of primary macrophages from the perspective that these may more closely mimic the in vivo environment of the pathogen in a more reproducible way. Within that context, the implementation of several ex vivo models in drug screening applications has been investigated [37–39]. Although peritoneal macrophages loaded with amastigotes do not fully represent the lesion cell, they are still considered the easiest, cheapest, quickest primary model to evaluate antileishmanial compounds. On the other hand, using primary cells is associated with ethical constraints, cells do not retain their in vivo functionality and morphology, cell storage is impossible and one cannot rule out the possible contamination by other cells. For example, when harvesting human monocyte-derived macrophages, only 10% of the peripheral blood monocytes will attach and transform into functional macrophages [17]. This factor combined with the difficult logistics of using human-derived primary cells do limit their use in large-scale evaluation campaigns. Although cell lines are physiologically less relevant to mimic the in vivo situation, they are a relatively cheap alternative to the use of primary cells with much less ethical restrictions. In addition, cell lines have a homogeneous background, stable phenotype and above all a long life-span allowing a high degree of reproducibility. However, repeated passage in vitro may alter their surface-structure, THP-1 cells require a lot of manipulation, immune activation is cell-type dependent [40] and their high replication potential complicates maintenance of a stable parasite infection over time (Fig 4), also leading to practical difficulties such as staining issues that obscure microscopic read-out.
Regardless of the cell type associated advantages and disadvantages (Table 5), identifying the better suited host cell for easy and reproducible detection of drug resistance in the field becomes a more urgent necessity given the growing incidence of therapy failure and drug resistance. Our starting approach was to comparatively evaluate drug susceptibility with the susceptible field isolate LEM3049 in each cell type and to determine the RI-values using experimentally generated resistant strains. Marginal host cell dependent variations in drug susceptibility could be noted, which is in agreement with another study comparing different cell types [21]. The observation of higher RI-values in the primary cells could imply higher sensitivity and specificity to detect resistance.
Table 5. Summary of advantages and disadvantages of the selected panel of host cells for Leishmania susceptibility testing.
| Cell type | Primary cells | Cell lines | ||||
|---|---|---|---|---|---|---|
| PEC | BMMφ | PBMC | THP-1 | J774 | RAW | |
| Peritoneal macrophages | Myeloid progenitor cell derived |
Monocyte derived macrophages |
human
monocyte derived |
BALB/c
monocyte derived |
BALB/c macrophage derived | |
| Source | Mouse | Mouse | Human blood | Commercial | Commercial | Commercial |
| Cell stimulation | starch or thioglycollate | L929 supernatant GM-CSF | NA | PMA, VitD3 or RA | NA | NA |
| Approximate yield | 107 cells/mouse | 107−108 cells/mouse | 105−106/mL (10% monocytes) |
NA | NA | NA |
| Ethical concerns | +++ | +++ | ++ | - | - | - |
| Biological relevance | ++ | ++ | +++ | - | - | - |
| Receptivity for infection | +++ | +++ | + | + | ++ | ++ |
| Intracellular multiplication | + | ++ | ++ | + | +/- | +/- |
| Reproducibility | +++ | +++ | +/- | +/- | ++ | ++ |
| Cost | ++ | ++ | +/- | +/- | +/- | +/- |
| Consumables needed | + | ++ | +++ | + | + | + |
| Technical complexity | + | +++ | ++ | +/- | +/- | +/- |
The study results also suggest that an exact IC50-value will to some extent depend on the nature of the host cell and the strain phenotype. Another question is whether drug susceptibility data collected on mice-derived host cells are fully translatable to human. The use of mouse-derived cells may indeed fail to demonstrate subtle variations in drug susceptibility, but their application in drug susceptibility campaigns has proven their value in identifying ‘clear resistant’ phenotypes of the parasite. In contrast to a previous study using spleen-derived amastigotes for infection [21], both BMMφ and PBMC-derived macrophages sustained intracellular replication for the whole panel of strains upon infection with metacyclic promastigotes. Earlier work with PECs already demonstrated different intracellular growth kinetics depending on the infecting parasite stage [25, 29].
Most laboratories prefer ex vivo amastigotes to infect cells as they provide higher and more consistent infection ratios in contrast to metacyclic promastigotes. Acceptable infections are generally obtained using an amastigote/macrophage ratio between 5 and 10 [14], but the amastigote quantification method as described by Stauber [14] may result in quite variable infection ratios in vitro. Particularly for clinical isolates, even upon exact microscopic counting of viable stages before infection, large fluctuations in infection ratios may occur due to the unpredictable intrinsic variation in parasite virulence and intracellular multiplication potential. Generating infective metacyclic promastigotes of clinical isolates remains a challenge since strain-specific differences will lead into different growth rates and ensuing metacyclogenesis rates. Promastigote preconditioning [9] has been proposed as an option to alleviate these variabilities to achieve more adequate infection ratios of clinical isolates. The promastigote medium is manually acidified 24h before infection enforcing metacyclogenesis in a more controlled way and thereby allowing simultaneous (comparative) infection of strains with different growth characteristics. The impact of preconditioning on the different strains was not part of the present study. Although HOMEM or RPMI-1640 are frequently used for promastigote cultivation, M199 medium was selected to cultivate the different clinical isolates as a cheaper alternative supporting the growth of most clinical isolates [22]. Attention should be given to keep the lag time between primary isolation and drug susceptibility assays to a minimum to avoid loss of virulence and other phenotypic alterations caused by long-term in vitro cultivation. Logically, the more rapid assay result will also benefit the proper therapeutic follow-up of the patient.
Comparison of the intracellular growth in different cell lines already demonstrated that LEM3049 is able to divide in PECs, while BH402/60 and ITMAP263 did not [25]. Quite similar trends were observed for the different cell types tested here: PBMC-derived macrophages and BMMφ show a rapid parasite internalization together with intracellular parasite multiplication, while phagocytosis and proliferation in PECs remains highly strain-specific, which again illustrates the complexity of the interaction between host cell and parasite.
It has already been demonstrated that the potency of SSG (SbV) decreased with increasing parasite burdens [14], explaining the higher IC50 observed for SbV in PECs. Here, a same trend can be observed for MIL. Similarly to the infection ratio, the percentage infected cells can also confound correct drug susceptibility determination as low infection levels will result in unreliable IC50 read-outs. A general rule should be that at least over 80% of the macrophages should be infected in the positive control before even considering defining susceptibility values. When taking this rule into account, the susceptibility read-out of the cell lines becomes severely compromised. While the infection ratio stays lower in THP-1 cells, the extensive cell proliferation of RAW and J774 causes a rapid decrease of the infection ratio (Fig 3). The intracellular replication of the four isolates in the present study was most evident in human PBMC-derived cells and BALB/c BMMφ (S2 Fig) with the highest infection ratio being reached at 24 hpi. While the exact causes of cell-dependent drug susceptibility still remain elusive, the cell differentiation rate may be one factor that can influence promastigote phagocytosis and subsequent promastigote-to-amastigote transformation. The time-dependent efficacy of the antileishmanial reference drugs advocates for treatment periods of at least 96 h in primary cells. Combining the need for prolonged drug exposure with the observed low infection rates obtained in the cell lines makes them less recommendable for drug susceptibility determination.
A final step in the procedure is the read-out, which for clinical isolates will generally remain dependent of light microscopy. Defining in vitro efficacy can in principle be done in two ways: (i) counting and calculating the percentage reduction of the average number of amastigotes per macrophage, or (ii) determining the reduction in the average percentage of infected cells. While the latter is simpler and less time-consuming, the former is more precise and can be particularly handy in case of near-toxicity [14].
In summary (Table 6), our results recommend the use of primary cells in drug susceptibility evaluation whenever possible. Although intracellular replication is more pronounced in PBMC-derived macrophages and BMMφ, their limited availability and technically challenging purification and transformation procedures will severely limit their application in large-scale settings. Given the narrow variation in IC50 values and RI-values between PBMCs-derived cells, BMMφ and PECs, we propose PECs as a pragmatic option. Intraperitoneal stimulation of outbred Swiss or CD-1 mice strongly enhances the macrophage yield and parasite phagocytosis [14].
Table 6. Summary of the proposed strategy for drug susceptibility testing of clinical isolates and/or laboratory-adapted strains.
| Leishmania strain | Field isolate | Laboratory strain Lab-adapted field isolate |
|---|---|---|
| Host cell types | Mouse PECs (after starch stimulation) THP-1 cells (after PMA stimulation) |
PEC (Swiss ~ BALB/c) BMMφ BALB/c: higher intracellular multiplication PBMC-derived macrophages (GM-CSF stimulation) |
| Parasite stage | Metacyclic promastigotes (preconditioning optional) | Ex vivo (spleen-derived) amastigotes |
| Multiplicity of infection* | 10–15 promastigotes/cell | 5–10 amastigotes/cell |
| Start drug exposure | 24h after infection | 2h after infection |
| Duration drug exposure | 120h | 120h |
* dependent on the intrinsic infectivity of the strain
If practical or ethical limitations would impose the application of a cell line, THP-1 cells are the best option (Table 5). Despite their relatively low susceptibility for infection and the need to apply exogenous stimuli for monocyte-to-macrophage transformation, they provide an easy accessible and reproducible alternative to primary cells. However, a standard stimulation procedure still needs to be decided to avoid inter-laboratory bias and to ensure adequate macrophage transformation before infection. Although J774 and RAW cells are more robust, require less manipulation and are less sensitive to all sorts of stress during cultivation, their continuous replication severely compromises evaluation of slower-acting drugs.
While the focus of this laboratory study was the comparative evaluation of cell-based drug susceptibility assays, evaluation of field isolates in disease-endemic countries may prove to be problematic because of the reliance on dedicated laboratory infrastructure and advanced technical expertise. On the other hand, the conclusions of this study will be practically useful for investigators (in academia, hospitals, central field labs) who have access to the appropriate lab infrastructure and logistics.
Supporting information
(DOCX)
Multiplication rates are determined by microscopically determining their average infection index every 24h after infection in relation to the initial infection index at 24h post infection. Results are expressed as average multiplication rate of the standard error of the mean (SEM) of two independent experiments run in duplicate.
(TIF)
Initial phagocytosis of 4 different L. infantum lines (A/ ITMAP263 laboratory reference strain and clinical isolates B/ LEM3049, C/ BH402/60 and L3015) is represented by microscopically determining their average infection index at 2h, 4h, 6h and 24hpi ± the standard error of the mean (SEM) of two independent experiments run in duplicate.
(TIF)
Intracellular amastigote proliferation of 4 different L. infantum strains (A/ ITMAP263 laboratory reference strain and clinical isolates B/ LEM3049, C/ BH402/60 and L3015) is measured by microscopically determining the average infection index ± SEM in the different cell types every 24h up to 168 hours post-infection (hpi) of two independent experiments run in duplicate.
(TIF)
Cell viability was determined by microscopic assessment of cell death upon trypan-blue staining. The average cell viability ± SEM is the result of two independent repeats run in duplicate.
(TIF)
Acknowledgments
The authors want to thank J. Moreno (WHOCC, Madrid, Spain) for the kind donation of the clinical L. infantum isolates, and Pim-Bart Feijens, Margot Desmet, Mandy Vermont and An Matheeussen for the excellent technical assistance. The clinical isolates LEM3049 and LEM3323 were kindly provided by Dr. L. Lachaud of the Laboratoire de Parasitologie-Mycologie et Centre National de Référence des Leishmanioses, Montpellier, France. LMPH is a partner of the Antwerp Drug Discovery Network (ADDN, www.addn.be) and the Excellence Centre ‘Infla-Med’ (www.uantwerpen.be/infla-med).
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was funded by the Research Fund Flanders (FWO) (L.M.: G051812N; S.H.: G013118N and 12I0317N) and the Research Council of the University of Antwerp: (G.C.: TT-ZAPBOF 33049) and (L.M.: TOP-BOF 35017). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kazemi-Rad E, Mohebali M, Khadem-Erfan MB, Saffari M, Raoofian R, Hajjaran H, et al. Identification of antimony resistance markers in Leishmania tropica field isolates through a cDNA-AFLP approach. ExpParasitol. 2013; 135: 344–349. [DOI] [PubMed] [Google Scholar]
- 2.Cojean S, Houze S, Haouchine D, Huteau F, Lariven S, Hubert V, et al. Leishmania resistance to miltefosine associated with genetic marker. Emerg Infect Dis. 2012; 18: 704–706. 10.3201/eid1804.110841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanaerschot M, Decuypere S, Downing T, Imamura H, Stark O, De DS, et al. Genetic markers for SSG resistance in Leishmania donovani and SSG treatment failure in visceral leishmaniasis patients of the Indian subcontinent. J Infect Dis. 2012; 206:752–755. 10.1093/infdis/jis424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ephros M, Bitnun A, Shaked P, Waldman E, Zilberstein D. Stage-specific activity of pentavalent antimony against Leishmania donovani axenic amastigotes. Antimicrobial agents and chemotherapy. 1999; 43: 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vermeersch M, da Luz RI, Tote K, Timmermans JP, Cos P, Maes L. In vitro susceptibilities of Leishmania donovani promastigote and amastigote stages to antileishmanial reference drugs: practical relevance of stage-specific differences. Antimicrob Agents Chemother. 2009; 53: 3855–3859. 10.1128/AAC.00548-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aulner N, Danckaert A, Rouault-Hardoin E, Desrivot J, Helynck O, Commere PH, et al. High Content Analysis of Primary Macrophages Hosting Proliferating Leishmania Amastigotes: Application to Anti-leishmanial Drug Discovery. PLoS Neglected Tropical Diseases. 2013; 7: e2154 10.1371/journal.pntd.0002154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Probst CM, Silva RA, B Menezes JP, Almeida TF, Gomes IN, Dallabona AC, et al. A comparison of two distinct murine macrophage gene expression profiles in response to Leishmania amazonensisinfection. BMC Microbiology. 2012; 12: 1–12. 10.1186/1471-2180-12-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paila YD, Saha B, Chattopadhyay A. Amphotericin B inhibits entry of Leishmania donovani into primary macrophages. Biochemical and Biophysical Research Communications. 2010; 399: 429–433. 10.1016/j.bbrc.2010.07.099 [DOI] [PubMed] [Google Scholar]
- 9.Inocencio da Luz RA, Vermeersch M, Dujardin JC, Cos P, Maes L. In vitro sensitivity testing of Leishmania clinical field isolates: preconditioning of promastigotes enhances infectivity for macrophage host cells. Antimicrob Agents Chemother. 2009; 53: 5197–5203. 10.1128/AAC.00866-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Looker DL, Martinez S, Horton JM, Marr JJ. Growth of Leishmania donovani amastigotes in the continuous human macrophage cell line U937: studies of drug efficacy and metabolism. The Journal of infectious diseases. 1986; 154: 323–327. 10.1093/infdis/154.2.323 [DOI] [PubMed] [Google Scholar]
- 11.Fata A, Mahmoudian M, Varasteh A, Sankian M. Monarch-1 Activation in Murine Macrophage Cell Line (J774 A.1) Infected with Iranian Strain of Leishmania major. Iranian Journal of Parasitology. 2013; 8: 207–211. [PMC free article] [PubMed] [Google Scholar]
- 12.Jain SK, Sahu R, Walker LA, Tekwani BL. A parasite rescue and transformation assay for antileishmanial screening against intracellular Leishmania donovani amastigotes in THP1 human acute monocytic leukemia cell line. J Vis Exp. 2012; 70: 4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dasgupta B, Roychoudhury K, Ganguly S, Akbar MA, Das P, Roy S. Infection of human mononuclear phagocytes and macrophage-like THP1 cells with Leishmania donovani results in modulation of expression of a subset of chemokines and a chemokine receptor. Scand J Immunol. 2003; 57: 366–374. 10.1046/j.1365-3083.2003.01227.x [DOI] [PubMed] [Google Scholar]
- 14.Neal RA, Croft SL. An in-vitro system for determining the activity of compounds against the intracellular amastigote form of Leishmania donovani. J Antimicrob Chemother. 1984; 14: 463–475. 10.1093/jac/14.5.463 [DOI] [PubMed] [Google Scholar]
- 15.Croft SL. In vitro screens in the experimental chemotherapy of leishmaniasis and trypanosomiasis. Parasitol Today. 1986; 2: 64–69. 10.1016/0169-4758(86)90157-2 [DOI] [PubMed] [Google Scholar]
- 16.Mans DR, Beerens T, Magali I, Soekhoe RC, Schoone GJ, Oedairadjsingh K, et al. In vitro evaluation of traditionally used Surinamese medicinal plants for their potential anti-leishmanial efficacy. Journal of ethnopharmacology. 2016; 180: 70–77. 10.1016/j.jep.2016.01.012 [DOI] [PubMed] [Google Scholar]
- 17.Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PloS one. 2010; 5: e8668 10.1371/journal.pone.0008668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croft SL. Monitoring drug resistance in leishmaniasis. Trop Med Int Health. 2001; 6: 899–905. 10.1046/j.1365-3156.2001.00754.x [DOI] [PubMed] [Google Scholar]
- 19.Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev. 2006; 19: 111–26. 10.1128/CMR.19.1.111-126.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendrickx S, Guerin PJ, Caljon G, Croft SL, Maes L. Evaluating drug resistance in visceral leishmaniasis: the challenges. Parasitology. 2018: 145: 453–463. 10.1017/S0031182016002031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seifert K, Escobar P, Croft SL. In vitro activity of anti-leishmanial drugs against Leishmania donovani is host cell dependent. J Antimicrob Chemother. 2010; 65: 508–511. 10.1093/jac/dkp500 [DOI] [PubMed] [Google Scholar]
- 22.Maes L, Cos P, Croft S. The relevance of susceptibility tests, breakpoints and markers In: Ponte-Sucre A, Diaz E, Padrón-Nieves M, editors. Drug Resistance in Leishmania Parasites: Springer; Vienna; 2013. p. 407–429. [Google Scholar]
- 23.da Costa Val A. Tratamento da leishmaniose visceral canina com antimonial pentavalente encapsulado em lipossomas. Tese de Doutorado em Ciência Animal, Escola de Veterinária, Universidade Federal de Minas Gerais, Belo Horizonte, 125p. 2004.
- 24.Inocencio da LR, Romero GA, Dorval ME, Cruz I, Canavate C, Dujardin JC, et al. Drug susceptibility of Leishmania infantum (syn. Leishmania chagasi) isolates from Brazilian HIV-positive and HIV-negative patients. J Antimicrob Chemother. 2011; 66: 677–679. 10.1093/jac/dkq508 [DOI] [PubMed] [Google Scholar]
- 25.Hendrickx S, Mondelaers A, Eberhardt E, Lachaud L, Delputte P, Cos P, et al. Intracellular amastigote replication may not be required for successful in vitro selection of miltefosine resistance in Leishmania infantum. Parasitol Res. 2015; 114: 2561–2565. 10.1007/s00436-015-4460-9 [DOI] [PubMed] [Google Scholar]
- 26.Arce A, Estirado A, Ordobas M, Sevilla S, Garcia N, Moratilla L, et al. Re-emergence of leishmaniasis in Spain: community outbreak in Madrid, Spain, 2009 to 2012. Euro surveillance: bulletin Europeen sur les maladies transmissible. European Communicable Disease Bulletin. 2013; 18: 20546. [DOI] [PubMed] [Google Scholar]
- 27.Moreira D, Santarem N, Loureiro I, Tavares J, Silva AM, Amorim AM, et al. Impact of continuous axenic cultivation in Leishmania infantum virulence. PLoS Negl Trop Dis. 2012; 6: e1469 10.1371/journal.pntd.0001469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van den Kerkhof M, Van Bockstal L, Gielis JF, Delputte P, Cos P, Maes L, et al. Impact of primary mouse macrophage cell types on Leishmania infection and in vitro drug susceptibility. Parasitology Research. 2018; 117: 3601–3612. 10.1007/s00436-018-6059-4 [DOI] [PubMed] [Google Scholar]
- 29.Hendrickx S, Leemans A, Mondelaers A, Rijal S, Khanal B, Dujardin JC, et al. Comparative Fitness of a Parent Leishmania donovani Clinical Isolate and Its Experimentally Derived Paromomycin-Resistant Strain. PloS one. 2015; 10: e0140139 10.1371/journal.pone.0140139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Hernandez R, Manzano JI, Castanys S, Gamarro F. Leishmania donovani develops resistance to drug combinations. PLoS Negl Trop Dis. 2012; 6: e1974 10.1371/journal.pntd.0001974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prajapati VK, Mehrotra S, Gautam S, Rai M, Sundar S. In vitro antileishmanial drug susceptibility of clinical isolates from patients with Indian visceral leishmaniasis—status of newly introduced drugs. Am J Trop Med Hyg. 2012; 87: 655–657. 10.4269/ajtmh.2012.12-0022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhandari V, Sundar S, Dujardin JC, Salotra P. Elucidation of cellular mechanisms involved in experimental paromomycin resistance in Leishmania donovani. Antimicrob Agents Chemother. 2014; 58: 2580–2585. 10.1128/AAC.01574-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadeghi S, Seyed N, Etemadzadeh MH, Abediankenari S, Rafati S, Taheri T. In Vitro Infectivity Assessment by Drug Susceptibility Comparison of Recombinant Leishmania major Expressing Enhanced Green Fluorescent Protein or EGFP-Luciferase Fused Genes with Wild-Type Parasite. The Korean Journal of Parasitology. 2015; 53: 385–394. 10.3347/kjp.2015.53.4.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rijal S, Ostyn B, Uranw S, Rai K, Bhattarai NR, Dorlo TP, et al. Increasing failure of miltefosine in the treatment of kala-azar in Nepal and the potential role of parasite drug resistance, reinfection, or noncompliance. Clin Infect Dis. 2013; 56: 1530–1538. 10.1093/cid/cit102 [DOI] [PubMed] [Google Scholar]
- 35.Chakravarty J, Sundar S. Drug resistance in leishmaniasis. J Glob Infect Dis. 2010; 2: 167–176. 10.4103/0974-777X.62887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purkait B, Kumar A, Nandi N, Sardar AH, Das S, Kumar S, et al. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob Agents Chemother. 2012; 56: 1031–1041. 10.1128/AAC.00030-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Terreros MJS, de Luna LAV, Giorgio S. Evaluation of antileishmanial drugs activities in an ex vivo model of leishmaniasis. Parasitology International. 2019; 71:163–166. 10.1016/j.parint.2019.04.011 [DOI] [PubMed] [Google Scholar]
- 38.Peniche AG, Osorio Y, Renslo AR, Frantz DE, Melby PC, Travi BL. Development of an ex vivo lymph node explant model for identification of novel molecules active against Leishmania major. Antimicrobial Agents and Chemotherapy. 2014; 58: 78–87. 10.1128/AAC.00887-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osorio Y, Travi BL, Renslo AR, Peniche AG, Melby PC. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Neglected Tropical Diseases. 2011; 5: e962 10.1371/journal.pntd.0000962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van den Bogaart E, Mens PF, Adams ER, Grobusch MP, Schallig HD. Phagocytosis of hemozoin by RAW 264.7 cells, but not THP-1 cells, promotes infection by Leishmania donovani with a nitric oxide-independent mechanism. Parasitology International. 2017; 66: 196–206. 10.1016/j.parint.2016.09.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
Multiplication rates are determined by microscopically determining their average infection index every 24h after infection in relation to the initial infection index at 24h post infection. Results are expressed as average multiplication rate of the standard error of the mean (SEM) of two independent experiments run in duplicate.
(TIF)
Initial phagocytosis of 4 different L. infantum lines (A/ ITMAP263 laboratory reference strain and clinical isolates B/ LEM3049, C/ BH402/60 and L3015) is represented by microscopically determining their average infection index at 2h, 4h, 6h and 24hpi ± the standard error of the mean (SEM) of two independent experiments run in duplicate.
(TIF)
Intracellular amastigote proliferation of 4 different L. infantum strains (A/ ITMAP263 laboratory reference strain and clinical isolates B/ LEM3049, C/ BH402/60 and L3015) is measured by microscopically determining the average infection index ± SEM in the different cell types every 24h up to 168 hours post-infection (hpi) of two independent experiments run in duplicate.
(TIF)
Cell viability was determined by microscopic assessment of cell death upon trypan-blue staining. The average cell viability ± SEM is the result of two independent repeats run in duplicate.
(TIF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.