

Abstract
Cardiac conduction defects decrease life expectancy in myotonic dystrophy type 1 (DM1), a CTG repeat disorder involving misbalance between two RNA binding factors, MBNL1 and CELF1. However, how DM1 condition translates into conduction disorders remains poorly understood. Here we simulated MBNL1 and CELF1 misbalance in the Drosophila heart and performed TU-tagging-based RNAseq of cardiac cells. We detected deregulations of several genes controlling cellular calcium levels, including increased expression of straightjacket/α2δ3, which encodes a regulatory subunit of a voltage-gated calcium channel. Straightjacket overexpression in the fly heart leads to asynchronous heartbeat, a hallmark of abnormal conduction, whereas cardiac straightjacket knockdown improves these symptoms in DM1 fly models. We also show that ventricular α2δ3 expression is low in healthy mice and humans, but significantly elevated in ventricular muscles from DM1 patients with conduction defects. These findings suggest that reducing ventricular straightjacket/α2δ3 levels could offer a strategy to prevent conduction defects in DM1.
Research organism: D. melanogaster
Introduction
Myotonic dystrophy type 1 (DM1), the most prevalent muscular dystrophy in adults (Theadom et al., 2014), is caused by a CTG triplet repeat expansion in the 3’ untranslated region of the dystrophia myotonica protein kinase (dmpk) gene. Transcripts of the mutated dmpk, containing expanded CUGs, form hairpin-like secondary structures in the nuclei, and sequester RNA-binding proteins with affinity to CUG-rich sequences. Among them, Muscleblind-like 1 (MBNL1) protein is trapped within the repeats, forming nuclear foci aggregates that hallmark the disease (Davis et al., 1997; Taneja et al., 1995). In parallel, the CUGBP- and ELAV-like family member 1 (CELF1) is stabilized (Kuyumcu-Martinez et al., 2007), creating misbalance between MBNL1 and CELF1. This leads to missplicing of several transcripts and a general shift from adult to fetal isoforms (Freyermuth et al., 2016; Kino et al., 2009; Savkur et al., 2001). In addition, repeat toxicity induces a range of splice-independent alterations including impaired transcript stability (Sicot et al., 2011). A combination of splice-dependent and splice-independent events thus underlies DM1 pathogenesis, with the latter remaining largely unexplored. DM1 affects mainly skeletal muscles and the heart, with about 80% of DM1 patients showing impaired heart function with arrhythmia and conduction disturbance, which can sometimes end in heart block and sudden death (de Die-Smulders et al., 1998; Groh et al., 2008; Mathieu et al., 1999). Cardiac symptoms, and particularly conduction defects, thus decrease life expectancy in DM1 (Wang et al., 2009). Data suggest that cardiac phenotypes, including conduction defects, are due to MBNL1/CELF1 misbalance. It was shown in a DM1 mouse model that PKC phosphorylates CELF1 leading to increased CELF1 levels, whereas PKC inhibition caused CELF1 reduction and amelioration of cardiac dysfunction (Wang et al., 2009). This suggests that increased CELF1 levels could cause heart phenotypes in DM1, a possibility supported by findings that heart-specific upregulation of CELF1 reproduces functional and electrophysiological cardiac changes observed in DM1 patients and mouse model (Koshelev et al., 2010). In parallel, analyses of Mbnl1 mutant mice (Dixon et al., 2015) and evidence that misregulation of MBNL1-splice target gene SCN5A encoding a cardiac sodium channel leads to cardiac arrhythmia and conduction delay (Freyermuth et al., 2016), indicate that Mbnl1 contributes to DM1 heart phenotypes. However, in spite of aberrant SCN5A splicing (Freyermuth et al., 2016) and downregulation of a large set of miRNAs (Kalsotra et al., 2014), gene deregulations causing cardiac dysfunctions in DM1 remain to be characterized.
To gain further insight into mechanisms underlying cardiac DM1 phenotypes, we used previously described Drosophila DM1 models (Picchio et al., 2013). The heart of the fruit fly is simple in structure, but like the human heart, it displays pacemaker-regulated rhythmic beating, involving functions of conserved ion channels (Ocorr et al., 2007; Taghli-Lamallem et al., 2016). We simulated pathogenic MBNL1/CELF1 misbalance specifically in the fly heart by attenuating the Drosophila MBNL1 ortholog Muscleblind (Mbl) and by overexpressing the CELF1 counterpart Bruno-3 (Bru-3) (Picchio et al., 2018). This caused asynchronous heartbeat (anterior and posterior heart segments beating at different rates), which in Drosophila results from partial conduction block (Birse et al., 2010). Using these two fly DM1 models, we hoped to identify molecular players involved in DM1-associated conduction defects. We did not observe asynchronous heartbeats in flies expressing in the heart 960CTG repeats. This DM1 model (Picchio et al., 2013) developed other cardiac phenotypes such as arrhythmia. To identify deregulated genes underlying conduction defects, we applied a heart-targeted TU-tagging approach (Miller et al., 2009) followed by RNA sequencing. This cardiac cell-specific genome-wide approach yielded a discrete number of evolutionarily conserved candidate genes with altered cardiac expression in both DM1 models used, including regulators of cellular calcium. Among them, we found increased transcript levels of straightjacket (stj)/CACNA2D3 (α2δ3), which encodes a major regulatory subunit of Ca-α1D/Cav1.2 voltage-gated calcium channel, and is a key regulator of calcium current density, which triggers cardiac contraction (Bodi, 2005; Dolphin, 2013; Hoppa et al., 2012; Mesirca et al., 2015). The role of stj transcript level in proper conduction is supported by cardiac-specific overexpression of stj, which leads to asynchronous heartbeat. Conversely, attenuating its expression by cardiac-specific knockdown in both DM1 fly heart models reverses cardiac asynchrony. Our hypothesis that stj contributes to the cardiac DM1-associated pathology is supported by our finding that ventricular α2δ3 expression level is low in healthy mouse and human hearts, but is significantly increased in DM1 patients with cardiac conduction defects. Hence lowering α2δ3 in ventricular cardiomyocytes could offer a potential treatment strategy for DM1-associated conduction defects and in particular intraventricular conduction delay (IVCD).
Results
Imbalance of MBNL1 and CELF1 counterparts in the Drosophila heart results in asynchronous heartbeat
The Drosophila counterparts of MBNL1 and CELF1, the Mbl and Bru-3 proteins, are both present in the fly heart cells, including contractile cardiomyocytes, and localize predominantly but not exclusively to the nuclei (Figure 1—figure supplement 1). To mimic the DM1-associated imbalance between these two RNA binding factors, we either knocked down Mbl, or overexpressed Bru-3. We used the GAL4/UAS system with a cardiac-specific GAL4 driver line, Hand-GAL4 to target UAS-driven transgene expression in the adult fly heart (Figure 1A). We found that the UAS-MblRNAi line efficiently attenuated Mbl expression, whereas UAS-Bru-3 generated Bru-3 overexpression in GAL4-expressing cardiac cells (Figure 1—figure supplement 1). The Hand-GAL4 driven overexpression of Bru-3 and attenuation of Mbl, both appeared homogenous along the heart tube.
Figure 1. Cardiac-specific knockdown of MBNL1 ortholog and overexpression of CELF1 counterpart in Drosophila lead to asynchronous heartbeats.
(A) The adult Drosophila heart expressing Hand-Gal4-driven GFP (Hand>GFP). Note that this Gal4 line drives expression exclusively in the heart. Arrows indicate cardiomyocytes (CM) and pericardial cells (PC) and A2–A5 denote abdominal segments. Scale bar, 150 μm. (B) Movie and M-modes views illustrating asynchronous heartbeats in Hand>MblRNAi flies registered in two adjacent heart segments (A3 and A4). Two-sided arrows indicate heart diameter in diastolic state. (C) Barplot graph showing percentage of flies with conduction defects in the different genetic contexts tested. Note the higher impact of attenuation of Mbl compared to overexpression of Bru-3. Number of fly hearts tested (n) is indicated and statistical significance (Fisher’s exact test) denoted by * (p<0.05) and *** (p<0.001). Below are examples of M-modes illustrating ‘partial’ and ‘total’ heart blocks observed in Hand>MblRNAi and Hand>Bru-3 flies developing conduction defects.
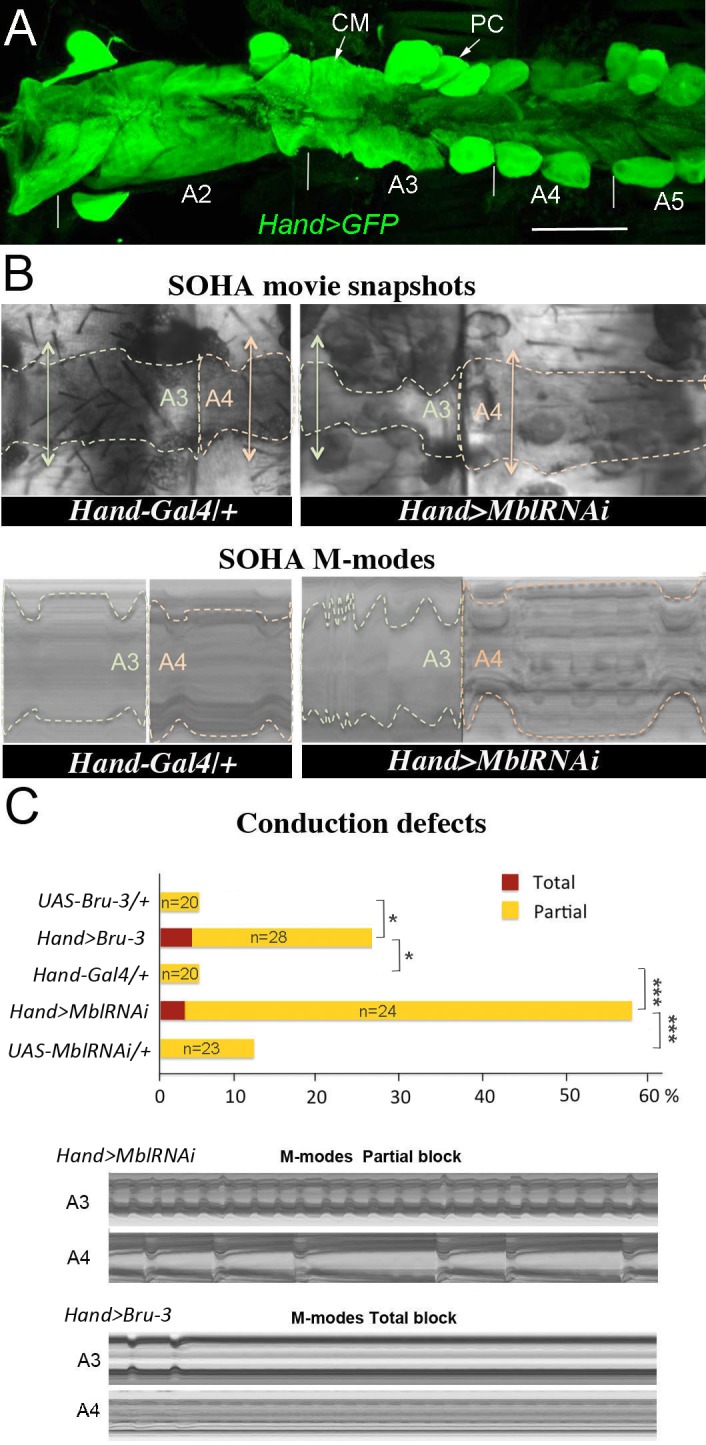
Figure 1—figure supplement 1. Bru-3 and Mbl are expressed in the adult fly heart and their transcript and protein levels are affected in Hand>Bru-3 and Hand>MblRNAi flies.

Figure 1—figure supplement 2. Effect of simultaneous cardiac attenuation of Mbl and overexpression of Bru-3 on conduction defects.
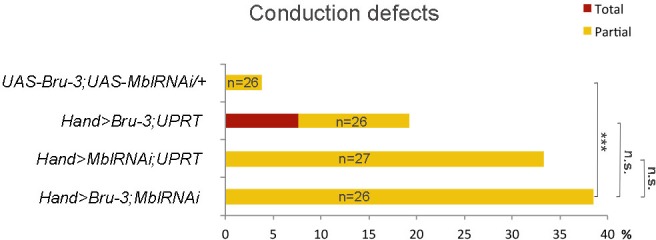
Figure 1—video 1. An example of asynchronous heartbeat phenotype observed in Hand>MblRNAi context.
To assess the cardiac function in these two DM1 models we used the semi-automated optical heartbeat analysis (SOHA) method, which consists in filming the adult beating heart from hemi-dissected flies and allows analysis of the contractile and rhythmic heart parameters (Fink et al., 2009; Ocorr et al., 2007). Besides frequently observed arrhythmia, about 58% of Hand>MblRNAi and 28% of Hand>Bru-3 flies displayed asynchronous heartbeat (Figure 1B,C and Figure 1—video 1), a hallmark of conduction disturbance (Birse et al., 2010). In some cases, asynchronous heartbeat can result in total heart block when at least one heart segment stops contracting. About 3% of Hand>MblRNAi and 4% of Hand>Bru-3 flies did develop a total heart block (Figure 1C). We also tested heartbeat in flies in which Mbl was attenuated in parallel to Bru-3 overexpression (Figure 1—figure supplement 2) and found a moderate additive effect on conduction defects of these two deregulations present in DM1. Taken together, these results show that the imbalance of Mbl/MBNL1 and Bru-3/CELF1 in the fly heart increases the occurrence of asynchronous heartbeat. This prompted us to perform transcriptional profiling of Hand>MblRNAi and Hand>Bru-3 in cardiac cells to identify misregulated gene expression underlying this phenotype.
Transcriptional profiling of cardiac DM1 Drosophila models using heart-specific TU-tagging approach
Transcriptional profiling of cardiac cells from Hand>MblRNAi and Hand>Bru-3 flies was performed using a cell-specific approach called TU-tagging (Miller et al., 2009), followed by RNA-seq. This method allows tissue-specific RNA isolation by expressing the uracil phosphoribosyltransferase (UPRT) enzyme cell-specifically and incorporation into neo-synthesized transcripts of a modified uracil, 4TU. Here, Hand-GAL4-driven UPRT expression in cardiac cells enabled us to isolate transcripts from adult heart cells only and we combined UAS-UPRT with UAS-MblRNAi and UAS-Bru-3 lines to perform TU-tagging in these two DM1 contexts (Figure 2A). Comparison of RNA-seq data from input RNA fraction and heart-specific TU-fraction from control Hand>LacZ;UPRT flies was used to assess the suitability of TU-tagging in low-abundance heart cells. We found that transcripts of cardiac-specific genes such as Hand and Tin were highly enriched in the cardiac-tagged TU fractions, whereas the transcripts of Npc1b and Obp19b genes expressed in midgut (Voght et al., 2007) and gustatory cells (Galindo and Smith, 2001), respectively, were depleted (Figure 2B). Thus the TU-tagging method proved well-suited to the transcriptional profiling of adult Drosophila cardiac cells, allowing the identification of both global gene deregulations (analyzed here) and isoform-specific differential gene expression (in a parallel study).
Figure 2. Cardiac-specific transcriptional profiling using TU-tagging method.

(A) Pipeline of heart-targeted transcriptional profiling using TU-tagging. Note that Gal4- inducible UPRT transgene (UAS-UPRT) has been combined with UAS-MblRNAi and with UAS-Bru-3 for the purpose of TU-tagging. Hand>UPRT;lacZ is the control line used to identify pathogenic gene deregulations in Hand>UPRT;MblRNAi and Hand>UPRT;Bru-3 contexts. Flies were starved at day 4 for 6 hr before being transferred to 4TU containing food for 12 hr. (B) Volcano plot and IGV tracks from control Hand>UPRT;lacZ flies show examples of enrichment of heart-specific genes (e.g. Hand, Tin) (red, right side) and depletion of non-heart-expressed genes (blue, left side), thus validating the specificity of heart targeting.
TU-tagging-based transcriptional profiling of DM1 models with heart asynchrony reveals aberrant expression of genes regulating cellular calcium transient
To select candidates with a potential role in conduction disturbance, we focused on a pool of genes commonly deregulated in Hand>MblRNAi and in Hand>Bru-3 contexts (Figure 3A, Venn diagram), comprising 118 genes, with 64 conserved in humans (Figure 3A, heatmap and Figure 3—source data 1). Among them, gene interaction network analysis (Figure 3B) identified four candidates involved in the regulation of cellular calcium level and known to be essential for proper heart function (van Weerd and Christoffels, 2016), namely inactivation no afterpotential D (inaD), Syntrophin-like 1 (Syn1), Rad, Gem/Kir family member 2 (Rgk2) and straightjacket (stj). Their respective human orthologs are FRMPD2, SNTA1, REM1 and CACNA2D3/α2δ3. They all show high DIOPT orthology scores with their Drosophila counterparts (Figure 3—source data 1), except for FRMPD2 encoding a scaffolding PDZ domain carrying protein structurally similar to inaD but with a low DIOPT score. As inaD, Syn1 and Rgk2 were downregulated in both DM1 contexts (Figure 3A,B) we assessed the effects of their attenuation. We observed that cardiac-specific knockdowns of inaD, Syn1 and Rgk2 overall led to an increase in diastolic and systolic diameters (Figure 3—figure supplement 1). However, we did not observe asynchronous heartbeat phenotypes, indicating that decrease in inaD, Syn1 and Rgk2 expression has no role in DM1-associated conduction defects. We also simulated increased cardiac expression of stj observed in both Hand>MblRNAi and in Hand>Bru-3 contexts and found that in addition to arrhythmia and asystoles, Hand>stj flies exhibited asynchronous heartbeat, reminiscent of conduction defects observed in DM1 (Figure 1C). This observation and the fact that the expression of two other Drosophila α2δ genes (CG42817 and CG16868) remained unchanged (Figure 3—figure supplement 2) prompted us to test whether elevated expression of stj could contribute to DM1 cardiac defects.
Figure 3. Heart-specific transcriptional profiling of DM1 flies identifies deregulation of genes controlling cellular calcium level.
(A) Venn diagrams of genes deregulated in Hand>UPRT;MblRNAi and in Hand>UPRT;Bru-3 contexts (FC > 1.7) followed by heatmap of commonly deregulated genes. (B) Genemania interaction network of conserved candidates including stj, Rgk2, Syn1 and inaD known to be involved in Ca2 regulation. A scheme presenting the structure of the voltage-gated calcium channel and its regulatory component Stj/α2δ3 is included. Color code in genemania network represents up and down regulation according to the heatmap. Cyan/Blue intensity code indicates fold change of genes expression in the heatmap.
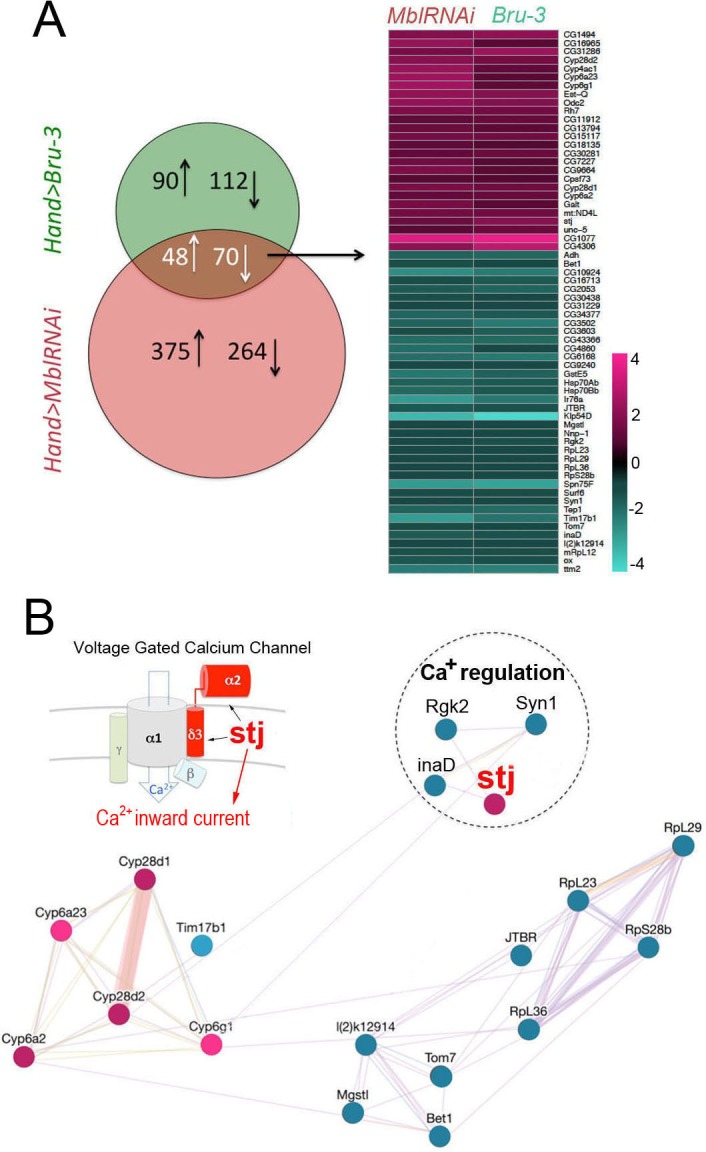
Figure 3—figure supplement 1. Attenuation of inaD, Syn1 and Rgk2 all lead to an increase of diastolic (DD) and systolic (SD) heart diameters.

Figure 3—figure supplement 2. The expression levels of CG42617 and CG16868, two additional α2δ protein-coding genes are not affected in the heart of DM1 fly models.

To link calcium transient to heart asynchrony, we first tested the propagation of calcium waves in normal and in asynchronously beating hearts. We applied the GCaMP3 fluorescent calcium sensor that allows detection of calcium waves in vivo (Limpitikul et al., 2018). As shown in Figure 4A, calcium waves perfectly shadow cardiac contractions in wild-type hearts, each calcium peak aligning with the beginning of contraction along the heart tube (here registered in segments A3 and A4). By contrast, in asynchronously beating Hand>MblRNAi hearts, calcium peaks are not always in perfect alignment with contractions in the anterior A3 segment and are not detectable in the A4 segment that does not beat (Figure 4A). Calcium waves thus correlate with cardiac contractions in normal fly hearts, and are affected in asynchronously beating hearts, linking calcium current regulation and DM1-associated conduction defects.
Figure 4. Calcium waves in asynchronous Drosophila heart.

In wild type hearts, calcium waves underlie cardiac contractions so that calcium peaks (arrows) align with the onset of contractions (arrowheads) in both A3 and A4 segments. In contrast, in asynchronously beating Hand>MblRNAi heart, in the anterior A3 segment, calcium peaks (arrows) are not always in perfect alignment with contractions (arrowheads) and could not be detected in A4 segment that does not beat. Notice that GFP signal in Hand>MblRNAi;GCaMP context is lower than in the control (Hand>GCaMP) most probably because of a lower Hand-Gal4 induction in two UAS transgene context.
Increased cardiac expression of Stj contributes to asynchronous heartbeats in DM1 flies
To determine the cardiac function of Stj, we first tested whether this protein actually accumulated in the adult fly heart. A weak cytoplasmic Stj signal associated with circular myofibers of cardiomyocytes and a strong signal in the myofibers of ventral longitudinal muscle (VLM) underlying the heart tube (Rotstein and Paululat, 2016) was visible in the adult Drosophila heart (Figure 5A and scheme Figure 5B) stained with anti-Stj antibody (Neely et al., 2010). This suggested that low stj levels in cardiac cells could have functional significance. To test this hypothesis, we overexpressed stj in the entire adult fly heart using Hand-Gal4 and in cardiomyocytes only using Tin-Gal4 (Figure 5C). When driven with Hand-Gal4, about 25% of individuals overexpressing stj displayed conduction defects, and the percentage of flies with asynchronous heartbeats increased to 30% in the Tin>stj context (Figure 5C). This suggests that in flies, a high Stj level in cardiomyocytes creates a risk of conduction disturbance.
Figure 5. Increased cardiac expression of Stj contributes to the conduction defects observed in DM1 fly models.
(A) Stj protein is detected at a low level in cardiomyocytes (CM) but at a higher level in the ventral longitudinal muscles (VLMs) that underlie adult heart tube. Stj in VLMs and in heart tube-attaching alary muscles (AMs) marks the network of T-tubules. PC denotes pericardial cells. Scale bars, 50 μm. (B) Scheme of the adult Drosophila heart with representation of Stj expression. (C) Both Hand-Gal4 (whole heart) and Tin-Gal4 (cardiomyocytes only) driven overexpression of Stj lead to asynchronous heartbeats similar to those observed in Hand>MblRNAi and Hand>Bru-3 contexts. (D) Barplot representing percentage of Hand>MblRNAi and Hand>Bru-3 flies displaying asynchronous heartbeats after reducing cardiac Stj expression via RNAi (in stj rescue conditions). Notice that lowering Stj expression efficiently reduces the risk of asynchronous heartbeats in Hand>Bru-3 context. Number of fly hearts tested (n) is indicated and statistical significance (Fisher’s exact test) denoted by ns (p>0.05) and * (p<0.05). (E) IGV tracks showing RNAseq peaks over the stj locus in healthy control and pathological contexts. Red boxes highlight stj-RC – specific exon 15 tracks enriched in Hand>MblRNAi and Hand>Bru-3 contexts and 3’UTR-specific tracks.

Figure 5—figure supplement 1. Stj protein levels in different genetic contexts visualized by immunostaining.

Figure 5—figure supplement 2. stj-RC transcript isoform carrying long 3’UTR and alternatively spliced exon 15 is up regulated in Hand>MblRNAi and in Hand>Bru-3 hearts.

To test whether stj is a required mediator of Bru-3-overexpression and/or MblRNAi-induced conduction defects, we performed genetic rescue experiments by attenuating stj expression via RNAi in Hand>Bru-3 and Hand>MblRNAi flies (Figure 5D). These experiments revealed reductions of asynchronous heartbeats, which were statistically significant for both DM1 contexts but non-significant when referenced to DM1/UPRT lines, showing lower conduction defects (Figure 5D). Intriguingly, lowering stj transcript levels in the heart rescued asynchronous heartbeat more efficiently in Hand>Bru-3 flies than in Hand>MblRNAi flies (Figure 5D). This suggests that fine-tuning of Ca2 level by stj, but potentially also other calcium regulators, may be at work to ensure synchronous heartbeat and avoid conduction block.
We then set out to correlate Stj protein expression level in Hand>Bru-3, Hand>MblRNAi and Hand>stj fly hearts with the extent of cardiac asynchrony in these contexts. We found that Stj signal in the cardiomyocytes was higher in Hand>stj flies than in Hand>MblRNAi and Hand>Bru-3 flies (Figure 5—figure supplement 1). However, the percentage of flies displaying conduction defects was higher in the Hand>MblRNAi context than in the Hand>stj context (Figure 5C,D) indicating that Stj was not the sole factor whose deregulation causes asynchronous heartbeat in the Hand>MblRNAi context. This observation is consistent with partial rescue of conduction defects in the Hand>MblRNAi;stjRNAi context (Figure 5D).
We also undertook to determine, which among three stj transcript isoforms (Figure 5E) is elevated in DM1 contexts. The analysis of RNAseq peaks over the stj locus (Figure 5E) indicated that transcripts harboring exon 15 (stj-RC) and long 3’UTR sequences (stj-RB and stj-RC) are enriched in both Hand>Bru-3 and Hand>MblRNAi fly hearts. The upregulation of stj-RC was validated by RT-qPCR on dissected wild type and DM1 hearts (Figure 5—figure supplement 2) showing that its expression level is equivalent to the expression of all stj transcripts thus providing evidence that stj-RC is the main stj isoform whose expression increases in pathological contexts.
Ventricular cardiomyocytes of DM1 patients with conduction disturbance show an increased expression of α2δ3
To determine whether our data from the fly model were relevant to DM1 patients, we first tested α2δ3 protein and transcript levels in ventricles and atria of normal mouse hearts. Like Stj, α2δ3 protein was also detected in the mouse cardiac cells, with high levels in the atrial and low levels in the ventricular cardiomyocytes (Figure 6A). This differential α2δ3 expression in atria and ventricles was also found at the transcript level (Figure 6B).
Figure 6. Increased cardiac expression of Straightjacket human ortholog α2δ3 is associated with conduction defects in DM1 patients.
(A) In mouse, α2δ3 protein is detected at a high level in atrial cardiac cells and at a low level in ventricles. LV denotes left ventricle. Scale bars, 100 mm. (B) RT-qPCR analysis of mouse α2δ3 transcripts confirms low expression level in ventricles and high expression level in atria. Statistical significance was determined by Mann-Whitney U test (*** p<0.001) (C, D) α2δ3 (C) transcript and (D) protein levels analysed by RT-qPCR (C) and western blot (D) in human ventricular muscle from normal controls and DM1 patients with conduction defects. Note the statistically relevant increase in both α2δ3 transcript and α2δ3 protein levels in ventricles of DM1 patients (Mann-Whitney U test). (E) A scheme illustrating normal and DM1 conditions with low and increased α2δ3 levels in ventricular muscle, respectively. In pathological context, the aberrant inward calcium current in ventricular cardiomyocytes could lead to conduction defects and in particular to the IVCD.

Figure 6—figure supplement 1. RT-qPCR analysis showing that Cav1.2 transcripts are elevated in human cardiac samples from DM1 patients with conduction defects.
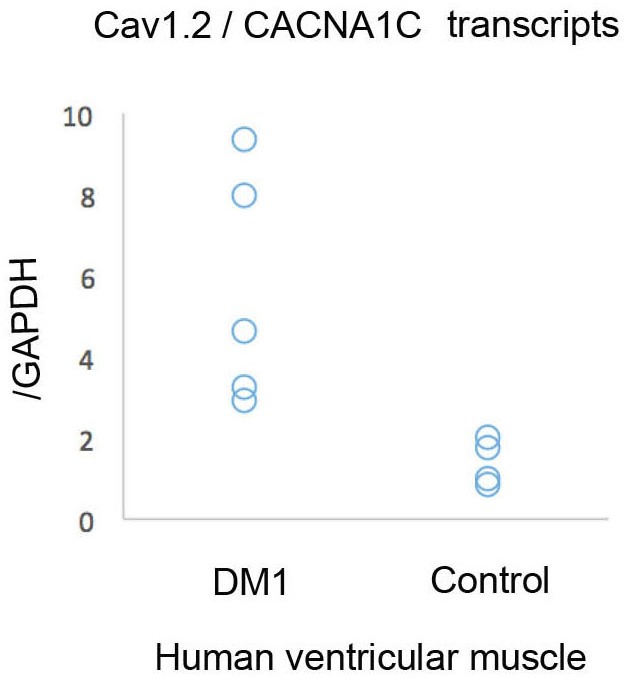
We then analyzed α2δ3 transcript and protein expression in a restricted number of human ventricular muscle samples from healthy donors and from DM1 patients with conduction defects. Ventricular α2δ3 transcript and protein levels were both significantly higher in DM1 patients with conduction disturbances than in controls (Figure 6C,D). Analyzing the same human samples, we also found a higher expression of the main calcium channel α1/Cav1.2 unit (Figure 6—figure supplement 1), further evidence (Figure 6E) that Ca2 inward current deregulation underlies DM1-associated conduction defects.
Discussion
Cardiac dysfunctions decrease life expectancy in DM1, and conduction disturbances affect up to 75% of DM1 patients (McNally and Sparano, 2011). They are diagnosed by a lengthened ECG PR segment indicating atrio-ventricular (AV) block or QRS complex widening (Groh et al., 2008) that might or might not be associated with bundle-branch blocks of the His-Purkinje conduction system. Lengthened QRS without complete or incomplete bundle-branch block results from IVCD. It involves affected conduction via Purkinje fibers but also via ventricular cardiomyocytes that could contribute to the QRS widening observed in DM1 patients.
To identify gene deregulations underlying conduction defects in DM1 patients, we used the Drosophila model, which harbors a simple heart tube without a specialized conduction system, implying that synchronous propagation of contraction waves involves cardiomyocytes only. We reasoned that this model could provide insights into IVCD associated with ventricular cardiomyocyte dysfunction.
We applied a heart cell-specific transcriptional profiling approach based on TU-tagging (Miller et al., 2009) by which we identified 64 conserved genes commonly deregulated in both DM1 contexts with asynchronous heartbeat. Applying a sensitive cell-specific approach followed by RNAseq was critical for revealing discrete gene deregulations, considering that cardiac asynchrony occurred only in a subset of DM1 flies. Also, Hand>MblRNAi and Hand>Bru-3 flies developed two other DM1-associated heart phenotypes: dilated cardiomyopathy and arrhythmias. To facilitate candidate gene selection, we generated gene interaction networks for deregulated candidates and found that four of them were involved in the regulation of cellular calcium level.
Calcium regulators and heart function
We previously reported (Picchio et al., 2013) that abnormal splicing of SERCA encoding the major SR calcium pump was involved in the myotonia phenotype in DM1 flies. Analyzing two heart-specific DM1 fly models, we found that four other conserved calcium regulatory genes, inaD/FRMPD2, Syn1/SNTA1, Rgk2/REM1 and stj/α2δ3, are commonly deregulated. Genes inaD and Syn, which were downregulated, encode scaffolding PDZ domain proteins with regulatory functions on TRP Ca2 channels (Shieh and Zhu, 1996; Ueda et al., 2008); their mutations cause cardiac hypertrophy, arrhythmia, and heart block (Spassova et al., 2004). Moreover, inaD was reported to prevent depletion of ER calcium store by inhibiting Ca2 release-activated Ca2 (CRAC) channels (Su et al., 2003). Rgk2, also downregulated, belongs to the Ras superfamily encoding small GTP-binding proteins (Puhl et al., 2014). Rgk proteins including Rgk2 interact with the α− or β-subunit of CaV1.2 calcium channel and negatively regulate its function (Puhl et al., 2014; Magyar et al., 2012). However, by interacting with CaMKII and with Rho kinase, both involved in cardiac hypertrophy and heart block, Rgk proteins could also influence heartbeat Cav1.2-independently. Finally, stj, whose transcript levels were significantly increased in both DM1 contexts, encodes an auxiliary subunit of this same CaV1.2 channel, and is known to positively regulate this channel’s abundance (Hoppa et al., 2012). CaV1.2 mutations are known to lead to diverse heartbeat dysfunctions (Splawski et al., 2004), and CaV1.2 deregulation is associated with DM1 (Rau et al., 2011). Importantly, the Drosophila counterpart of Cav1.2 (Ca-1αD) also ensures cardiac contractions and calcium transients in the fly heart (Limpitikul et al., 2018), suggesting a conserved role of Cav1.2/Ca-1αD in heartbeat. Here, by following a fluorescent calcium sensor (GCaMP3), we found that calcium waves shadowed synchronous cardiac contractions but were disrupted in asynchronously beating DM1 fly hearts, implying that aberrant calcium transient in cardiomyocytes is associated with heart asynchrony. Thus even if no specialized conduction system is present in the Drosophila heart, propagation of calcium waves along the cardiac tube is finely regulated to ensure synchronous heartbeat. Among selected calcium regulators, only cardiac overexpression of stj resulted in asynchronous heartbeat, so we focused our study on this candidate gene.
Stj/α2δ3, new candidate gene for DM1-associated conduction defects
A previous large study dedicated to identifying genes involved in heat nociception (Neely et al., 2010) identified stj/α2δ3 as an evolutionarily conserved pain gene. Here, we found stj transcripts upregulated in cardiomyocytes of two DM1 fly models Hand>Bru-3 and Hand>MblRNAi exhibiting asynchronous heartbeat. We also show that reducing stj transcript levels significantly ameliorated heart asynchrony in Hand>Bru-3 hearts while improving the conduction defect phenotype of Hand>MblRNAi flies. Thus additional gene deregulations contribute to asynchronous heartbeat in Hand>MblRNAi context, attesting the complexity of heart dysfunction in DM1. At the protein level, Stj is also more abundant in cardiomyocytes of two Drosophila DM1 contexts with cardiac asynchrony, while at a very low level in the wild-type cardiomyocytes. This suggests Stj protein involvement in the DM1-associated asynchronous heartbeat in flies. The precise mechanisms causing stj transcript elevation in DM1 contexts remain to be defined, but our data suggest that a combination of splice-dependent and 3’UTR-dependent mechanisms could be in play.
Intriguingly, vertebrate stj counterpart α2δ3 also displays low transcript and protein levels in ventricular cardiomyocytes and a distinctively higher expression in atrial cardiac cells. We hypothesize that this differential expression could be related to the mechanisms regulating cardiomyocyte contraction in atria and in ventricles. The high α2δ3 expression in atria is consistent with cardiomyocyte-dependent conduction in atria that do not harbor a specialized conduction system. By contrast, its low expression in ventricles could be correlated with the presence of His-Purkinje fibers facilitating conduction in the large ventricular muscles. We were, however, unable to test α2δ3 involvement in cardiac conduction defects in a DMSXL mouse DM1 model carrying large numbers of repeats (Huguet et al., 2012) as it displays only mild cardiac involvement without conduction disturbances. We thus tested α2δ3 expression directly in human cardiac samples. Ventricular cardiomyocytes from healthy donors showed, like the control mice, low α2δ3 transcript expression, which was significantly elevated in ventricular cardiac cells from DM1 patients with conduction defects. DM1-associated increase of expression also occurs for α2δ3 protein and is accompanied by high transcript levels of the main cardiac channel unit Cav1.2/α1, suggesting that the α2δ3 elevation leads to a higher channel density and increased calcium entry to the cardiomyocytes. This could contribute to IVCD affecting the ventricular cardiomyocyte conduction rate and thereby the synchrony of cardiac contraction.
Taken together, with evidence based on heart-specific transcriptional profiling of DM1 Drosophila models and on gene deregulation in cardiac samples from DM1 patients, we propose that the conduction disturbance observed in DM1, and in particular IVCD, arises from likely impacted Ca-α1D/Cav1.2 calcium channel function in ventricular cardiomyocytes in which regulatory subunit stj/α2δ3 plays a central role.
Materials and methods
Key resources table.
| 1 | Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|---|
| 2 | Gene (D. melanogaster) |
stj | FLYB: FBgn0261041 | ||
| 3 | Gene (D. melanogaster) |
mbl | FLYB: FBgn0265487 | ||
| 4 | Gene (D. melanogaster) |
bru3 | FLYB: FBgn0264001 | ||
| 5 | Genetic reagent (D. melanogaster) |
UAS-stj | FlyORF Zurich ORFeome Project | FlyORF ID: F001252 | |
| 6 | Genetic reagent (D. melanogaster) |
stj TRIP | Bloomington Drosophila Stock Center | RRID:BDSC25807 | RNAi line |
| 7 | Genetic reagent (D. melanogaster) |
Mbl RNAi | VDRC Vienna Drosophila Resource Center | VDRC: GD 28732 | RNAi line |
| 8 | Genetic reagent (D. melanogaster) |
P{XP}bru3d09837 | Exelixis at Harvard Medical School | Harvard: d09837 | UAS carrying P element insertion line |
| 9 | Genetic reagent (D. melanogaster) |
UAS-GFP | Bloomington Drosophila Stock Center | RRID:BDSC_32201 | |
| 10 | Genetic reagent (D. melanogaster) |
UAS-HA-UPRT | Bloomington Drosophila Stock Center | RRID:BDSC_27604 | |
| 11 | Genetic reagent (D. melanogaster) |
UAS-lacZ | Bloomington Drosophila Stock Center | RRID:BDSC_1776 | |
| 12 | Genetic reagent (D. melanogaster) |
UAS-GCaMP3 | Bloomington Drosophila Stock Center | RRID:BDSC_32116 | |
| 13 | Genetic reagent (D. melanogaster) |
Ca-alpha1D TRIP | Bloomington Drosophila Stock Center | RRID:BDSC_25830 | RNAi line |
| 14 | Genetic reagent (D. melanogaster) |
Rgk2 TRIP | Bloomington Drosophila Stock Center | RRID:BDSC_65932 | RNAi line |
| 15 | Genetic reagent (D. melanogaster) |
Rgk2 RNAi | VDRC Vienna Drosophila Resource Center | VDRC: GD44611 | RNAi line |
| 16 | Genetic reagent (D. melanogaster) |
Syn1 RNAi | VDRC Vienna Drosophila Resource Center | VDRC: GD27893 | RNAi line |
| 17 | Genetic reagent (D. melanogaster) |
Syn1 RNAi | Bloomington Drosophila Stock Center | RRID: BDSC_27504 | RNAi line |
| 18 | Genetic reagent (D. melanogaster) |
inaD RNAi | VDRC Vienna Drosophila Resource Center | VDRC: GD26211 (unavailable) | RNAi line |
| 19 | Genetic reagent (D. melanogaster) |
inaD RNAi | VDRC Vienna Drosophila Resource Center | VDRC: 330227 | RNAi line |
| 20 | Genetic reagent (D. melanogaster) |
Hand-GAL4 | Laurent Perrin, TAGC, Marseille, France |
GAL4 driver line | |
| 21 | Genetic reagent (D. melanogaster) |
TinCΔ4-GAL4 | Rolf Bodmer, SBPMD Institute, San Diego, US | GAL4 driver line | |
| 22 | Antibody | anti-Stj | Hugo Bellen, Baylor College of Medicine, Huston, USA | guinea pig polyclonal | Use 1:500 (IHC) |
| 23 | Antibody | anti-α2δ3 | Greg Neely, University of Sidney, Australia | rabbit polyclonal | Use 1:200 (IHC) |
| 24 | Antibody | anti-α2δ3 | GeneTex | GTX16618 - rabbit polyclonal | Use 1:500 (WB) |
| 25 | Antibody | anti-Bru3 | Millegen, Toulouse, France | rabbit polyclonal | Use 1:1000 (IHC) |
| 26 | Antibody | anti-Mbl | Darren Monckton, Glasgow, UK | sheep polyclonal | Use 1:200 (IHC) |
| 27 | Antibody | anti-GFP | Abcam | ab5450 – goat polyclonal | Use 1:500 (IHC) |
| 28 | Antibody | Anti-GAPDH | Sigma Aldrich | SAB2108266 – rabbit polyclonal | Use 1:3000 (WB) |
Drosophila stocks
All fly stocks were maintained at 25°C on standard fly food in a 12 hr:12 hr light-dark cycling humidified incubator. To generate the DM1 Drosophila models, we used the inducible lines UAS-MblRNAi (v28732, VDRC, Vienna, Austria) and UAS-Bru3 37 (bru-3d09837, BDSC, Bloomington, USA) and crossed them with the driver line, Hand-Gal4 (Han and Olson, 2005; Sellin et al., 2006) (kindly provided by L. Perrin, TAGC, Marseille), to target transgene expression to the adult fly heart (cardiomyocytes and pericardial cells). To examine the expression pattern of the Hand-Gal4 driver line, we crossed them to UAS-GFP line (#32201, BDSC). Control lines were generated by crossing the above-cited lines with w1118 flies. For TU-tagging experiments, the UPRT line (UAS-HA-UPRT 3.2, #27604, BDSC) was combined with the previously cited UAS lines and with UAS-LacZ (#1776, BDSC) as a control, generating UAS-LacZ;UAS-HA-UPRT, UAS-MblRNAi;UAS-HA-UPRT and UAS-Bru3;UAS-HA-UPRT stocks. For functional analyses of straightjacket, the lines used were UAS-Stj (FlyORF, F 001252) and UAS-stj RNAi (#25807, BDSC) and tinCΔ4-Gal4 cardiomyocyte-specific driver (Lo and Frasch, 2001; Perrin et al., 2004) (kindly provided by R. Bodmer, SBPMD Institute, San Diego). For functional analyses of the candidate genes, we used the following UAS-RNAi lines: #26211 (VDRC) and #330227 (VDRC) for inaD; #27893 (VDRC) and #27504 (BDSC) for Syn1 and #44611 (VDRC) and #65932 (BDSC) for Rgk2. The UAS-GCaMP3 line (#32116, BDSC) was crossed to Hand-Gal4 driver and applied as a sensor of calcium waves in the cardiac tube.
Optical heartbeat analyses of adult Drosophila hearts
To assess cardiac physiology in adult flies, we used the method previously described (Fink et al., 2009; Ocorr et al., 2007). Briefly, 1 week old flies were anesthetized using FlyNap (# 173025, Carolina), and then dissected in artificial hemolymph solution, removing head, legs, wings, gut, ovaries and fat body. The hearts were allowed to equilibrate with oxygenation for 15–20 min prior to filming 30 s movies with a Hamamatsu camera (>100 frames/s) under a Zeiss microscope (10X magnification). Movie analysis was performed with SOHA (semi-automatic optical heartbeat analysis) based on using Matlab R2009b (Mathworks, Natick, MA, USA) to collect contractility (diastolic and systolic diameters, fractional shortening) and rhythmicity (heart period, diastolic and systolic intervals and arrhythmicity index) parameters.
Immunofluorescence on Drosophila heart
Briefly, the fly hearts were dissected as described above and fixed for 15 min in 4% formaldehyde. The immunostaining procedure was performed as described elsewhere (Picchio et al., 2013). The following primary antibodies were used: sheep anti-Mbl antibody (1:200, kindly provided by Darren Monckton), rabbit anti-Bru-3 (1:1000; Millegen, Toulouse, France), rabbit anti-Stj (1:500; kindly provided by Hugo Bellen), rabbit anti-α2δ3 (1:200, gift of Greg Neely) and goat anti-GFP (1:500, Abcam, ab 5450). Rhodamine phalloidin (ThermoFischer Scientific) was used to reveal actin filaments. Fluorescent secondary antibodies were from Jackson ImmunoResearch. For Mbl, we used a biotinylated anti-sheep antibody (Biotin-SP-AffiniPure Donkey Anti-Sheep IgG (H L), Jackson ImmunoResearch) combined with a DTAF-conjugated streptavidin (Jackson ImmunoResearch).
RNA extraction and RT-qPCR on adult fly heart samples
Total RNA was isolated from about 10–15 hearts from 1 week old flies, using TRIzol reagent (Invitrogen) combined with the Quick-RNA MicroPrep Kit (Zymo Research) following the manufacturer’s instructions. RNA quality and quantity were respectively assessed using Agilent RNA 6000 Pico kit on an Agilent 2100 Bioanalyzer (Agilent Technologies) and Qubit RNA HS assay kit on a Qubit 3.0 Fluorometer (Thermo Fischer Scientific). RT-qPCR (three biological replicates) was performed as previously described (Picchio et al., 2013), using Rp49 as a reference gene. The following pairs of primers were used: Rp49: forward GCTTCAAGGGACAGTATCTG and reverse AAACGCGGTTCTGCATGAG; Mbl: forward CGTGGAGGTCCAGAACGG and reverse AATATCAGGTCAATATAAAGCG; Bru-3: forward CCGAAGGTTGAGTTTGCTC and reverse CTTCAGCTGTAAAGCACGG; total stj transcripts: forward AGGCTCGCGAGTTCAACC and reverse AAGGCCTGCTCCGTGTAC and exon 15 carrying transcripts: forward GACGCCCTTTACTCTGGTCA and reverse ATAAAGGGCACAAACTTGCG.
Immunofluorescence on murine heart
Mouse hearts were dissected and fixed for 2 hr in paraformaldehyde (4% in 1X PBS) at 4°C and processed for cryosectioning as described elsewhere (Beyer et al., 2011). Frozen sections were incubated overnight at 4°C with anti-α2δ3 (1:200) (Neely et al., 2010). Fluorescent images were obtained using a Zeiss AxioimagerZ1 with an Apotome module and an AxioCamMRm camera.
RNA extraction and RT-qPCR on murine heart samples
RNA extraction was performed as described elsewhere (Huguet et al., 2012). Transcripts were amplified in a 7300 real-time PCR system (Applied Biosystems) using Power SybrGreen detection (Life Technologies). Standard curves were established with mouse tissues expressing the tested genes. Samples were quantified in triplicate, and experiments were repeated twice. α2δ3 transcripts were amplified using forward primer GGGAACCAGATGAGAATGGAGTC and reverse primer TTTGGAGAAGTCGCTGCCTG. Polr2a was used as internal control (forward primer GGCTGTGCGGAAGGCTCTG and reverse primer TGTCCTGGCGGTTGACCC).
RNA extraction, RT-qPCR, and immunoblot on DM1 patient cardiomyocytes
Human ventricular cardiac muscle tissues were obtained at autopsy from five DM1 patients and four normal controls after informed consent was obtained. All experimental protocols were approved by the Institutional Review Board at Osaka University, and carried out in accordance with the approved guidelines. Total mRNA was extracted and first-strand complementary DNA synthesized using protocols described previously (Nakamori et al., 2008). RT-qPCR was performed using TaqMan Gene Expression assays (Hs01045030_m1 and 4333760F, Applied Biosystems) on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems), as described previously (Nakamori et al., 2011). Level of CACNA2D3/α2δ3 mRNA and Cav1.2/α1 mRNA were normalized to 18S rRNA. For protein analysis, cardiac muscle tissues were homogenized in a 10x volume of radioimmunoprecipitation assay buffer (25 mM Tris-HCl; pH 7.5; 150 mM NaCl; 1% NP-40; 1% sodium deoxycholate; and 0.1% sodium dodecyl sulfate) containing a protein inhibitor cocktail (Sigma-Aldrich). The homogenate was centrifuged for 10 min at 10,000 × g and the supernatant was collected. Equal amounts of protein (30 µg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto Immobilon-P membranes (Millipore), as previously described (Nakamori et al., 2008). Blots were blocked for nonspecific protein binding with 5% (w/v) nonfat milk and then incubated with a 1:500-diluted antibody against CACNA2D3/α2δ3 (Gene Tex) or 1:3000-diluted antibody against GAPDH (glyceraldehyde 3-phosphate dehydrogenase) (Sigma-Aldrich). After repeated washings, the membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (Life Technologies). The ECL Select Western Blotting Detection Reagent (GE Healthcare) and ChemiDoc Touch Imaging System (Bio-Rad) were used to detect the proteins. Only the upper α2δ3 band (Figure 6D) corresponding to predicted protein size was quantified.
TU-tagging experiments
The TU-tagging protocol was adapted from previously published studies (Gay et al., 2014; Miller et al., 2009).
4TU treatment, fly collection, and total RNA extraction
TU-tagging experiments were performed on the following 1-week-old adult flies: Hand>LacZ;HA-UPRT, Hand>MblRNAi;HA-UPRT and Hand>Bru3;HA-UPRT. Briefly, flies were starved for 6 hr at 25°C and transferred to fresh food media containing 4TU (Sigma) at 1 mM. After 12 hr incubation at 29°C, about 30–40 flies of the described genotypes were dissected (removing head, wings, legs, ovaries and gut) in DPBS 1X (Gibco, Life Technologies), immediately transferred to Eppendorf tubes, and snap-frozen in liquid nitrogen. Total RNA isolation was performed as described above in TRIzol following the manufacturer’s instructions (ThermoFischer Scientific).
Purification of TU-tagged RNA
Bio-thio coupling was performed on about 30 µg of total RNA with 1 mg/mL biotin-HPDP (Pierce) for 3 hr followed by two chloroform purification steps to eliminate the unbound biotin, as previously described (Miller et al., 2009). To verify the efficiency of the biotinylation step, an RNA dot blot was performed using streptavidin-HRP antibody. The streptavidin purification step then served to collect the thiolated-RNA fraction (cardiac-specific 4TU) and the unbound fraction (non-cardiac RNAs) using a µMACS streptavidin kit (MACS, Miltenyi Biotec) following the manufacturer’s instructions. The purified fraction was precipitated as previously described (Miller et al., 2009). RNA quality and quantity were assessed using Bioanalyzer and Qubit systems according to the manufacturer’s instructions. RT-qPCR was performed to check for enrichment of cardiac-specific transcripts (Hand and H15) by comparing 4TU fraction relative to input fraction (i.e. the biotinylated fraction containing cardiac and non-cardiac RNAs), with Rp49 gene used as reference.
RNA sequencing
Library preparation
Library preparation was performed on about 50 ng of cardiac-specific RNA from the previously described conditions, using the Ovation RNA-Seq Systems 1–16 for Model Organisms adapted to Drosophila (NuGEN, #0350–32) as per the manufacturer’s instructions with a few modifications. First-strand synthesis was performed with the integrated DNAse treatment (HL-dsDNAse, ArcticZymes) and cDNA was fragmented using a Bioruptor sonication system (Diagenode) via 30 cycles of 30 s ON/OFF on low power. 15 PCR library amplification cycles were performed on 50 ng of starting RNA, and a size-selective bead purification step was done using RNAClean XP beads (Agencourt AMPure, Beckman Coulter). Quantitative and qualitative assessment of the library was performed using an Agilent dsDNA HS kit on an Agilent 2100 Bioanalyzer (Agilent Technologies).
Sequencing parameters
The indexed Illumina libraries prepared from the cardiac-specific Hand>LacZ;HA-UPRT, Hand>MblRNAi;HA-UPRT and Hand>Bru3;HA-UPRT RNA fractions were mixed at equal concentrations and sequenced (100 bp paired-end reads) on the same lane of a HiSeq 2000 system (EMBL Gene Core Illumina Sequencing facility, Heidelberg, Germany). All four genotype sets of 1-week-old flies were sequenced in duplicate to give a total of eight samples sequenced.
Data analysis
The RNA-sequencing data were checked for good quality using the FastQC package (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Each sample was aligned to the reference Dmel genome release 5.49 using Bowtie2 (Langmead and Salzberg, 2012). The aligned reads were sorted and indexed using SAMtools (Li et al., 2009). Pearson correlation was determined for each duplicate of each condition. Differential gene expression was obtained using a pipeline based on the Deseq2 package. Rlog transformation was applied on raw count data obtained for the duplicates of each condition before computing differential expression. Genes were tested individually for differences in expression between conditions. We set a fold-change threshold at 1.7 and p-value threshold at 0.05 for meaningful differential up-expression, and fold-change threshold at 0.59 and p-value threshold at 0.05 for meaningful differential down-expression.
In vivo calcium transient analyses
We performed short (10 s) time lapse recording of beating hearts, dissected as for SOHA experiments, and mounted inverted on 35 mm glass-bottom dishes (IBIDI ref 81218) for imaging on LEICA SP8 confocal microscope. Twenty control hearts (Hand>GCaMP) and 35 DM1 hearts dissected from Hand>MblRNAi;GCaMP 1-week-old flies were analyzed. Two channel films recorded from a single optical level (GCaMP-GFP channel representing calcium waves and transmitted light channel representing contractions) were analyzed using Image J. For each film, we first transformed Frames on Image stacks and used ‘Plot Z axis profile’ function to generate graphs.
Statistical analyses and access to RNAseq data
All statistical analyses were performed using GraphPad Prism v5.02 software (GraphPad Inc, USA). Fisher’s exact test was applied to analyze statistical significance of conduction defect phenotypes, and Mann-Whitney U test to analyze RT-qPCR data from mouse and human heart samples and Western blot α2δ3 expression data in human samples. We also applied one-way ANOVA, Kruskal-Wallis Dunn’s multiple comparison post-test to statistically analyze RT–qPCR data for stj isoforms and changes in diastolic and systolic heart diameters. All RNAseq data reported here were deposited with the GEO-NCBI tracking system under accession number #18843071.
Acknowledgements
This work was supported by AFM-Téléthon (MyoNeurAlp Strategic Program), Agence Nationale de la Recherche (Tefor Infrastructure Grant), Fondation pour la Recherche Médicale (Equipe FRM Award) and the Intramural Research Grant for Neurological and Psychiatric Disorders (NCNP grant to MN).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Monika Zmojdzian, Email: monika.zmojdzian@uca.fr.
Krzysztof Jagla, Email: christophe.jagla@udamail.fr.
Margaret Buckingham, CNRS UMR 3738, Institut Pasteur, France.
Utpal Banerjee, University of California, Los Angeles, United States.
Funding Information
This paper was supported by the following grants:
AFM-Téléthon MyoNeurAlp strategic program to Krzysztof Jagla.
Agence Nationale de la Recherche Tefor Infrastructure to Krzysztof Jagla.
Fondation pour la Recherche Médicale Equipe FRM to Krzysztof Jagla.
National Center of Neurology and Psychiatry Intramural Research Grant for Neurological and Psychiatric Disorders to Masayuki Nakamori.
Additional information
Competing interests
No competing interests declared.
Author contributions
Formal analysis, Investigation, Methodology, Writing—original draft.
Funding acquisition, Investigation, Methodology, Writing—original draft.
Formal analysis, Investigation, Methodology, Writing—original draft.
Formal analysis, Investigation, Methodology.
Formal analysis, Investigation, Methodology.
Methodology.
Formal analysis, Methodology, Writing—review and editing.
Supervision, Funding acquisition.
Supervision, Investigation, Methodology, Writing—review and editing.
Supervision, Methodology, Writing—review and editing.
Formal analysis, Investigation, Methodology.
Conceptualization, Formal analysis, Investigation, Visualization, Methodology, Writing—review and editing.
Conceptualization, Formal analysis, Supervision, Funding acquisition, Investigation, Methodology, Writing—review and editing.
Ethics
Human subjects: Human ventricular cardiac muscle tissues were obtained at autopsy from five DM1 patients and four normal controls after informed consent was obtained. All experimental protocols were approved by the Institutional Review Board at Osaka University (Protocol Number 14183-6) and carried out in accordance with the approved guidelines.
Additional files
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files. Sequencing data have been deposited with the GEO-NCBI tracking system under accession number GSE109370.
The following dataset was generated:
Plantie E, Picchio L, Renaud Y. 2018. Deregulation associated with cardiac conduction defects in Myotonic Dystrophy Type 1 using TU-Tagging. NCBI Gene Expression Omnibus. GSE109370
References
- Beyer S, Kelly RG, Miquerol L. Inducible Cx40-Cre expression in the cardiac conduction system and arterial endothelial cells. Genesis. 2011;49:83–91. doi: 10.1002/dvg.20687. [DOI] [PubMed] [Google Scholar]
- Birse RT, Choi J, Reardon K, Rodriguez J, Graham S, Diop S, Ocorr K, Bodmer R, Oldham S. High-fat-diet-induced obesity and heart dysfunction are regulated by the TOR pathway in Drosophila. Cell Metabolism. 2010;12:533–544. doi: 10.1016/j.cmet.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodi I. The L-type calcium channel in the heart: the beat Goes on. Journal of Clinical Investigation. 2005;115:3306–3317. doi: 10.1172/JCI27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbé J-C, Lorca T, Castro A. Loss of human greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. PNAS. 2010;107:12564–12569. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3' untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. PNAS. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Die-Smulders CE, Höweler CJ, Thijs C, Mirandolle JF, Anten HB, Smeets HJ, Chandler KE, Geraedts JP. Age and causes of death in adult-onset myotonic dystrophy. Brain. 1998;121:1557–1563. doi: 10.1093/brain/121.8.1557. [DOI] [PubMed] [Google Scholar]
- Dixon DM, Choi J, El-Ghazali A, Park SY, Roos KP, Jordan MC, Fishbein MC, Comai L, Reddy S. Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Scientific Reports. 2015;5:9042. doi: 10.1038/srep09042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. The α2δ subunits of voltage-gated calcium channels. Biochimica Et Biophysica Acta (BBA) - Biomembranes. 2013;1828:1541–1549. doi: 10.1016/j.bbamem.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Fink M, Callol-Massot C, Chu A, Ruiz-Lozano P, Izpisua Belmonte JC, Giles W, Bodmer R, Ocorr K. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. BioTechniques. 2009;46:101–113. doi: 10.2144/000113078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyermuth F, Rau F, Kokunai Y, Linke T, Sellier C, Nakamori M, Kino Y, Arandel L, Jollet A, Thibault C, Philipps M, Vicaire S, Jost B, Udd B, Day JW, Duboc D, Wahbi K, Matsumura T, Fujimura H, Mochizuki H, Deryckere F, Kimura T, Nukina N, Ishiura S, Lacroix V, Campan-Fournier A, Navratil V, Chautard E, Auboeuf D, Horie M, Imoto K, Lee KY, Swanson MS, de Munain AL, Inada S, Itoh H, Nakazawa K, Ashihara T, Wang E, Zimmer T, Furling D, Takahashi MP, Charlet-Berguerand N. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nature Communications. 2016;7:11067. doi: 10.1038/ncomms11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo K, Smith DP. A large family of divergent Drosophila odorant-binding proteins expressed in gustatory and olfactory sensilla. Genetics. 2001;159:1059–1072. doi: 10.1093/genetics/159.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay L, Karfilis KV, Miller MR, Doe CQ, Stankunas K. Applying thiouracil tagging to mouse transcriptome analysis. Nature Protocols. 2014;9:410–420. doi: 10.1038/nprot.2014.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV, Marashdeh MM, Zipes DP, Pascuzzi RM. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. New England Journal of Medicine. 2008;358:2688–2697. doi: 10.1056/NEJMoa062800. [DOI] [PubMed] [Google Scholar]
- Han Z, Olson EN. Hand is a direct target of tinman and GATA factors during Drosophila cardiogenesis and hematopoiesis. Development. 2005;132:3525–3536. doi: 10.1242/dev.01899. [DOI] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. α2δ expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486:122–125. doi: 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, Mohr SE. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguet A, Medja F, Nicole A, Vignaud A, Guiraud-Dogan C, Ferry A, Decostre V, Hogrel JY, Metzger F, Hoeflich A, Baraibar M, Gomes-Pereira M, Puymirat J, Bassez G, Furling D, Munnich A, Gourdon G. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLOS Genetics. 2012;8:e1003043. doi: 10.1371/journal.pgen.1003043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ, Cooper TA. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Reports. 2014;6:336–345. doi: 10.1016/j.celrep.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino Y, Washizu C, Oma Y, Onishi H, Nezu Y, Sasagawa N, Nukina N, Ishiura S. MBNL and CELF proteins regulate alternative splicing of the skeletal muscle chloride channel CLCN1. Nucleic Acids Research. 2009;37:6477–6490. doi: 10.1093/nar/gkp681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Human Molecular Genetics. 2010;19:1066–1075. doi: 10.1093/hmg/ddp570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Molecular Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpitikul WB, Viswanathan MC, O'Rourke B, Yue DT, Cammarato A. Conservation of cardiac L-type Ca2 channels and their regulation in Drosophila: a novel genetically-pliable channelopathic model. Journal of Molecular and Cellular Cardiology. 2018;119:64–74. doi: 10.1016/j.yjmcc.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo PC, Frasch M. A role for the COUP-TF-related gene seven-up in the diversification of cardioblast identities in the dorsal vessel of Drosophila. Mechanisms of Development. 2001;104:49–60. doi: 10.1016/s0925-4773(01)00361-6. [DOI] [PubMed] [Google Scholar]
- Magyar J, Kiper CE, Sievert G, Cai W, Shi GX, Crump SM, Li L, Niederer S, Smith N, Andres DA, Satin J. Rem-GTPase regulates cardiac myocyte L-type calcium current. Channels. 2012;6:166–173. doi: 10.4161/chan.20192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu J, Allard P, Potvin L, Prévost C, Bégin P. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology. 1999;52:1658. doi: 10.1212/WNL.52.8.1658. [DOI] [PubMed] [Google Scholar]
- McNally EM, Sparano D. Mechanisms and management of the heart in myotonic dystrophy. Heart. 2011;97:1094–1100. doi: 10.1136/hrt.2010.214197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesirca P, Torrente AG, Mangoni ME. Functional role of voltage gated Ca(2 ) channels in heart automaticity. Frontiers in Physiology. 2015;6:19. doi: 10.3389/fphys.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MR, Robinson KJ, Cleary MD, Doe CQ. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nature Methods. 2009;6:439–441. doi: 10.1038/nmeth.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamori M, Kimura T, Kubota T, Matsumura T, Sumi H, Fujimura H, Takahashi MP, Sakoda S. Aberrantly spliced alpha-dystrobrevin alters alpha-syntrophin binding in myotonic dystrophy type 1. Neurology. 2008;70:677–685. doi: 10.1212/01.wnl.0000302174.08951.cf. [DOI] [PubMed] [Google Scholar]
- Nakamori M, Gourdon G, Thornton CA. Stabilization of expanded (CTG)•(CAG) repeats by antisense oligonucleotides. Molecular Therapy. 2011;19:2222–2227. doi: 10.1038/mt.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely GG, Hess A, Costigan M, Keene AC, Goulas S, Langeslag M, Griffin RS, Belfer I, Dai F, Smith SB, Diatchenko L, Gupta V, Xia CP, Amann S, Kreitz S, Heindl-Erdmann C, Wolz S, Ly CV, Arora S, Sarangi R, Dan D, Novatchkova M, Rosenzweig M, Gibson DG, Truong D, Schramek D, Zoranovic T, Cronin SJ, Angjeli B, Brune K, Dietzl G, Maixner W, Meixner A, Thomas W, Pospisilik JA, Alenius M, Kress M, Subramaniam S, Garrity PA, Bellen HJ, Woolf CJ, Penninger JM. A genome-wide Drosophila screen for heat nociception identifies α2δ3 as an evolutionarily conserved pain gene. Cell. 2010;143:628–638. doi: 10.1016/j.cell.2010.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocorr K, Reeves NL, Wessells RJ, Fink M, Chen HS, Akasaka T, Yasuda S, Metzger JM, Giles W, Posakony JW, Bodmer R. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. PNAS. 2007;104:3943–3948. doi: 10.1073/pnas.0609278104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin L, Monier B, Ponzielli R, Astier M, Semeriva M. Drosophila cardiac tube organogenesis requires multiple phases of hox activity. Developmental Biology. 2004;272:419–431. doi: 10.1016/j.ydbio.2004.04.036. [DOI] [PubMed] [Google Scholar]
- Picchio L, Plantie E, Renaud Y, Poovthumkadavil P, Jagla K. Novel Drosophila model of myotonic dystrophy type 1: phenotypic characterization and genome-wide view of altered gene expression. Human Molecular Genetics. 2013;22:2795–2810. doi: 10.1093/hmg/ddt127. [DOI] [PubMed] [Google Scholar]
- Picchio L, Legagneux V, Deschamps S, Renaud Y, Chauveau S, Paillard L, Jagla K. Bruno-3 regulates sarcomere component expression and contributes to muscle phenotypes of myotonic dystrophy type 1. Disease Models & Mechanisms. 2018;11:dmm031849. doi: 10.1242/dmm.031849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhl HL, Lu VB, Won YJ, Sasson Y, Hirsch JA, Ono F, Ikeda SR. Ancient origins of RGK protein function: modulation of Voltage-Gated calcium channels preceded the protostome and deuterostome split. PLOS ONE. 2014;9:e100694. doi: 10.1371/journal.pone.0100694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, Wahbi K, Day JW, Fujimura H, Takahashi MP, Auboeuf D, Dreumont N, Furling D, Charlet-Berguerand N. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nature Structural & Molecular Biology. 2011;18:840–845. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- Rotstein B, Paululat A. On the morphology of the Drosophila heart. Journal of Cardiovascular Development and Disease. 2016;3:15. doi: 10.3390/jcdd3020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nature Genetics. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- Sellin J, Albrecht S, Kölsch V, Paululat A. Dynamics of heart differentiation, visualized utilizing heart enhancer elements of the Drosophila Melanogaster bHLH transcription factor hand. Gene Expression Patterns. 2006;6:360–375. doi: 10.1016/j.modgep.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Shieh BH, Zhu MY. Regulation of the TRP Ca2 channel by INAD in Drosophila photoreceptors. Neuron. 1996;16:991–998. doi: 10.1016/S0896-6273(00)80122-1. [DOI] [PubMed] [Google Scholar]
- Sicot G, Gourdon G, Gomes-Pereira M. Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: new findings and future challenges. Human Molecular Genetics. 2011;20:R116–R123. doi: 10.1093/hmg/ddr343. [DOI] [PubMed] [Google Scholar]
- Spassova MA, Soboloff J, He L-P, Hewavitharana T, Xu W, Venkatachalam K, van Rossum DB, Patterson RL, Gill DL. Calcium entry mediated by SOCs and TRP channels: variations and enigma. Biochimica Et Biophysica Acta (BBA) - Molecular Cell Research. 2004;1742:9–20. doi: 10.1016/j.bbamcr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 Calcium Channel Dysfunction Causes a Multisystem Disorder Including Arrhythmia and Autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Su Z, Barker DS, Csutora P, Chang T, Shoemaker RL, Marchase RB, Blalock JE. Regulation of Ca2 release-activated Ca2 channels by INAD and Ca2 influx factor. American Journal of Physiology. Cell Physiology. 2003;284:C497–C505. doi: 10.1152/ajpcell.00183.2002. [DOI] [PubMed] [Google Scholar]
- Taghli-Lamallem O, Plantié E, Jagla K. Drosophila in the heart of understanding cardiac diseases: modeling channelopathies and cardiomyopathies in the fruitfly. Journal of Cardiovascular Development and Disease. 2016;3:7. doi: 10.3390/jcdd3010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. The Journal of Cell Biology. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theadom A, Rodrigues M, Roxburgh R, Balalla S, Higgins C, Bhattacharjee R, Jones K, Krishnamurthi R, Feigin V. Prevalence of muscular dystrophies: a systematic literature review. Neuroepidemiology. 2014;43:259–268. doi: 10.1159/000369343. [DOI] [PubMed] [Google Scholar]
- Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. PNAS. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weerd JH, Christoffels VM. The formation and function of the cardiac conduction system. Development. 2016;143:197–210. doi: 10.1242/dev.124883. [DOI] [PubMed] [Google Scholar]
- Voght SP, Fluegel ML, Andrews LA, Pallanck LJ. Drosophila NPC1b promotes an early step in sterol absorption from the midgut epithelium. Cell Metabolism. 2007;5:195–205. doi: 10.1016/j.cmet.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Wang GS, Kuyumcu-Martinez MN, Sarma S, Mathur N, Wehrens XH, Cooper TA. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. Journal of Clinical Investigation. 2009;119:3797–3806. doi: 10.1172/JCI37976. [DOI] [PMC free article] [PubMed] [Google Scholar]