

Abstract
The molecular mechanisms underlying inclination responses in trees are unclear. In this study, we identified a MADS-box transcription factor differentially expressed early after inclination in the stems of Pinus radiata D. Don. PrMADS10 has a CDS of 582 bp and encodes a group II MADS-box transcription factor. We measured highest accumulation of this transcript on the lower side of inclined pine stems. In an effort to identify putative targets, we stably transformed Arabidopsis thaliana with a 35S::PrMADS10 construct. Transcriptome analysis revealed 1,219 genes differentially-expressed, with 690 and 529 genes up- and down-regulated respectively, when comparing the transgenic and wild-type. Differentially-expressed genes belong to different biological processes, but were enriched in cell wall remodeling and phenylpropanoid metabolic functions. Interestingly, lignin content was 30% higher in transgenic as compared to wild-type plants consistent with observed changes in gene expression. Differentially expressed transcription factors and phenylpropanoid genes were analyzed using STRING. Several MYB and NAC transcription factors showed interactions with genes of the phenylpropanoid pathway. Together, these results implicate PrMADS10 as a regulatory factor, triggering the expression of other transcription factors and genes involved in the synthesis of lignin.
Subject terms: Transcriptomics, Plant molecular biology
Introduction
The loss of verticality in trees triggers morphological changes in the cell wall, which affect wood quality. The underlying molecular response involves the expression of particular genes, calcium signaling, and the synthesis of hormones like auxins and ethylene1–7.
Remodeling of the secondary cell wall (SCW) is regulated by two large families of transcription factors (TFs): R2R3-MYB and NAC5,8–13. For example, the synthesis of the major secondary wall components such as cellulose, hemicelluloses and lignins, was regulated by TFs expressed in differentiating xylem of Eucalyptus. EgMYB1 acts as repressor14 and EgMYB2 as activator15 in this process. Both regulate the entire development of SCW, acting therefore as a second level of “master switch” due to the fact that NAC TFs like SND1 (Secondary Wall-associated NAC Domain Protein16) is at the top level of the hierarchical network. Apparently, AtMYB46, the orthologue of EgMYB2 is a direct target of SND18. Similarly, the expression of two MYB TFs from Populus trichocarpa are also activated by PtrWND2, which is an orthologue of SND1 in Populus trichocarpa17.
PtrWNDs are expressed in stem structure of P. trichocarpa18 which could be either fiber- or vessel-specific expression. Ohtani19 described 12 PtrWND genes that redundantly modulate the differentiation of vessels and fiber cells during xylem formation. These TFs modulate the expression of genes involved in SCW formation and programmed cell death, and also other TFs18,19.
Other NACs and MYBs which participate in the regulation of SCW dynamics have also been identified both in Arabidopsis and trees. These include PtrMYB289 and PtrMYB120 potential orthologues of lignin-specific MYB activators21, as well as PtrNAC118, 122, 128 and 129 from P. trichocarpa22 which are potential orthologues of Xylem NAC Domain 1 (XND1), a transcriptional repressor that regulates the expression of genes involved in programmed cell death and SCW formation23. Interestingly, the overexpression of SND2 is associated to particular phenotypes in woody and herbaceous stems24.
Other TF families, apart from NAC and MYB, have been involved in regulating the biosynthesis of SCW. For example, KNOTTED1-Like Homeodomain Protein7 targeted directly SND125. This protein negatively regulates secondary wall biosynthesis in poplar and Arabidopsis, functioning as a negative feedback loop that represses metabolically SCW formation, and maintaining homeostasis26. Another TF, NtLIM1 regulates lignin biosynthesis pathway in tobacco and E. camaldulensis transformants27,28.
A partial sequence corresponding to a MADS-box TF was identified in a subtractive suppressed hybridization library (SSH) of one-year old radiata pine seedlings exposed to inclination29. The transcription factor MADS-box gene belongs to a highly-conserved multigene family previously identified in a wide range of eukaryotic genomes30. These proteins are major regulators of plant development, and the expression of the gene has been described in roots, stems, abscission zones, leaves, developing ovules and embryos31–33. Also, the formation of higher order MADS-box complexes is a means by which they obtain their diverse functions34,35. These TFs are known to be involved in flowering36, ripening processes in fruit37,38 and anthocyanin biosynthesis39–41.
MADS-box proteins share a highly-conserved DNA-binding domain, with a length of 56–60 amino acid residues. MADS-box proteins recognize a CC(A/T)6GG DNA sequence known as the CArG-box element, and the functional role is performed as a protein dimer42,43. Even if most reports have related their role in flowering, several authors have shown that MADS-box genes are expressed in differentiating primary/secondary xylem and phloem during wood formation in poplar44. In eucalyptus, MADS-box TFs have been detected in vegetative tissues45, and xylem tissue of white spruce6. Nine MADS-box genes have been identified in P. radiata (PrMADS 1 to 9), which have been detected in vegetative outbreak, floral organs and roots, all of them orthologues of TOMATO MADS3 (TM3). PrMADS 4 to 9 were more abundantly-expressed in young flowering tissue than in adult tissue46 and are members of the TM3 clade. But PrMADS1 is orthologue to AGL2 clade and, PrMADS2, PrMADS3 are members to AGL6 clade, playing a possible roles in regulation of reproductive development47–49.
Functional analysis of TFs from trees is not an easy task, so the use of the model plant Arabidopsis thaliana can help to provide clues concerning their functional role. For example, the expression of Populus tremuloides MADS-box 3 and 4 (PTM3/4) genes in Arabidopsis shows that they take part in floral development50, whilst, PTM5 is involved in vegetative development51. The constitutive expression of two MADS-box TFs members in Arabidopsis, SHP1 and SHP2 genes (previously known as AtAGL1 and AtAGL5, respectively), promotes the lignification of cells adjacent to reproductive organs52.
The present report shows the identification and characterization of PrMADS10. This MADS-box gene has greater expression levels in the stems of inclined radiata pine, tissues which also rapidly-accumulate lignin29. Is PrMADS10 a regulator for the synthesis of lignin? What other genes are modulated by the over expression of PrMADS10? These questions were answered by heterologously-overexpressing full length PrMADS10 in Arabidopsis, carrying out a microarray assay and performing a MapMan53,54 analysis to obtain a metabolic overview of differentially expressed genes.
Results
Sequence and phylogenetic analysis of PrMADS10
PrMADS10 full-length cDNA sequence was obtained using a partial EST sequence as template from the SSH library29, followed by 5′- and 3′-RACE-PCR. The sequence of 943 bp long contains 111 and 250 bp of 5′- and 3′-UTRs, respectively. PrMADS10 has a CDS of 582 bp, encoding a deduced protein of 193 amino acids and 22 kDa (pI 9.42; GenBank accession number, KM887510; Fig. 1A). The predicted PrMADS10 protein has the typical conserved structural features of MADS-box TFs and possesses a MIKC type protein structure (Fig. 1A). A phylogenetic analysis was performed with 48 MADS-box amino acid sequences, including proteins from poplar, pine and spruce. PrMADS10 is classified in group II (MIKCc) according to MADS sub classification, and very close to orphans genes like AtSVP and StMADS16, genes that are expressed in vegetative tissue (Fig. 1B).
Figure 1.

Sequence analysis of the deduced PrMADS10 protein from radiata pine with other MADS-box proteins. (A) Multiple alignment of the deduced PrMADS10 sequence with Arabidopsis thaliana, Oryza sativa, Paulownia kawakamii, Picea abies, Pinus tabuliformis, Populus trichocarpa, and Solanum tuberosum MADS-box TFs was performed using Clustal W and BioEdit Sequence Alignment Editor v7.0 software. Gaps are indicated by dashes, letters with a black background are identical amino acids, and letters with a gray background are similar amino acids. The box indicate M, I, K and C domains shared between MADS-box proteins, the double arrow and lines are DNA binding, thin arrow is dimerization interface; the filled arrow is a putative phosphorylation site. (B) Phylogenetic analysis was performed using MEGA X software, with Neighbor-joining, bootstrap consensus tree inferred from 10000 replicate. The evolutionary distances were computed using the Poisson correction method and are in the units of the number of amino acid substitutions per site. This analysis involved 48 amino acid sequences: Arabidopsis thaliana AtAGL1 (AAA32730), AtAGL2 (BAC43207), AtAGL3 (NP_849930), AtAGL6 (NP_182089), AtAGL12 (AEE35216), AtAGL14 (AEE83062), AtAGL24 (AEE84922), AtAGL28 (AEE27300), AtAGL30 (AEC05661), AtAGL32 (NP_974823) AtAGL33 (AEC07824), AtAGL34 (AED93593), AtAGL36 (AED93581), AtAGL38 (AEE34356), AtAGL39 (AED93653), AtAGL51 (AEE82144), AtAGL57 (AEE74037), AtAGL64 (AEE31158), AtAGL75 (AED94653), AtAGL94 (AEE34947), AtSVP (NP_179840), AtFLC (NP_196576), Coffea arabica CaS07 (ADU56825), Ginkgo biloba GbMADS1 (AIC79629), Ipomoea batatas IbMADS10 (ABD66305), Malus domestica MdMADS16 (BAG48168), Oryza sativa OsMADS3 (Q40704), OsMADS47 (Q5K4R0), Paulownia kawakamii PkMADS1 (AAF22455), Picea abies PaDAL10 (AAQ13443), Pinus pinaster PpiMADS5 (est_pipn_28509135_001R), Pinus radiata PrMADS1 (AAD09206), PrMADS2 (AAD09207), PrMADS3 (AAB58907), PrMADS4 (AAB80807), PrMADS5 (AAB80808), PrMADS6 (AAB80809), PrMADS7 (AAB80810), PrMADS8 (AAC27353), PrMADS9 (AAC80806), PrMADS10 (AKC96434), Pinus tabuliformis PtabMADS1 (AJP06319), Pinus taeda PtaMADS20 (est_pita_11126880, est_pita_11604453, est_pita_9457518), Prunus avium PaMADS1 (ABW82563), Populus tomentosa PtMADS10 (AAR92206), Populus tremuloides PtreMADS (AAP46287), Populus trichocarpa PtrSVP (XP_002310310), Solanum tuberosum StMADS16 (AAV65504).
PrMADS10 transcripts are preferentially accumulated in pine stems and its protein has nuclear localization
RT-qPCR analysis shows a higher transcript accumulation of PrMADS10 in the lower side of inclined stems, reaching a maximal difference between upper and lower sides after 24 h (Fig. 2). A four-fold transcript increase was detected on the lower side of inclined seedling stems 24 h after the onset of the inclination response in comparison to non-inclined control. In addition, transcript levels were strongly downregulated in needles and roots after inclination.
Figure 2.
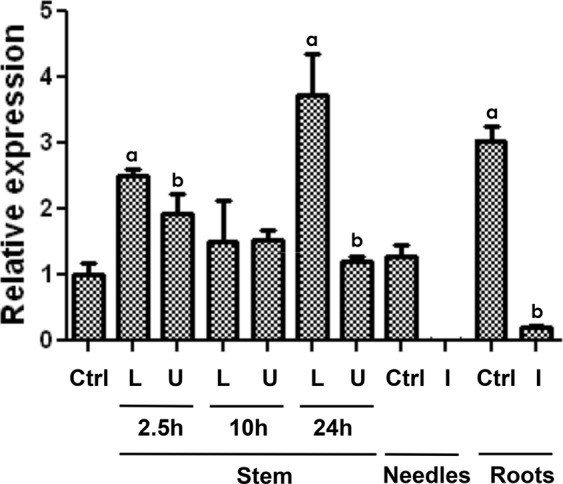
Transcripts levels of PrMADS10 in young radiata pine seedlings after inclination stimuli. Stem samples were taken at different times of inclination either on the lower stem side (L) or upper stem side (U) of the stem. Roots and needles were obtained from the same inclined (I) seedlings. Ctrl (control) means non-inclined seedlings. Data correspond to mean ± SE of three biological replicates and different letters indicate statistical differences (p ≤ 0.05).
To determine the subcellular localization of PrMADS10 tobacco leaves were agroinfiltrated55 (Fig. 3). PrMADS10 fused to GFP co-localizes with SYTO83, a nuclear marker. This data reveals PrMADS10 is a nuclear localized protein, consistent with its predicted function as a transcription factor based on protein sequence.
Figure 3.

Subcellular localization of PrMADS10 fused to GFP (35 S::PrMADS10-GFP) in Nicotiana benthamiana leaves. SYTO83 was used to stain the nuclei. Merge: merging of GFP, SYTO83, and phase contrast image (PCI) images. Bar = 10 μm.
Overexpression of PrMADS10 in Arabidopsis alters lignin and flavonoids accumulation
The CDS of radiata pine MADS10 gene was isolated and introduced into pBI121 binary vector under the control of CMV35S promoter. Homozygous Arabidopsis T3 transgenic lines were obtained and used for performing morphological and transcriptional analyses. Four different PrMADS10 over-expressing lines were obtained (Suppl. Fig. 1). The evaluation of plant morphology and the weight of one hundred seeds indicated no changes between transgenic and control lines, however the length of rosette leaves were smaller in the transgenic lines (Suppl. Fig. 2). At the same time, lignin content in three of the transgenic lines was 30% higher than wild-type plants and Arabidopsis plants transformed with empty-vector (control), while one line showed no difference compared to controls (Fig. 4A). In addition, similar total area was observed when stem cross-sections from transgenic Arabidopsis and WT plants were stained with phloroglucinol-HCl. But, in the case of hypocotyl, two of three transgenic lines showed smaller total area compared to wild-type. On the other hand, qualitative difference was found in stem and hypocotyl when lignin was detected by using Phloroglucinol-HCl (Suppl. Fig. 3A,B). Besides, anthocyanin content was significantly-reduced in two transgenic lines (Fig. 4B).
Figure 4.
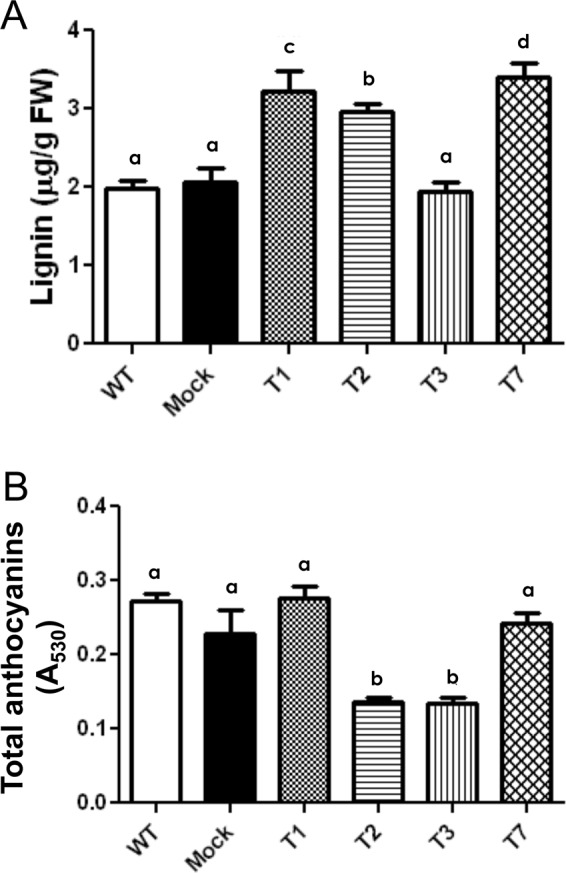
Quantification of lignin (A) and anthocyanins (B) in T3 Arabidopsis plants transformed with 35 S::PrMADS10. Data correspond to mean ± SE of three biological replicates per transgenic line of plants overexpressing PrMADS10, and different letters indicate statistical differences (p ≤ 0.05). Analysis of variance (ANOVA) and the t-student test was used.
Gene modulation in Arabidopsis overexpressing PrMADS10
PrMADS10 is differentially-expressed in inclined pine stems29, and could modulate the expression of a series of genes. Microarray analysis was used to determine global changes in gene expression resulting from constitutive over-expression of PrMADS10. The AraGene-1_0-ST 90k chip, with 28,501 annotated genes was used for the analysis. The raw data was normalized and differential gene expression was determined using standard statistical procedures (see Materials and Methods). We identified 1,219 differentially expressed genes when comparing transgenic and wild-type plants (Suppl. dataset 1). PrMADS10 over-expression induces 690 and represses 529 genes. All 1,219 differentially expressed genes were analyzed using hieratical clustering (Suppl. Fig. 4). Three sub-clusters are shown in Suppl. Fig. 4. A high consistency is observed both within each group of plants (either control or transgenic), and within the branches generated. Unsupervised grouping chooses clustering by k-means, and three clusters were resolved having the first 339 genes, the second 383, and the third 498 genes. The first cluster has 235 up-regulated genes as compared to wild-type plants, 219 of which could be classified using the Panther database. 84 genes have an annotation in the molecular function gene ontology, with the following categories found: catalytic activity (53% of the genes), transporter activity (23%), binding (14%), transcription regulator activity (9%), structural molecule activity (4%), and both molecular transducer activity and molecular function as regulator (1%). A total of 104 genes were down-regulated, 52 of which had gene ontology annotations in similar categories for the up-regulated genes, except for the absence of molecular transducer activity category (Fig. 5A). Additionally, GOrilla56 was used with all the data from the microarray and it was determined that molecular function was the most significant category for this cluster (Suppl. Fig. 5A).
Figure 5.

Unsupervised clustering with k-means and GO for molecular function of differentially expressed genes. (A) Cluster one has 339 genes in total with an homogeneity of 0.313, with 235 genes up and 104 down-regulated compared to control, and gene ontology classification for molecular function. (B) Cluster two has 383 genes in total with an homogeneity of 0.122, with 167 genes up and 207 down-regulated compared to control, and gene ontology classification for molecular function. C. Cluster three has 498 genes in total with an homogeneity of 0.040, with 280 genes up and 218 down-regulated compared to control, and gene ontology classification for molecular function. Sub-clustering considering the analysis of the 1219 differentially expressed genes (absolute expression), where red is 0~2 and green 0~−2, K-Means from Expander was used.
The second cluster contained 167 up-regulated genes, 52 of which were annotated as follows: 57% were classified to catalytic activity, 30% to binding, 9% to transporter activity, and 4% to structural molecule activity (Fig. 5B). 207 genes were down-regulated, with 110 annotated in different gene ontology categories: catalytic activity (43%), binding (32%), molecular function regulator (8%), transporter activity (5%), transcription regulator activity (5%), molecular transducer activity (5%), and structural molecule activity (2%) (Fig. 5B). When GOrilla was used in this cluster the best significant group was biological process category (Suppl. Fig. 5B).
The third cluster grouped 255 up-regulated genes, where 99 had gene ontology annotations: 47% classified to catalytic activity, 25% to binding, 19% to transporter activity, 3% to transcription regulator activity, 2% to structural molecule activity, 2% to molecular transducer activity and 2% to molecular function regulator. Finally, this cluster contains 213 down-regulated genes and 116 showed annotations in the molecular function ontology: most of them were related to catalytic activity (50%) and binding (30%), followed by molecular transducer activity (9%), transporter activity (3%), transcription regulator activity (3%), molecular function regulator (3%), and structural molecule activity (2%) (Fig. 5C). When GOrilla was used in this cluster the best significant group was biological process category (Suppl. Fig. 5C). Interestingly, in this cluster several sub-categories associated to stress and secondary metabolite are the most representatives.
The fifty most-up regulated genes were listed in Table 1 (those with >2.6 fold change). Phosphate starvation1 showed the highest differential expression (over 5-fold change), and other genes related to phosphate metabolism or transport are higher and within the most-up regulated genes. Interestingly, other genes and TFs are also up regulated by the over-expression of PrMADS10 in Arabidopsis. TFs like basic leucine-zipper 48, homeobox protein 16 are predominant, as well as, MIR399C, MIR399D and MIR827a. Other proteins like Ring U/Box superfamily, glutathione-S-transferase, alpha/beta hydrolases, rhamnose biosynthesis, phospholipase D, and phosphoenolpyruvate carboxylase kinase are all more abundant when PrMADS10 is over-expressed.
Table 1.
List of 50 most up-regulated genes in 35S::PrMADS10 transgenic Arabidopsis plants using microarray analysis.
| AGI number | Exp log2 | Gene description |
|---|---|---|
| AT3G09922 | 5.237 | induced by phosphate starvation1 |
| AT4G24890 | 5.107 | purple acid phosphatase 24 |
| AT1G17710 | 4.609 | Pyridoxal phosphate phosphatase-related protein |
| AT5G20790 | 4.160 | Unknown |
| AT3G61410 | 4.097 | Unknown |
| AT1G23110 | 3.997 | Unknown |
| AT5G17220 | 3.879 | glutathione S-transferase phi 12 |
| AT2G11810 | 3.879 | monogalactosyldiacylglycerol synthase type C |
| AT3G44510 | 3.823 | alpha/beta-Hydrolases superfamily protein |
| AT5G62162 | 3.822 | MIR399C; miRNA |
| AT1G08310 | 3.748 | alpha/beta-Hydrolases superfamily protein |
| AT3G14790 | 3.628 | rhamnose biosynthesis 3 |
| AT2G34202 | 3.591 | MIR399D; miRNA |
| AT5G08030 | 3.577 | PLC-like phosphodiesterases superfamily protein |
| AT1G73220 | 3.515 | organic cation/carnitine transporter1 |
| AT2G04038 | 3.475 | basic leucine-zipper 48 |
| AT2G45135 | 3.473 | RING/U-box superfamily protein |
| AT2G45130 | 3.428 | SPX domain gene 3 |
| AT3G25240 | 3.409 | Protein of unknown function (DUF506) |
| AT4G40060 | 3.372 | homeobox protein 16 |
| AT3G05630 | 3.344 | phospholipase D P2 |
| AT3G04530 | 3.306 | phosphoenolpyruvate carboxylase kinase 2 |
| AT2G34210 | 3.276 | Transcription elongation factor Spt5 |
| AT1G67600 | 3.263 | Acid phosphatase/vanadium-dependent haloperoxidase-related protein |
| AT3G59884 | 3.258 | MIR827a; miRNA |
| AT4G36350 | 3.246 | purple acid phosphatase 25 |
| AT3G02040 | 3.226 | senescence-related gene 3 |
| AT5G03545 | 3.134 | Unknown |
| AT4G12090 | 3.096 | Cornichon family protein |
| AT1G19200 | 3.078 | Protein of unknown function (DUF581) |
| AT3G03530 | 3.052 | non-specific phospholipase C4 |
| AT4G01380 | 3.036 | plastocyanin-like domain-containing protein |
| AT5G44562 | 3.019 | other RNA |
| AT3G25233 | 3.014 | Unknown |
| AT4G36850 | 3.008 | PQ-loop repeat family protein / transmembrane family protein |
| AT4G17220 | 2.982 | microtubule-associated proteins 70-5 |
| AT3G09285 | 2.939 | Unknown |
| AT3G02550 | 2.928 | LOB domain-containing protein 41 |
| AT5G53048 | 2.913 | other RNA |
On the other hand, terpenoid cyclase, O-acyltransferase, and fatty acid reductase are the most down-regulated genes. Interestingly, several TFs like WRKY, MYB, and B-box type zinc finger proteins are also down-regulated, as are other genes like expansins, pectin lyase, small auxin up RNA (SAUR), S-adenosyl-L-methionine (SAM) dependent methyltransferase and 2-oxoglutarate dependent oxygenase (Table 2).
Table 2.
List of 50 most down-regulated genes in 35 S::PrMADS10 transgenic Arabidopsis using microarray analysis.
| AGI number | Exp log 2 | Gene description |
|---|---|---|
| AT1G78950 | −3.176 | Terpenoid cyclases family protein |
| AT5G22490 | −3.128 | O-acyltransferase (WSD1-like) family protein |
| AT3G57460 | −3.068 | catalytics;metal ion binding |
| AT3G56700 | −3.022 | fatty acid reductase 6 |
| AT2G40080 | −2.912 | Protein of unknown function (DUF1313) |
| AT5G22570 | −2.837 | WRKY DNA-binding protein 38 |
| AT2G24850 | −2.734 | tyrosine aminotransferase 3 |
| AT5G42900 | −2.690 | cold regulated gene 27 |
| AT1G57750 | −2.643 | cytochrome P450, family 96, subfamily A, polypeptide 15 |
| AT5G13330 | −2.555 | related to AP2 6 l |
| AT4G33790 | −2.551 | Jojoba acyl CoA reductase-related male sterility protein |
| AT1G57560 | −2.547 | myb domain protein 50 |
| AT1G02450 | −2.530 | NIM1-interacting 1 |
| AT5G28080 | −2.521 | Protein kinase superfamily protein |
| AT2G21140 | −2.516 | proline-rich protein 2 |
| AT1G07050 | −2.501 | CCT motif family protein |
| AT1G17665 | −2.482 | Unknown |
| AT5G38000 | −2.481 | Zinc-binding dehydrogenase family protein |
| AT1G30040 | −2.469 | gibberellin 2-oxidase |
| AT5G26220 | −2.457 | ChaC-like family protein |
| AT2G23910 | −2.353 | NAD(P)-binding Rossmann-fold superfamily protein |
| AT3G44860 | −2.328 | farnesoic acid carboxyl-O-methyltransferase |
| AT3G28220 | −2.299 | TRAF-like family protein |
| AT4G33980 | −2.276 | Unknown |
| AT2G21660 | −2.271 | cold, circadian rhythm, and rna binding 2 |
| AT3G07650 | −2.258 | CONSTANS-like 9 |
| AT5G44568 | −2.254 | Unknown |
| AT5G20630 | −2.206 | germin 3 |
| AT1G66380 | −2.183 | myb domain protein 114 |
| AT4G38825 | −2.174 | SAUR-like auxin-responsive protein family |
| AT1G68050 | −2.169 | flavin-binding, kelch repeat, f box 1 |
| AT5G45960 | −2.143 | GDSL-like Lipase/Acylhydrolase superfamily protein |
| AT3G46490 | −2.137 | 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenase superfamily protein |
| AT3G05770 | −2.108 | Unknown |
| AT4G25860 | −2.097 | OSBP(oxysterol binding protein)-related protein 4 A |
| AT3G20810 | −2.095 | 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenase superfamily protein |
| AT5G37970 | −2.073 | S-adenosyl-L-methionine-dependent methyltransferases superfamily protein |
| AT5G37940 | −2.070 | Zinc-binding dehydrogenase family protein |
| AT3G60160 | −2.068 | multidrug resistance-associated protein 9 |
| AT2G40610 | −2.064 | expansin A8 |
| AT1G55525 | −2.063 | other RNA |
| AT5G48250 | −2.051 | B-box type zinc finger protein with CCT domain |
| AT4G17470 | −2.032 | alpha/beta-Hydrolases superfamily protein |
| AT1G28050 | −2.016 | B-box type zinc finger protein with CCT domain |
| AT1G14250 | −1.992 | GDA1/CD39 nucleoside phosphatase family protein |
| AT5G01900 | −1.965 | WRKY DNA-binding protein 62 |
| AT3G59270 | −1.964 | FBD-like domain family protein |
| AT5G25460 | −1.949 | Protein of unknown function, DUF642 |
| AT5G63180 | −1.902 | Pectin lyase-like superfamily protein |
| AT1G75780 | −1.882 | tubulin beta-1 chain |
Networking interaction analysis in Arabidopsis overexpressing PrMADS10
MapMan53,54 was used to visualize genes modulated by 35S::PrMADS10 in Arabidopsis. The terms for MapMan were assigned for 166 differentially-expressed genes; but due to a lack of matching MapMan terms, no functional assignment was done for the other 1,055 genes. The results obtained show a general view of different functional categories, which are being affected by 35S::PrMADS10, such as major and minor carbohydrate, amino acid, nucleotide, fermentation, lipids, secondary metabolisms and cell wall (Fig. 6). Metabolism-related genes are further divided as 56 genes for cell wall, 30 for lipid, 29 for secondary, 12 for amino acid, 5 for light, 9 for major carbohydrate, 7 for minor carbohydrate, and a few for glycolysis, fermentation, tricarboxylic acid (TCA) cycle, S-assimilation, nucleotide metabolism, tetrapyrrole synthesis, or mitochondrial electron transport. When the phenylpropanoid pathway was analyzed, several genes were positively- or negatively-regulated (Fig. 6).
Figure 6.

Metabolic overview using MapMan. The 1219 differentially-expressed genes were used to visualize the changes in metabolic pathways. One gene can have more than one classification. Up-regulated steps are represented in red and down-regulated steps in blue.
Differentially expressed TFs and phenylpropanoid pathway genes were selected from the microarray assay to perform a MapMan classification. The list of TFs and phenylpropanoid pathway´s genes were introduced in STRING in order to determine network interactions (Fig. 7). Genes without interactions were erased from the image. The interactions observed can be of different types, depending on the color of the line joining the spheres, and recognized as co-expression, co-localization and text-mining, and therefore it cannot be taken strictly as a direct protein-protein interaction. It can be observed that genes were clustered into different groups. TFs like MYB and NAC are included in one group (MYB50, MYB52, MYB42, NAC010, NAC073, MYB43, MYB83, HB16, MYB85, MYB32, NAC012 and NST2), and PAL, 4-coumarate-CoA ligase (4CL), and caffeoyl-CoA O-methyltransferase (CCoAOMT) are in a different one. Interestingly, seven genes from the phenylpropanoid pathway are grouped and interconnected. MapMan showed that these genes (PAL4, 4CL8, 4CL2, CCOAMT, 4CL5, CCR2; AT4G26220; UDP glucosyl transferase 72E1) are differentially expressed. The gene AT4G26220 has been reported as a probable CCoAOMT7 (amplified view of Fig. 7).
Figure 7.

Network of gene interactions among genes differentially expressed using STRING. Genes used in the analysis were previously classified within the phenylpropanoid metabolic pathway and a series of TFs differentially expressed. Each node represents the interaction, described in the literature, between the proteins differentially-expressed in Arabidopsis over-expressing PrMADS10.
In a small network interaction, MYC4 and MYB76 are grouped to other TFs. At the same time, LHY is related to AGL24, TOC1 and CCA, and indirectly to DOF, PRR5 or PRR9. Finally, a zinc transporter protein correlates with WRKY70, which has direct interactions with WRKY38, WRKY51 and WRKY54.
Discussion
The importance in flower development was the initial interest to study MADS-box genes in plants33. However, a MADS-box gene (PdPI) was isolated from Populus deltoides, and it is expressed during flower development and in different vegetative organs. The evidence suggests that PdPI plays multiple functions in the development of this species57. In radiata pine, these TFs are expressed in male and female strobiles, for example PrMADS3 (AGL6-like) is transcribed in the primordium of the acule47. Even though evidence defines roles for MADS-box TFs in flowering and other reproductive processes, their expression in roots, leaves and stems33, like in the case of PrMADS10, suggest another type of function in these other organs, perhaps expressed in response to stress.
In a first effort to understand the role that this MADS-box gene plays in radiata pine, the full length of PrMADS10 was obtained by RACE. Deduced amino acid sequence was aligned with MADS-box proteins from different species available in Genbank database. PrMADS10 contains the typical MADS-box conserved domains: MADS-box, Intermittent, K-box and C-terminal58. According to Shore and Sharrocks43, a conserved sequence of 56 amino acids is characteristic in this domain, where 16 of which are identical in all family members. Structural features can also be observed in the MADS-box domain44, and the region of DNA responsible for protein interactions and dimerization43.
Phylogenetic analysis of PrMADS10 showed a distribution of sequences grouped into two main clades, with the sequences of interest being clustered in clade II. In this clade, all proteins have a MIKC-like structure59,60. In the phylogenetic tree, PrMADS10 is closely related to PaMADS1 from P. abies (Fig. 1B). PaMADS1 is expressed mainly in female and male pine cones, controlling the development of these tissues61. In addition, protein sequences of interest are also associated with a sub-clade classified as similar to SVP, AGL and MADS, such as PtSVP (P. trichocarpa) and AtSVP (A. thaliana), AtAGL24 (A. thaliana), StMADS16 (S. tuberosum) and PtMADS1 (P. tomentosa). PtSVP is a protein from the SVP (Short Vegetative Phase) family, and is very closely-related to AtSVP, which is a negative floral regulator62. This sequence is conserved in both angiosperms and gymnosperms. Similarly, AtAGL24 promotes the identity of the inflorescence in Arabidopsis whose expression occurs at the apex of the buds at the time of floral transition63. Most of the genes near to PrMADS10 were named orphan genes, because StMADS11, StMADS16, AtAGL24, and AtSVP are expressed in vegetative tissues, like vascular cambium region64, and having the same expression pattern than PrMADS10. Thus, the phylogenetic evidence suggests that PrMADS10 could be involved in floral development, but not observable in one-year-old seedlings. Nevertheless, this sequence was isolated from the lower side of pine stems, shortly after tilting29. This implies that its role in pine is related to the loss of verticality, modulating gene expression in vegetative tissues rather than in floral development.
PrMADS10 was differentially expressed 2.5 hours after tilting and accumulation of transcripts was preferentially observed on the lower side of bent young pine stems29. The result suggests that expression of PrMADS10 is temporally and spatially regulated in pine stem sections. The change in expression levels for this transcription factor in pine stems may be important for control of metabolic pathways that modify cell wall structure and properties which impacts on wood anatomy. Different molecular events related to cell wall modifications in response to verticality loss, including changes in a large number of TFs have been reported1,29. The work of Allona1 and Ramos29 were complementary, as both report new genes that respond to tilt, albeit there is no mention concerning the differential expression of MADS-box TFs in the former study.
Several different MADS-boxes have been described in radiata pine; PrMADS4 to 9 were detected in vegetative shoots, floral organs and roots, with greater expression in young floral tissue compared to adult tissue46. PrMADS1 to 3 could be involved in regulating female reproductive structures47,48 and cone development49. The expression of MADS-box genes during flower development and vegetative organs have also been reported in P. deltoides57. In contrast, Cseke44 reported PTM5 which is specific to vascular tissue and expressed in differentiated primary-secondary xylem and phloem. Therefore, although MADS-box genes are associated with flowering, other tissues like stem can be the tissue where this transcription factor can be expressed, which seems to be the case of PrMADS10, and more likely involved in stems modulating the response to inclination.
The AraGene microarray shows that 35S::PrMADS10 differentially regulates the expression of 1,219 genes when heterologously-expressed in Arabidopsis. PrMADS10 induces the expression of 690 different genes and down-regulates that of 529. Aswath and Kim33 state that the constitutive expression of MADS-box genes into tobacco or Arabidopsis plants has proved to be a useful tool to analyze gene functions, as shown by the characterization of SAG1 from the conifer black spruce (Picea mariana), an homologous to AGAMOUS65. The overexpression of SAG1 produces homeotic floral conversion in transgenic Arabidopsis (from sepal to carpel and petal to stamen). On the other hand, overexpression of a MADS-box from P. tomentosa (PtAP3) in tobacco plants causes a fast growth and early flowering phenotype66.
The 35S::PrMADS10 transgenic lines of Arabidopsis did not show any discernible phenotypic changes, including flowering time, even though the transgene was over-expressed by at least 20,000 fold in all 4 lines obtained. The first 50 most-overexpressed genes were those involved in phosphate starvation, as well as, TFs and sucrose synthase 4 (SUS4) (Table 1). Unexpectedly, genes for phosphorous starvation were the most differentially-expressed. Pi was not a limiting factor in our assays, yet some reports indicate that Pi starvation is under the control of TFs required to maintain phosphorus homeostasis, which in turn is affected by environmental stress67–71.
Two genes, a multigenic AtSUS isoforms (SUS1–6) that synthesize monosaccharides for cellulose and starch biosynthesis71, and rhamnose biosynthesis 3 (RHM3), providing rhamnose for the synthesis of the cell wall72, are also up-regulated by PrMADS10. Basic chitinase is also up-regulated, which is a protein leading to inhibition of seedling growth73. In addition, the gene microtubule-associated protein 70-5 is positively-regulated by PrMADS10. This gene is essential for defining SCW polymers and is expressed in the cellular cortex of wood-forming cells, influencing the pattern of SCW thickenings in tracheary elements74. These results suggest that PrMADS10 positively regulates several genes involved in wood formation, such as secondary metabolism, sugar synthesis or remodeling of cell wall components, as shown in the MapMan analysis (Fig. 7).
The accumulation of lignin is increased by 30% in the T3 transgenic lines, concomitant with the differential expression of genes involved in phenylpropanoid pathway, where a few genes from the pathway are positively-regulated by PrMADS10. Interestingly, Pinus taeda MYB4 has been reported as a positive regulator in lignifying cells75. Moreover, MYBs have also been related to cell wall metabolism, and have been shown to be the main master switches of SCW biosynthesis in different species13,76,77. More recently, the biosynthesis of cellulose/xylan/lignin is triggered by MYB46/83 and MYB8578. Both MYBs (MYB83 and MYB85) genes are up-regulated in our experiment, which indicates that PrMADS10 can induce the expression of both TFs, thus enhancing the accumulation of lignin. A full network of protein-protein relationships reported by STRING indicates that these TFs are also associated with NAC. Moreover, the network correlates the interaction between MYBs and NACs with phenylpropanoid genes, such as CCoAOMT, 4CL2, 4CL5, PAL4 and 4CL8. The co-expression of these groups of genes was reported when transcriptomic analysis was performed in Arabidopsis stem during development79. Additionally, in woody plants like eucalyptus and poplar, the over-expression of MYB216 increases the accumulation of transcripts of 4CL5 and PAL480,81. The knock-out for PtMYB156 in poplar showed an increment in the accumulation of 4CL5 transcripts indicating a selective regulation of phenylpropanoid genes82.
TFs like NAC and MYB are preferentially co-expressed in xylem in Picea glauca83 and Pinus pinaster84, suggesting that both integrate a network which takes part in the development of complex wood traits, and as regulator for the synthesis of phenylalanine. In our assay, NAC10, NAC073 and MYB52, MYB42, MYB85 TFs were differentially expressed, and also showed relationships with other NACs and MYBs in the STRING network. Coincidently, several MYB TFs were characterized from radiata pine seedlings and are differentially-expressed on both sides of an inclined stem, yet preferentially on the upper side85. Whether NAC is modulating the expression of MYB and NAC genes, or if PrMADS10 could play a central role in this interaction in response to bending in pine, is an open question that requires further analysis.
The most regulated genes in the compression side of adult radiata pine tree trunks are those like: cell division, cellulose biosynthesis, lignin deposition and microtubules86. Genes like tubulin beta, sucrose synthase, proline-rich protein, and pectin lyase-like were differentially-expressed in 35S::PrMADS10 Arabidopsis, as well as, in adult pine trees86. Also, tubulin alpha chain, the cellulose synthase gene family, sucrose synthase and expansin were preferentially-expressed in Pinus taeda differentiated xylem87. These genes are active in some way during the process of cell wall remodeling, and are also differentially-expressed in 35S::PrMADS10 transgenic Arabidopsis.
Genes from the phenylpropanoid pathway are also differentially-regulated in 35S::PrMADS10 plants. The increase in COMT transcript levels was similar to that observed for PAL. If COMT levels increase, the amounts of synapyl alcohol should be modified, which is the last step for lignin (syringyl) synthesis. Similarly, if COMT and CAD transcript levels increase, they could modify the amounts of synapyl and p-coumaryl alcohols, the final step for the synthesis of lignin (syringyl and p-hydroxyphenyl, respectively). This is corroborated by previous studies, which suggest that the level of C3H transcripts could be determinant in the flow of metabolites towards lignin production, and the transcripts of CCoAOMT, CCR and CAD are modulated according to the metabolic demand88. Similar findings were reported in inclined radiata pine stems29. The net result of increasing the level of transcripts of these genes is an increment in flux through the monolignol portion for the lignin biosynthesis pathway75. Patzlaff75 reported that PtMYB4 recognizes AC elements of the PAL promoter and genes encoding lignin biosynthesis enzymes are altered after overexpressing the PtMYB4 gene in transgenic tobacco plants. In addition, the deposition of lignin is increased, and spreads to other types of cells that normally do not lignify, showing that PtMYB4 was sufficient to induce lignification in a heterologous system. In contrast, overexpressing PtrMYB3 from populus in Arabidopsis led to an increase in the deposition of the three main polymers of cell wall89. A 30% increment in lignin was observed in 35S::PrMADS10 lines, suggesting stimulation towards the lignin biosynthesis pathway. Besides, when one-year old pine seedlings were tilted, lignin accumulation and wall thickening were observed after 15 days90. This suggests that remodeling of the cell wall could initially be regulated by PrMADS10 after tilting.
Methods
Materials and Methods
One year-old half-sib Pinus radiata D. Don (radiata pine) seedlings were grown in a local nursery from seeds obtained from an open-pollinated population (half sibs). Seedlings of around 30 cm tall were maintained at 20 °C, and following the protocol established previously29,91. Nine inclined seedlings were collected at each sampling time; their stems were cut into different parts along the longitudinal axis into lower and upper sides, pooled, immediately frozen in liquid nitrogen and stored at −80 °C until RNA extraction. Additionally, ‘non-inclined’ seedlings were sampled as control. Previous inclination procedure was followed29.
Gene cloning and vector construction
Pinus radiata MADS10 (PrMADS10) was cloned from a cDNA library constructed from the stems of young seedlings exposed to inclination at different times29. The PrMADS10 ORF was amplified and cloned following the RACE strategy, and using RNA from inclined stems (BD Smart RACE cDNA Amplification Kit, Clontech, USA). The following primers: PrMADS10-F 5′-CAGATCTCCGTCGGCAGTTAAAGGAAC-3′ and PrMADS10-R 5′-GGCTGCGAAGATAACCCTAGATGCAAG-3′ were used to obtain 3′ and 5′ sequences, respectively. PCR conditions for amplification was: 30 cycles each of 1 min at 94 °C, 1 min at 66 °C, and 3 min at 72 °C. A final 20-extension step of min at 72 °C was performed.
PrMADS10 was cloned into the pBI121 binary vector, using forward (5′-GGATCCATGGCCGGCGAGAAAAGAAAGAT-3′) and reverse (5′-GAGCTCAATCTTTGATTCGGACGACTGT-3′) primers, designed to include BamHI and SacI restriction sites, respectively. The reverse primer had the stop codon deleted. The final construction, denominated 35S::PrMADS10, was confirmed by sequencing.
For the construction of the PrMADS10-GFP fusion, the PrMADS10 CDS was amplified with PrMADS10FL-F 5′-CACCATGGCCGGCGAGAAAAGAAAGAT-3′ and PrMADS10FL-R 5′-AATCTTTGATTCGGACGACTGT-3′ primers, without the stop codon and inserted into pENTR/D-TOPO vector (Invitrogen). Subsequently, the insert was transferred to the plant binary vector pKF7WG2 by recombination using Gateway® LR ClonaseTM II Enzyme Mix kit (Invitrogen) following manufacturer’s instruction. The 35 S::PrMADS10-GFP construct was used for subcellular localization studies. All clones used were confirmed by sequencing.
Subcellular localization in tobacco leaves
Gateway® LR ClonaseTM II Enzyme Mix kit (Invitrogen) was used to perform a 35S:PrMADS10-GFP construction. Manufacturer’s instruction was followed for recombination.
After two days of cultures Agrobacterium was collected and centrifuged at 6000 x g for 10 min at 4 °C. The LB medium was supplemented with three antibiotics: gentamycin (100 μg/ml), rifampicin (10 μg/ml) and spectinomycin (50 μg/ml). Pellet was re-suspended in distilled and sterile water. Young tobacco leaves were infiltrated on the abaxial side, and were analyzed after three days post-infiltration. Syto® 84 Orange Fluorescent Nucleic Acid stain (Thermo Scientific) was used to label the nucleus. Subcellular localization of PrMADS10 in transient transformed leaves were analyzed through tissue visualization under a confocal fluorescence microscope (Carl Zeiss Confocal microscopy LMS 700) employing phase contrast image. Bar represent 10 μm.
Stable arabidopsis transformation
Columbia ecotype (Col-0) of Arabidopsis thaliana (L.) Heynh plants were transformed using floral dip method92. Germinated seeds were placed in vessels containing rock wool and embedded in a hydroponic medium. The plants were maintained at 25 °C in the growth chamber with a long day photoperiod regime (16 h light/8 h dark). In the T2 generation, lines with single T-DNA insertion site were selected based on the segregation of resistant and sensitive seedlings to glufosinate ammonium containing medium and verified by PCR with specific primers. Then, T3 homozygous lines for PrMADS10 construct were selected and used in the analyses.
RNA extraction and quantitative RT-PCR (RT-qPCR)
Total RNA was extracted from radiata pine seedlings following the procedure described by Le Provost93. Integrity of RNAs was checked on agarose gels stained with GelRed (Biotium Inc.), and their concentration determined by a ND-1000 UV spectrophotometer (Nanodrop Technologies, Montchanin, DE, USA). cDNA synthesis was performed using First Strand cDNA Synthesis Kit (Fermentas Life Science, Glen Burnie, MD, USA).
Total RNA was isolated from 35S::PrMADS10 transgenic Arabidopsis, using the SV total RNA isolation system (Promega). Primers for quantitative real time-PCR (RT-qPCR) were designed using Beacon Designer v 2.0 software (Premier Biosoft, Palo Alto, CA, USA). All primers used in this work are listed in Table 1. YBR Green/ROX quantitative PCR (qPCR) Master Mix (2 × ; Fermentas Life Science) was used for all qPCR quantifications in a final volume of 20 μL following the manufacturer’s protocol. All experiments were run on a real-time Mx3000P PCR detection system (Stratagene, Cedar Creek, TX, USA). The instrument was set to measure SYBR green dye fluorescence at the end of each cycle. Initial primer concentrations were 250 nM for all reactions, and the cDNA template for each sample was synthesized using 1 μg of DNase-treated total RNA using a first-strand cDNA synthesis kit (Fermentas Life Science) according to the manufacturer’s instructions. The first-strand RT reaction product was diluted ten-fold, and 2 μL was used for each qPCR reaction. The cycle threshold (Ct) line was determined manually as the point where the R2 value for the standard curve reached its highest point94. Standard curves were determined in duplicate reactions from the dilution series of each amplicon. qPCR determinations were run in duplicate and values of each sample corresponded to a mean ± SE of three biological replicates. A melting curve analysis was performed for each set of primers in order to avoid non-specific amplification. The expression levels were normalized with the stable expression level of three housekeeping genes (Suppl. Table 1). The overexpression of the PrMADS10 gene was obtained from 3 biological replicates. Relative expression was calculated using primers for AtFbox, AtUbi1095 and AtPP2 as normalizing genes. Data were analyzed using the methods derived from the algorithm of Vandesompele96. Two-way ANOVA-LSD post hoc was used to determine the main effects of inclination and time of inclination exposure effect for each gene using Statistica for Windows (v. 7.0; StatSoft, Tulsa, OK, USA). Significant differences were inferred at P ≤ 0.05. Differences in PrMADS10 transgenic expression, lignin and anthocyanin quantification was analyzed by one-way ANOVA-LSD post hoc and the significant differences inferred at P ≤ 0.05.
Gene expression using Affymetrix ATH1 microarrays
For microarray hybridizations, total RNA was processed using the GeneChip one-cycle target-labeling kit (Affymetrix). Biotinylated cRNA was synthesized from 5 μg of total RNA from Arabidopsis stems (three months old) using the Affymetrix IVT kit according to the manufacturer’s instructions. cRNA was used to hybridize ATH1 GeneChip expression microarrays. Three un-transformed plants were used as controls and four transgenic lines were considered as biological replicate. Affymetrix data were normalized in R (http://www.r-project. org/) using RMA97. For detecting differentially regulated genes, normalized log2-transformed data were analyzed using Rank product statistic, as described before98–100. The data was processed with bioinformatics tools available at the VirtualPlant web site (http://www.virtualplant.org).
For the heatmap, Expander 7.11 (http://acgt.cs.tau.ac.il/expander/) was used. K-means of 3 clustering were obtained from microarray data. The clusters were obtained with 50 max iteration used distance metric with Pearson correlation. The GO pie charts were obtained using PANTHER14.1 DB and graphic genic onthology was obtained using GOrilla56.
Microarray data
Raw signal intensity values were first normalized with RMA method using affy package in R language101,102 and probes were mapped to Locus ids of Arabidopsis genome. Genes with at least 2-fold change and p-value < 0.05 in Rank product analysis were considered as differentially expressed. Microarray data was analyzed using K-means clustering from Expander 7.11 (http://acgt.cs.tau.ac.il/expander/).
Functional classification based on MapMan
Gene expression data in a context of metabolic overview was visualized using MapMan (version 3.6.0RC) software53,54. MapMan uses a plant-specific ontology that classifies genes into well-defined hierarchical categories, denominated BINs and suBINS103. The diagram shows positively and negatively-regulated genes in red and blue, respectively. The data set obtained from microarray analysis (AraGene-1_0-ST) where compared to Ath_AGI_TAIR9_Jan2010 integrated in MapMan from TAIR.
STRING interaction network
String is a database where known and predicted direct (physical) interactions, as well as, indirect (functional) interactions can be established based on co-expression, co-localization, or text-mining and others104–106. Differentially-expressed genes related to transcription factors and those genes involved in the synthesis of lignin were picked and the web server was interrogated in order to uncover potential protein-protein association networks. The database was interrogated for the last time on 28th March (2019).
Anthocyanin and lignin content
Leaf and stem samples from three months-old Arabidopsis were ground in liquid nitrogen and extracted overnight in 1.0 mL of 1% (v/v) HCl in methanol at 4 °C following a previous report107. Relative anthocyanin levels were determined by measuring the absorbance at 530 nm of the aqueous phase108. Lignin was extracted as described by Campbell and Ellis109. Samples from whole Arabidopsis stems were diluted in 1 M NaOH (1/3, v/v) and hydrolyzed. A colorimetric assay was performed using thioglycolic acid (Sigma-Aldrich), and the absorbance was measured at 280 nm. The results were expressed as μg lignin per gram of fresh weight (FW).
Supplementary information
Acknowledgements
This work was supported by FONDECYT project N° 1150964. NC acknowledges Beca-Conicyt and TM acknowledges Universidad de Talca for their Doctoral scholarships. Authors declare no competing interests. PR acknowledges ‘Núcleo Científico Multidisciplinario’ from Universidad de Talca. NC acknowledges Dr. Ricardo Pérez, for his collaboration and support in the generation of transgenic plants. We thank Michael Handford (Universidad de Chile) for language support.
Author contributions
R.H., N.C., M.A.M.-L. and R.G. conceived and designed the experiments. N.C., T.M., A.V., D.U., T.M. and P.R. performed the experiments. The manuscript was written by N.C., T.M. and R.H. and critically reviewed by M.A.M.-L. and R.G. Each author participated sufficiently in the work to take public responsibility for appropriate portions of the content. All authors read, edited and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Nicolás Cruz and Tamara Méndez.
Supplementary information
is available for this paper at 10.1038/s41598-019-55276-7.
References
- 1.Allona I, et al. Analysis of xylem formation in pine by cDNA sequencing. Proceedings of the National Academy of Sciences USA. 1998;95:9693–9698. doi: 10.1073/pnas.95.16.9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hertzberg M, et al. A transcriptional roadmap to wood formation. Proceedings of the National Academy of Sciences USA. 2001;98:14732–14737. doi: 10.1073/pnas.261293398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whetten R, Sun Y, Zhang Y, Sederoff R. Functional genomics and cell wall biosynthesis in loblolly pine. Plant Molecular Biology. 2001;47:275–291. doi: 10.1023/A:1010652003395. [DOI] [PubMed] [Google Scholar]
- 4.Andersson-Gunneras S, et al. Asymmetric expression of a poplar ACC oxidase controls ethylene production during gravitational induction of tension wood. The Plant Journal. 2003;34:339–349. doi: 10.1046/j.1365-313X.2003.01727.x. [DOI] [PubMed] [Google Scholar]
- 5.Demura T, Fukuda H. Transcriptional regulation in wood formation. Trends in Plant Sciences. 2007;12:64–70. doi: 10.1016/j.tplants.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Pavy N, et al. Identification of conserved core xylem gene sets: conifer cDNA microarray development, transcript profiling and computational analyses. New Phytologist. 2008;180:766–786. doi: 10.1111/j.1469-8137.2008.02615.x. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Wu HX, Dillon SK, Southerton SG. Generation and analysis of expressed sequence tags from six developing xylem libraries in Pinus radiata D. Don. BMC Genomics. 2009;10:41. doi: 10.1186/1471-2164-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong R, Richardson EA, Ye ZH. The MYB46 transcription factor is a direct target of SND1 and regulates secondary wall biosynthesis in Arabidopsis. The Plant Cell. 2007;19:2776–2792. doi: 10.1105/tpc.107.053678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong R, Ye ZH. Transcriptional regulation of lignin biosynthesis. Plant Signaling and Behavior. 2009;4:1028–1034. doi: 10.4161/psb.4.11.9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong R, Lee C, Ye ZH. Evolutionary conservation of the transcriptional network regulating secondary cell wall biosynthesis. Trends in Plant Science. 2010;15:625–632. doi: 10.1016/j.tplants.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Q, Dixon RA. Transcriptional networks for lignin biosynthesis: more complex than we thought? Trends in Plant Science. 2011;16:227–233. doi: 10.1016/j.tplants.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Grima-Pettenati, J., Soler, M., Camargo, E. & Wang, H. Transcriptional Regulation of the Lignin Biosynthetic Pathway Revisited: New Players and Insights. In Advances in Botanical Research. (Jouanin, L. and Lapierre, C., editors) p 173–218 (2012).
- 13.Wang H, Dixon R. On-off switches for secondary cell wall biosynthesis. Molecular Plant. 2012;5:297–303. doi: 10.1093/mp/ssr098. [DOI] [PubMed] [Google Scholar]
- 14.Legacy S, et al. EgMYB1, an R2R3 MYB transcription factor from eucalyptus negatively regulates secondary cell wall formation in Arabidopsis and poplar. The New Phytologist. 2010;188:774–786. doi: 10.1111/j.1469-8137.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- 15.Goicoechea M, et al. EgMYB2, a new transcriptional activator from Eucalyptus xylem, regulates secondary cell wall formation and lignin biosynthesis. The Plant Journal. 2005;43:553–567. doi: 10.1111/j.1365-313X.2005.02480.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhong R, Demura T, Ye ZH. SND1, a NAC domain transcription factor, is a key regulator of secondary wall synthesis in fibers of Arabidopsis. The Plant Cell. 2006;18:3158–3170. doi: 10.1105/tpc.106.047399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarthy RL, et al. The poplar MYB transcription factors, PtrMYB3 and PtrMYB20, are involved in the regulation of secondary wall biosynthesis. Plant Cell Physiology. 2010;51:1084–1090. doi: 10.1093/pcp/pcq064. [DOI] [PubMed] [Google Scholar]
- 18.Zhong R, Lee C, Ye ZH. Functional characterization of poplar wood-associated NAC domain transcription factors. Plant Physiology. 2010;152:1044–1055. doi: 10.1104/pp.109.148270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohtani M, et al. A NAC domain protein family contributing to the regulation of wood formation in poplar. The Plant Journal. 2011;67:499–512. doi: 10.1111/j.1365-313X.2011.04614.x. [DOI] [PubMed] [Google Scholar]
- 20.Bomal C, et al. Involvement of Pinus taeda MYB1 and MYB8 in phenylpropanoid metabolism and secondary cell wall biogenesis: a comparative in planta analysis. Journal of Experimental Botany. 2008;59:3925–3939. doi: 10.1093/jxb/ern234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou J, Lee C, Zhong R, Ye ZH. MYB58 and MYB63 are transcriptional activators of the lignin biosynthetic pathway during secondary cell wall formation in Arabidopsis. The Plant Cell. 2009;21:248–266. doi: 10.1105/tpc.108.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant EH, Fujino T, Beers EP, Brunner AM. Characterization of NAC domain transcription factors implicated in control of vascular cell differentiation in Arabidopsis and Populus. Planta. 2010;232:337–352. doi: 10.1007/s00425-010-1181-2. [DOI] [PubMed] [Google Scholar]
- 23.Zhao C, Avci U, Grant EH, Haigler CH, Beers EP. XND1, a member of the NAC domain family in Arabidopsis thaliana, negatively regulates lignocellulose synthesis and programmed cell death in xylem. Plant Journal. 2008;53:425–436. doi: 10.1111/j.1365-313X.2007.03350.x. [DOI] [PubMed] [Google Scholar]
- 24.Hussey SG, et al. SND2, a NAC transcription factor gene, regulates genes involved in secondary cell wall development in Arabidopsis fibres and increases fibre cell area in Eucalyptus. BMC Plant Biology. 2011;11:173. doi: 10.1186/1471-2229-11-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong R, Lee C, Zhou J, McCarthy RL, Ye ZH. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. The Plant Cell. 2008;20:2763–2782. doi: 10.1105/tpc.108.061325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li E, et al. The Class II KNOX gene KNAT7 negatively regulates secondary wall formation in Arabidopsis and is functionally conserved in Populus. The New Phytologist. 2012;194:102–115. doi: 10.1111/j.1469-8137.2011.04016.x. [DOI] [PubMed] [Google Scholar]
- 27.Kawaoka A, Ebinuma H. Transcriptional control of lignin biosynthesis by tobacco LIM protein. Phytochemistry. 2001;57:1149–1157. doi: 10.1016/S0031-9422(01)00054-1. [DOI] [PubMed] [Google Scholar]
- 28.Kawaoka A, Nanto K, Ishii K, Ebinuma H. Reduction of lignin content by suppression of expression of the LIM domain transcription factor in Eucalyptus camaldulensis. Silvae Genetica. 2007;6:269–277. [Google Scholar]
- 29.Ramos P, Le Provost G, Plomion C, Gantz C, Herrera R. Transcriptional analysis of differential expressed genes in response to stem inclination in young seedlings of pine. Plant Biology. 2012;14:923–933. doi: 10.1111/j.1438-8677.2012.00572.x. [DOI] [PubMed] [Google Scholar]
- 30.Messenguy F, Dubois E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene. 2003;316:1–21. doi: 10.1016/S0378-1119(03)00747-9. [DOI] [PubMed] [Google Scholar]
- 31.Alvarez-Buylla ER, et al. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc Natl Acad Sci USA. 2000;97:5328–5333. doi: 10.1073/pnas.97.10.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Bodt S, Raes J, Van de Peer Y, Theißen G. 2003. And then there were many: MADS goes genomic. Trends in Plant Sciences. 2016;8:475–483. doi: 10.1016/j.tplants.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Aswath C, Kim S. Another story of MADS-Box genes – their potential in plant biotechnology. Plant Growth Regulation. 2005;46:177–188. doi: 10.1007/s10725-005-8482-7. [DOI] [Google Scholar]
- 34.Egea-Cortines M, Saedler H, Sommer H. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. The EMBO Journal. 1999;18:5370–5379. doi: 10.1093/emboj/18.19.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honma T, Goto K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature. 2001;409:525–529. doi: 10.1038/35054083. [DOI] [PubMed] [Google Scholar]
- 36.Theissen G, Saedler H. Plant biology. Floral quartets. Nature. 2001;409:469–471. doi: 10.1038/35054172. [DOI] [PubMed] [Google Scholar]
- 37.Vrebalov J, et al. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (Rin) locus. Science. 2002;296:343–346. doi: 10.1126/science.1068181. [DOI] [PubMed] [Google Scholar]
- 38.Manning K, et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nature Genetics. 2006;38:948–952. doi: 10.1038/ng1841. [DOI] [PubMed] [Google Scholar]
- 39.Nesi N, Jond C, Debeaujon I, Caboche M, Lepiniec L. The Arabidopsis TT2 gene encodes an R2R3 MYB domain protein that acts as a key determinant for proanthocyanidin accumulation in developing seed. The Plant Cell. 2001;13:2099–2114. doi: 10.1105/TPC.010098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lalusin A, Nishita K, Kim S, Ohta M, Fujimura T. A new MADS-box gene (IbMADS10) from sweet potato (Ipomoea batatas (L.) Lam) is involved in the accumulation of anthocyanin. Molecular Genetics and Genomics. 2006;275:44–54. doi: 10.1007/s00438-005-0080-x. [DOI] [PubMed] [Google Scholar]
- 41.Jaakola L, et al. A SQUAMOSA MADS box gene involved in the regulation of anthocyanin accumulation in bilberry fruits. Plant Physiology. 2010;153:1619–1629. doi: 10.1104/pp.110.158279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riechmann JL, Krizek BA, Meyerowitz EM. Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS6. Proceedings of the National Academy of Sciences USA. 1996;93:4793–4798. doi: 10.1073/pnas.93.10.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shore P, Sharrocks A. The MADS-box family of transcription factors. European Journal of Biochemistry. 1995;229:1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x. [DOI] [PubMed] [Google Scholar]
- 44.Cseke LJ, Zheng J, Podila GK. Characterization of PTM5 in aspen trees: a MADS-box gene expressed during woody vascular development. Gene. 2003;318:55–67. doi: 10.1016/S0378-1119(03)00765-0. [DOI] [PubMed] [Google Scholar]
- 45.Dias DO, et al. Unravelling MADS-box gene family in Eucalyptus spp.: A starting point to an understanding of their developmental role in trees. Genetics and Molecular Biology. 2005;18(28):501–510. doi: 10.1590/S1415-47572005000400004. [DOI] [Google Scholar]
- 46.Walden A, Wang D, Walter C, Gardner R. A large family of TM3 MADS-box cDNAs in Pinus radiata includes two members with deletions of the conserved K domain. Plant Science. 1998;138:167–176. doi: 10.1016/S0168-9452(98)00160-5. [DOI] [Google Scholar]
- 47.Mouradov A, et al. Family of MADS-box genes expressed early in male and female reproductive structures of Monterey pine. Plant Physiology. 1998;117:55–61. doi: 10.1104/pp.117.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mouradov A, et al. DEF/GLO-like MADS-Box Gene From a Gymnosperm: Pinus radiata Contains an Ortholog of Angiosperm B Class Floral Homeotic Genes. Developmental Genetics. 1999;25:245–6 252. doi: 10.1002/(SICI)1520-6408(1999)25:3<245::AID-DVG7>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 49.Mouradov A, Glassick T, Hamdorf B, Teasdale R. Molecular control of early cone development in Pinus radiata. Protoplasma. 1999;208:3–12. doi: 10.1007/BF01279069. [DOI] [Google Scholar]
- 50.Cseke L, et al. SEP-class genes in P. tremuloides and their likely role in reproductive survival of poplar trees. Gene. 2005;358:1–16. doi: 10.1016/j.gene.2005.05.035. [DOI] [PubMed] [Google Scholar]
- 51.Cseke L, Cseke S, Ravinder N, Pandey A, Podila G. Identification of PTM5 protein interaction partners, a MADS-box gene involved in aspen tree vegetative development. Gene. 2007;391:209–222. doi: 10.1016/j.gene.2006.12.033. [DOI] [PubMed] [Google Scholar]
- 52.Liljegren SJ, et al. SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature. 2000;404:766–770. doi: 10.1038/35008089. [DOI] [PubMed] [Google Scholar]
- 53.Thimm O, et al. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. The Plant Journal. 2004;37:914–939. doi: 10.1111/j.1365-313X.2004.02016.x. [DOI] [PubMed] [Google Scholar]
- 54.Usadel B, et al. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiology. 2005;138:1195–1204. doi: 10.1104/pp.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matsuo Kouki, Fukuzawa Noriho, Matsumura Takeshi. A simple agroinfiltration method for transient gene expression in plant leaf discs. Journal of Bioscience and Bioengineering. 2016;122(3):351–356. doi: 10.1016/j.jbiosc.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 56.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: A Tool For Discovery And Visualization of Enriched GO Terms in Ranked Gene Lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang B, Su X, Zhou X. A MADS-box gene of Populus deltoides expressed during flower development and in vegetative organs. Tree Physiology. 2008;28:929–934. doi: 10.1093/treephys/28.6.929. [DOI] [PubMed] [Google Scholar]
- 58.Kaufmann K, Melzer R, Theißen G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene. 2005;347:183–198. doi: 10.1016/j.gene.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 59.Gramzow L, Ritz MS, Theissen G. On the origin of MADS-domain transcription factors. Trends in Genetics. 2010;26:149–153. doi: 10.1016/j.tig.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 60.Smaczniak C, Immink R, Angenent G, Kaufmann K. Developmental and evolutionary diversity of plant MADS-domain factors: insight from recent studies. Development. 2012;139:3081–3098. doi: 10.1242/dev.074674. [DOI] [PubMed] [Google Scholar]
- 61.Tandre K, Svenson M, Svensson M, Engström P. Conservation of gene structure and activity in the regulation of reproductive organ development of conifers and angiosperms. The Plant Journal. 1998;15:615–623. doi: 10.1046/j.1365-313x.1998.00236.x. [DOI] [PubMed] [Google Scholar]
- 62.Silva C, et al. Evolution of the Plant Reproduction Master Regulators LFY and the MADS Transcription Factors: The Role of Protein Structure in the Evolutionary Development of the Flower. Frontiers in Plant Science. 2015;6:1193. doi: 10.3389/fpls.2015.01193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu C, et al. Direct interaction of AGL24 and SOC1 integrates flowering signals in Arabidopsis. Development. 2008;135(11):1481–1491. doi: 10.1242/dev.020255. [DOI] [PubMed] [Google Scholar]
- 64.Kim. S-H, Mizuno K, Fujimura T. Isolation of MADS-box Genes from Sweet Potato (Ipomoaea batatas (L.) Lam.) Expressed Specifically in Vegetative Tissues. Plant Cell Physiol. 2002;43(3):314–322. doi: 10.1093/pcp/pcf043. [DOI] [PubMed] [Google Scholar]
- 65.Rutledge R, et al. Characterization of an AGAMOUS homologue from the conifer black spruce (Picea mariana) that produces floral homeotic conversions when expressed in Arabidopsis. The Plant Journal. 1998;15:625–634. doi: 10.1046/j.1365-313x.1998.00250.x. [DOI] [PubMed] [Google Scholar]
- 66.An X, et al. Ectopic expression of a poplar APETALA3-like gene in tobacco causes early flowering and fast growth. Biotechnology Letters. 2011;33:1239–1247. doi: 10.1007/s10529-011-0545-4. [DOI] [PubMed] [Google Scholar]
- 67.Dai, X., Wang, Y. & Zhang, W.H. OsWRKY74, a WRKY transcription factor, modulates tolerance to phosphate starvation in rice. Journal of Experimental Botany67, 947–960. [DOI] [PMC free article] [PubMed]
- 68.Baek D, Chun HJ, Yun DJ, Kim MC. Cross-talk between phosphate starvation and other environmental stress signaling pathways in plants. Molecules and Cells. 2017;40:13 697–705. doi: 10.14348/molcells.2017.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gu M, et al. Maintenance of phosphate homeostasis and root development are coordinately regulated by MYB1, an R2R3-tye MYB transcription factor in rice. Journal of Experimental Botany. 2017;68:360–3615. doi: 10.1093/jxb/erx174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pal S, et al. Transdetect identifies a new regulatory module controlling phosphate accumulation. Plant Physiology. 2017;175:916–926. doi: 10.1104/pp.17.00568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baroja E, et al. Sucrose synthase activity in the sus1/sus2/sus3/sus4 Arabidopsis mutant is sufficient to support normal cellulose and starch production. Proceedings of the National Academy of Sciences USA. 2012;109:321–326. doi: 10.1073/pnas.1117099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Madduri K, Waldron C, Merlo D. Rhamnose Biosynthesis Pathway Supplies Precursors for Primary and Secondary Metabolism in Saccharopolyspora spinosa. Journal of Bacteriology. 2001;183:5632–5638. doi: 10.1128/JB.183.19.5632-5638.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Q, Bleecker A. Analysis of Ethylene Signal-Transduction Kinetics Associated with Seedling-Growth Response and Chitinase lnduction in Wild-Type and Mutant Arabidopsis. Plant Physiology. 1995;108:597–607. doi: 10.1104/pp.108.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pesquet E, Korolev A, Calder G, Lloyd C. The microtubule-associated protein AtMAP70-5 regulates secondary wall patterning in Arabidopsis wood cells. Current Biology. 2010;30:744–749. doi: 10.1016/j.cub.2010.02.057. [DOI] [PubMed] [Google Scholar]
- 75.Patzlaff A, et al. Characterisation of a pine MYB that regulates lignification. The Plant Journal. 2003;36:743–754. doi: 10.1046/j.1365-313X.2003.01916.x. [DOI] [PubMed] [Google Scholar]
- 76.Patzlaff A, et al. Characterisation of PtMYB1, an R2R3-MYB from pine xylem. Plant Molecular Biology. 2003;53:597–608. doi: 10.1023/B:PLAN.0000019066.07933.d6. [DOI] [PubMed] [Google Scholar]
- 77.Zhao K, Bartley L. Comparative genomic analysis of the R2R3 MYB secondary cell wall regulators of Arabidopsis, poplar, rice, maize, and switchgrass. BMC Plant Biology. 2014;14:135. doi: 10.1186/1471-2229-14-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tang X, et al. Poplar PdMYB221 is involved in the direct and indirect regulation of secondary wall biosynthesis during wood formation. Scientific Report. 2015;5:12240. doi: 10.1038/srep12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Minic Z, et al. Transcriptomic analysis of Arabidopsis developing stems: a close-up on cell wall genes. BMC Plant Biology. 2009;9:6. doi: 10.1186/1471-2229-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Salazar MM, et al. Xylem transcription profiles indicate potential metabolic 22 responses for economically relevant characteristics of Eucalyptus species. BMC 23 Genomics. 2013;14:201. doi: 10.1186/1471-2164-14-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tian Q, et al. Functional characterization of the poplar R2R3-MYB transcription factor PtoMYB216 involved in the regulation of lignin biosynthesis during wood formation. PLoS One. 2013;8:e76369. doi: 10.1371/journal.pone.0076369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang L, et al. PtoMYB156 is involved in negative regulation of phenylpropanoid metabolism and secondary cell wall biosynthesis during wood formation in poplar. Scientific Report. 2017;7:41209. doi: 10.1038/srep41209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lamara M, et al. Genetic architecture of wood properties based on association analysis and co-expression networks in white spruce. The New Phytologist. 2016;210:240–255. doi: 10.1111/nph.13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pascual M, et al. PpNAC1, a main regulator of the phenilalanine biosynthesis and utilization in maritime pine. Plant Biotechnol J. 2018;16:1094–1104. doi: 10.1111/pbi.12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gomez R, Gonzalez J, Herrera R, Ramos P. MYB Transcription Factors and a Putative Flavonoid Transporter ABCC-Like are Differentially Expressed in Radiata Pine Seedlings Exposed to Inclination. Journal of Plant Growth Regululation. 2018;37:64–3 75. doi: 10.1007/s00344-017-9707-5. [DOI] [Google Scholar]
- 86.Li X, Yang X, Wu H. Transcriptome profiling of radiate pine branches reveals new insights into reaction wood formation with implications in plant gravitropism. BMC Genomics. 2013;14:768. doi: 10.1186/1471-2164-14-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang S, Zyl L, No E, Loopstra C. Microarray analysis of genes preferentially expressed in differentiating xylem of loblolly pine (Pinus taeda) Plant Science. 2004;166:1185–1195. doi: 10.1016/j.plantsci.2003.12.030. [DOI] [Google Scholar]
- 88.Anterola AM, Lewis NG. Trends in lignin modification: a comprehensive analysis of the effects of genetic manipulation/mutations on lignification and vascular integrity. Phytochemistry. 2002;61:221–294. doi: 10.1016/S0031-9422(02)00211-X. [DOI] [PubMed] [Google Scholar]
- 89.Zhang J, Elo A, Helariutta Y. Arabidopsis as a model for wood formation. Current Opinion in Biotechnology. 2010;22:1–7. doi: 10.1016/j.copbio.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 90.Ramos P, Herrera R. Anatomical changes of xylem cells in stem of Pinus radiata seedlings exposed to inclination and ethylene. Biologia Plantarum. 2013;57:525–530. doi: 10.1007/s10535-013-0321-5. [DOI] [Google Scholar]
- 91.Ramos P, et al. ACC oxidase and ACC synthase expression profiles after leaning of young radiata (P. radiata D. Don) and maritime pine (P. pinaster Ait.) seedlings. Journal of Plant Growth Regulation. 2012;31:382–391. doi: 10.1007/s00344-011-9248-2. [DOI] [Google Scholar]
- 92.Clough S, Bent A. Floral dip: a simplified method for Agrobacterium mediated transformation of Arabidopsis thaliana. Plant Journal. 1998;16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- 93.Le Provost G, et al. A micromethod for high throughput RNA extraction in forest trees. Biological Research. 2007;40:291–21 297. doi: 10.4067/S0716-97602007000400003. [DOI] [PubMed] [Google Scholar]
- 94.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucl Acids Research. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Remans T, et al. Normalisation of real-time RT-PCR gene expression measurements in Arabidopsis thaliana exposed to increased metal concentrations. Planta. 2008;227:1343–1349. doi: 10.1007/s00425-008-0706-4. [DOI] [PubMed] [Google Scholar]
- 96.Vandesompele J, et al. Accurate normalization of realtime quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology. 2002;3:1–11. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Irizarry RA, et al. Summaries of Affymetrix GeneChip probe-level data. Nucleic Acids Research. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hong FX, et al. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006;22:2825–2827. doi: 10.1093/bioinformatics/btl476. [DOI] [PubMed] [Google Scholar]
- 99.Jeffrey IB, Higgins DG, Culhane AE. Comparison and evaluation of methods for generating differentially expressed gene lists from microarray data. BMC Bioinformatics. 2006;7:359. doi: 10.1186/1471-2105-7-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koziol J. The rank product method with two samples. FEBS Lett. 2010;584:4481–4484. doi: 10.1016/j.febslet.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 102.Campain A, Yang YH. Comparison study of microarray meta-analysis methods. BMC Bioinformatics. 2010;11:408. doi: 10.1186/1471-2105-11-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Teixeira PJPL, et al. High-Resolution Transcript Profiling of the Atypical Biotrophic Interaction between Theobroma cacao and the Fungal Pathogen Moniliophthora perniciosa. The Plant Cell. 2014;26:4245–4269. doi: 10.1105/tpc.114.130807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Szklarczyk D, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Research. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Szklarczyk D, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Research. 2017;45:D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Szklarczyk D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. Jan. 2019;47:D607–613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bieza K, Lois R. An Arabidopsis mutant tolerant to lethal ultraviolet-B levels shows constitutively elevated accumulation of flavonoids and other phenolics. Plant physiology. 2001;126:1105–1115. doi: 10.1104/pp.126.3.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mancinelli AL, Hoff AM, Cottrell M. Anthocyanin production in Chl-rich and Chl poor seedlings. Plant Physiology. 1988;86:652–654. doi: 10.1104/pp.86.3.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Campbell MM, Ellis BE. Fungal elicitor-mediated response in pine cell cultures. Induction of phenylpropanoid metabolism. Planta. 1992;186:409–417. doi: 10.1007/BF00195322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.