

Abstract
Islet amyloid is a pathologic feature of type 2 diabetes (T2D) that is associated with β-cell loss and dysfunction. These amyloid deposits form via aggregation of the β-cell secretory product islet amyloid polypeptide (IAPP) and contain other molecules including the heparan sulfate proteoglycan perlecan. Perlecan has been shown to bind amyloidogenic human IAPP (hIAPP) via its heparan sulfate glycosaminoglycan (HS GAG) chains and to enhance hIAPP aggregation in vitro. We postulated that reducing the HS GAG content of perlecan would also decrease islet amyloid deposition in vivo. hIAPP transgenic mice were crossed with Hspg2Δ3/Δ3 mice harboring a perlecan mutation that prevents HS GAG attachment (hIAPP;Hspg2Δ3/Δ3), and male offspring from this cross were fed a high fat diet for 12 months to induce islet amyloid deposition. At the end of the study body weight, islet amyloid area, β-cell area, glucose tolerance and insulin secretion were analyzed. hIAPP;Hspg2Δ3/Δ3 mice exhibited significantly less islet amyloid deposition and greater β-cell area compared to hIAPP mice expressing wild type perlecan. hIAPP;Hspg2Δ3/Δ3 mice also gained significantly less weight than other genotypes. When adjusted for differences in body weight using multiple linear regression modeling, we found no differences in islet amyloid deposition or β-cell area between hIAPP transgenic and hIAPP;Hspg2Δ3/Δ3 mice. We conclude that loss of perlecan exon 3 reduces islet amyloid deposition in vivo through indirect effects on body weight and possibly also through direct effects on hIAPP aggregation. Both of these mechanisms may promote maintenance of glucose homeostasis in the setting of T2D.
Keywords: perlecan, heparan sulfate proteoglycan, islet amyloid, type 2 diabetes
Introduction
Islet amyloid deposits are pathologic protein aggregates found in the majority of individuals with type 2 diabetes (T2D) (Clark et al., 1988; Westermark, 1972; Hull et al., 2004). In humans, the degree of islet amyloid deposition is directly correlated with the rate of β-cell apoptosis and inversely correlated with β-cell mass (Jurgens et al., 2011). Islet amyloid polypeptide (IAPP, also known as amylin), a normal β-cell secretory product (Kahn et al., 1990), is a unique protein constituent of islet amyloid deposits (Westermark et al., 1987; Cooper et al., 1987). Human IAPP (hIAPP) and rodent IAPP (rIAPP) differ in a few critical amino acid residues, which results in hIAPP being amyloidogenic and cytotoxic, while rIAPP is not (Westermark et al., 1990; Lorenzo et al., 1994). Thus, hIAPP transgenic mice have been developed to facilitate in vitro and in vivo studies of cytotoxic hIAPP aggregation and β-cell loss (D’Alessio et al., 1994; Matveyenko and Butler, 2006). Numerous studies have examined approaches to decrease hIAPP aggregation or increase its clearance in order to reduce hIAPP-induced β-cell cytotoxicity in T2D (Meng et al., 2010; Zraika et al., 2010; Hopping et al., 2014; Oskarsson et al., 2018).
Relatively few studies have focused on the non-hIAPP components of islet amyloid deposits, such as serum amyloid P (Pepys et al., 1994), apolipoprotein E (Chargé et al., 1996; Vidal et al., 2003) and the heparan sulfate proteoglycan (HSPG) perlecan (Young et al., 1992). However, reducing the level of these molecules may be another approach to lessen the degree of amyloid formation and decrease the severity of this pathology. Although other HSPG molecules may play a role, perlecan is well-placed to participate in the process of islet amyloid formation in vivo as it is localized to the peri-capillary extracellular matrix in islets (Cross et al., 2017; Irving-Rodgers et al., 2008), and this is the site of islet amyloid deposition (Hull et al., 2004; Verchere et al., 1996; de Koning et al., 1994). In vitro studies have shown perlecan binds amyloidogenic hIAPP, but not non-amyloidogenic rIAPP (Castillo et al., 1998; Potter-Perigo et al., 2003). This high affinity binding was found to occur through interaction of hIAPP with heparan sulfate glycosaminoglycan (HS GAG) chains attached to the perlecan core protein (Castillo et al., 1998; Potter-Perigo et al., 2003; Abedini et al., 2006; Hull et al., 2012) and to result in increased amyloid formation (Castillo et al., 1998; Abedini et al., 2006; Hull et al., 2012). Further, heparin, a naturally occurring glycosaminoglycan, has been shown to increase hIAPP aggregation in cell-free systems and to increase amyloid deposition in hIAPP expressing islets (Potter et al., 2015). Conversely, heparinase treatment, which reduces HS content (Potter et al., 2015), and interventions that block HS synthesis (Hull et al., 2007; Oskarsson et al., 2015) or increase HS GAG chain degradation (Oskarsson et al., 2015) have been shown to decrease islet amyloid formation in cultured islets. Additionally, it has been shown that HS deficient cells are protected from hIAPP-induced cell death (Oskarsson et al., 2015). However, no studies to date have determined whether decreased expression of perlecan HS GAGs can reduce islet amyloid deposition in vivo.
Several mice with deletion mutations at different sites in the perlecan gene (Hspg2) have been developed. Deletion of either exon 6 or exon 7 prevents perlecan core protein formation, and mice homozygous for either of these mutations demonstrate embryonic or early neonatal lethality (Arikawa-Hirasawa et al., 1999; Costell et al., 1999), consistent with the critical role of perlecan during embryogenesis and early development. Another mouse model was developed that lacks exon 3 of Hspg2 (Hspg2Δ3/Δ3), resulting in production of a nearly full-length perlecan core protein deficient in HS GAG attachment sites and loss of HS GAG chains on the perlecan core protein (Rossi et al., 2003). In contrast to the other models, homozygous Hspg2Δ3/Δ3 mice survive to adulthood and have no major developmental abnormalities aside from mild ophthalmic deformities and some changes in their skin and cartilage (Walz et al., 1997; Rossi et al., 2003; Shu et al., 2016). With the knowledge that perlecan HS GAGs enhance hIAPP fibril formation in vitro (Castillo et al., 1998;Abedini et al., 2006), and that they are a component of islet amyloid in vivo (Young et al., 1992;Kahn et al., 1999), we generated mice with β-cell hIAPP expression and the Hspg2Δ3/Δ3 mutation (hIAPP;Hspg2Δ3/Δ3) to examine for the first time whether loss of perlecan HS GAGs reduces islet amyloid formation and β-cell loss in vivo.
Materials and Methods
Animals
Homozygous Hspg2Δ3/Δ3 mice on a C57BL/6 background were obtained from Dr. Karl Tryggvason (Karolinska Institutet, Stockholm) and bred with hemizygous hIAPP mice (hIAPP+/0) on a DBA/2 background at the VA Puget Sound Health Care System (VAPSHCS). hIAPP transgene expression was driven by the rat insulin II promoter to express hIAPP specifically in β cells. This initial cross generated heterozygous Hspg2+/Δ3 mice with and without the hIAPP transgene. The hIAPP+/0;Hspg2+/Δ3 animals were then crossed to produce siblings harboring the four study genotypes: wild type, WT (hIAPP0/0; Hspg2+/+, n = 12), Hspg2Δ3/Δ3 (hIAPP0/0;Hspg2Δ3/Δ3, n = 10), hIAPP (hIAPP+/0;Hspg2+/+, n = 10) and hIAPP;Hspg2Δ3/Δ3 (hIAPP+/0;Hspg2Δ3/Δ3, n = 6). All mice underwent pancreas histology evaluation, glucose tolerance testing and body weight determination. Mice were housed and studied at VAPSHCS following approval by the Institutional Animal Care and Use Committee.
Genotyping
Presence of the hIAPP transgene was determined by polymerase chain reaction of tail DNA using specific primers as previously described (Andrikopoulos et al., 2000). For Hspg2 Δ3 mice, the wild type Hspg2 gene was amplified with a 5′-GGG GAC ACT TGT CAT CCT CT-3′ sense primer and an 5′-GCC GAG GCC ATC TGC AAG AA-3′ antisense primer. The mutant Hspg2 Δ3 allele was amplified with a 5′-TGT CAT CTC ACC TTG CTC CTG-3′ sense primer and a 5′-TCA AGA AGG CGA TAG AAG GCG-3′ antisense primer.
In vivo studies
We previously showed that 81% of male hIAPP transgenic mice develop amyloid in vivo compared to only 11% of females (Verchere et al., 1996). Therefore, only male mice were used in this study. At 8–10 weeks of age, male mice from all four study genotypes were placed on a high fat diet (HFD) containing 45% of calories from fat, 35% from carbohydrates and 20% from protein (D12290; Research Diets, New Brunswick, NJ). Mice were fed HFD for 1 year to induce amyloid formation in vivo as we have done previously (Hull et al., 2003). Body weight was determined at the beginning and the end of the study.
IPGTT, glucose and insulin assays
After 12 months of HFD feeding, mice were fasted overnight and then anesthetized with sodium pentobarbital. A baseline blood sample was taken via the retro-orbital sinus and an intraperitoneal glucose bolus (1 g/kg body weight) was administered. Blood samples were then drawn at 5, 15, 30, 60, 90 and 120 min after glucose administration. Centrifugation for plasma separation was then performed and samples were stored at −20°C for subsequent analysis.
Plasma glucose concentration was determined using a plate based colorimetric assay utilizing the glucose oxidase method, and plasma insulin was measured using an ultrasensitive mouse insulin enzyme-linked immunosorbent assay (Alpco, Salem, NH) (Aston-Mourney et al., 2013).
Histology
Pancreata were fixed in 4% (wt/vol) phosphate-buffered paraformaldehyde, processed and embedded in paraffin. Four micrometer pancreas sections were cut and stained with insulin antibody (I-2018, 1:2000, Sigma-Aldrich, St. Louis, MO) followed by Cy3-conjugated anti-mouse IgG to visualize β cells, and thioflavin S (T-1892, diluted 0.5% wt/vol; Sigma-Aldrich, St. Louis, MO) counterstain to visualize amyloid deposits (Hull et al., 2003; Aston-Mourney et al., 2013; Wang et al., 2001). Perlecan was visualized with an antibody against perlecan core protein (ab2501, 1:150, Abcam, Cambridge, MA) followed by Cy3-conjugated anti-rat IgG. Histological assessment was performed using an epifluorescence microscope (Eclipse NiE or Eclipse E800; Nikon, Japan), and images were analyzed using computer-based quantitative imaging software (NIS Elements, Nikon). Islet area, thioflavin S-positive (amyloid) and insulin-positive (β cell) areas were determined as described previously (Hull et al., 2003; Aston-Mourney et al., 2013; Wang et al., 2001). The number of islets analyzed for each group is as follows [mean ± SEM, range]: WT, 38.1 ± 3.3, 24–59; Hspg2Δ3/Δ3, 46.8 ± 2.2, 34–58; hIAPP, 40.7 ± 5.9, 23–71; hIAPP;Hspg2Δ3/Δ3, 38.7 ± 6.1, 23–61.
Calculations and statistical analysis
Amyloid prevalence was defined as the proportion of islets per mouse containing amyloid. Amyloid severity was defined as (Σ amyloid area/Σ islet area) × 100% for all islets from each mouse (Hull et al., 2003; Aston-Mourney et al., 2013; Wang et al., 2001). β-cell area was calculated as (Σ β-cell area/Σ islet area) × 100% for each mouse. Plasma glucose and insulin concentrations during the intraperitoneal glucose tolerance test (IPGTT) were analyzed by general linear model. β-cell secretory function was assessed by calculating the change in insulin from time 0 to 30 min, divided by the change in glucose from time 0 to 30 min. Comparisons of changes between groups were performed by Student’s t-test for parametric data (body weight), or by Mann–Whitney U test for non-parametric data (amyloid deposition, β-cell area, plasma glucose and plasma insulin), and Bonferroni post-test was applied where appropriate. Multiple linear regression was performed to assess whether changes in the variables of interest (amyloid prevalence, amyloid severity and β-cell area) were related to the observed changes in body weight. Square root transformation was performed to satisfy normality assumptions for linear regression. Data are presented as mean ± SEM with P ≤ 0.05 was considered significant.
Results
Perlecan is a component of islet amyloid deposits in hIAPP transgenic mice
To confirm that perlecan is a component of islet amyloid deposits in hIAPP transgenic mice in vivo, as has been shown in other models, we analyzed islet amyloid deposits in these mice following 1 year of HFD feeding (Fig. 1a). Islet perlecan staining (Fig. 1b) was found to colocalize with amyloid staining (Fig. 1c) in hIAPP mouse islets, again suggesting a role for perlecan HS GAGs in hIAPP aggregation and islet amyloid deposition in vivo.
Fig. 1.
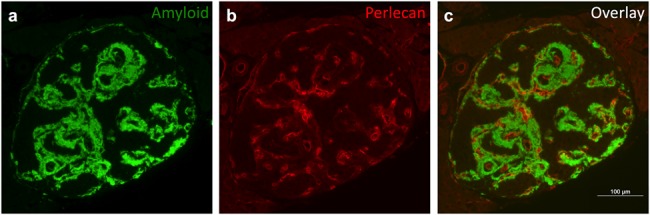
Perlecan colocalizes with islet amyloid deposits in hIAPP transgenic mice a) islet amyloid deposits were visualized in hIAPP transgenic mouse pancreata by thioflavin S staining (green) following 1 year of HFD feeding. b) Perlecan immunostaining (red) is present in the islet peri-capillary space, and c) colocalizes with amyloid deposits in hIAPP transgenic mouse islets. Representative images are shown. Scale bar equals 100 μm
Loss of perlecan heparan sulfate glycosaminoglycans reduces islet amyloid deposition and ameliorates amyloid-induced β-cell loss
In total, 8 out of 10 male hIAPP mice developed islet amyloid after 1 year of HFD feeding, compared with only two out of six hIAPP;Hspg2Δ3/Δ3 mice (Fig. 2a). As expected, WT and Hspg2Δ3/Δ3 mice did not develop islet amyloid since they express mouse IAPP that is not amyloidogenic, with hIAPP mice exhibiting significantly higher islet amyloid prevalence (Fig. 2b) and amyloid severity (Fig. 2c) than WT mice. In contrast, hIAPP;Hspg2Δ3/Δ3 mice lacking perlecan HS GAGs exhibited significantly lower islet amyloid prevalence (Fig. 2b) and amyloid severity (Fig. 2c) compared to hIAPP mice. Furthermore, β-cell area (Fig. 2d) was decreased in islet amyloid-laden hIAPP compared to WT mice, and this reduction in β-cell area was rescued in hIAPP;Hspg2Δ3/Δ3 mice. In WT and Hspg2Δ3/Δ3 mice lacking hIAPP expression, β-cell area was not different, indicating that Hspg2Δ3/Δ3deletion alone does not affect β-cell area (Fig. 2d). Representative immunohistochemistry images illustrate the amyloid deposition and reduced β-cell area observed in hIAPP islets (Fig. 2e).
Fig. 2.
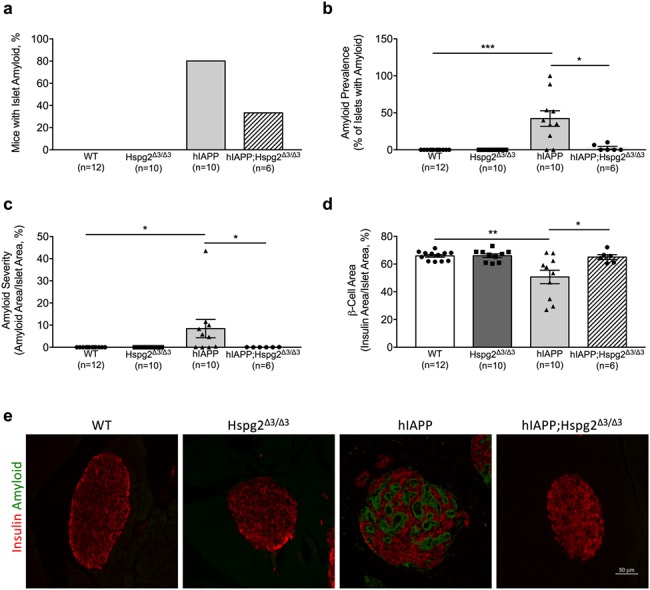
Hspg2Δ3/Δ3 genotype is associated with reduced islet amyloid deposition and β-cell loss in hIAPP transgenic mice a) Expression of perlecan lacking exon 3 reduces incidence of islet amyloid in hIAPP;Hspg2Δ3/Δ3 vs hIAPP transgenic mice. b) Islet amyloid prevalence (% of islets containing amyloid) and c) islet amyloid severity (% of islet area occupied by amyloid) were significantly higher in hIAPP mice compared to both WT and hIAPP;Hspg2Δ3/Δ3 mice after 1 year of HFD feeding; *P < 0.05, ***P < 0.001. d) β-cell area was significantly reduced in hIAPP mice compared to WT mice and hIAPP;Hspg2Δ3/Δ3 mice after 1 year of HFD feeding; *P < 0.05, **P < 0.01. e) Representative images are shown from each genotype (insulin immunostaining, red; amyloid staining with thioflavin S, green). Scale bar equals 50 μm. n = 6–12
Loss of perlecan heparan sulfate glycosaminoglycans does not alter glucose tolerance or insulin secretion
As expected, hIAPP mice with islet amyloid deposition displayed impaired glucose tolerance (Fig. 3a) and reduced insulin secretion (Fig. 3b and c) in response to an intraperitoneal glucose bolus compared to WT mice. Loss of perlecan HS GAGs did not alter glucose tolerance (Fig. 3a) or insulin secretion (Fig. 3b and c) in the absence of β-cell hIAPP expression. Despite our observation that loss of perlecan HS GAGs reduces amyloid deposition in hIAPP transgenic islets, hIAPP;Hspg2Δ3/Δ3 mice did not display improved glucose tolerance (Fig. 3a) or insulin secretion (Fig. 3b and c) compared to hIAPP mice.
Fig. 3.
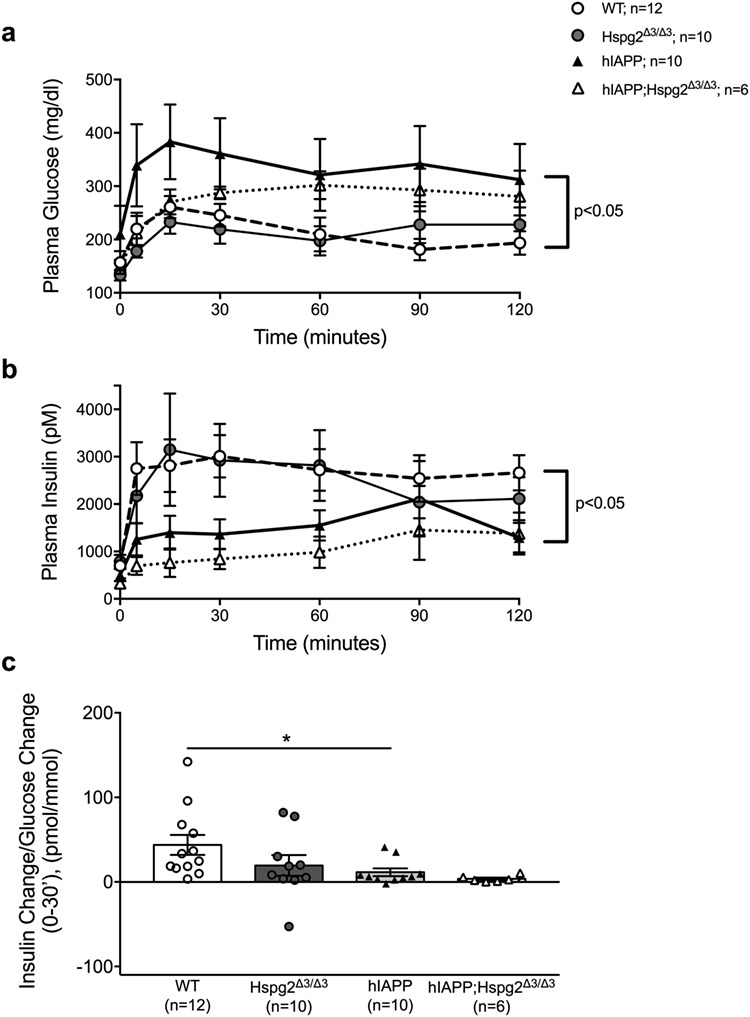
Hspg2Δ3/Δ3 genotype does not alter glucose tolerance or insulin secretion assessed by an IPGTT a) Glucose tolerance in response to intraperitoneal glucose was significantly poorer in hIAPP mice (filled triangle) vs WT mice (open circle) over 120 min; P < 0.05. Glucose tolerance was not different between hIAPP mice (filled triangle) and hIAPP;Hspg2Δ3/Δ3 mice (open triangle). b) Insulin response to intraperitoneal glucose was decreased in hIAPP (filled triangle) compared to WT (open circle) mice; P ≤ 0.05. Insulin response was not different between hIAPP mice (filled triangle) and hIAPP;Hspg2Δ3/Δ3 mice (open triangle. c) Insulin secretion was decreased in hIAPP mice (filled triangle) compared to WT mice (open circle); *P < 0.05. Insulin secretion was not different between hIAPP mice (filled triangle) and hIAPP;Hspg2Δ3/Δ3 mice (open triangle). n = 6–12
Loss of perlecan heparan sulfate glycosaminoglycans leads to decreased weight gain in hIAPP mice fed a HFD
At 8–10 weeks of age, body weight was not significantly different among study groups regardless of genotype (Fig. 4a). At the end of the study, however, hIAPP;Hspg2Δ3/Δ3 mice weighed significantly less than hIAPP mice (Fig. 4b). hIAPP;Hspg2Δ3/Δ3 mice also exhibited significantly decreased body weight gain in response to HFD feeding compared to hIAPP mice (Fig. 4c).
Fig. 4.
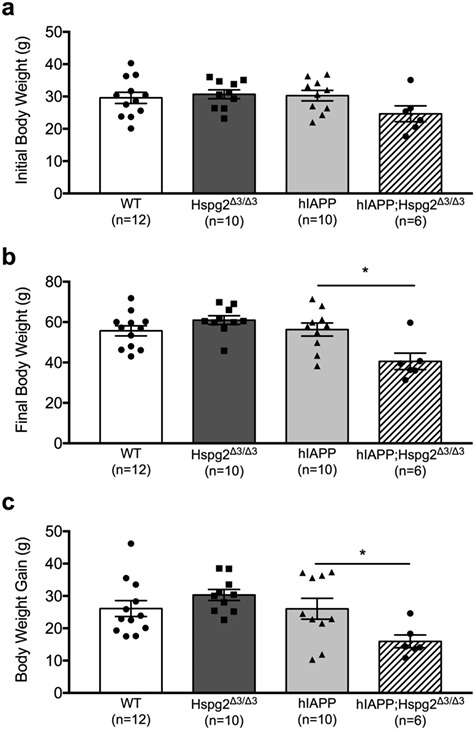
Hspg2Δ3/Δ3 genotype is associated with decreased weight gain in hIAPP transgenic mice fed a HFD a) Body weight was not different among WT, Hspg2Δ3/Δ3, hIAPP and hIAPP;Hspg2Δ3/Δ3 groups at the beginning of the study. b) Following 1 year of HFD feeding, hIAPP;Hspg2Δ3/Δ3 mice displayed lower body weight compared to hIAPP mice; *P < 0.05. c) hIAPP;Hspg2Δ3/Δ3 mice also exhibited less weight gain compared to hIAPP mice after 1 year of HFD feeding; *P < 0.05. n = 6–12
When adjusted for body weight, islet amyloid deposition and β-cell area are not different between groups
Because we observed lower weight gain and final body weight in hIAPP;Hspg2Δ3/Δ3 compared to hIAPP mice, we adjusted the associations between genotype and islet morphology parameters for body weight differences using multiple linear regression analysis. After this adjustment, amyloid prevalence, amyloid severity and β-cell areas were no longer significantly different between hIAPP and hIAPP;Hspg2Δ3/Δ3 mice (Table I).
Table I.
Unadjusted and body weight-adjusted P-values for islet morphological parameters
| hIAPP vs hIAPP; Hspg2Δ3/Δ3 comparison | Unadjusted P-value | Body weight-adjusted P-value |
|---|---|---|
| Amyloid prevalence | 0.01 | 0.40 |
| Amyloid severity | 0.01 | 0.20 |
| β-Cell area | 0.05 | 0.99 |
Discussion
The heparan sulfate proteoglycan perlecan is known to be a component of islet amyloid in humans with T2D (Young et al., 1992; Kahn et al., 1999), and we found this is also the case in islet amyloid deposits of hIAPP transgenic mice (Fig. 1). Each perlecan molecule contains up to three HS GAG chains attached to its N-terminal region (Castillo et al., 1998; Abedini et al., 2006), and previous work has revealed that HS GAGs enhance hIAPP amyloid formation in vitro (Castillo et al., 1998; Potter-Perigo et al., 2003; Hull et al., 2012; Potter et al., 2015). In this study, we determined for the first time whether perlecan HS GAG deficiency decreases islet amyloid formation and β-cell loss in vivo.
In line with our hypothesis, we found that genetic deletion of the perlecan HS GAG attachment site in hIAPP,Hspg2Δ3/Δ3 mice was associated with decreased islet amyloid deposition compared to hIAPP mice expressing wild type perlecan in vivo. This finding is consistent with previous in vitro work showing that the interaction between hIAPP and perlecan is mediated through HS GAGs (Castillo et al., 1998; Potter-Perigo et al., 2003) and that inhibition of GAG synthesis decreases islet amyloid deposition (Hull et al., 2007). Other work observed decreased islet amyloid deposition in vitro in hIAPP transgenic islets with heparinase treatment (Potter et al., 2015) or overexpression of heparanase (Oskarsson et al., 2015), an endoglycosidase that cleaves HS chains. In the present study, we found that the decreased amyloid deposition observed in hIAPP;Hspg2Δ3/Δ3 versus hIAPP mice was also associated with increased β-cell area. This is consistent with the well-known cytotoxic nature of oligomeric hIAPP aggregates, and the inverse relationship between amyloid deposition and β-cell area that has been described in numerous studies and in multiple amyloid-prone species (Jurgens et al., 2011; Wang et al., 2001; Guardado-Mendoza et al., 2009; Howard, 1986). Our data suggest that loss of perlecan HS GAGs reduces the aggregation of hIAPP species into cytotoxic structures, thereby reducing amyloid deposition and ameliorating β-cell loss. It is unlikely that the increase in β-cell area observed in hIAPP;Hspg2Δ3/Δ3 compared to hIAPP mice occurred due a direct effect of loss of perlecan HS GAGs on the β cell, as we observed no difference in β-cell area between WT and perlecan HS GAG deficient Hspg2Δ3/Δ3 mice.
Consistent with the known association between islet amyloid formation, glucose intolerance and impaired insulin secretion (Jurgens et al., 2011; Verchere et al., 1996; Hull et al., 2003; Howard, 1986; Montane et al., 2017; Westwell-Roper et al., 2015), we observed glucose intolerance and impaired insulin secretion in islet amyloid-prone hIAPP mice compared to amyloid-free WT mice. However, we found no difference in glucose tolerance or insulin secretion between hIAPP and hIAPP;Hspg2Δ3/Δ3 mice. Given the significant decrease in islet amyloid observed in hIAPP;Hspg2Δ3/Δ3 mice, improvements in these parameters were anticipated in this group. One potential explanation for this finding could be the presence of a hypovolemic response during the IPGTT in hIAPP;Hspg2Δ3/Δ3 mice, which weighed significantly less than their hIAPP littermates and may have resulted in increased hepatic glucose production, decreased insulin secretion, and/or decreased insulin-mediated glucose utilization (Sherwin et al., 1980). Our failure to observe improvements in glucose tolerance and insulin secretion in hIAPP;Hspg2Δ3/Δ3 compared to hIAPP mice may also be explained by the variability in plasma glucose and insulin observed within groups during the IPGTT.
As discussed, hIAPP;Hspg2Δ3/Δ3 mice exhibited significantly less weight gain and thus lower body weight compared to other study groups after 1 year of HFD feeding. We did not collect data on food intake or energy expenditure during this study, so the decreased body weight observed in hIAPP;Hspg2Δ3/Δ3 mice could be due to decreased food intake, increased energy expenditure or other factors. This difference in body weight could be due to mutation of the perlecan gene in a tissue other than the pancreas since we used global Hspg2 exon 3 knockout mice in this study. For example, previous studies have shown that perlecan is required for fibroblast growth factor 2 (FGF2) signaling in the central nervous system (Kerever et al., 2014; Ornitz and Itoh, 2015) and FGF2 levels are positively associated with fat mass (Hao et al., 2006). However, we did not observe decreased body weight in Hspg2Δ3/Δ3 compared to WT mice, indicating that an interaction between hIAPP and loss of perlecan HS GAGs likely underlies this observation.
Interventions that are associated with reduced body weight have previously been shown to decrease β-cell insulin/IAPP secretion and islet amyloid deposition (Hull et al., 2005; Aston-Mourney et al., 2013). In this study, the reduced body weight and lower weight gain observed in hIAPP;Hspg2Δ3/Δ3 mice was again associated with reduced islet amyloid deposition. Consistent with a weight driven phenotype, when we adjusted measures of amyloid deposition and β-cell area for body weight using multiple linear regression modeling, we found that the observed differences in islet morphology between hIAPP and hIAPP;Hspg2Δ3/Δ3 mice were confounded by the lower final body weight observed in hIAPP;Hspg2Δ3/Δ3 mice. Given that linear regression modeling is a statistical correction, this result suggests that our findings are related to changes in body weight, but does not rule out the possibility that perlecan HS GAGs directly promote hIAPP aggregation in vivo.
We believe it is unlikely that the reduced body weight observed in these mice resulted from a toxic effect of combining hIAPP transgene expression and the Hspg2Δ3/Δ3 mutation per se. Mice with the hIAPP;Hspg2Δ3/Δ3 genetic modification appeared healthy, gained weight during the study (albeit to a lesser extent than the other genotypes), and all hIAPP;Hspg2Δ3/Δ3 mice enrolled completed the 1 year in vivo study. Additionally, both hIAPP and Hspg2Δ3/Δ3 mouse models have previously been crossed with several other genetically altered mouse models without adverse effects (Andrikopoulos et al., 2000; Celie et al., 2007; Wijesekara et al., 2016). This includes the crossing of hIAPP transgenic mice with mice that overexpresses heparanase, resulting in a model similar to the one in the present study in its reduced HS content and hIAPP expression (Oskarsson et al., 2015).
In summary, we have shown that loss of perlecan HS GAGs in hIAPP;Hspg2Δ3/Δ3 mice significantly reduces islet amyloid deposition, and that this is accompanied by an increase in β-cell area compared to hIAPP mice. These effects appear to be related to the lower body weight and weight gain observed in hIAPP;Hspg2Δ3/Δ3 mice. Given the known role of HS GAGs to bind hIAPP (Castillo et al., 1998; Potter-Perigo et al., 2003) and promote amyloid aggregation (Abedini et al., 2006; Hull et al., 2012), the effects of perlecan HS GAGs on islet amyloid formation in vivo may also include direct effects related to hIAPP aggregation in addition to indirect effects related to body weight. Therapeutics targeting these effects may reduce islet amyloid deposition in the setting of T2D.
Acknowledgements
We thank Karl Tryggvason (Karolinska Institutet, Stockholm) for providing the homozygous Hspg2Δ3/Δ3 mice used in this study, and Edward Boyko (University of Washington, Seattle) for expert assistance with multiple linear regression modeling.
PEDS Board Member Responsible for Editing: Daniel Raleigh
Funding
This study was supported by funding from the Department of Veterans Affairs (I01-BX001060 to S.E.K.) and the National Institutes of Health (K01-DK074404 to R.L.H., F32-DK107022 to A.T.T., T32-DK007247 and P30-DK017047).
Conflict of Interest
The authors declare that they have no relevant conflicts of interest.
Ethical Approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted.
References
- Abedini A., Tracz S.M., Cho J.-H., Raleigh D.P. (2006) Characterization of the heparin binding site in the N-terminus of human pro-islet amyloid polypeptide: implications for amyloid formation. Biochemistry, 45, 9228–9237. [DOI] [PubMed] [Google Scholar]
- Andrikopoulos S., Verchere C.B., Terauchi Y., Kadowaki T., Kahn S.E. (2000) Beta-cell glucokinase deficiency and hyperglycemia are associated with reduced islet amyloid deposition in a mouse model of type 2 diabetes. Diabetes, 49, 2056–2062. [DOI] [PubMed] [Google Scholar]
- Arikawa-Hirasawa E., Watanabe H., Takami H., Hassell J.R., Yamada Y. (1999) Perlecan is essential for cartilage and cephalic development. Nat. Genet., 23, 354–358. [DOI] [PubMed] [Google Scholar]
- Aston-Mourney K., Subramanian S.L., Zraika S., Samarasekera T., Meier D.T., Goldstein L.C. et al. (2013) One year of sitagliptin treatment protects against islet amyloid-associated β-cell loss and does not induce pancreatitis or pancreatic neoplasia in mice. Am. J. Physiol. Endocrinol. Metab., 305, E475–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo G.M., Cummings J.A., Yang W., Judge M.E., Sheardown M.J., Rimvall K. et al. (1998) Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan’s enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes, 47, 612–620. [DOI] [PubMed] [Google Scholar]
- Chargé S.B., Esiri M.M., Bethune C.A., Hansen B.C., Clark A. (1996) Apolipoprotein E is associated with islet amyloid and other amyloidoses: implications for Alzheimer’s disease. J. Pathol., 179, 443–447. [DOI] [PubMed] [Google Scholar]
- Celie JW, Rutjes NW, Keuning ED, Soininen R, Heljasvaara R, Pihlajaniemi T. et al. (2007) Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am J Pathol., 170, 1865–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A., Wells C.A., Buley I.D., Cruickshank J.K., Vanhegan R.I., Matthews D.R. et al. (1988) Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res., 9, 151–159. [PubMed] [Google Scholar]
- Cooper G.J., Willis A.C., Clark A., Turner R.C., Sim R.B., Reid K.B. (1987) Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. U. S. A., 84, 8628–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costell M., Gustafsson E., Aszódi A., Mörgelin M., Bloch W., Hunziker E. et al. (1999) Perlecan maintains the integrity of cartilage and some basement membranes. J. Cell Biol., 147, 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross S.E., Vaughan R.H., Willcox A.J., McBride A.J., Abraham A.A., Han B. et al. (2017) Key matrix proteins within the pancreatic islet basement membrane are differentially digested during human islet isolation. Am. J. Transplant., 17, 451–461. [DOI] [PubMed] [Google Scholar]
- D’Alessio D.A., Verchere C.B., Kahn S.E., Hoagland V., Baskin D.G., Palmiter R.D. et al. (1994) Pancreatic expression and secretion of human islet amyloid polypeptide in a transgenic mouse. Diabetes, 43, 1457–1461. [DOI] [PubMed] [Google Scholar]
- Guardado-Mendoza R., Davalli A.M., Chavez A.O., Hubbard G.B., Dick E.J., Majluf-Cruz A. et al. (2009) Pancreatic islet amyloidosis, β-cell apoptosis, and α-cell proliferation are determinants of islet remodeling in type-2 diabetic baboons. Proc. Natl. Acad. Sci., 106, 13992–13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao R.-H., Guo Y., Dong S.-S., Weng G.-Z., Yan H., Zhu D.-L. et al. (2016) Associations of plasma FGF2 levels and polymorphisms in the FGF2 gene with obesity phenotypes in Han Chinese population. Sci. Rep., 6, 19868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopping G., Kellock J., Barnwal R.P., Law P., Bryers J., Varani G. et al. (2014) Designed α-sheet peptides inhibit amyloid formation by targeting toxic oligomers. Elife, 3, e01681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard C.F. (1986) Longitudinal studies on the development of diabetes in individual Macaca nigra. Diabetologia, 29, 301–306. [DOI] [PubMed] [Google Scholar]
- Hull R.L., Andrikopoulos S., Verchere C.B., Vidal J., Wang F., Cnop M. et al. (2003) Increased dietary fat promotes islet amyloid formation and beta-cell secretory dysfunction in a transgenic mouse model of islet amyloid. Diabetes, 52, 372–379. [DOI] [PubMed] [Google Scholar]
- Hull R.L., Westermark G.T., Westermark P., Kahn S.E. (2004) Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J. Clin. Endocrinol. Metab., 89, 3629–3643. [DOI] [PubMed] [Google Scholar]
- Hull R.L., Shen Z.-P., Watts M.R., Kodama K., Carr D.B., Utzschneider K.M. et al. (2005) Long-term treatment with rosiglitazone and metformin reduces the extent of, but does not prevent, islet amyloid deposition in mice expressing the gene for human islet amyloid polypeptide. Diabetes, 54, 2235–2244. [DOI] [PubMed] [Google Scholar]
- Hull R.L., Zraika S., Udayasankar J., Kisilevsky R., Szarek W.A., Wight T.N. et al. (2007) Inhibition of glycosaminoglycan synthesis and protein glycosylation with WAS-406 and azaserine result in reduced islet amyloid formation in vitro. Am. J. Physiol. Cell Physiol., 293, C1586–C1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull R.L., Peters M.J., Perigo S.P., Chan C.K., Wight T.N., Kinsella M.G. (2012) Overall sulfation of heparan sulfate from pancreatic islet β-TC3 cells increases maximal fibril formation but does not determine binding to the amyloidogenic peptide islet amyloid polypeptide. J. Biol. Chem., 287, 37154–37164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving-Rodgers H.F., Ziolkowski A.F., Parish C.R., Sado Y., Ninomiya Y., Simeonovic C.J. et al. (2008) Molecular composition of the peri-islet basement membrane in NOD mice: a barrier against destructive insulitis. Diabetologia, 51, 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens C.A., Toukatly M.N., Fligner C.L., Udayasankar J., Subramanian S.L., Zraika S. et al. (2011) β-cell loss and β-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am. J. Pathol., 178, 2632–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn S.E., D’Alessio D.A., Schwartz M.W., Fujimoto W.Y., Ensinck J.W., Taborsky G.J. et al. (1990) Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes, 39, 634–638. [DOI] [PubMed] [Google Scholar]
- Kahn S.E., Andrikopoulos S., Verchere C.B. (1999) Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes, 48, 241–253. [DOI] [PubMed] [Google Scholar]
- Kerever A., Mercier F., Nonaka R., de Vega S., Oda Y., Zalc B. et al. (2014) Perlecan is required for FGF-2 signaling in the neural stem cell niche. Stem Cell Res., 12, 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koning E.J., Höppener J.W., Verbeek J.S., Oosterwijk C., van Hulst K.L., Baker C.A. et al. (1994) Human islet amyloid polypeptide accumulates at similar sites in islets of transgenic mice and humans. Diabetes, 43, 640–644. [DOI] [PubMed] [Google Scholar]
- Lorenzo A., Razzaboni B., Weir G.C., Yankner B.A. (1994) Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature, 368, 756–760. [DOI] [PubMed] [Google Scholar]
- Matveyenko A.V. and Butler P.C. (2006) Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J., 47, 225–233. [DOI] [PubMed] [Google Scholar]
- Meng F., Abedini A., Plesner A., Middleton C.T., Potter K.J., Zanni M.T. et al. (2010) The sulfated triphenyl methane derivative acid fuchsin is a potent inhibitor of amyloid formation by human islet amyloid polypeptide and protects against the toxic effects of amyloid formation. J. Mol. Biol., 400, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montane J., de Pablo S., Castaño C., Rodríguez-Comas J., Cadavez L., Obach M. et al. (2017) Amyloid-induced β-cell dysfunction and islet inflammation are ameliorated by 4-phenylbutyrate (PBA) treatment. FASEB J., 31, 5296–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D.M. and Itoh N. (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol., 4, 215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson M.E., Singh K., Wang J., Vlodavsky I., Li J.-P., Westermark G.T. (2015) Heparan sulfate proteoglycans are important for islet amyloid formation and islet amyloid polypeptide-induced apoptosis. J. Biol. Chem., 290, 15121–15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson M.E., Hermansson E., Wang Y., Welsh N., Presto J., Johansson J. et al. (2018) BRICHOS domain of Bri2 inhibits islet amyloid polypeptide (IAPP) fibril formation and toxicity in human beta cells. Proc. Natl. Acad. Sci. U. S. A., 115, E2752–E2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepys M.B., Rademacher T.W., Amatayakul-Chantler S., Williams P., Noble G.E., Hutchinson W.L. et al. (1994) Human serum amyloid P component is an invariant constituent of amyloid deposits and has a uniquely homogeneous glycostructure. Proc. Natl. Acad. Sci. U. S. A., 91, 5602–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter K.J., Werner I., Denroche H.C., Montane J., Plesner A., Chen Y. et al. (2015) Amyloid formation in human islets is enhanced by heparin and inhibited by heparinase. Am. J. Transplant., 15, 1519–1530. [DOI] [PubMed] [Google Scholar]
- Potter-Perigo S., Hull R.L., Tsoi C., Braun K.R., Andrikopoulos S., Teague J. et al. (2003) Proteoglycans synthesized and secreted by pancreatic islet beta-cells bind amylin. Arch. Biochem. Biophys., 413, 182–190. [DOI] [PubMed] [Google Scholar]
- Rossi M., Morita H., Sormunen R., Airenne S., Kreivi M., Wang L. et al. (2003) Heparan sulfate chains of perlecan are indispensable in the lens capsule but not in the kidney. EMBO J., 22, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwin R.S., Shamoon H., Hendler R., Saccà L., Eigler N., Walesky M. (1980) Epinephrine and the regulation of glucose metabolism: effect of diabetes and hormonal interactions. Metabolism, 29, 1146–1154. [DOI] [PubMed] [Google Scholar]
- Shu C., Smith S.M., Melrose J. (2016) The heparan sulphate deficient Hspg2 exon 3 null mouse displays reduced deposition of TGF-β1 in skin compared to C57BL/6 wild type mice. J. Mol. Histol., 47, 365–374. [DOI] [PubMed] [Google Scholar]
- Verchere C.B., D’Alessio D.A., Palmiter R.D., Weir G.C., Bonner-Weir S., Baskin D.G. et al. (1996) Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proc. Natl. Acad. Sci. U. S. A., 93, 3492–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal J., Verchere C.B., Andrikopoulos S., Wang F., Hull R.L., Cnop M. et al. (2003) The effect of apolipoprotein E deficiency on islet amyloid deposition in human islet amyloid polypeptide transgenic mice. Diabetologia, 46, 71–79. [DOI] [PubMed] [Google Scholar]
- Walz A., McFarlane S., Brickman Y.G., Nurcombe V., Bartlett P.F., Holt C.E. (1997) Essential role of heparan sulfates in axon navigation and targeting in the developing visual system. Development, 124, 2421–2430. [DOI] [PubMed] [Google Scholar]
- Wang F., Hull R.L., Vidal J., Cnop M., Kahn S.E. (2001) Islet amyloid develops diffusely throughout the pancreas before becoming severe and replacing endocrine cells. Diabetes, 50, 2514–2520. [DOI] [PubMed] [Google Scholar]
- Westermark P. (1972) Quantitative studies on amyloid in the islets of Langerhans. Ups. J. Med. Sci., 77, 91–94. [DOI] [PubMed] [Google Scholar]
- Westermark P., Wilander E., Westermark G.T., Johnson K.H. (1987) Islet amyloid polypeptide-like immunoreactivity in the islet B cells of type 2 (non-insulin-dependent) diabetic and non-diabetic individuals. Diabetologia, 30, 887–892. [DOI] [PubMed] [Google Scholar]
- Westermark P., Engström U., Johnson K.H., Westermark G.T., Betsholtz C. (1990) Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. U. S. A., 87, 5036–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwell-Roper C.Y., Chehroudi C.A., Denroche H.C., Courtade J.A., Ehses J.A., Verchere C.B. (2015) IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia, 58, 575–585. [DOI] [PubMed] [Google Scholar]
- Wijesekara N, Kaur A, Westwell-Roper C, Nackiewicz D, Soukhatcheva G, Hayden MR. et al. (2016). ABCA1 deficiency and cellular cholesterol accumulation increases islet amyloidogenesis in mice. Diabetologia, 59, 1242–6. [DOI] [PubMed] [Google Scholar]
- Young I.D., Ailles L., Narindrasorasak S., Tan R., Kisilevsky R. (1992) Localization of the basement membrane heparan sulfate proteoglycan in islet amyloid deposits in type II diabetes mellitus. Arch. Pathol. Lab. Med., 116, 951–954. [PubMed] [Google Scholar]
- Zraika S., Aston-Mourney K., Marek P., Hull R.L., Green P.S., Udayasankar J. et al. (2010) Neprilysin impedes islet amyloid formation by inhibition of fibril formation rather than peptide degradation. J. Biol. Chem., 285, 18177–18183. [DOI] [PMC free article] [PubMed] [Google Scholar]