

Abstract
The polypeptide amylin is responsible for islet amyloid in type 2 diabetes, a process which contributes to β-cell death in the disease. The role of the N-terminal region of amylin in amyloid formation is relatively unexplored, although removal of the disulfide bridged loop between Cys-2 and Cys-7 accelerates amyloid formation. We examine the des Lys-1 variant of human amylin (h-amylin), a variant which is likely produced in vivo. Lys-1 is a region of high charge density in the h-amylin amyloid fiber. The des Lys-1 polypeptide forms amyloid on the same time scale as wild-type amylin in phosphate buffered saline, but does so more rapidly in Tris. The des Lys-1 variant is somewhat less toxic to cultured INS cells than wild type. The implications for the in vitro mechanism of amyloid formation and for comparative analysis of amyloidogenicity are discussed.
Keywords: amylin, amyloid, davalintide, IAPP, type-2 diabetes
Introduction
Islet amyloid formation contributes to pancreatic β-cell dysfunction and death in type 2 diabetes (T2D) (Cooper et al., 1987; Westermark et al., 1987; Cooper et al., 1988; Opie 1901; Hull et al., 2004; Westermark et al., 2011; Cao et al., 2012a; Abedini and Schmidt, 2013; Raleigh et al., 2017) and to the failure of islet transplants, potential limiting their use in the treatment of type 1 diabetes (Udayasankar et al., 2009; Potter et al., 2010; Westermark et al., 2012). The 37-residue neuro pancreatic polypeptide hormone amylin (also known as islet amyloid polypeptide or IAPP) is responsible for islet amyloid formation in vivo. Amylin is produced in the pancreatic β-cells, stored together with insulin in the insulin secretory granule and co-secreted with insulin (Lukinius et al., 1989; Stridsberg et al., 1993; Cao et al., 2013). Amylin normally plays a role in regulating glucose metabolism, but forms islet amyloid in T2D by an unknown mechanism (Westermark et al., 2011; Lutz 2013; Akter et al., 2016; Raleigh et al., 2017). The human amylin (h-amylin) polypeptide is one of the most amyloidogenic naturally occurring sequences and forms amyloid in vitro more rapidly than the Aβ peptide of Alzheimer’s disease (Cao et al., 2013).
Amylin is 37-resides in length, contains a strictly conserved disulfide bridge between residues two and seven and an amidated C-terminal Tyr (Fig. 1). There are no acidic residues in h-amylin and the C-terminus is amidated, and thus the polypeptide has a net positive charge at all physiologically relevant pH values (Abedini and Raleigh, 2005a; Marek et al., 2012). The human peptide contains one Lys at position 1, an Arg at position 11 and a His at position 18. Arg-11 and Lys-1 are protonated at pH 7.4., and thus the net charge at pH 7.4 is between +2 and +4 depending upon the exact pKa of His-18 and the N-terminus. This suggests that charge repulsion could play a role in modulating the rate of h-amylin formation; however, this is a relatively unexplored area. Linearized Poisson–Boltzmann (PB) calculations on structural models of the h-amylin amyloid fibril indicate that Lys-1 makes significant unfavorable electrostatic interactions in the amyloid fibril (Marek et al., 2012).
Fig. 1.
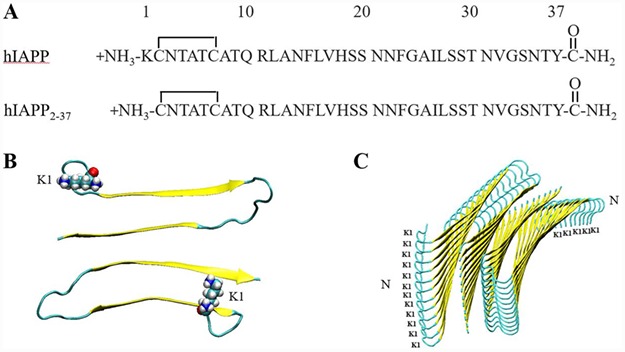
The primary structure of h-amylin and des Lys-1 h-amylin and models of the h-amylin amyloid fibril. (A) Primary sequences. (B) A top-down view of the high-resolution model of the h-amylin amyloid fiber derived from the studies of steric zipper fragments of h-amylin (Wiltzius et al., 2008). The β-strands are colored yellow. One layer of the fiber structure is shown and Lys-1 is depicted in space filling format. (C) A side view of the same model of the amyloid fiber. Two stacks of 12 peptides per stack are shown, the position of Lys-1 is indicated and the N-termini of the chains are labeled.
Not all organisms develop islet amyloid and its presence or absence correlates with the propensity for different organisms to develop diabetes and with the in vitro amyloidogenicity of the polypeptide sequence (Betsholtz et al., 1989; Westermark et al., 1990; Ashburn and Lansbury Jr, 1993; Westermark et al., 2011; Akter et al., 2016). In particular, rodents do not form islet amyloid in vivo and have a form of amylin which is non-amyloidogenic in vitro except at very high concentration (Westermark et al., 1990). These observations have led to interest in comparative studies of amylins from different species and studies of amylin variants in an attempt to deduce the features which control amyloidogenicity and toxicity (Marek et al., 2007; Fox et al., 2010; Westermark et al., 2011; Cao et al., 2012a; Fernandez, 2014; Akter et al., 2017; Akter et al., 2018). Part of the motivation for these investigations has been to aid in the design of non-toxic, non-amyloidogenic variants of amylin for possible therapeutic use (Kruger and Gloster, 2004; Akkati et al., 2011; Wu and Shea, 2013; Wang et al., 2014). Much of this work has focused on the region between residues 20 and 29 as human and rat amylin differ the most in this segment including three proline substitutions (Betsholtz et al., 1989; Westermark et al., 1990; Ashburn and Lansbury Jr, 1993). In contrast, the role of the N-terminal region of amylin in modulating amyloidogenicity is relatively unexplored. This is likely due in part to the fact that the disulfide bond in amylin constraints the chain in a conformation which is believed to be incompatible with the cross β-sheet structure of the amyloid fiber and the first seven residues are thought to be disordered or only partially ordered in the h-amylin amyloid fiber, nevertheless removal of the first seven residues does affect the rate of amyloid formation (Koo and Miranker, 2005; Luca et al., 2007; Wiltzius et al., 2009; Ilitchev et al., 2016; Rodriquez et al., 2017; Quittot et al., 2018; Ridgway et al., 2018). Truncated analogues of h-amylin that lack residues one to seven form amyloid faster than wild-type h-amylin (Quittot et al., 2018; Ridgway et al., 2018). This may be due to the removal of the disulfide and/or removal of the charged Lys-1. Removal of Lys-1 to generate des-Lys-1 amylin will reduce unfavorable charge–charge interactions, even if the residue is not part of the cross β-core, since the parallel arrangement of the peptide chains in h-amylin amyloid will lead to the close proximity of the N-terminal and ε-amino groups between adjacent peptides within one stack of the amyloid fiber (Fig. 1) (Luca et al., 2007; Wiltzius et al., 2009). pH- and salt-dependent studies indicate that electrostatic interactions play a role in amyloid formation by h-amylin, but the role of specific charge residues is unexplored with the exception of His-18 (Charge et al., 1995; Abedini and Raleigh 2005a; Marek et al., 2012; Li et al., 2013; Jha et al. 2014; Tu et al., 2014; Patil and Alexandrescu, 2015). Analysis of the des-Lys analogue will help to shed light on the importance of repulsive charge–charge interactions in the N-terminus in modulating amyloidogenicity.
The des-Lys-1 variant is physiological relevant as it has been reported to be present in vivo and to be bio-active as an amylin agonist (Hay et al. 2015). Furthermore, a designed variant of h-amylin denoted Davalintide has been explored for potential clinical use. Like h-amylin, Davalintide contains an N-terminal Lys residue and a disulfide bridge between residues two and seven and the two sequences are identical over this region. There is strong evidence that a des Lys-1 product of Davalintide is produced in vivo by proteolytic cleavage and is bio-active, consistent with the des-Lys-1 variant of h-amylin being produced in vivo (Mack et al., 2010; Wang et al., 2012). A number of cleavage products of h-amylin have been found in vivo or postulated to be found in vivo, and they have different amyloidogenicities relative to wild type (Nilsson and Raleigh, 1999; Guan et al., 2012; Aston-Mourney et al., 2013; Hay et al., 2015; Meier et al., 2015); however, the amyloidogenicity and toxicity of des-Lys-1 amylin have not been reported.
Results
Removal of Lys-1 does not significantly affect the rate of amylin amyloid formation in phosphate buffered saline
The primary sequence of h-amylin is displayed in Fig. 1. Lys-1 is believed to not participate in the cross β-sheet core of the h-amylin amyloid fiber, as the disulfide bridge between Cys two and Cys seven constrains this region of the polypeptide chain in a fashion which is incompatible with the cross β-structure of high-resolution models of the h-amylin fiber (Luca et al., 2007; Wiltzius et al., 2008). Nevertheless, the presence of Lys-1 leads to a high positive charge density due to the N-terminus and ε-amino groups. Indeed, this region of h-amylin has the highest charge density in models of the h-amylin amyloid fibers as judged by linearized PB calculations conducted on a molecular dynamics minimized model of the h-amylin amyloid fiber derived from crystallographic studies of steric zipper peptides. In this model, there are no close contacts between charged residues in the two chains belonging to the same layer (i.e. chains located in the same layer, but in different stacks), but there are charge–charge contacts between the different layers along the fiber axis (Fig. 1), and Lys-1 is predicted to make significant unfavorable electrostatic interactions (Marek et al., 2012).
We first compared the ability of wild-type and des-Lys-1 h-amylin to form amyloid at pH 7.4 in phosphate buffered saline, (PBS, 10-mM sodium phosphate and 140-mM KCl). The time scale of amyloid formation was followed using fluorescence monitored thioflavin-T binding assays. Thioflavin-T is a small dye whose fluorescence increases upon binding to amyloid fibers. The dye is an extrinsic probe and there are examples of thioflavin-T assays giving false negatives (Wong et al., 2016), but it has been shown that thioflavin-T assays accurately report on amyloid formation by h-amylin under the condition of our studies (Tu et al., 2014). Nevertheless, we also used transmission electron microscopy (TEM) to confirm the presence of amyloid fibers. Both polypeptides exhibit sigmoidal thioflavin-T fluorescence curves that are typical of h-amylin amyloid formation (Figs 2 and S1).
Fig. 2.
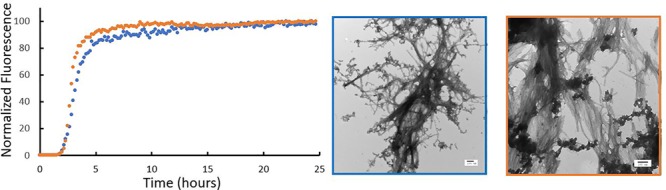
Removal of Lys-1 does not significantly alter the rate of amyloid formation by h-amylin in PBS. (A) Thioflavin-T assays were conducted using wild-type h-amylin (leftmost) and des Lys-1 h-amylin (righmost). (B) TEM images recorded for samples collected at the end of the kinetic experiments: wild-type h-amylin (left) and des Lys-1 h-amylin (right). Scale bars represent 100 nm.
The time scale of amyloid formation is similar for the two polypeptides, as judged by the similar T50 values. T50 is defined as the time required to reach 50% of the fluorescence changes in a thioflavin-T experiment. The values observed for wild-type and des Lys-1 h-amylin are 3.8 ± 0.5 and 3.1 ± 0.3 h in PBS, respectively. The experiments were repeated four separate times with three repeats per experiments (12 measurements in total) and included two separately prepared batches of wild-type amylin. The differences are modest at best, and moderately statistically significant, as judged by the P-value (P = 0.028). The differences are actually smaller than reported for batch-to-batch variations in the T50 value of wild-type h-amylin. Thus, we do not view the time scale of amyloid formation between these two peptides as being fundamentally different in PBS. The shape of the curves is also similar. Aliquots were removed from each sample at the end of the thioflavin-T experiments and examined using TEM. Both polypeptides gave rise to typical amylin amyloid fibers (Fig. 2). It is difficult to detect changes in the structure of fibers with the resolution offered by TEM, but the TEM images do appear to differ somewhat between the polypeptides. This might reflect different polymorphs of the amylin amyloid structure formed by the two polypeptides. Collectively, these studies show that the removal of Lys-1 has no significant effect on the time scale of amylin amyloid formation in PBS. The results are somewhat surprising since the removal of Lys-1 is expected to reduce electrostatic repulsions during amyloid assembly. However, removal of Lys-1 may also weaken the interactions of phosphate anions with h-amylin. Thus, we also compared the relative time course of amyloid formation in a different buffer.
Removal of Lys-1 does affect the rate of amylin amyloid formation in Tris buffer
We next compared the time scale of amyloid formation of the two peptides in Tris buffer. Our motivation was two-fold: first, while PBS is a more physiologically relevant buffer, numerous biophysical studies of amylin have been reported in Tris buffer, and second, the time scale of amyloid formation by wild-type h-amylin is known to be longer in Tris than in PBS and to be sensitive to ionic strength (Marek et al., 2012; Tu et al., 2014; Wong et al., 2016). In addition, Tris is believed to interact with h-amylin more weakly than does phosphate (Marek et al. 2012). Furthermore, comparative studies of h-amylin and puffer fish amylin have shown that the relative time scale of amyloid formation of these two polypeptides is reversed in Tris buffer and PBS even though the pH is the same. Puffer fish amylin has a larger T50 than h-amylin in PBS, but a smaller T50 than h-amylin in Tris buffer (Wong et al., 2016), highlighting the fact that comparative amyloid formation kinetics can depend on modest changes in solution conditions.
Both polypeptides form amyloid in Tris buffer as expected (Figs 3 and S1). The T50 value for the des Lys-1 variant is slightly larger in Tris, 4.1 ± 0.2 h, compared with the value for this polypeptide in PBS, 3.1 ± 0.3 h. In contrast, the T50 value of h-amylin is noticeably larger in Tris buffer than in PBS, 20.1 ± 0.8 h vs 3.8 h, consistent with earlier studies of wild-type h-amylin. We also tested if the change in T50 is associated with a change in the shape of the curve by plotting the data on a reduced time scale in which each individual curve is normalized by its own T50. Changes in shape can reflect changes in the relative importance of primary, nucleation, elongation and secondary nucleation (Ariso et al. 2014). The normalized curves for wild-type and Des Lys-1 amylin have similar shapes in Tris buffer (Fig. S2).
Fig. 3.
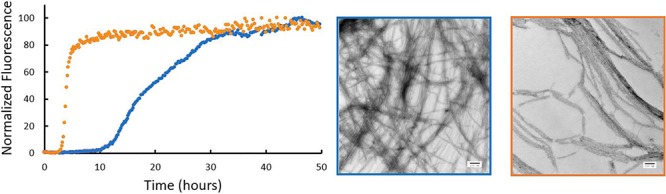
Removal of Lys-1 alters the rate of amyloid formation by h-amylin in Tris. (A) Thioflavin-T assays in Tris buffer (20 mM, pH = 7.4) were conducted using wild-type h-amylin (righmost) and des Lys-1 h-amylin (right). (B) TEM images recorded for samples collected at the end of the kinetic experiments: wild-type h-amylin (left) and des Lys-1 h-amylin (leftmost). Scale bars represent 100 nm.
To test if the different relative behavior observed in Tris vs PBS is primarily due to the change of buffer rather than the additional salt present in PBS (i.e. the change in ionic strength), we repeated the measurements in phosphate buffer with no added salt. The T50 values in phosphate are 6.7 ± 0.2 h for h-amylin and 4.1 ± 0.1 h for des Lys-1 amylin, both of which are larger than the respective T50 values measured in PBS but notably smaller than the value measured in Tris for wild type (Fig. S3). The comparative measurements in PBS and phosphate indicate that there is a contribution due to the change in ionic strength between PBS and Tris. However, this is not the dominate effect since the difference caused by changing the buffer from phosphate to Tris is noticeable larger than the change in going from phosphate to PBS. The ratio of the T50 values of the two polypeptides (T50 wild type/T50 des-Lys-1) is 1.2 in PBS, 1.6 in phosphate and 4.9 in Tris.
Removal of Lys-1 modestly impacts h-amylin toxicity toward cultured cells
We next examined the effect of the removal of Lys-1 on the toxicity of h-amylin toward a cultured pancreatic β-cell line (INS-1 cells). Side-by-side concentration-dependent assays were conducted to examine the effect of the two polypeptides on cell viability. We assessed the toxic effect of each of the polypeptides using three independent viability assays: one which monitored the integrated redox status of the cells via NAD(P)H-dependent resazurin reduction (Alamar Blue), a second which measured net cellular ATP levels via luciferase (CellTiter-Glo 2.0) and a third that monitored plasma membrane integrity/permeability using a membrane impermeable DNA intercalating fluorescent probe (CellTox Green). The concentration of polypeptide required to achieve 50% of the effect, EC50, was approximately 2.1- to 2.3-fold higher for the des Lys-1 variant relative to wild type (Fig. 4, Table 1), indicating that the removal of Lys-1 modestly, but consistently, reduces the toxicity of h-amylin in cell culture. The differences between wild-type amylin and de Lys-1 amylin are moderate but are statistically significant. It is interesting to note that the EC50 values determined from the Alamar Blue and CellTiter-Glo assays for a given polypeptide are the same, but the values deduced from the CellTox Green assay are apparently lower. This is true for both wild-type and des-Lys-1 amylin. The observed differences are not statistically significant for the number of repeats measured here (n = 3, P > 0.5); however, we have observed the same trend with other studies of wild-type h-amylin, i.e. the EC50 measured from the cell permeability assay is lower than measured by Alamar Blue or CellTiter-Glo assays. This argues that modest changes in cell permeability (as measured by the small molecule dye employed in the assay) can occur before significant changes in global ATP levels (CellTiter-Glo assay) or changes in cellular redox balance (Alamar Blue assay).
Fig. 4.
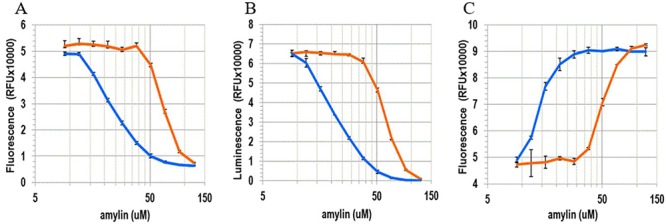
Removal of Lys-1 modestly alters the toxicity of h-amylin toward cultured INS-1 cells. The panels represent dose-dependent response of the cells to wild-type h-amylin (leftmost curves) and des Lys-1 h-amylin (righmost curves). The data are plotted on a log scale. Dose responses were assessed for each peptide using three different cytotoxicity assays: (A) Alamar Blue, (B) CellTiter-Glo 2.0 and (C) CellTox Green. The data are plotted as the mean ± SD, n = 3. The horizontal axis is plotted on a log scale.
Table I.
EC50 values for cytotoxicity toward cultured INS-1 cells measured for wild-type h-amylin and des Lys-1 h-amylin from six independent experiments using three different cell viability assays.
| Assay | EC50 ± SEM (μM) | |
|---|---|---|
| WT | des Lys-1 | |
| Alamar Blue | 31 ± 3 | 73 ± 5 |
| CellTiter-Glo 2.0 | 35 ± 5 | 74 ± 5 |
| CellTox Green | 24 ± 4 | 56 ± 9 |
Statistical analysis was completed using Graph Pad Prism 5. All data are presented as mean ± standard error of mean (SEM).
Conclusions
The analysis presented here shows that the removal of Lys-1 led to an approximately 2.1- to 2.3-fold reduction of in vitro toxicity. The effect of removing Lys-1 on amyloid kinetics is more complicated: no significant effect is observed in PBS, but a clear difference is detected in Tris and the difference is largely due to the choice of buffer rather than the change in ionic strength. The buffer-dependent effect of the removal of Lys-1 is particular noteworthy as Lys-1 is predicted to destabilize the amyloid fiber via repulsive electrostatic interactions as judged by PB calculations (Marek et al., 2012). Thus, its removal might be expected to accelerate amyloid formation, and this is observed in Tris but not in PBS. However, prior experimental work has also shown that the time course of h-amylin amyloid formation is strongly influenced by the choice of anions in solution, even when they are present at modest concentration. The observed effects correlate with the ion selectivity series; anions which bind more strongly to ion exchange resins have a larger accelerating effect upon h-amylin amyloid formation than anions which interact more weakly (Marek et al., 2012). Phosphate interacts more strongly with ion exchange resins than Tris, (it is positioned higher in the ion selectivity series), and phosphate is well known to interact with cationic groups in proteins. Based on these observations, a plausible explanation for the different relative effects observed in Tris vs PBS is that the removal of Lys-1 reduces electrostatic repulsion and, hence, accelerates amyloid formation in a buffer which does not interact significantly with h-amylin, i.e. in Tris. In contrast, removal of Lys-1 weakens the interactions of phosphate with h-amylin leading to two competing effects in PBS. Removing the positively charged Lys sidechain accelerates amyloid formation by reducing electrostatic repulsion on the one hand, but removal of Lys-1 lessens interactions with phosphate, which slows amyloid formation. In this scenario, the two effects compete leading to a modest overall effect of the removal of Lys-1 in PBS. Irrespective of the detailed molecular explanation for the difference behavior in Tris vs PBS, the results indicate that ranking of amyloidgenicity by aggregation times can be strongly buffer dependent and, hence, should be treated with caution. The results also show the importance of examining multiple solution conditions.
Materials and Methods
Synthesis and purification of the h-amylin peptide
Human IAPP and the des Lys-1peptide were synthesized using a CEM Liberty microwave peptide synthesizer on a 0.01 mmol scale with 9-fluorenylmethyloxycarbonyl (Fmoc) protected amino acids. Pseudoproline dipeptide (oxazolidine) derivatives of Ala-Thr and Leu-Ser were used as previously described to improve the yield (Abedini and Raleigh, 2005b; Marek et al., 2010). A 5-(4’-Fmoc-aminomethyl-3′,5′-dimethoxyphenol) valeric acid (PAL-PEG) resin was used to provide an amidated C-terminus. All β-branched residues, pseudoproline dipeptides and the first amino acid (Tyr) attached to the resin were double coupled. Coupling reactions were carried out for 2 min at 90°C except for Cys and His which were coupled at 55°C to minimize the risk of racemization. Peptides were cleaved using a cleavage cocktail made of 92.5, 2.5, 2.5 and 2.5% of trifluoroacetic acid, triisopropylsilane, 3.6-dioxa-1,8-octanedithiol and H2O, respectively. Cleaved peptides were dissolved in 20% (v/v) acetic acid and lyophilized prior to the formation of the disulfide. The disulfide bond between the residues two and seven in wild-type h-amylin and des lys-1 amylin was formed by dissolving the dry peptide in 100% dimethyl sulfoxide at a concentration of 10 mg/ml and incubating at room temperature for 3 days. Peptides were purified using reverse-phase high-performance liquid chromatography (RP-HPLC) with a Higgins Analyzed Proto 300 C18 preparatory column. Residual scavengers were removed using a1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) extraction procedure. Purified peptide was dissolved in neat HFIP, and the solution was incubated at room temperature for 4 h, then filtered with a 0.45-μM GHP membrane filter, and purified via RP-HPLC. Masses of purified peptides were measured using a matrix-assisted laser desorption time-of-flight mass spectrometer; hIAPP expected: 3903.3, observed: 3902.9; hIAPP2–37 expected: 3775.1, observed: 3774.8.
Peptide stock and sample preparation
Stock solutions were prepared by dissolving purified peptides into neat HFIP to a concentration of 1.6 mM. The concentration was determined by lyophilizing a 20-μL aliquot of the HFIP stock for at least 12 h, dissolving in 120 μl of 20-mM Tris buffer (pH 7.4) and measuring the absorbance at 280 nm. An extinction coefficient of 1600 M−1 cm−1 was used for h-amylin and des-Lys-1 amylin.
Thioflavin-T fluorescence assays
Aliquots of the HFIP stock solutions were lyophilized overnight (at least 12 h) and reconstituted in buffer at pH 7.4 for kinetic assays. The final concentrations of the peptides and thioflavin-T were 16 and 32 μM, respectively. Black Corning 96-well non-binding plates were used for thioflavin-T kinetic assays. Plates were sealed with polyethylene sealing tape, and a clear lid was used. Wells at the edge of the plate were not used, but unused wells were filled with buffer. Fluorescence measurements were taken on a Beckmann Coulter DTX 880 plate reader and a Spectramax Gemini EM plate reader, with a multimode detector using an excitation wavelength of 450 nm and an emission of 485 nm. Readings were taken from the bottom of the wells in 10-min intervals without agitation. Three parallel samples using different solutions of h-amylin and des Lys-1 h-amylin were tested for each experiment. Kinetic experiments were repeated using two different plate readers on different days and average T50 values are reported. The quoted uncertainties are the estimated standard deviations. All fluorescence assays were performed at 25°C.
Transmission electron microscopy
TEM images were recorded using a FEI Bio TwinG2 transmission electron microscope at the Life Science Microscopy Center at Stony Brook University. Samples for TEM measurements were collected from the samples used for the kinetic assays at the end of the kinetic experiments. About 15 μl of peptide solution was collected at the end of the kinetic assays and blotted on a carbon-coated Formvar 300-mesh copper grid for 1 min and then negatively stained with 2% uranyl acetate for 1 min.
Cytotoxicity assays
INS-1 cells were purchased from AddexBio and cultivated in optimized RPMI-1640 (AddexBio, #C0004–02) supplied with 10% ultra-low IgG FBS (Gibco, #16250078). Alamar Blue (Generon, #MBS238967), CellTiter-Glo 2.0 (Promega, #G9242) and CellTox Green (Promega, # G8741) assays were used to evaluate the cytotoxicity of the peptides on INS-1 cells. Briefly, evaluation of the cytotoxicity of the peptides toward INS-1 cells was performed as follows. The cells were seeded at ~ 50% confluence on a 96-well half-area clear bottom white plates (Greiner, #675083) and incubated for 36 h in 5% CO2 humidified incubator at 37°C. Cultured cells were exposed to the peptide diluted in the fresh complete medium for further 24 h. Serial dilutions of the peptides were freshly prepared before use from lyophilized peptide aliquots. For the Alamar Blue assays, an equal volume of 20% of Alamar Blue reagent in complete medium was added to the treated cells and the plates were further incubated at 37°C for ~ 1 h. Fluorescence of the Alamar Blue reduction product was measured using excitation/emission wavelengths of 550 and 590 nm, respectively. For the CellTiter-Glo 2.0 assay, the culture plates were cooled to room temperature and an equal volume of the assay reagent was added to the treated cells. The plates were vigorously (700 rpm) shaken for 1 min and luminescence intensity was measured using a Clariostar plate reader. For the CellTox Green assay, cells were exposed to peptide in the presence of the assay dye (1:5000 dilution) for 24 h before the fluorescence intensity was measured using excitation at 480 nm and emission at 525 nm. Statistical analysis and calculation of EC50 values were completed using Graph Pad Prism 5.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the members of the Raleigh lab for helpful discussions and Mr. Lakshan Manathunga for experimental assistance.
Funding
This work was supported by the National Institutes of Health grant [GM078114].
References
- Abedini A. and Raleigh D.P. (2005a) Biochemistry, 44, 16284–16291. [DOI] [PubMed] [Google Scholar]
- Abedini A. and Raleigh D.P. (2005b) Org. Lett., 7, 693–696. [DOI] [PubMed] [Google Scholar]
- Abedini A. and Schmidt A.M. (2013) FEBS Lett., 587, 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkati S., Sam K.G., Tungha G. (2011) J. Clin. Pharmacol., 51, 796–804. [DOI] [PubMed] [Google Scholar]
- Akter R., Abedini A., Ridgway Z., Zhang X., Kleinberg J., Schmidt A.M., Raleigh D.P. (2017) Israel J. Chem., 57, 750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akter R., Bower R.L., Abedini A., Schmidt A.M., Hay D.L., Raleigh D.P. (2018) ACS Chem. Biol., 13, 2747–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akter R., Cao P., Noor H. et al. (2016) J. Diabetes Res., 2798269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariso P., Vendruscolo M., Dobson C.M., Knowles P.J. (2014) Trends Pharmacol. Sci., 35, 127–135. [DOI] [PubMed] [Google Scholar]
- Ashburn T.T. and Lansbury P.T. Jr. (1993) J. Am. Chem. Soc., 115, 11012–11013. [Google Scholar]
- Aston-Mourney K., Zrika S., Udayasankar J., Subramanian S.L., Green P.S., kahn S.E. (2013) J. Biol. Chem., 288, 3555–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betsholtz D., Christmansson L., Engstrom U., Rorsman F., Svensson V., Johnson K.H., Westermark P. (1989) FEBS Lett., 251, 261–264. [DOI] [PubMed] [Google Scholar]
- Cao P., Abedini A., Raleigh D.P. (2012a) Curr. Opin. Struct. Biol., 23, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P., Marek P., Noor H., Patsalo V., Tu L.-H., Wang H., Abedini A., Raleigh D.P. (2013) FEBS Lett., 587, 1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P., Tu L.-H., Abedini A., Levsh O., Akter R., Ratsalo V., Schmidt A.M., Raleigh D.P. (2012b) J. Mol. Biol., 421, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charge S.B., de Koning E.J., Clark A. (1995) Biochemistry, 34, 14588–14593. [DOI] [PubMed] [Google Scholar]
- Cooper G.J.S., Leighton B., Dimitriadis G.D., Parry-billingst M., Kowalchuk J.M., Howland K., Rothbard J.B., Willis A.C., Reid K.B.M. (1988) Proc. Natl. Acad. Sci. U. S. A., 85, 7763–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G.J.S., Willis A.C., Clark A., Turner R.C., Sim R.B., Reid K.B.M. (1987) Proc. Natl. Acad. Sci. U. S. A., 84, 8628–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez M.S. (2014) Cell Calcium, 56, 416–427. [DOI] [PubMed] [Google Scholar]
- Fox A., Snollaerts T., Errecart C.C., Calciano A., Nogaj L.A., Moffet D.A. (2010) Biochemsitry, 49, 7783–7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H., Chow K.M., Shah R., Rhodes C.J., Hersh L.B. (2012) Diabetologia, 55, 2989–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D.L., Shen C., Lutz T.A., Parkers D.G., Roth J.D. (2015) Pharmacol. Rev., 67, 564–600. [DOI] [PubMed] [Google Scholar]
- Hull R.L., Westermark G.T., Wesermark R., Kahn S.E. (2004) J. Clin. Endocrinol. Metab., 89, 3629–3643. [DOI] [PubMed] [Google Scholar]
- Ilitchev A.I., Giammona M.J., Do T.D., Wong A.G., Buratto S.K., Shea J.-E., Raleigh D.P., Bowers M.T. (2016) J. Am. Soc. Mass Spectrom., 27, 1010–1018. [DOI] [PubMed] [Google Scholar]
- Jha S., Snell J.M., Sheftic S.R., Patil S.M., Daniels S.B., Kolling F.W., Alexandrescu A.T. (2014) Biochemistry, 53, 300–310. [DOI] [PubMed] [Google Scholar]
- Koo B.W. and Miranker A.D. (2005) Protein Sci., 14, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger D.F. and Gloster M.A. (2004) Drugs, 64, 1419–1432. [DOI] [PubMed] [Google Scholar]
- Li Y., Xu W., Mu Y., Zhang J.Z.H. (2013) J. Chem. Phys., 139, 055102. [Google Scholar]
- Luca S., Yau W.-M., Leapman R., Tycko R. (2007) Biochemistry, 139, 13505–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinius A., Wilander E., Westermark G.T., Engstrom U., Westermark P. (1989) Diabetologia, 32, 240–244. [DOI] [PubMed] [Google Scholar]
- Lutz T.A. (2013) Obes. Metab., 15, 99–111. [DOI] [PubMed] [Google Scholar]
- Mack C.M., Soares C.J., Wilson J.K. et al. (2010) Int. J. Obes., 34, 385–395. [DOI] [PubMed] [Google Scholar]
- Marek P., Abedini A., Song B., Kanungo M., Johnson M.E., Gupta R.I., Zaman W., Wong S.S., Raleigh D.P. (2007) Biochemistry, 46, 3255–3261. [DOI] [PubMed] [Google Scholar]
- Marek P., Woys A.M., Sutton K., Zanni M.T., Raleigh D.P. (2010) Org. Lett., 12, 4848–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek P.J., Patsalo V., Green D.F., Raleigh D.P. (2012) Biochemistry, 51, 8478–8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier D.T., Tu L.H., Zraika S., Hogan M.F., Templin A.T., Hull R.L., Kahn S.E. (2015) J. Biol. Chem., 290, 30475–30485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M.R. and Raleigh D.P. (1999) J. Mol. Biol., 294, 1375–1385. [DOI] [PubMed] [Google Scholar]
- Opie E.L. (1901) J. Exp. Med., 5, 517–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S.M. and Alexandrescu A.T. (2015) J. Diabetes Res., 2015, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter K.J., Abedini A., Marek P.J. et al. (2010) Proc. Natl. Acad. Sci. U. S. A., 107, 4305–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quittot N., Sebastiao M., Al-Halifa S., Bourgault S. (2018) Bioconjug. Chem., 29, 517–527. [DOI] [PubMed] [Google Scholar]
- Raleigh D.P., Zhang X., Hastoy B., Clark A. (2017) J. Mol. Endocrinol., 59, R121–R140. [DOI] [PubMed] [Google Scholar]
- Ridgway Z., Zhang X., Wong A.G., Abedini A., Schmidt A.M., Raleigh D.P. (2018) Biochemistry, 57, 3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriquez C., Diana C., Tripsianes K. et al. (2017) Si Rep., 7, 44041. [Google Scholar]
- Stridsberg M., Sandler S., Wilander E. (1993) Regul. Pept., 45, 363–370. [DOI] [PubMed] [Google Scholar]
- Tu L.-H., Serrano A.L., Zanni M.T., Raleigh D.P. (2014) Biophys. J., 106, 1520–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udayasankar J., Kodama K., Hull R.L., Zraika S., Aston-Mourney K., Subramanian S.L., Tong J., Faulenbach M.V., Vidal J., Kahn S.E. (2009) Diabetologia, 52, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Abedini A., Ruzxicska B., Raleigh D.P. (2014) Biochemistry, 53, 5876–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.I., Bellows C.L., Ahn J.S., Burkey J.L., Taylor S.W. (2012) Bioanalysis, 4, 2141–2152. [DOI] [PubMed] [Google Scholar]
- Westermark G.T., Davalli A.M., Secchi A., Folli F., Kin T., Toso C., Shapiro A.J., Korsgren O. (2012) Transplantation, 93, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P., Andersson A., Westermark G.T. (2011) Physiol. Rev., 91, 795–826. [DOI] [PubMed] [Google Scholar]
- Westermark P., Engstrom U., Johnson K.H., Westermark G.T., Betsholtz C. (1990) Proc. Natl. Acad. Sci. U. S. A., 87, 5036–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P., Wernstedt C., Wilander E., Hayden D.W., Obrien T.D., Johnson K.H. (1987) Proc. Natl. Acad. Sci. U. S. A., 84, 3881–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltzius J.J., Sievers S.A., Sawaya M.R., Cascio D., Popov D., Riekel C., Eisenberg D. (2008) Protein Sci., 17, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltzius J.J., Sievers S.A., Sawaya M.R., Eisenberg D. (2009) Protein Sci., 18, 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A.G., Wu C., Hannaberry E., Watson M.D., Shea J.E., Raleigh D.P. (2016) Biochemistry, 55, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. and Shea J.E. (2013) PLoS Comput. Biol., 9, e1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.