

Abstract
α-Synuclein misfolding results in the accumulation of amyloid fibrils in Parkinson’s disease. Missense protein mutations (e.g. A53T) have been linked to early onset disease. Although α-synuclein interacts with synaptic vesicles in the brain, it is not clear what role they play in the protein aggregation process. Here, we compare the effect of small unilamellar vesicles (lipid composition similar to synaptic vesicles) on wild-type (WT) and A53T α-synuclein aggregation. Using biophysical techniques, we reveal that binding affinity to the vesicles is similar for the two proteins, and both interact with the helix long axis parallel to the membrane surface. Still, the vesicles affect the aggregation of the variants differently: effects on secondary processes such as fragmentation dominate for WT, whereas for A53T, fibril elongation is mostly affected. We speculate that vesicle interactions with aggregate intermediate species, in addition to monomer binding, vary between WT and A53T, resulting in different consequences for amyloid formation.
Keywords: amyloid fibrils, kinetic profiles, membrane-bound α-synuclein, protein aggregation, small unilamellar vesicles
Introduction
The intrinsically disordered protein α-synuclein is expressed ubiquitously in the brain (Kim et al., 2014). Proteinaceous deposits composed of amyloid fibrils of α-synuclein have been identified as the major constituent of Lewy bodies, intracellular neuronal inclusions that are the hallmark of a related group of neurodegenerative disorders including Parkinson’s disease (PD) (Spillantini et al., 1998; Kim et al., 2014). A series of mutations in the α-synuclein gene linked to early-onset forms of the disorder, including A53T, have been identified in families with PD (Polymeropoulos et al., 1997; Kruger et al., 1998). The kinetics of the aggregation process of mutants have been extensively studied but the mechanistic effects of the mutations on α-synuclein amyloid formation have yet to be understood (Conway et al., 2000; Li et al., 2001, 2002; Lemkau et al., 2012). In most reported studies, A53T variant aggregates at faster rates compared to the wild-type (WT) protein. Interestingly, single-molecule Förster resonance energy transfer studies showed that WT and A53T α-synuclein variants form similar concentrations of oligomers during the lag phase of the aggregation but the conversion from initially formed oligomers to more compact aggregates, rich in extended β-sheet structures, varies considerably between the variants (Tosatto et al., 2015). A more detailed investigation of the effect of lipid membranes on the amyloid fibril formation by α-synuclein is physiologically relevant considering that the protein is present in presynaptic nerve termini and appears to interact with synaptic vesicles (Maroteaux et al., 1988; Nakajo et al., 1990; Iwai et al., 1995). α-Synuclein membrane binding is driven by the N-terminus of the protein and leads to α-helix structure formation (Bartels et al., 2010). It has been established that the effect of membranes on the aggregation of α-synuclein is dependent on the lipid-to-protein (L/P) ratio, the chemical structure of the lipids and the lipid molecule ordering (Pfefferkorn et al., 2012; Galvagnion et al., 2016; Kiskis et al., 2017). Studies have shown that lipid membranes can both speed up and slow down the aggregation of α-synuclein (Zhu et al., 2003; Martinez et al., 2007; Reynolds et al., 2011; Pfefferkorn et al., 2012; Hellstrand et al., 2013; Galvagnion et al., 2015, 2016; Kiskis et al., 2017). Furthermore, single point mutations in the sequence of α-synuclein can affect the protein–membrane interaction. The aggregation of the A53T mutant, for example, was found to be enhanced in the presence of DMPS (1,2-dimyristoyl-sn-glycero-3-phospho-l-serine) vesicles when compared to the WT, again suggesting differences between the two variants with respect to mechanisms and/or intermediate species formation during aggregation (Flagmeier et al., 2016). Many questions regarding lipid–α-synuclein interactions remain, especially with respect to modulation of amyloid formation pathways. Here, we used biophysical methods and purified proteins to study the consequences on protein aggregation upon interaction of WT and A53T α-synuclein with small unilamellar lipid vesicles (SUVs) composed of phosphatidylcholine, phosphatidylethanolamine (zwitterionic lipids), phosphatidylserine (negatively charged lipid) and cholesterol to mimic synaptic lipid membranes (Lim and Wenk, 2010).
Materials and Methods
Chemicals
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and 1,2-dioleoyl-sn-glycero-3-phospho-L-serine, sodium salt (DOPS) chloroform solutions were purchased from Avanti Polar Lipids. Cholesterol (≥99%), sodium phosphate monobasic (NaH2PO4; ≥99%), sodium phosphate dibasic (Na2HPO4; ≥99.0%), ethylenediaminetetraacetic acid (EDTA) (≥99%), sodium azide (≥99.5%) and thioflavin T (ThT) UltraPure Grade (≥95%) were purchased from Sigma-Aldrich.
Protein expression and purification
Human WT and A53T α-synuclein were expressed in Escherichia coli. The proteins were prepared by transforming their constructs into BL21 (DE3) (Novagen) cells. The bacteria were first grown to an OD600 of 0.6 in Luria broth (LB) containing 100 μg/ml carbenicillin at 37°C and then induced with 1 mM IPTG (isopropyl β-d-1-thiogalactopyranoside) followed by expression overnight at 25°C. The cells were harvested and then lysed by sonication on an ice bath through a sonicator probe in pulse mode in 20 mM Tris-HCl buffer pH 8.0 in the presence of protease inhibitor cocktail (Roche). After the sonication, the lysate was treated with a universal nuclease (Pierce) for 15 min at room temperature. The lysate was then heated at 90°C for 10 min and centrifuged for 30 min at 15 000g. The supernatant was filtered through 0.2 μm filter, loaded on to a pre-equilibrated 5 ml HiTrap Q FF anion exchange column (GE Healthcare) and eluted by linear gradient with 1 M NaCl in 20 mM Tris-HCl buffer pH 8.0. Fractions containing WT or A53T α-synuclein were combined and concentrated with Ultra-15 Ultracel 10K centrifugal filter devices (Millipore). The concentrate was loaded on to Hiload 16/600 Superdex 75 pg column (GE Healthcare) and retrieved in 20 mM Tris-sulfate buffer pH 7.4. For all purified proteins, the sample purity was confirmed by a single band on SDS-PAGE gel and a single elution peak in size exclusion chromatography. Fractions containing pure protein were pooled, frozen in liquid nitrogen and stored at −80°C. The concentration of WT and A53T α-synuclein was determined using ε280 = 5960 M−1 cm−1.
Seed fibril formation
Seed fibrils of WT and A53T α-synuclein were produced by incubating 500 μl of 200 μM of each protein in 20 mM Tris-sulfate buffer, pH 7.4, for 48 h at 37°C under agitation with three glass beads of 2 mm using an Eppendorf ThermoMixer® C (speed set to 800 rpm). Fibrils were diluted with 20 mM phosphate buffer, pH 6.5, to a monomer equivalent concentration of 50 μM and sonicated for 10 s using a probe sonicator (stepped microtip and Ultrasonic Processor Sonics Vibra-Cell) at an amplitude of 20% and alternating cycle of 5 s on and 5 s off.
Lipid vesicle preparation
DOPC:DOPS:DOPE:cholesterol vesicles with a molar ratio of 30:20:25:25 were prepared by the lipid film hydration method. Appropriate volumes of chloroform solution of the lipids DOPC, DOPS, DOPE and cholesterol were transferred to a round bottom flask, and the organic solvent was removed by rotary evaporation. The resultant film was further dried under vacuum for at least 3 h and then hydrated with 20 mM phosphate buffer, pH 6.5, and with 1 mM EDTA. The size of the liposomes was reduced by sonication on ice for 12 min (ultrasonic probe with an amplitude of 40%, pulse on 10 s, off 50 s). The size of the liposomes was measured by dynamic light scattering using a Zetasizer Nano ZS instrument (Malvern Instruments Ltd, UK).
Circular dichroism spectroscopy
Far-UV circular dichroism (CD) spectra of WT and A53T α-synuclein (5 μM) titrated with liposomes were recorded at 37°C on a Jasco J-810 spectropolarimeter. CD spectra between 260 and 190 nm were obtained using a quartz cuvette with a path length of 1 mm, a bandwidth of 1 nm, a step size of 1 nm and a scanning speed of 50 nm/min. Three individual spectra were acquired and averaged for each condition. A mock titration of liposomes into buffer without protein was used to obtain control curves, which were subtracted from the CD signal of the protein samples. Corrections for dilution during the titration were made, and the resulting intensities in millidegrees were converted to mean residue molar ellipticity. CD titration curves were fitted using a one-step binding model: F + LipidL ⇆ B (Lipid)L, where F and B are the fractions of α-synuclein (αS) protein free in solution and bound to the vesicles, respectively, and L is the number of lipid molecules per one molecule of protein (Galvagnion et al., 2015; Kiskis et al., 2017). Then,
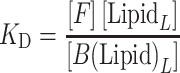 |
(1) |
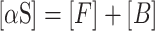 |
(2) |
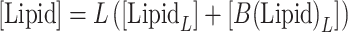 |
(3) |
CD signals can be expressed as a sum of contributions from α-synuclein (αS) protein bound to the vesicles, 𝑥B = [B]/[αS], and free in solution, 𝑥F = [F]/[αS]:
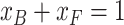 |
(4) |
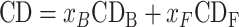 |
(5) |
where CDB and CDF are the CD signals of the protein bound and free, respectively.
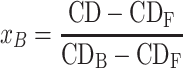 |
(6) |
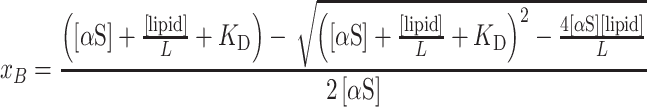 |
(7) |
The CD titration curves were fitted using equations (6) and (7) and Matlab software.
Linear dichroism spectroscopy
Linear dichroism (LD) spectra were recorded on a Chirascan CD spectrometer between 200 and 350 nm in 1 nm increments at a time per point of 0.7 s and a bandwidth of 1 nm. The alignment of the liposomes was achieved by a custom-made outer-cylinder-rotation Couette flow cell with a path length of 1 mm. The shear rate was 3100 s−1. Baselines at zero shear gradients were collected and subtracted from all spectra. The macroscopic orientation of the membrane was probed using curcumin absorption band at 424 nm (Rocha et al., 2016). Stock solutions of curcumin (Sigma-Aldrich) were prepared with ethanol (99.7%), and a specific volume (less than 0.5% of the total sample volume) was added to the vesicles in order to have a final concentration of about 2 μM. The measurements were done in a high viscosity buffer (containing 50 wt% sucrose) to reduce the light scattering of the liposomes by matching their refractive index (Ardhammar et al., 2002; Rocha et al., 2016).
Measurement of aggregation kinetics
Each protein was loaded on a gel filtration column prior to setting up the kinetic measurements and eluted with 20 mM phosphate buffer, pH 6.5, and with 1 mM EDTA. Samples containing WT or A53T variant (30 or 50 μM), 12 mM sodium azide, 20 μM ThT and vesicles at lipid concentrations of 0.75, 1.25, 2 and 3 mM were prepared. A volume of 50 μl/well of each sample was incubated in non-binding polystyrene 96-well half-area plates with clear bottom (Corning Life Sciences) at 37°C in the presence of a glass bead under agitation (5 min before each measurement). For the seeding experiment, preformed fibrils were added to the samples and the plates were incubated under quiescent conditions. The ThT fluorescence signal was monitored every 20 min (excitation wavelength 440 nm, emission wavelength 480 nm) using a microplate reader (FLUOstar Optima, BMG Labtech).
Atomic force microscopy
For atomic force microscopy (AFM) imaging, samples were diluted with Milli-Q water (10–20 times) and deposited on freshly cleaved mica. After 15 min, the mica was rinsed with Milli-Q water and dried under a gentle nitrogen stream. Images were recorded in intermittent contact mode in air using an NTEGRA Prima setup (NT-MDT) and a gold-coated single crystal silicon cantilever (NT-MDT, NSG01, spring constant of ∼5.1 N/m) and a resonance frequency of ∼180 kHz. Images were analyzed using the Gwyddion software.
Results
Binding of WT and A53T α-synuclein to synaptic-like vesicles
We measured the far-UV CD spectra of WT and A53T α-synuclein in the presence of SUVs composed of DOPC:DOPS:DOPE:cholesterol (at a molar ratio of 30:20:25:25), and we find the α-helix content of the proteins to increase as a function of [lipid]:[protein] (L/P) ratios (Fig. 1). As previously observed for other lipid vesicles, the CD signal at 222 nm of the proteins varies almost linearly with increasing SUV concentration until saturation is attained. The data are well described by a one-step binding model similar to previous studies (Galvagnion et al., 2015; Kiskis et al., 2017). The number of lipid molecules associated with each protein, L, and the lipid–protein dissociation constants KD, obtained from the fits, are similar for WT (L = 143, KD = 2.5 × 10−8 M) and A53T (L = 137, KD = 6.5 × 10−8 M).
Fig. 1.
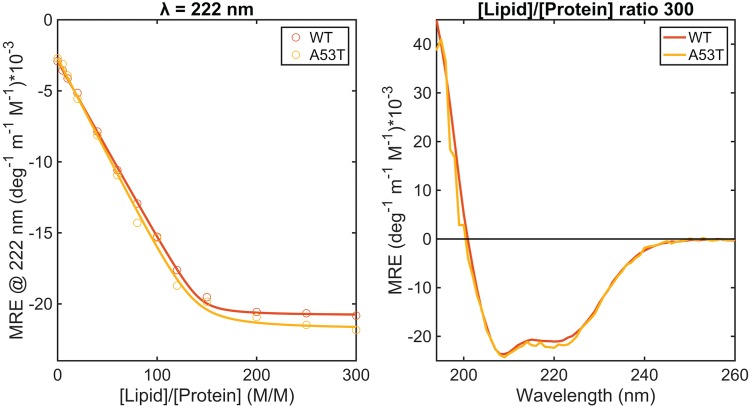
Structural changes of WT and A53T α-synuclein at pH 6.5 in the presence of DOPC:DOPS:DOPE:cholesterol SUVs at different L/P ratios. CD signal at 222 nm (left graph; the fit to a single-step binding model is represented by the lines) and CD spectra of the proteins in the presence of vesicles at a L/P ratio of 300 (on the right). The protein concentration was 5 μM.
Orientation of WT and A53T helices at synaptic-like membrane
We probe the orientation of WT and A53T α-synuclein at the membrane by flow LD, which is defined as the difference in the absorption between parallel and perpendicularly polarized light and reports on the orientation of the electronic transition moments. Thus, the technique requires samples to have a net orientation, which here is attained by subjecting the vesicles to shear flow in a Couette cell. If protein molecules are randomly oriented, then LD = 0. However, proteins bound to the vesicles are aligned relative to the flow and expected to give an LD signal at their absorption bands (Caesar et al., 2009). The geometry of the experiment is as follows: the flow direction creates the long axis of the vesicle but the membrane normal (which is perpendicular to the vesicle long axis) is the molecular orientation direction (Nordén et al., 2010; Rocha et al., 2016). The alignment of the DOPC:DOPS:DOPE:cholesterol SUV vesicles was confirmed by adding the probe curcumin, which binds to the membrane surface (Fig. S1) (Rocha et al., 2016). The LD spectrum of WT and A53T proteins bound to vesicles at a L/P ratio of 28 is dominated by a negative LD band at around 225 nm and a strong positive maximum at ∼208 nm (Fig. 2). Typical absorption spectra of α-helices in the region 180–240 nm region show features of the n–π* amide transition at ∼220 nm, which is perpendicularly polarized to the helix axis, and the exciton coupling of the π–π* transition that is split into two orthogonally polarized bands at 195 and 208 nm, polarized perpendicular and along the helix axis, respectively. Thus, LD experiments indicate that WT and A53T α-helices are oriented with their long axis perpendicular to the membrane normal. Though, the conformation of the membrane-bound helical segment of each variant is slightly different as the ratio between the 208 and 225 nm bands is not the same and the bands of WT are marginally shifted to higher wavelengths, which could be an indication of a more hydrophobic environment for the A53T helix. The absolute value of the LD signal at 208 and 225 nm decreases over time, suggesting a rearrangement of the orientation of the helices at the membrane (Fig. 2). The weak band at around 280 nm in the LD spectrum of WT is attributed to the Lb π–π* transition of the aromatic residue tyrosine present in the N-terminal. There are four tyrosine residues in WT and A53T variants, one in the N-terminal and three in the C-terminal. The UV transition polarization at 280 nm arises probably from the tyrosine present in the N-terminal (at position 39) since α-synuclein binds to the membranes through the residues 1–100. The transition moment of the UV absorption Lb (centered at 280 nm) of the benzene ring of tyrosine is perpendicular to the symmetry axis (C1–C4) and to the transition moment of the UV absorption La (around 230 nm), polarized parallel with the symmetry axis (Fornander et al., 2014). The La band is obscured due to overlapping absorption of the protein backbone, whereas the negative LD in the Lb band region 250–290 nm reveals that the Lb transition moment is oriented parallel to the membrane normal. This band is barely visible in the A53T LD spectra, supporting the hypothesis that the membrane-bound structures of the two variants are somewhat different.
Fig. 2.
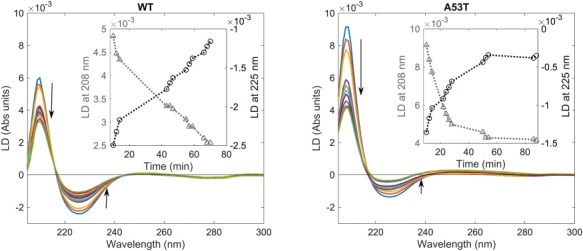
Orientation of WT and A53T α-synuclein at the vesicle surface monitored by LD in high viscosity buffer (20 mM phosphate buffer, 1 mM EDTA and 50% wt % sucrose). The spectra were measured over 10 min (arrows indicate time in increasing order). The protein concentration was 30 μM and the L/P ratio was 28. Inset shows the signal at 225 (circles) and 208 nm (triangles) as a function of time.
Aggregation of α-synuclein variants in the presence of synaptic-like vesicles
We monitored, using the ThT fluorescence assay, the kinetics of aggregation of WT and A53T α-synuclein in the presence of SUVs at 37°C under agitation with a borosilicate glass bead of 2 mm. The presence of only protein monomers at the starting point was ensured by performing gel filtration chromatography of the samples directly prior to each aggregation experiment. The concentration of the proteins was 50 μM and the L/P ratios (M/M) were 15 (0.75 mM total lipids), 25 (1.25 mM lipids), 40 (2 mM lipids) and 60 (3 mM lipids). Agitation in combination with a glass bead is used to increase the reproducibility of fibrillation onset and the dominant nucleation mechanism is thought to be fragmentation of the initially formed fibrils (Giehm et al., 2011). We first confirmed that the integrity of the liposomes is not affected by the presence of a glass bead (Fig. S2).
The ThT fluorescence results of the proteins incubated with a glass bead in the absence of SUVs show sigmoidal aggregation profiles with a lag phase of around 23 h for WT and 7 h for the A53T mutant, a growth phase until about 40 h for WT and 14 h for A53T followed by a plateau (Fig. S3). Under shaking and glass bead conditions, we observed that the WT aggregation rates are affected by SUVs at a L/P ratio of 25 or higher. For A53T, the aggregation rate in the presence of SUVs remains fast and comparable to that of the protein alone with higher final ThT fluorescence intensity (Fig. 3). The normalized profiles show a small increase in the lag phase from 7 to 11 h at the L/P ratio of 60 (lipid concentration of 3 mM) for A53T (Fig. 4). Maintaining the lipid concentrations (0.75, 1.25, 2, 3 mM) but decreasing the protein concentration from 50 to 30 μM, we observed that the α-synuclein aggregation rates slowed down markedly in the presence of SUVs in a concentration-dependent manner even for A53T (L/P ratios were 25, 42, 67, 100) (Fig. 4 and Fig. S4). We note that in the absence of shaking and glass beads, and regardless of the presence or absence of vesicles, the variants did not show any change in the ThT fluorescence within the time frame that our studies were conducted (75 h).
Fig. 3.
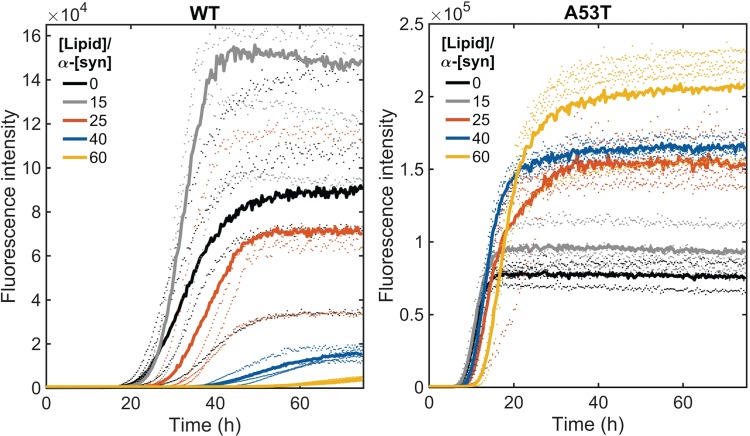
Protein aggregation under agitation in the presence of a glass bead of 2 mm at pH 6.5 and 37°C. ThT fluorescence when WT and A53T were incubated in the absence and presence of SUVs at L/P ratios of 15, 25, 40 and 60. The protein concentration was 50 μM. The averages of four replicate traces are shown in bold with individual traces dotted.
Fig. 4.
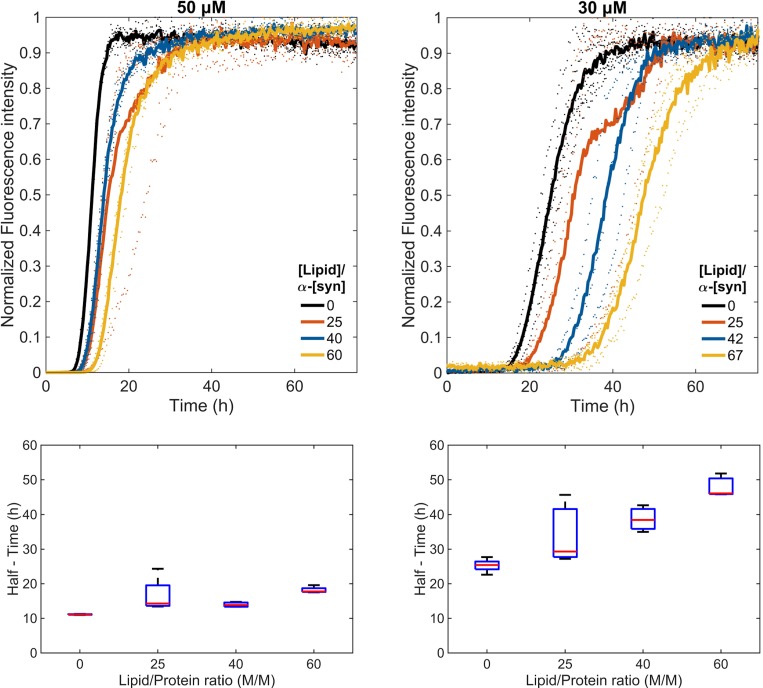
Normalized ThT fluorescence (top) and boxplot of half times (bottom) of A53T incubated at 30 and 50 μΜ in the absence and presence of SUVs at different L/P ratios.
AFM analysis was performed for the end point samples containing 50 μM protein. Imaging of the two protein variants aggregated under agitation with beads in the absence of vesicles shows fibrils with heights of 5–7 nm. For L/P ratios of 60 for WT protein, condition in which the ThT plateau phase was not yet reached, spheroid structures are also present (Fig. 5). In these cases, fibrils appear to be attached to some of the spheroids, which are attributed to SUVs as they appear similar to AFM analysis of vesicles alone (Fig. S2). It is not possible to assess whether the fibrils are formed first in solution and then bind to the vesicles or are formed at the membrane surface. No vesicles/spheroids were detected by AFM in A53T samples of any L/P ratio.
Fig. 5.
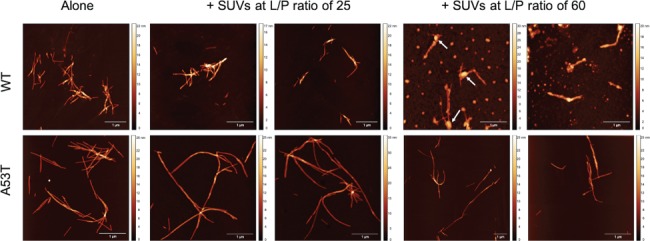
AFM images of WT and A53T fibrils obtained after incubation of 50 μM of protein monomers at pH 6.5 and 37°C for 75 h in the absence and presence of SUVs at L/P ratios of 25 and 60. Spheroid structures bound to fibrils (white arrows) at L/P ratios of 60 for WT protein are attributed to vesicles.
Effect of synaptic-like vesicles on the fibril elongation of α-synuclein variants
We studied the influence of SUVs on fibril elongation by measuring the aggregation kinetics of monomeric WT and A53T α-synuclein (50 μM) in the presence of preformed seed fibrils at a concentration of 5 μM (monomer equivalent) under quiescent conditions at pH 6.5 and 37°C (Fig. 6). At high seed concentration (≥5% of the concentration of monomeric protein), the kinetics of the amyloid formation by α-synuclein are dominated by elongation of the seed fibrils, and thus, the aggregation profile is expected to be a single exponential function as observed for WT and A53T variants at 50 μM incubated with 10% of seed fibrils (Buell et al., 2014; Flagmeier et al., 2016). Though there is a decrease in the final ThT intensity over seeded samples in the presence of SUVs for both variants (Fig. S5), the elongation rate seems to increase for WT, whereas it decreases for A53T with increasing concentrations of vesicles (Fig. 6).
Fig. 6.
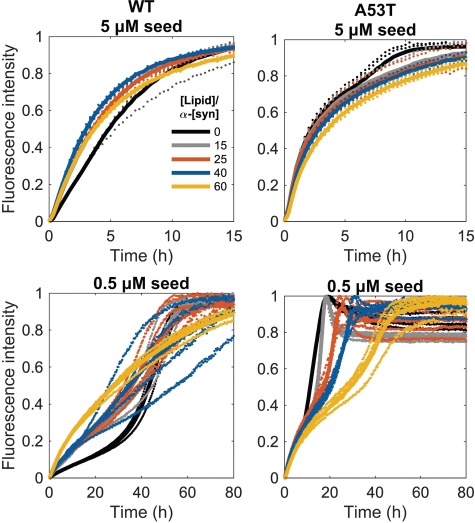
Seeded aggregation kinetics of 50 μM WT and A53T α-synuclein monomers at pH 6.5 and 37°C in the presence of SUVs at L/P ratios of 15, 25, 40 and 60. The seed fibril concentrations are 10 and 1% of the monomer concentration at time 0. The kinetic curves for L/P ratios of 15 and 25 for the WT protein with 5 μM seeds overlap. The data are presented as normalized ThT fluorescence intensity versus time, with four replicates shown as dotted traces and their averages represented by a continuous line. These data are shown non-normalized in Fig. S7.
We then decreased the concentration of seed fibrils to 0.5 μM, reducing therefore the rate of consumption of monomer by fibril elongation. At this lower seed concentration, the time-resolved ThT profiles of the proteins in the absence of SUVs become steeper with time, indicating a growing (with time) contribution of secondary processes (Buell et al., 2014). The presence of SUVs decreases the overall intensity of the ThT fluorescence for aggregation of WT in the presence of 1% seeds, but most notably, it changes the shape of the kinetic trace from convex to concave, in particular, at L/P ratios of 60 (Fig. 6). In contrast, the kinetic profiles of A53T aggregation in the presence of 1% seed fibrils simply shift to the right with the presence of SUVs (i.e. similar to at 10% seed fibrils), indicating an overall delay in aggregation (Fig. 6).
Discussion
The first 25 residues of α-synuclein sequence are described to be crucial for the membrane binding and for the initiation of the folding of the protein (Bartels et al., 2010; Fusco et al., 2014). The removal of only the two first residues is sufficient to change the protein interaction with vesicles of a mixed lipid composition, decreasing significantly both the affinity and the α-helix content when compared to the full-length protein. Therefore, initial binding has been attributed to the first 25 residues of the protein (Bartels et al., 2010). Subsequently to the binding of the first 25 residues, the proposed two-step interaction model states that residues 26–100 will next cooperatively bind and fold (Bartels et al., 2010). Adopting this model, it is not surprising that A53T mutant binds to vesicles and forms α-helix structure much like as the WT protein and with similar affinity. The number of lipid molecules interacting with the proteins in the case of DOPC:DOPS:DOPE:cholesterol SUVs was found to be higher than the numbers obtained previously for purely negatively charged vesicles (Kiskis et al., 2017). The number of DOPS lipid molecules per WT protein was 83, compared to 143 obtained here for DOPC:DOPS:DOPE:cholesterol SUVs, which could be attributed to reduction of the charge density of the membrane in the latter. The electrostatic nature of the interaction between the α-synuclein variants and SUVs is supported by the fact that, in the presence of 150 mM NaCl, the α-helix content of the proteins induced by the vesicles is considerably lower than that in the absence of salt (Fig. S6).
The amphipathic helices of vesicle-bound WT and A53T variants are oriented parallel to the membrane surface similar to what has been described in the literature for WT α-synuclein (Ulmer et al., 2005; Jao et al., 2008; Bodner et al., 2009; Lokappa and Ulmer, 2011; Robotta et al., 2011). However, the LD measurements showed that the orientation and/or conformation of the membrane-bound WT and A53T at L/P ratios of 28 is not the same as the ratio of the 208 and 225 nm bands differs between the proteins. The LD signal from the polypeptide backbone of the membrane-bound variants decreases in magnitude over time, which is an indication of protein orientation changes and/or membrane changes. Membrane disruption would result in a poor vesicle alignment and consequently in the drop of the protein LD signal. The LD changes could also be due to insertion of parts of the helix in the membrane. It is interesting to note that the Lb transition moment of tyrosine (280 nm band) observed for the WT, though weak, does not change over time, strongly suggesting that the changes observed in the α-helix absorption bands are not due to membrane disruption. The variations in the LD spectra are likely a result of overall alterations in the orientation and/or conformation of the proteins combined with local lipid ordering changes triggered by the variants since α-synuclein is known to induce lateral expansion of lipids, thinning of the bilayer and positive mean curvature as well as local negative Gaussian curvature in membranes (Braun et al., 2012; Ouberai et al., 2013).
A quantitative understanding of the aggregation of WT and A53T α-synuclein is difficult to attain due to the complexity and heterogeneity of the process. In this study, similarly to other works, we observed that the overall rate of α-synuclein aggregation increases in the case of the A53T mutation. However, how this single-point mutation changes the kinetics of the amyloid fibril formation is not completely understood (Conway et al., 2000; Lemkau et al., 2012; Flagmeier et al., 2016). Here, the aggregation of WT and A53T α-synuclein occurred in the presence of a glass bead under shaking conditions, which induce bubble formation leading to an increase of the area of the air–water interface, which in turn enhances nucleation (Giehm et al., 2011; Campioni et al., 2014). In addition, under agitation, the fragmentation is favored as it is a more effective source of additional fibrils than primary nucleation. Therefore, the lag time and maximal growth rate will depend primarily on amyloid fibril breakage rates (Knowles et al., 2009). Initially, in the absence of fibrils, oligomers are formed by primary nucleation, but once a critical concentration of amyloid fibrils is exceeded, the secondary process will dominate (Knowles et al., 2009). The critical concentration of monomers at equilibrium with fibrils for A53T was found to be similar to that for WT, and thus, the faster aggregation of A53T was attributed to different nucleation rates rather than a decrease of monomer solubility or increase of fibril stability (Wood et al., 1999). The mutation of alanine at position 53 to threonine seems to increase the β-sheet structure formation and reduce the intramolecular long-range interactions in the N-terminal region in comparison to the WT protein, which could explain the higher propensity of A53T to aggregate (Bussell and Eliezer, 2001; Bertoncini et al., 2005; Camilloni and Vendruscolo, 2013; Coskuner and Wise-Scira, 2013).
Interestingly, under shaking conditions (i.e. secondary processes dominate), we observe that synaptic vesicle mimics significantly reduce the aggregation rate of WT already at 50 μM at the L/P ratios of 40 and 60, but not that of A53T α-synuclein. However, the overall aggregation rate of A53T was decreased by vesicles when the protein concentration was reduced to 30 μM. Assuming that the fraction of protein bound is about 40% at L/P ratios of 60 (determined from the CD data), the amount of free monomers in samples containing 50 μM A53T variant would be ~30 μM. We observe that the aggregation rates of samples with 50 μM protein and SUVs at L/P ratios of 60 are not comparable to those obtained at a monomer concentration of 30 μM (Fig. S7). Thus, these results cannot be explained simple by depletion of monomers from the solution due to the interaction with SUVs, as previously suggested (Galvagnion et al., 2015). Instead, our findings indicate that protein assemblies such as oligomers or fibrils might interact with the vesicles, in addition to monomers. Previous studies demonstrated that dimers of WT and A53T variants bind to lipid vesicles with higher affinity than monomers (Giannakis et al., 2008). Moreover, there are structural variations in oligomeric species between WT and A53T variants, and conversion between different types of oligomers is much faster for A53T mutant than for WT (Tosatto et al., 2015). Thus, variations in oligomer structures and interconversion rates between A53T and WT may explain the observed data in the presence of SUVs.
Aggregation experiments performed in the presence of 10% seed fibrils (i.e. condition where elongation dominates) under quiescent conditions demonstrated that the vesicles slow down elongation of A53T but accelerate that of WT. At conditions where the rate of consumption of monomer by fibril elongation is reduced (1% seed fibrils), the decrease of overall aggregation by SUVs remains for A53T, whereas the aggregation mechanism of WT appears to change. The aggregation of WT at 1% seed fibrils is still accelerated by SUVs, but the shape of the curves is distinctly altered when SUVs are added. We previously described a hidden amyloid formation pathway involving oligomeric WT α-synuclein species that, when bound to membranes, acted as templates for linear growth of thin fibrils and inhibit the typical cooperative aggregation seen for monomers (Kiskis et al., 2017). Something similar may take place in the current experiments: in fact, the shapes of the ThT curves for WT with 1% seed fibrils in the presence of SUVs are similar to the curves found for 10% seed fibrils. This implies that, in the presence of SUV, elongation becomes highly favored despite a low seed level.
Taken together, our study shows that the overall aggregation rate of the two α-synuclein variants WT and A53T decreases in the presence of vesicles, but the microscopic steps in the aggregation mechanisms are clearly affected differently. We speculate that if monomers and early-formed oligomers are depleted above a critical concentration (by vesicle binding) then the protein aggregation is delayed. In contrast, if the conversion to oligomers occurs fast enough and they reach a critical concentration in solution, fibrils will be formed rapidly independently of the presence of vesicles, which will be most likely disrupted by the aggregates. The presence of seeds eliminates the need for primary nucleation, and in this case, SUVs are able to prevent the growth of A53T fibrils, whereas the elongation mechanism is altered for the WT variant. AFM imaging showed that amyloid fibrils can also bind to the vesicles, and this might depend on their morphology and could affect their breakage properties. Accordingly, α-synuclein mutants are known to form amyloids that are morphologically distinct from WT amyloids (Heise et al., 2008; Lemkau et al., 2013; Nielsen et al., 2013; Tosatto et al., 2015). The A53T mutation was found to result in significant structural rearrangements near the mutation site in the amyloid fibrils (Nielsen et al., 2013). As a result A53T oligomers and amyloid fibers may interact differently with vesicles as compared to WT α-synuclein.
To conclude, the present work shows that vesicles with synaptic-like lipid membrane composition delay amyloid formation of WT and A53T α-synuclein when the process is dominated by fibril fragmentation, but the impact of the vesicles on aggregation mechanisms differs between the variants. On the one hand, the aggregation rate of A53T is less reduced by vesicles than that of WT when incubated under shaking conditions. On the other hand, vesicles decrease the elongation rate of A53T but increase this rate for WT. The observed differences are not due to differences in protein–vesicle binding affinity; instead, they appear related to different rates of interconversion and structural variations of intermediate, partially assembled species of WT versus A53T. The results call for further time-resolved studies focusing on the early steps of aggregation of WT and A53T. Differences in aggregation mechanism, and thereby different effects on cellular membranes, may be related to early onset of PD in the case of A53T. From a technical point of view, we want to highlight the use of LD as a complementary method to analyze protein orientation on membranes.
Funding
This work was supported by the Swedish Parkinson Foundation (Parkinsonfonden), the Knut and Alice Wallenberg Foundation, the Swedish Research Council and the Swedish Dementia Association (Demensförbundet).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The work presented here started in the framework of a bachelor’s thesis completed by Agnes Björkman, Johanna Gustafsson, Emma Lorentzon, Anna Olsson, Nathalie Sundvall, Frida Vånder, to whom we are grateful in particular for conducting the first CD measurements of WT α-synuclein and different mutants in the presence of small unilamellar vesicles.
References
- Ardhammar M., Lincoln P., Norden B. (2002) Proc. Natl. Acad. Sci. USA, 99, 15313–15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T., Ahlstrom L.S., Leftin A., Kamp F., Haass C., Brown M.F. et al. (2010) Biophys. J., 99, 2116–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoncini C.W., Fernandez C.O., Griesinger C., Jovin T.M., Zweckstetter M. (2005) J. Biol. Chem., 280, 30649–30652. [DOI] [PubMed] [Google Scholar]
- Bodner C.R., Dobson C.M., Bax A. (2009) J. Mol. Biol., 390, 775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A.R., Sevcsik E., Chin P., Rhoades E., Tristram-Nagle S., Sachs J.N. (2012) J. Am. Chem. Soc., 134, 2613–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buell A.K., Galvagnion C., Gaspar R., Sparr E., Vendruscolo M., Knowles T.P.J. et al. (2014) Proc. Natl. Acad. Sci. USA, 111, 7671–7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussell R. and Eliezer D. (2001) J. Biol. Chem., 276, 45996–46003. [DOI] [PubMed] [Google Scholar]
- Caesar C.E.B., Esbjorner E.K., Lincoln P., Norden B. (2009) Biophys. J., 96, 3399–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilloni C. and Vendruscolo M. (2013) J. Phys. Chem. B, 117, 10737–10741. [DOI] [PubMed] [Google Scholar]
- Campioni S., Carret G., Jordens S., Nicoud L., Mezzenga R., Riek R. (2014) J. Am. Chem. Soc., 136, 2866–2875. [DOI] [PubMed] [Google Scholar]
- Cohen S.I., Vendruscolo M., Dobson C.M., Knowles T.P. (2011) Int. J. Mol. Sci., 12, 5844–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway K.A., Lee S.J., Rochet J.C., Ding T.T., Williamson R.E., Lansbury P.T. (2000) Proc. Natl. Acad. Sci. USA, 97, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskuner O. and Wise-Scira O. (2013) ACS Chem. Neurosci., 4, 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagmeier P., Meisl G., Vendruscolo M., Knowles T.P.J., Dobson C.M., Buell A.K. et al. (2016) Proc. Natl. Acad. Sci. USA, 113, 10328–10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornander L.H., Feng B., Beke-Somfai T., Norden B. (2014) J. Phys. Chem. B, 118, 9247–9257. [DOI] [PubMed] [Google Scholar]
- Fusco G., De Simone A., Gopinath T., Vostrikov V., Vendruscolo M., Dobson C.M. et al. (2014) Nat. Commun., 5, 3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvagnion C., Brown J.W.P., Ouberai M.M., Flagmeier P., Vendruscolo M., Buell A.K. et al. (2016) Proc. Natl. Acad. Sci. USA, 113, 7065–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvagnion C., Buell A.K., Meisl G., Michaels T.C., Vendruscolo M., Knowles T.P.J. et al. (2015) Eur. Biophys. J. Biophy., 44, S101-S. [Google Scholar]
- Giannakis E., Pacifico J., Smith D.P., Hung L.W., Masters C.L., Cappai R. et al. (2008) Biochim. Biophys. Acta Biomembr., 1778, 1112–1119. [DOI] [PubMed] [Google Scholar]
- Giehm L., Lorenzen N., Otzen D.E. (2011) Methods, 53, 295–305. [DOI] [PubMed] [Google Scholar]
- Heise H., Celej M.S., Becker S., Riede D., Pelah A., Kumar A. et al. (2008) J. Mol. Biol., 380, 444–450. [DOI] [PubMed] [Google Scholar]
- Hellstrand E., Nowacka A., Topgaard D., Linse S., Sparr E. (2013) PLoS One, 8, e77235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai A., Masliah E., Yoshimoto M., Ge N.F., Flanagan L., Desilva H.A.R. et al. (1995) Neuron, 14, 467–475. [DOI] [PubMed] [Google Scholar]
- Jao C.C., Hegde B.G., Chen J., Haworth I.S., Langen R. (2008) Proc. Natl. Acad. Sci. USA, 105, 19666–19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W.S., Kagedal K., Halliday G.M. (2014) Alzheimers Res. Ther., 6, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskis J., Horvath I., Wittung-Stafshede P., Rocha S. (2017) Q. Rev. Biophys., 50, 1–9. [DOI] [PubMed] [Google Scholar]
- Knowles T.P.J., Waudby C.A., Devlin G.L., Cohen S.I.A., Aguzzi A., Vendruscolo M. et al. (2009) Science, 326, 1533–1537. [DOI] [PubMed] [Google Scholar]
- Kruger R., Kuhn W., Muller T., Woitalla D., Graeber M., Kosel S. et al. (1998) Nat. Genet., 18, 106–108. [DOI] [PubMed] [Google Scholar]
- Lemkau L.R., Comellas G., Kloepper K.D., Woods W.S., George J.M., Rienstra C.M. (2012) J. Biol. Chem., 287, 11526–11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkau L.R., Comellas G., Lee S.W., Rikardsen L.K., Woods W.S., George J.M. et al. (2013) Plos One, 8, e49750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Uversky V.N., Fink A.L. (2001) Biochemistry USA, 40, 11604–11613. [DOI] [PubMed] [Google Scholar]
- Li J., Uversky V.N., Fink A.L. (2002) Neurotoxicology, 23, 553–567. [DOI] [PubMed] [Google Scholar]
- Lim L. and Wenk M.R. (2010) Handbook of Neurochemistry and Molecular Neurobiology: Neural Lipids, Lajtha A., Tettamanti G., Goracci G. (eds). Springer US, Boston, MA, pp. 223–238. [Google Scholar]
- Lokappa S.B. and Ulmer T.S. (2011) J. Biol. Chem., 286, 21450–21457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L., Campanelli J.T., Scheller R.H. (1988) J. Neurosci., 8, 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Z., Zhu M., Han S., Fink A.L. (2007) Biochemistry USA, 46, 1868–1877. [DOI] [PubMed] [Google Scholar]
- Nakajo S., Omata K., Aiuchi T., Shibayama T., Okahashi I., Ochiai H. et al. (1990) J. Neurochem., 55, 2031–2038. [DOI] [PubMed] [Google Scholar]
- Nielsen S.B., Macchi F., Raccosta S., Langkilde A.E., Giehm L., Kyrsting A. et al. (2013) PLoS One, 8, e67713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordén B., Rodger A., Dafforn T. (2010) Linear Dichroism and Circular Dichroism: A Textbook on Polarized-light Spectroscopy. Royal Society of Chemistry, Cambridge. [Google Scholar]
- Ouberai M.M., Wang J., Swann M.J., Galvagnion C., Guilliams T., Dobson C.M. et al. (2013) J. Biol. Chem., 288, 20883–20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferkorn C.M., Jiang Z.P., Lee J.C. (2012) Biochim. Biophys. Acta Biomembr., 1818, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A. et al. (1997) Science, 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Reynolds N.P., Soragni A., Rabe M., Verdes D., Liverani E., Handschin S. et al. (2011) J. Am. Chem. Soc., 133, 19366–19375. [DOI] [PubMed] [Google Scholar]
- Robotta M., Braun P., van Rooijen B., Subramaniam V., Huber M., Drescher M. (2011) ChemPhysChem, 12, 267–269. [DOI] [PubMed] [Google Scholar]
- Rocha S., Kogan M., Beke-Somfai T., Norden B. (2016) Langmuir, 32, 2841–2846. [DOI] [PubMed] [Google Scholar]
- Spillantini M.G., Crowther R.A., Jakes R., Hasegawa M., Goedert M. (1998) Proc. Natl. Acad. Sci. USA, 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosatto L., Horrocks M.H., Dear A.J., Knowles T.P.J., Dalla Serra M., Cremades N. et al. (2015) Sci. Rep. UK, 5, 16696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer T.S., Bax A., Cole N.B., Nussbaum R.L. (2005) J. Biol. Chem., 280, 9595–9603. [DOI] [PubMed] [Google Scholar]
- Wood S.J., Wypych J., Steavenson S., Louis J.C., Citron M., Biere A.L. (1999) J. Biol. Chem., 274, 19509–19512. [DOI] [PubMed] [Google Scholar]
- Zhu M., Li J., Fink A.L. (2003) J. Biol. Chem., 278, 40186–40197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.