

Abstract
Esophageal squamous cell carcinoma (ESCC) is a common type of esophageal cancer and is prevalent worldwide. Understanding the mechanisms underlying its formation and the search for more effective therapeutic strategies are critical due to the occurrence of chemotherapeutic drug resistance. The aim of the present study was to determine the functional relevance and therapeutic potential of carbohydrate sulfotransferase 15 (CHST15) in ESCC. CHST15 levels were measured in different ESCC cell lines and evaluated in ESCC tissues using tissue chip immunohistochemistry. Cell growth and apoptosis assays, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays, and clonogenic assays were conducted using TE-1 cells and lenti-shCHST15 virus constructs were used to investigate the function of CHST15 in cell proliferation and apoptosis. mRNA microarray analysis was performed to determine the underlying mechanism of CHST15 regulation in TE-1 cell proliferation and apoptosis. The results showed that knockdown of CHST15 inhibited TE-1 cell growth and proliferation, but induced cell apoptosis. CHST15 was more frequently detected in ESCC tissue compared with that in normal esophageal tissue. Microarray data analysis indicated that the inhibition of cell proliferation and activation of cell apoptosis in CHST15-knockdown cells may be caused by altered CHST15/ILKAP/CCND1 and CHST15/RABL6/PMAIP1 signaling axes, respectively.
Keywords: esophageal squamous cell carcinoma, esophageal cancer, carbohydrate sulfotransferase 15, CHST15, cell proliferation, cell apoptosis, shRNA
Introduction
Esophageal squamous cell carcinoma (ESCC) is a predominant type of esophageal cancer (EC) that is among the leading causes of cancer-related death worldwide (1). China has a very high incidence of ESCC, with more than 100 cases per 100,000 population annually. Studies have shown that certain factors are associated with increased risk of ESCC, such as alcohol and tobacco consumption, genetic mutation, diet and nutrition deficiency (2,3).
Similar to other types of cancer, surgery, chemotherapy, radiotherapy, or a combination of these methods are the main treatments for patients with EC. However, with the increasing incidence of esophageal cancer and the poor 5-year survival rate, more research is needed to investigate the underlying mechanisms underlying EC, to determine how to prevent esophageal cancer development, and to discover more effective treatment strategies for patients with EC.
Studies have identified many prognostic markers related to cell proliferation, cell apoptosis, and metastasis in regards to ESCC, e.g., epidermal growth factor receptor (EGFR) has been reported to be associated with the clinical outcome of many types of cancer, including ESCC. EGFR overexpression is positive in most patients with EC (4). In addition, the level of phosphorylated mammalian target or rapamycin (mTOR), which has an important role in intracellular metabolic and anabolic processes, is associated with the poor prognosis of patients with EC (5). B-cell CLL/Lymphoma 2 (BCL2) family proteins, which are regulators of programmed cell death, such as Fas cell surface death receptor (FAS), BCL2 associated X, apoptosis regulator (BAX) and, especially, BCL2 like 1 (BCL-X) were reported to contribute to ESCC progression (6). Octamer-binding protein 4 (OCT4) and SRY-box 2 (SOX2) also have a high prevalence in ESCC (7). Recently, Cheng et al found that solute carrier family 39 member 6 (SLC39A6), a zinc transporter, is associated with ESCC invasiveness (8). Non-coding RNAs have also been identified as essential players in ESCC development (9,10). In most cases, ESCC formation and progression is a complex result of multiple factors, for example, genetic alterations and risk factors of lifestyle. Very recently, Yokoyama et al reported that heavy smoking and drinking substantially accelerate the remodeling process of the esophageal epithelium via numerous driver-mutated clones in ESCC development (11). Overall, ESCC is a heterogeneous disease with variable outcomes. However, there are no widely accepted biomarkers for ESCC screening, treatment response, and recurrence prediction.
Carbohydrate sulfotransferase 15 (CHST15), is a type II transmembrane glycoprotein that acts as a sulfotransferase and participates in chondroitin sulfate E (CS-E) biosynthesis (12). It is widely reported that CS-E plays a pivotal role in tumor progression (13). CHST15 is also expressed in B cells as a membrane-integrated glycoprotein disulfide-linked dimer (14). CHST15 was previously reported to be associated with bone marrow-derived mast cell and pulmonary cell metastasis (15,16), as well as tissue fibrosis formation (17–19). In addition, CHST15 correlates with cancer clinical relevance (20–23). For example, Nishimura et al evaluated the safety and efficacy of a double-stranded RNA oligonucleotide that specifically represses CHST15 for use in patients with pancreatic cancer. The results showed that CHST15 reduction could predict tumor progression and overall survival (20). Ito et al indicated significant associations between CHST15 overexpression and disease-free survival and overall survival of patients with pancreatic ductal adenocarcinoma (21).
In the present study, we investigated the correlation between CHST15 expression and proliferation or apoptosis or both in esophageal cancer cells. We further performed gene chip microarray analysis to elucidate the underlying molecular mechanisms in the regulation of esophageal tumor formation or progression by CHST15.
Materials and methods
Construction of a recombinant lentiviral vector
The target sequence (ACAGCATCACAACTAGGAT) from human CHST15 mRNA (NM_015892) was selected for the knockdown experiment. The sequence of the control short hairpin RNA (shRNA) was TTCTCCGAACGTGTCACGT. The CHST15 shRNA and control shRNA oligonucleotides were designed as stem-loop structures and inserted into vector lenti-GV115-EGFP (GeneChem, Shanghai, China) at the AgeI/EcoRI sites. Recombinant lentiviruses were produced by co-transfection of the shRNA vector and helper vectors (pHelper1.0 and pHelper2.0) into 293T cells. The medium was replaced with fresh medium 6 h post-transfection. After 48 h post-transfection, cell debris was removed from conditioned medium by centrifugation at 4,000 × g and 4°C for 10 min. The conditioned medium was then filtered through a 0.45-µm pore-size filter and centrifuged at 4°C and 100,000 × g for 2 h. Lentiviruses particles were resuspended in fresh medium and stored at −80°C. The lentivirus titer was calculated using a fluorescence titering assay.
Cell culture and recombinant lentivirus infection
Human ESCC cell lines TE-1, Eca-109, and EC9706 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured in CM2-1 medium comprised of 90% Roswell Park Memorial Institute (RPMI)-1640 (Corning, Inc.), 10% fetal bovine serum (FBS; Ausbian) and 1% penicillin-streptomycin (Beyotime Institute of Biotechnology) at 37°C in a 5% CO2 incubator. Cells were subjected to mycoplasma testing before experiments. TE-1 cells were plated on 6-well plates and infected with lenti-shCtrl or lenti-shCHST15 at a multiplicity of infection (MOI) of 10. At 72 h post-infection, the cells were observed under a fluorescence microscope (magnification, ×200) and harvested to determine knockdown efficiency using quantitative RT-PCR or for other purposes.
Target validation of lenti-shCHST15 in 293T cells and western blotting
293T cells were transfected with the CHST15 plasmid (GV143-hCHST15) using Lipofectamine 2000 (Thermo Fisher Scientific, Inc.) and then infected with lenti-shCtrl or lenti-shCHST15, respectively. Briefly, 293T cells were plated on 24-well plates and transfected with 0.5 µg of the constructs and 1 µl of Lipofectamine 2000 when cells reached 80–90% confluence. After 36–48 h of transfection, the cells were rinsed twice with ice-cold phosphate-buffered saline (PBS), collected by scrapping, and lysed using Radioimmunoprecipitation assay (RIPA) buffer (cat. no. P0013B; Beyotime Institute of Biotechnology) supplemented with 1% protease inhibitor cocktail on ice for 10–15 min. Cells were further lysed by sonication at 200 W four times, for 5 sec each time, with a 2-sec interval between pulses. Supernatants were collected after centrifugation at 12,000 × g, at 4°C for 15 min. Protein concentrations were determined using the bicinchoninic acid assay (BCA assay). An amount of 20 µg protein of each sample were loaded onto 10% SDS-PAGE gel. After electrophoresis, the proteins were transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked in 5% non-fat milk at room temperature for 1 h. The blots were then incubated with anti-Flag antibodies (dilution 1:2,000, cat. no. F1804; Sigma-Aldrich;Merck KGaA) or anti-β-GAPDH antibody (dilution 1:2,000; cat. no. sc-32233; Santa Cruz Biotechnology, Inc.) overnight at 4°C followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (dilution 1:2,000; cat. no. sc-2005; Santa Cruz Biotechnology, Inc.) for 2 h at room temperature. Blots were visualized using the enhanced chemiluminescent detection method (GE Healthcare).
RNA extraction and cDNA synthesis
Cells were collected and resuspended in 1 ml TRIzol reagent (cat. no. 3101-100; Pufei). RNA was precipitated using isopropanol and dissolved in RNase-free water. The RNA concentration was measured using a NanoDrop 2000 instrument (Thermo Fisher Scientific, Inc.). cDNA for each sample was obtained via a reverse transcription reaction using a Promega M-MLV kit (cat. no. M1705; Promega). All the above steps were performed according to the manufacturer's instruction.
Quantitative real-time reverse transcription PCR (RT-qPCR)
The qPCR reaction was set up by mixing primers, SYBR TAQ, and cDNA at the proportion according to the manufacturer's instructions (cat. no. DRR041B, SYBR Master Mixture; Takara). The primer sequences for GAPDH were: 5′-TGACTTCAACAGCGACACCCA-3′ (forward) and 5′-CACCCTGTTGCTGTAGCCAAA-3′ (reverse). The primer sequences for CHST15 were: 5′-AACACCACCGACCCCTAC-3′ (forward) and 5′-TGATGGCGGAGAACTTGA-3′ (reverse); the product sizes for GAPDH and CHST15 were 121 and 232 bp, respectively. The qPCR reactions were performed utilizing the Mx3000P qPCR System (Agilent) and at 95°C for 15 sec; followed by 45 cycles at 95°C for 5 sec and 60°C for 30 sec. To compare mRNA levels between different samples, the 2−ΔΔCq method (24) was employed to analyze the data.
Cell growth assay
TE-1 cells infected with lenti-shCtrl or lenti-shCHST15 were plated at 800 cells/well onto a 96-well plate and cultured at 37°C in a 5% CO2 incubator. Cells with enhanced green fluorescent protein (EGFP) fluorescence in each well were counted daily using a Celigo imaging cytometer (Nexcelom) for 5 days. A cell growth curve was drawn (based on cell numbers) by plotting the numbers of fluorescent-positive cells and time-points. For each cell type, the cell proliferation rates were calculated by dividing the cell number at each time-point by the cell number at day 1.
Cell apoptosis assay
Cell apoptosis was assessed using an Annexin V Apoptosis Detection Kit APC (cat. no. 88-8007; eBioscience). TE-1 cells were seeded on a 6-well plate and infected with lenti-shCtrl or lenti-shCHST15. Four days later, the cells were trypsinized and resuspended in fresh complete medium. The cells were washed with pre-cooled D-Hanks and 1X binding buffer. The cells were suspended in 1X binding buffer, stained with Annexin V-APC, and analyzed using a flow cytometer (Guava easyCyte HT; Milllipore). All samples were examined in triplicate.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cell viability after CHST15 knockdown was measured using an MTT assay. Cells were seeded onto 96-well plates at 2,000 cells/well. At each time point, 20 µl of 5 mg/ml MTT was added to each well and incubated with the cells for 4 h. Thereafter, the medium was completely removed and 100 µl of dimethyl sulfoxide (DMSO) was added into each well. After brief shaking, the absorbance at 570 nm of each well was obtained using a microplate reader (M2009PR; Tecan Infinite). Blank wells without cells were included in this experiment. All samples were examined in quintuplicate.
Clonogenic assay
Cells infected with shCtrl or shCHST15 were seeded into 6-well plates at 400–1,000 cells/well. After 10 days of culture, the colonies were washed with PBS and fixed in 4% paraformaldehyde (PFA) for 30–60 min. Colonies were stained with 0.25% crystal violet and counted for analysis. Colonies were counted manually without an instrument. Experiments were performed in triplicate.
Tissue chip immunohistochemistry
The tissue chip used in this experiment included 20 paraffin-embedded sections of human esophageal squamous cell carcinoma and four adjacent tissue sections. Briefly, the tissue chip was incubated in a 60°C incubator for 30 min. It was then soaked in xylene and serial gradient ethanol from 100–75% for rehydration, incubated with 3% H2O2 for 10 min, and stored in distilled water. Antigens in the specimens were retrieved using the sodium citrate (10 mM, pH 6.0) heat-induction method. The tissue chip was blocked in 10% FBS and incubated with anti-CHST15 antibodies (dilution 1:50, cat. no. SAB2701480; Sigma-Aldrich;Merck KGaA) overnight at 4°C. A Vulcan Fast Red Chromogen Kit 2 (Vector Laboratories) was used as the secondary detection system. The slide was then stained with 3,3′-diaminobenzidine (DAB) and hematoxylin. Samples were then dehydrated in gradient ethanol from 75–100% and xylene. The slide was mounted in neutral mounting medium and observed under a microscope (XDS-100; Carl Zeiss). CHST15 expression in each specimen was evaluated based on the percentage of immunopositivity and immunointensity. Each sample was scored based on a range of 0 (non-immunostaining), 1 (weak staining), 2 (moderate staining), 3 (strong staining) based on its immunointensity and scored at the range of 0 (0%), 1 (1–25%), 2 (26–50%), 3 (51–75%), 4 (76–100%) based on its percentage of immunopositivity. The final score of each sample was calculated by multiplying the immunointensity score and the immunopositivity score. Three researchers independently evaluated the chip result to avoid possible biased outcome.
TCGA database and bioinformatic analysis of CHST15 expression
The CHST15 gene expression data sets for esophageal squamous cell carcinoma and adjacent normal tissue were obtained from The Cancer Genome Atlas (TCGA) database (http://cancergenome.nih.gov), containing a total of 185 carcinoma and 13 normal samples. Comparison of the relative mRNA expression levels between two groups of samples was conducted based on the ‘genomic Matrix’ file.
Gene microarray and data analysis
Total RNA was isolated from the shCHST15 and control TE-1 cells and processed via hybridization to the GeneChip Primeview Human gene array (901838; Affymetrix) using a GeneChip® 3′ IVT Plus Kit (Affymetrix) following manufacturer's protocol. Three biologically independent assays were performed. Washing and staining were performed on the chip using a GeneChip hybridization wash and stain kit. The array was processed using the Affymetrix Genechip Fluidics Station 450 system, after which they were imaged using an Affymetrix Genechip Scanner 3000 7G for subsequent generation of cell intensity (.CEL) files. The raw data were evaluated for quality control using signal histogram analysis, relative signal box plot, Pearson's correlation analysis, and principal component analysis. Qualified data were then used for further analysis. After data cleaning, normalized probe sets were subjected to variance analysis, Bayesian P-value computation, and Benjamini-Hochberg false discovery rate (FDR) for multiple testing correction. Differentially expressed genes based on the comparison of CHST15 knockdown (KD) and control cells were selected if |fold change|>1.5 and the FDR was <0.05.
Gene Ontology and pathway analysis of differentially expressed genes were conducted using Ingenuity Pathway Analysis (IPA) software (www.qiagen.com/ingenuity, Qiagen) and Databases for Annotation, Visualization, and Integrated Discovery (https://david.ncifcrf.gov/), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (https://www.genome.jp/kegg/pathway.html).
Gene Set Enrichment Analysis (GSEA) was performed using GSEA software v2.2.1 from the Broad Institute (25) to detect if a series of pre-defined biological processes or gene sets were enriched in the gene rank derived from differentially expressed genes between CHST15-KD and control cells. Gene sets were judged as significantly enriched if P<0.05 and the false discovery rates (FDR) was <0.25 in GSEA.
Statistical analysis
Statistical analysis was performed, and graphs were plotted using Microsoft Excel 2013. P-values were determined using two-tailed Student's t-test in at least three biological replicated experiments. Significance was defined as P<0.05. The results are presented as mean ± standard deviation (SD).
Results
CHST15 expression in ESCC cell lines and CHST15 knockdown in TE-1 cells
We examined CHST15 expression in three different ESCC cell lines: TE-1, Eca-109, and EC9706. Real-time PCR was performed and the ΔCq value (CHST15-GAPDH) of each cell line was calculated for fold change analysis. The data suggested that CHST15 is expressed in these cells, among which TE-1 has higher CHST15 expression than Eca-109 and EC9706 (Fig. 1). To investigate the function of CHST15 in TE-1 cells, a lentivirus CHST15 shRNA construct was designed and produced. CHST15-knockdown (KD) experiments were performed in TE-1 cells. First, we conducted target validation of lenti-CHST15 shRNA (shCHST15). 293T cells overexpressing CHST15 were infected with shCHST15. Western blot analysis showed a significantly decreased CHST15 protein level, which confirmed the targeting of CHST15 by lenti-shCHST15 (Fig. 2A). After 72 h of lenti-shCHST15 infection, CHST15 mRNA expression was significantly reduced by 78% (Fig. 2B) in the shCHST15 group compared with that in the Lenti-shCtrl control cells. Fig. 2C shows the TE-1 cells infected with Lenti-shCHST15 and Lenti-shCtrl constructs, respectively.
Figure 1.
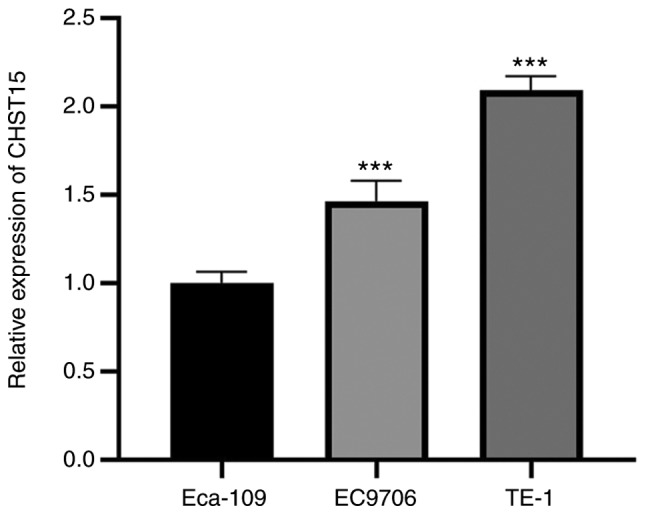
Relative CHST15 expression in three ESCC cell lines. CHST15 mRNA levels were measured using RT-qPCR in TE-1, Eca-109 and EC9706 cell lines. GAPDH gene expression was used as an internal control. The fold change in CHST15 expression in the three different cell lines is presented. n=3, ***P<0.001 (TE-1 vs. Eca-109, EC9706 vs. Eca-109). CHST15, carbohydrate sulfotransferase 15; ESCC, esophageal squamous cell carcinoma.
Figure 2.

Target validation of lenti-shCHST15 virus in 293T cells and CHST15 knockdown in TE-1 cells. (A) CHST15-overexpressing 293T cells were infected with lenti-shCHST15 or lenti-shCtrl. Cell lysates were subjected to western blotting analysis. (B) CHST15 knockdown in TE-1 cells was confirmed by RT-qPCR. n=3, **P<0.01. (C) TE-1 cells were infected with lenti-shCHST15 or lenti-shCtrl viruses and imaged by fluorescence microscope at day 3 after infection. Magnification, ×200. CHST15, carbohydrate sulfotransferase 15.
Identification of CHST15 as a critical gene regulating cell proliferation and apoptosis of TE-1 cells
Three days after shCHST15 infection of TE-1 cells, cell proliferation rates were evaluated and compared between shCHST15 and shCtrl cells. Cells were counted and images were taken daily from both groups for 5 days. Compared with the substantial increase in cell numbers of the shCtrl cells, the shCHST15 KD cells exhibited a significantly reduced proliferation rate (Fig. 3A and B), which indicated that deprivation of CHST15 inhibited TE-1 cell proliferation. This result was further confirmed using MTT assays, which assessed the viability of shCHST15 KD and control cells at different time points (Fig. 3C).
Figure 3.

Inhibition of cell proliferation by CHST15 knockdown (KD) in TE-1 cells. (A) TE-1 cells were infected with lenti-shCHST15 or lenti-shCtrl and imaged using a Celigo imaging cytometer. (B) A cell proliferation curve was made by calculating the fold change of cell numbers at each time-point. (C) Cell viability of CHST15 KD and control TE-1 cells was assessed using the MTT assay. n=3, ***P<0.001 (shCHST15 vs. shCtrl). CHST15, carbohydrate sulfotransferase 15.
In addition, we determined whether CHST15 KD would affect the cell colony formation capacity. As expected, the shCHST15 group formed fewer colonies compared with the shCtrl cells (12 vs. 140) at 10 days after plating the cells on 6-well plates (Fig. 4). These results revealed that CHST15 KD significantly inhibited the colony formation capacity of the TE-1 cells.
Figure 4.

Reduction in cell colony-formation ability by CHST15 knockdown. shCHST15 and shCtrl cells were plated in 6-well plates and after 10 days, colonies in each well were counted for analysis. The experiment was performed in triplicate. **P<0.01. CHST15, carbohydrate sulfotransferase 15.
We speculated that cell apoptosis is also altered in the CHST15 KD cells. At day 4 after infection, shCHST15 and shCtrl cells were stained with Annexin V-APC and analyzed by fluorescence activated cell sorting. The data revealed that the percentage of apoptotic cells was significantly higher in the shCHST15 infected cells (10%) compared with that in the shCtrl cells (2%) (Fig. 5). Taken together, the results suggested that CHST15 has a dual role of promoting proliferation and inhibiting apoptosis in TE-1 cells.
Figure 5.

Enhanced cell apoptosis by CHST15-knockdown (KD) in TE-1 cells. Cell apoptosis was determined by Annexin-V staining and flow cytometry. Increased cell apoptosis was found in CHST15 KD cells compared with the control cells. The experiment was performed in triplicate. **P<0.01. CHST15, carbohydrate sulfotransferase 15.
Significant CHST15 overexpression in esophageal squamous cell carcinoma of patients
These in vitro data prompted us to hypothesize that CHST15 may also be expressed in esophageal squamous cell carcinoma and play a possible role in carcinoma formation and progression in vivo. To determine the clinical significance of CHST15 in ESCC samples, in situ evaluation of CHST15 was conducted using an esophageal squamous cell carcinoma tissue chip which included 20 tissue sections from carcinoma specimens and 4 sections from adjacent normal tissue. Patients and sample information are shown in Table I. Immunohistochemistry for CHST15 in this tissue array is presented in Fig. 6A and B. The immunostaining signal of CHST15 was found only in the cytoplasm, but not in the cell membrane or nuclei. The CHST15 expression level in each tumor tissue specimen was evaluated by a score based on the percentage of immunopositivity and immunointensity. Statistical analysis showed that esophageal squamous cell carcinoma presented a 3.5-fold higher CHST15 level than that noted in the adjacent tissues. The relative CHST15 mRNA expression level in ESCC samples was calculated by retrieving data sets of 185 ESCC samples and 13 adjacent normal samples from the TCGA database (http://cancergenome.nih.gov), which showed that ESCC samples had higher CHST15 expression (Fig. 6C). Overall, these data indicated that CHST15 is overexpressed in esophageal squamous cell carcinoma and may have an important role in ESCC formation or progression.
Table I.
Characteristics of the patients with ESCC (N=20).
| Characteristics | Data |
|---|---|
| Age (mean ± SD) in years | 56.6±7.0 |
| Sex, n (%) | |
| Female | 10 (50) |
| Male | 10 (50) |
| Tumor grade, n (%) | |
| G1 | 8 (40) |
| G2 | 10 (50) |
| GX | 2 (10) |
GX, undetermined grade; ESCC, esophageal squamous cell carcinoma.
Figure 6.

CHST15 overexpression in ESCC tissue specimens. (A) Tissue array consisting of paraffin-embedded sections (a1-d6) of both ESCC and adjacent tissues (shown in the rectangle) were incubated with anti-CHST15 antibodies and stained with DAB and hematoxylin (original magnification, ×40). (B) Higher magnification of representative carcinoma tissue (a2 in the array) and normal adjacent tissue (d3 in the array) with CHST15 staining (magnification, ×400; scale bar, 50 µm). CHST15 expression in each specimen was evaluated and scored based on the percentage of immunopositivity and immunointensity. (C) The TCGA database indicated higher CHST15 mRNA expression in tumor tissues (n=185) when compared with that in normal tissues (n=13). *P<0.05. SD, standard deviation. CHST15, carbohydrate sulfotransferase 15.
Analysis of the mRNA profiles of CHST15 KD and control TE-1 cells
To understand the underlying molecular mechanism of the function of CHST15 in TE-1 cell proliferation and apoptosis, as well as its possible role in ESCC formation and proliferation, we performed genome-wide mRNA microarray analysis to compare the mRNA profiles of the shCHST15 and shCtrl TE-1 group cells. CHST15 KD cells were prepared by lentivirus-shCHST15 infection. Real-time PCR was used to detect the CHST15 knockdown efficiency. The CHST15 mRNA level was reduced by 58.3% compared with that in the control cells. A gene microarray experiment was conducted using shCHST15 and shCtrl cells with three biological repeats. Only qualified RNA samples (1.7< A260/A280 <2.2, RNA Integrity Number ≥7.0) were processed by hybridization to the GeneChip Primeview human gene array. We evaluated the raw data from the microarray by signal histogram analysis, relative signal box plot analysis, Pearson's correlation analysis, and principal component analysis. All of these analyses suggested that the microarray data were suitable for next-step analysis. After data filtering and cleaning, 554 differentially expressed gene transcripts were identified in this gene microarray, among which 188 genes were upregulated and 366 genes were downregulated in the siCHST15 group compared with the shCtrl control group (Fig. 7A and B).
Figure 7.

Differentially expressed genes and pathways in CHST15-knockdown (KD) TE-1 cells. (A) Volcano plot of differentially expressed genes. Red dots represent differentially expressed genes, which were selected with |fold change| >1.5 and false discovery rate (FDR) <0.05. (B) Hierarchical clustering heatmap of differentially expressed genes. Rows represent individual genes (188 upregulated genes and 366 downregulated genes in CHST15-KD cells). Each column shows the gene expression level of each sample from the shCHST15 group and the shCtrl group. (C) Canonical pathways differentially regulated in CHST15-knockdown TE-1 cells were generated by gene enrichment analysis and IPA software. All signaling pathways were ranked by -log10 (P-value). An orange bar indicates an activated pathway. Purple bars represent inhibited pathways. A darker color shade reflects a higher degree of activation or inhibition. CHST15, carbohydrate sulfotransferase 15; IPA, Ingenuity Pathway Analysis.
To identify pathways related to transcriptome changes, we categorized these genes by their associated canonical signaling pathways using Ingenuity Pathway Analysis (IPA) software. Enrichment analysis of differentially expressed genes revealed that the integrin-linked kinase (ILK) and transforming growth factor-β (TGF-β) signaling pathways were inhibited in the CHST15 KD cells, whereas, check point kinase (CHK) proteins in the cell cycle checkpoint control and p53 signaling pathways were activated in the CHST15 KD cells (Fig. 7C). We found that genes encoding proteins involved in the ILK signaling network, such as integrin subunit β6 (ITGB6), phosphatidylinositol- 4-phosphate 3-kinase catalytic subunit type 2β (PIK3C2B), protein tyrosine kinase 2 (PTK2), fermitin family member 2 (FERMT2), and cyclin D1 (CCND1), were downregulated in the shCHST15 TE-1 cells according to the dataset.
We also performed a GSEA analysis using the microarray dataset to gain a further insight into the biological processes that CHST15 may be involved in. This analysis was performed to enrich gene sets from differentially expressed genes between CHST15-KD cells and control cells that share common biological function, chromosomal location, and regulation. GSEA revealed that genes associated with cell growth, proliferation, blood vessel morphogenesis, and tissue development were markedly enriched in the dataset of differentially expressed genes (Fig. 8), suggesting that CHST15 may be involved in these biological processes, which are also features of cancer progression.
Figure 8.

Identification of CHST15-associated biological processes by GSEA. The analysis showed that genes related to cell growth, cell proliferation, cell development, and blood vessel morphogenesis were significantly enriched in the dataset of differentially expressed genes. CHST15, carbohydrate sulfotransferase 15; GO, Gene Ontology; GSEA, Gene Set Enrichment Analysis.
Differentially expressed genes (DEGs) identified in the CHST15 knockdown cells were analyzed according to several functional network criteria within IPA. We generated a network with 22 DEGs related to cell proliferation and apoptosis processes (Table II), showing interactions with CHST15. This network diagram indicated a molecular mechanism by which CHST15 regulates the proliferation and apoptosis of TE-1 cells (Fig. 9).
Table II.
Network of 22 differentially expressed genes identified in the CHST15 knockdown cells related to cell proliferation and apoptosis processes showing interactions with CHST15.
| Gene symbol | Fold change | Location | Family |
|---|---|---|---|
| APCDD1 | −2.249056649 | Plasma membrane | Other |
| BMP2 | 2.552303689 | Extracellular space | Growth factor |
| BTG2 | −1.801089658 | Nucleus | Transcription regulator |
| CCND1 | −1.574172163 | Nucleus | Transcription regulator |
| CHST15 | −2.578621457 | Plasma membrane | Enzyme |
| CLU | −1.726946433 | Cytoplasm | Other |
| CREM | 1.573881221 | Nucleus | Transcription regulator |
| EEF1A1 | −1.600578387 | Cytoplasm | Translation regulator |
| HSPA5 | −1.588890827 | Cytoplasm | Enzyme |
| LEPR | 1.966722394 | Plasma membrane | Transmembrane receptor |
| MIR17HG | 1.509101665 | Other | Other |
| NF1 | 1.510434197 | Cytoplasm | Other |
| PLAU | 1.906482559 | Extracellular space | Peptidase |
| PMAIP1 | 1.714217298 | Cytoplasm | Other |
| PTHLH | 2.256082774 | Extracellular space | Other |
| PTK2 | −1.699664502 | Cytoplasm | Kinase |
| SAE1 | −1.840426328 | Cytoplasm | Enzyme |
| TGFA | −1.540818218 | Extracellular space | Growth factor |
| TIMP3 | −1.590628214 | Extracellular space | Other |
| TPM1 | 1.923481561 | Cytoplasm | Other |
| USO1 | −2.010930656 | Cytoplasm | Transporter |
| WNT5A | −1.526294473 | Extracellular space | Cytokine |
CHST15, carbohydrate sulfotransferase 15; APCDD1, APC down-regulated 1; BMP2, bone morphogenetic protein 2; BTG2, BTG anti-proliferation factor 2; CCND1, cyclin D1; CLU, clusterin; CREM, cAMP responsive element modulator; EEF1A1, eukaryotic translation elongation factor 1 α 1; HSPA5, heat shock protein family A (Hsp70) member 5; LEPR, leptin receptor; MIR17HG, miR-17-92a-1 cluster host gene; NF1, neurofibromin 1; PLAU, plasminogen activator, urokinase; PMAIP1, phorbol-12-myristate-13-acetate-induced protein 1; PTHLH, parathyroid hormone like hormone; PTK2, protein tyrosine kinase 2; SAE1, SUMO1 activating enzyme subunit 1; TGFA, transforming growth factor-α; TIMP3, TIMP metallopeptidase inhibitor 3; TPM1, tropomyosin 1; USO1, USO1 vesicle transport factor; WNT5A, Wnt family member 5A.
Figure 9.

Gene network describing the possible mechanism of the regulation by CHST15 of cell proliferation and apoptosis as determined by IPA. The key to network is indicated in the rectangular box. IPA, Ingenuity Pathway Analysis.
Discussion
Esophageal squamous cell carcinoma (ESCC), a common type of esophageal cancer, is considered a serious malignancy, with a low 5-year overall patient survival rate. Despite the current availability of multiple treatment strategies, the survival rate for ESCC has not significantly improved as many patients present with local advanced disease at the time of diagnosis (3,26). Determining the molecular mechanism underlying ESCC development would be beneficial to identify new diagnostic approaches and therapeutic targets.
In the present study, we showed that carbohydrate sulfotransferase 15 (CHST15) is highly expressed in ESCC cell lines and ESCC tissues. We designed lenti-shCHST15 virus constructs and performed CHST15 knockdown experiments on TE-1 cells. Silencing of CHST15 inhibited TE-1 cell proliferation and promoted TE-1 cell apoptosis, suggesting that CHST15 contributes to the pathogenesis of ESCC. Tissue array immunostaining and bioinformatic analysis of TCGA data sets of ESCC and adjacent normal tissues both showed that CHST15 is overexpressed in ESCC samples, indicating that CHST15 may play an essential role in mediating the tumorigenicity of ESCC cells.
To gain a deeper insight into the molecular function of CHST15 in ESCC cells, a gene microarray assay was conducted to compare the mRNA profiles of CHST15-knockdown cells and control cells. Subsequent Gene Ontology (GO) and KEGG enrichment analysis of the identified differentially expressed genes (DEGs) indicated that CHST15 may be involved in integrin-linked kinase (ILK) and p53 signaling, which regulate cell proliferation and cell apoptosis, respectively. ILKs are important regulators of integrin-mediated signaling. The main function of ILK is to connect integrins to the cytoskeleton. ILK recruits other adaptor molecules into a large complex to regulate actin dynamics and integrin function (27). Overexpression or activation of ILK leads to increased tumor cell proliferation, motility and invasion; thus, ILK may be a promising therapeutic target in many types of cancer (28,29). Downregulated ILK signaling in CHST15-knockdown cells may contribute to the inhibition of cell proliferation. P53 is a well-known tumor suppressor that can inhibit cancer progression by provoking cell growth arrest, by enabling DNA repair, or by advancing cellular death programs (30). In this study, the increased apoptosis of TE-1 cells may be attributable to activated p53 signaling induced by CHST15 knockdown. The results of GSEA also suggested that CHST15 is significantly associated with cell growth and proliferation processes.
By analyzing DEGs and mapping using IPA software, we identified two possible signaling axes, CHST15/ILK associated serine/threonine phosphatase (ILKAP)/CCND1 and CHST15/RAB, member RAS oncogene family like 6 (RABL6)/phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1), which regulate ESCC cell proliferation and apoptosis, respectively. CHST15 is reported to interact with RABL6 (31). RABL6 is a small GTPase belonging to the Ras superfamily, which mainly relays signals from receptors at the cell plasma membrane and modulates many cellular signaling pathways that regulate cell proliferation, differentiation and survival (32). Tang et al demonstrated that knockdown of RABL6 upregulated retinoblastoma 1 (Rb) expression and thus downregulated Rb inhibitory downstream targets, such as cyclin A2, cyclin D1, c-Myc, and cyclin-dependent kinase 2 (33). RABL6 is also a binding partner of PMAIP1, which is a pro-apoptotic member of the BCL2 protein family, but only contains a BH3 domain. PMAIP1 expression was found to activate the mitochondrial apoptotic cascade and induce increased oxidative stress and calcium release, resulting in the activation of apoptosis signal-regulating kinase 1 (ASK1) and its downstream effectors JUN N-terminal kinase (JNK) and mitogen-activated protein kinase 14 (MAPK14, also known as p38) (34). PMAIP1 was also found to regulate p53-induced apoptosis (35). According to the microarray dataset, PMAIP1 expression was increased in the CHST15-knockdown TE-1 cells (Table II). Therefore, CHST15 may promote TE-1 cell apoptosis by interacting with RABL6 and affecting PMAIP1 expression and their subsequent downstream molecules.
CHST15 was also indicated to interact with ILKAP (31), which is a serine/threonine protein phosphatase associated with ILK. It selectively inhibits the ILK-mediated glycogen synthase kinase 3β (GSK3β) signaling pathway and further regulates the Wnt signaling pathway by modulating GSK3β phosphorylation. ILK-mediated inhibition of GSK3β was found to induce the expression of cyclin D1 (36,37). Cyclin D1 (encoded by CCND1) plays a central role in the regulation of proliferation, linking the extracellular signaling environment to cell cycle progression (38). In the CHST15-knockdown TE-1 cells, reduced cyclin D1 levels might be the main reason leading to the inhibition of cell proliferation through the CHST15/ILKAP/CCND1 signaling axis. These observations and predictions provide valuable clues for future detailed investigation of possible CHSR15-associated intracellular signaling pathways.
In summary, the present study investigated the role of CHST15 in cell growth and apoptosis of ESCC and demonstrated the clinical implication of CHST15 in ESCC. CHST15 could be a promising diagnostic marker or therapeutic target for this disease.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China Grants (21335007) and Fundamental Research Funds for the Central Universities (3332018071).
Availability of data and materials
The microarray dataset in this study is not publicly available since we are using this dataset for further studies. However, it is still available from the corresponding author upon reasonable request.
Authors' contributions
LW and NB designed the study and analyzed the data. XW conducted the majority of experiments and wrote the manuscript. QF, DC and ZZ were involved in construction of the recombinant lentiviral vector. WW and LD conducted the cell growth and apoptosis assays and collected the data. XW, KX, XX, and GC conducted the gene chip microarray experiment and bioinformatic analyses. TZ and XW carried out the tissue chip immunohistochemistry experiment. All authors read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics approval and consent to participate
The use of human tissue specimen was approved by the Ethics Committee of National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. The reference number is 19/140-1924. Twenty patients signed consent forms prior to participation.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Napier KJ, Scheerer M, Misra S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol. 2014;6:112–120. doi: 10.4251/wjgo.v6.i5.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Domper Arnal MJ, Ferrández Arenas Á, Lanas Arbeloa Á. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J Gastroenterol. 2015;21:7933–7943. doi: 10.3748/wjg.v21.i26.7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400–412. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Yu JM, Jing SW, Guo Y, Wu YJ, Li N, Jiao WP, Wang L, Zhang YJ. Relationship between EGFR over-expression and clinicopathologic characteristics in squamous cell carcinoma of the esophagus: A meta-analysis. Asian Pac J Cancer Prev. 2014;15:5889–5893. doi: 10.7314/APJCP.2014.15.14.5889. [DOI] [PubMed] [Google Scholar]
- 5.Hirashima K, Baba Y, Watanabe M, Karashima R, Sato N, Imamura Y, Hiyoshi Y, Nagai Y, Hayashi N, Iyama K, Baba H. Phosphorylated mTOR expression is associated with poor prognosis for patients with esophageal squamous cell carcinoma. Ann Surg Oncol. 2010;17:2486–2493. doi: 10.1245/s10434-010-1040-1. [DOI] [PubMed] [Google Scholar]
- 6.Takayama T, Nagao M, Sawada H, Yamada Y, Emoto K, Fujimoto H, Ueno M, Hirao S, Nakajima Y. Bcl-X expression in esophageal squamous cell carcinoma: Association with tumor progression and prognosis. J Surg Oncol. 2001;78:116–123. doi: 10.1002/jso.1130. [DOI] [PubMed] [Google Scholar]
- 7.Nagaraja V, Eslick GD. Forthcoming prognostic markers for esophageal cancer: A systematic review and meta-analysis. J Gastrointest Oncol. 2014;5:67–76. doi: 10.3978/j.issn.2078-6891.2013.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng X, Wei L, Huang X, Zheng J, Shao M, Feng T, Li J, Han Y, Tan W, Tan W, et al. Solute carrier family 39 member 6 gene promotes aggressiveness of esophageal carcinoma cells by increasing intracellular levels of zinc, activating phosphatidylinositol 3-kinase signaling, and up-regulating genes that regulate metastasis. Gastroenterology. 2017;152:1985–1997.e1912. doi: 10.1053/j.gastro.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Jang HJ, Lee HS, Burt BM, Lee GK, Yoon KA, Park YY, Sohn BH, Kim SB, Kim MS, Lee JM, et al. Integrated genomic analysis of recurrence-associated small non-coding RNAs in oesophageal cancer. Gut. 2017;66:215–225. doi: 10.1136/gutjnl-2015-311238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Yu X, Zhang Z, Pang L, Xu J, Jiang J, Liang W, Chai Y, Hou J, Li F. Linc-ROR promotes esophageal squamous cell carcinoma progression through the derepression of SOX9. J Exp Clin Cancer Res. 2017;36:182. doi: 10.1186/s13046-017-0658-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, Shiozawa Y, Sato Y, Aoki K, Kim SK, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019;565:312–317. doi: 10.1038/s41586-018-0811-x. [DOI] [PubMed] [Google Scholar]
- 12.Ohtake S, Ito Y, Fukuta M, Habuchi O. Human N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase cDNA is related to human B cell recombination activating gene-associated gene. J Biol Chem. 2001;276:43894–43900. doi: 10.1074/jbc.M104922200. [DOI] [PubMed] [Google Scholar]
- 13.Takakura K, Shibazaki Y, Yoneyama H, Fujii M, Hashiguchi T, Ito Z, Kajihara M, Misawa T, Homma S, Ohkusa T, Koido S. Inhibition of cell proliferation and growth of pancreatic cancer by silencing of carbohydrate sulfotransferase 15 in vitro and in a xenograft model. PLoS One. 2015;10:e0142981. doi: 10.1371/journal.pone.0142981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verkoczy LK, Guinn Ba, Berinstein NL. Characterization of the human B cell RAG-associated gene, hBRAG, as a B cell receptor signal-enhancing glycoprotein dimer that associates with phosphorylated proteins in resting B cells. J Biol Chem. 2000;275:20967–20979. doi: 10.1074/jbc.M001866200. [DOI] [PubMed] [Google Scholar]
- 15.Ohtake-Niimi S, Kondo S, Ito T, Kakehi S, Ohta T, Habuchi H, Kimata K, Habuchi O. Mice deficient in N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase are unable to synthesize chondroitin/dermatan sulfate containing N-acetylgalactosamine 4,6-bissulfate residues and exhibit decreased protease activity in bone marrow-derived mast cells. J Biol Chem. 2010;285:20793–20805. doi: 10.1074/jbc.M109.084749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizumoto S, Watanabe M, Yamada S, Sugahara K. Expression of N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase involved in chondroitin sulfate synthesis is responsible for pulmonary metastasis. Biomed Res Int. 2013;2013:656319. doi: 10.1155/2013/656319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato H, Sagara S, Nakajima N, Akimoto T, Suzuki K, Yoneyama H, Terai S, Yahagi N. Prevention of esophageal stricture after endoscopic submucosal dissection using RNA-based silencing of carbohydrate sulfotransferase 15 in a porcine model. Endoscopy. 2017;49:491–497. doi: 10.1055/s-0042-123189. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki K, Arumugam S, Yokoyama J, Kawauchi Y, Honda Y, Sato H, Aoyagi Y, Terai S, Okazaki K, Suzuki Y, et al. Pivotal role of carbohydrate sulfotransferase 15 in fibrosis and mucosal healing in mouse colitis. PLoS One. 2016;11:e0158967. doi: 10.1371/journal.pone.0158967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kai Y, Tomoda K, Yoneyama H, Kitabatake M, Nakamura A, Ito T, Yoshikawa M, Kimura H. Silencing of carbohydrate sulfotransferase 15 hinders murine pulmonary fibrosis development. Mol Ther Nucleic Acids. 2017;6:163–172. doi: 10.1016/j.omtn.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishimura M, Matsukawa M, Fujii Y, Matsuda Y, Arai T, Ochiai Y, Itoi T, Yahagi N. Effects of EUS-guided intratumoral injection of oligonucleotide STNM01 on tumor growth, histology, and overall survival in patients with unresectable pancreatic cancer. Gastrointest Endosc. 2018;87:1126–1131. doi: 10.1016/j.gie.2017.10.030. [DOI] [PubMed] [Google Scholar]
- 21.Ito Z, Takakura K, Suka M, Kanai T, Saito R, Fujioka S, Kajihara M, Yanagisawa H, Misawa T, Akiba T, et al. Prognostic impact of carbohydrate sulfotransferase 15 in patients with pancreatic ductal adenocarcinoma. Oncol Lett. 2017;13:4799–4805. doi: 10.3892/ol.2017.6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Steen SC, van Tilborg AA, Vallen MJ, Bulten J, van Kuppevelt TH, Massuger LF. Prognostic significance of highly sulfated chondroitin sulfates in ovarian cancer defined by the single chain antibody GD3A11. Gynecol Oncol. 2016;140:527–536. doi: 10.1016/j.ygyno.2015.12.024. [DOI] [PubMed] [Google Scholar]
- 23.ten Dam GB, van de Westerlo EM, Purushothaman A, Stan RV, Bulten J, Sweep FC, Massuger LF, Sugahara K, van Kuppevelt TH. Antibody GD3G7 selected against embryonic glycosaminoglycans defines chondroitin sulfate-E domains highly up-regulated in ovarian cancer and involved in vascular endothelial growth factor binding. Am J Pathol. 2007;171:1324–1333. doi: 10.2353/ajpath.2007.070111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, Xie X, Zhou C, Peng S, Rao D, Fu J. Which factors are associated with actual 5-year survival of oesophageal squamous cell carcinoma? Eur J Cardiothorac Surg. 2012;41:e7–e11. doi: 10.1093/ejcts/ezr240. [DOI] [PubMed] [Google Scholar]
- 27.Dedhar S. Cell-substrate interactions and signaling through ILK. Curr Opin Cell Biol. 2000;12:250–256. doi: 10.1016/S0955-0674(99)00083-6. [DOI] [PubMed] [Google Scholar]
- 28.Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: A cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5:51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- 29.Shirley LA, McCarty S, Yang MC, Saji M, Zhang X, Phay J, Ringel MD, Chen CS. Integrin-linked kinase affects signaling pathways and migration in thyroid cancer cells and is a potential therapeutic target. Surgery. 2016;159:163–170. doi: 10.1016/j.surg.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stegh AH. Targeting the p53 signaling pathway in cancer therapy-the promises, challenges and perils. Expert Opin Ther Targets. 2012;16:67–83. doi: 10.1517/14728222.2011.643299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 32.Montalbano J, Lui K, Sheikh MS, Huang Y. Identification and characterization of RBEL1 subfamily of GTPases in the Ras superfamily involved in cell growth regulation. J Biol Chem. 2009;284:18129–18142. doi: 10.1074/jbc.M109.009597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang H, Ji F, Sun J, Xie Y, Xu Y, Yue H. RBEL1 is required for osteosarcoma cell proliferation via inhibiting retinoblastoma 1. Mol Med Rep. 2016;13:1275–1280. doi: 10.3892/mmr.2015.4670. [DOI] [PubMed] [Google Scholar]
- 34.Hassan M, Alaoui A, Feyen O, Mirmohammadsadegh A, Essmann F, Tannapfel A, Gulbins E, Schulze-Osthoff K, Hengge UR. The BH3-only member Noxa causes apoptosis in melanoma cells by multiple pathways. Oncogene. 2008;27:4557–4568. doi: 10.1038/onc.2008.90. [DOI] [PubMed] [Google Scholar]
- 35.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 36.Kumar AS, Naruszewicz I, Wang P, Leung-Hagesteijn C, Hannigan GE. ILKAP regulates ILK signaling and inhibits anchorage-independent growth. Oncogene. 2004;23:3454–3461. doi: 10.1038/sj.onc.1207473. [DOI] [PubMed] [Google Scholar]
- 37.Leung-Hagesteijn C, Mahendra A, Naruszewicz I, Hannigan GE. Modulation of integrin signal transduction by ILKAP, a protein phosphatase 2C associating with the integrin-linked kinase, ILK1. EMBO J. 2001;20:2160–2170. doi: 10.1093/emboj/20.9.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang K, Hitomi M, Stacey DW. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 2006;1:32. doi: 10.1186/1747-1028-1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The microarray dataset in this study is not publicly available since we are using this dataset for further studies. However, it is still available from the corresponding author upon reasonable request.