

Abstract
Doxorubicin (DOX), or Adriamycin, an anthracycline antibiotic discovered serendipitously as a chemotherapeutic drug several decades ago, is still one of the most effective drugs for treating various adult and pediatric cancers (breast cancer, Hodgkin's disease, lymphoblastic leukemia). However, one of the major side effects of the continuous use of DOX is dose-dependent, long-term, and potentially lethal cardiovascular toxicity (congestive heart failure and cardiomyopathy) in cancer survivors many years after cessation of chemotherapy. In addition, predisposition to cardiotoxicity varied considerably among individuals. The long-held notion that DOX cardiotoxicity is caused by reactive oxygen species formed from the redox-cycling of DOX semiquinone lacks rigorous proof in a chronic animal model, and administration of reactive oxygen species detoxifying agents failed to reverse DOX-induced cardiac problems. In this review, I discuss the pros and cons of the reactive oxygen species pathway as a primary or secondary mechanism of DOX cardiotoxicity, the role of topoisomerases, and the potential use of mitochondrial-biogenesis-enhancing compounds in reversing DOX-induced cardiomyopathy. New approaches for well-designed clinical trials that repurpose FDA-approved drugs and naturally occurring polyphenolic compounds prophylactically to prevent or mitigate cardiovascular complications in both pediatric and adult cancer survivors are needed. Essentially, the focus should be on enhancing mitochondrial biogenesis to prevent or mitigate DOX-induced cardiotoxicity.
Keywords: Chemotherapy, Reactive oxygen species, Topoisomerase, Mitochondrial biogenesis, Cardio-oncology, Cardioprotection
Graphical abstract

Highlights
-
•
Evidence linking ROS to DOX-induced cardiomyopathy is lacking.
-
•
Iron-chelating mechanism of cardioprotection by dexrazoxane is not established.
-
•
Mitochondrial biogenesis-inducing drugs may enhance protection against DOX toxicity.
1. Are today's cancer patients tomorrow's cardiac patients
Thanks to dramatic improvements in cancer treatment, many more cancer patients are living longer, and the number of cancer survivors will continue to increase in the future. In 10 years, it is expected that nearly 70% of adults will live at least five years after being diagnosed with breast cancer [1]. Clearly, the tremendous advances made in new drug discovery and treatment modalities are extending and saving lives. Although this is welcome news, many cancer survivors develop cardiomyopathy (heart muscle dysfunction) years after treatment that undoubtedly decreases the quality of life and often causes life-threatening problems [2]. The side effects from cytotoxic and radiation therapies used to treat cancer cause cardiovascular problems and, in many instances, mortality in cancer survivors stems from such cardiovascular complications [3].
Cardiomyocytes have a lower ability to regenerate and, thus, are potentially more susceptible to the long-term adverse effects of doxorubicin (DOX), one of the most widely used chemotherapeutic drugs [4,5]. Is it possible to delay or prevent such damage? Clearly, eradication of cancer in patients is of primary importance, and the best treatment option is the most effective combination cytotoxic chemotherapy prescribed by oncologists. Co-administering drugs that have been shown to be cardioprotective should not compromise the efficacy of chemotherapy. Discovering new approaches to delay or prevent cardiotoxicity prophylactically in cancer survivors using a post-chemotherapeutic intervention strategy is urgently needed.
This review focuses on the need for potentially new cardiotoxicity preventive strategies and uses DOX as an example, although most cancer treatments are multifaceted and consist of multiple chemotherapeutics, targeted therapies, and radiation.
2. DOX chemotherapy: still going strong after years of cardiotoxicity
DOX, also known as Adriamycin (Fig. 1), is a cytotoxic chemotherapeutic drug that is most widely used as the first line of defense either in combination with other antitumor drugs or in combination with surgery and radiation. DOX is a highly potent and effective cytotoxic drug, either alone or in combination with other cytotoxic antitumor drugs, for eradication of tumors [4]. However, DOX elicits dose-dependent acute and chronic toxic side effects [[6], [7], [8], [9], [10], [11], [12]]. The life-threatening toxic side effects are related to cardiotoxicity (heart failure, cardiomyopathy) that occurs many years after the cessation of cancer treatment and remission of tumor. Children treated for lymphoblastic leukemia with DOX therapy develop cardiac complications in their adulthood (nearly eight times more susceptible), nearly 15–20 years after DOX chemotherapy [11,12]. Most patients who are fully recovered from cancer lead very normal lives until they experience shortness of breath, chest pain, and other signs of cardiovascular dysfunction. When this happens, recovering cancer patients are treated by cardiologists. Currently, there is much interest in understanding the mechanisms of drug-induced cardiotoxicity. Because so much basic mechanistic research has already been done with respect to the DOX mechanism of cardiotoxicity, DOX is chosen as a representative chemotherapeutic drug. Most cytotoxic chemotherapy or radiation therapy and immunotherapy could ultimately result in cardiotoxicity in cancer patients [[13], [14], [15]]; therefore, the more we understand about the basic mechanisms of cardiotoxicity in cancer patients after the cessation of treatment, the better will we be able to mitigate and delay the occurrence of cardiovascular complications.
Fig. 1.
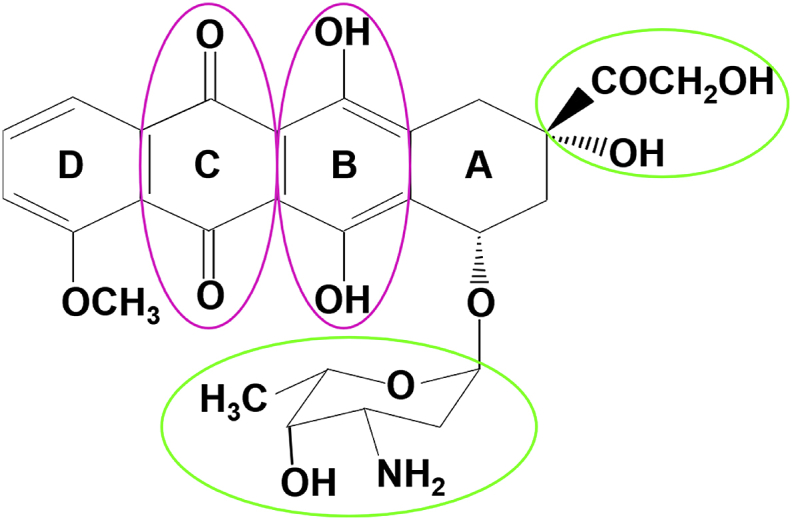
Chemical structure of DOX/Adriamycin. The functional groups that are thought to be critical to the DOX mechanism of action are marked.
3. Redox cycling, the iron story, ICRF-187, and cardioprotection
DOX is a quinone-containing anthracycline family of drugs. Early on, it was shown that several enzymes including the cytochrome P450 reductase, NADH dehydrogenase, and xanthine oxidoreductase could reduce DOX via a one-electron reduction mechanism [[16], [17], [18], [19], [20], [21], [22], [23], [24], [25]], giving rise to the semiquinone intermediate that can rapidly reduce oxygen to superoxide (O2•–) via a futile redox-cycling mechanism (Fig. 2) [16,17,24,25]. In the presence of a redox active metal ion such as iron (III), hydroxyl radicals are formed via the Fenton mechanism. Chelators such as desferrioxamine inhibit the formation of hydroxyl radicals through inhibition of the Haber-Weiss mechanism. Based on these in vitro studies, redox activation of DOX to O2•–, hydrogen peroxide (H2O2), and iron-catalyzed hydroxyl radical formation was suggested to be the predominant mechanism of DOX toxicity [[26], [27], [28], [29], [30]]. Oxidative stress is thought to be primarily responsible for DOX cardiotoxicity because the myocardial tissues lack sufficient antioxidant mechanisms [31]. Targeting ferroptosis (non-apoptotic cell death induced by iron and lipid hydroperoxides) was recently proposed as a strategy for treating DOX-induced cardiomyopathy [32].
Fig. 2.

Redox-cycling of DOX semiquinone and ROS-induced mechanism of mitochondrial oxidation and cardiotoxicity. Reprinted by permission from Springer Nature Customer Service Centre GmbH: Springer Nature Molecular and Cellular Biochemistry (Doxorubicin-induced apoptosis: Implications in cardiotoxicity Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S), Kluwer Academic Publishers (2002).
The target organ of DOX toxicity is the myocardium enriched with mitochondria, and mitochondrial dysfunction was linked to reactive oxygen species (ROS) formation from DOX [33]. DOX accumulates into the mitochondria of cardiomyocytes. In vitro experiments using endothelial cells and cardiomyocytes revealed the redox cycling mechanism of DOX as monitored by inactivation of aconitase, redox dye oxidation, and inhibition by superoxide dismutase mimetics, as well as by overexpression of manganese superoxide dismutase (SOD) and other redox modulators including N-acetyl cysteine [34]. Again, these results supported the redox mechanisms and catalytic role of iron in DOX-induced oxidative damage.
In an acute model of DOX toxicity where DOX was used in much higher concentrations, antioxidants and iron chelators afforded protection against acute damage [35,36]. However, to our knowledge, there exists no experimental proof connecting DOX redox cycling and ROS to enhanced cardiomyopathy or to reversal of cardiomyopathy by established bonafide iron chelators in a chronic animal model.
Dexrazoxane (DXR) is the only FDA-approved cardioprotective drug for treating anthracycline cardiotoxicity and extravasation injury [25,[37], [38], [39], [40]]. DXR (ICRF-187, ZINECARD®, or Cardioxane®) has been shown to provide cardioprotection in DOX-treated children with acute lymphoblastic leukemia (ALL) [[41], [42], [43]]. DXR did not compromise the effectiveness of DOX [44]. A majority of survivors of childhood cancers are at increased risk of cardiovascular complications in their adulthood [43]. Thus, prophylactic intervention is even more critical to mitigate and prevent cardiotoxicity in this group of cancer survivors.
4. Time to rethink redox cycling of DOX and ROS involvement as the primary mechanism of DOX-induced cardiotoxicity?
Despite the many publications [21,45] suggesting that the ROS generated from the redox cycling of DOX in mitochondria is responsible for DOX cardiotoxicity, the rat model designed to test the chronic toxicity of DOX revealed that the ROS mechanism is unlikely to be the key mechanism of cardiotoxicity and that the widely used mitochondria targeted co-enzyme Q (Mito-Q) [46,47] is cardioprotective by inducing other mitochondrial redox signaling mechanisms that are still not yet fully understood [48]. Also, other reports exist that suggest ROS is not involved as a primary mechanism of DOX cardiotoxicity [[49], [50], [51]]. It is conceivable that inhibiting endothelial toxicity and endothelial dysfunction could mitigate DOX-induced cardiomyopathy [52].
5. A rat model of DOX-induced cardiomyopathy
We used a comprehensive DOX-induced cardiomyopathy rat model that closely mimics DOX-induced cardiomyopathy in the clinic [53]. The experimental protocol is shown in Fig. 3. Low doses of DOX were chronically administered to Sprague-Dawley rats once a week (2.5 mg/kg) for 12 weeks and two-dimensional echocardiography was used to assess the morphologic and functional changes in the left ventricle [48]. Animals were randomly assigned to four different treatment groups: vehicle alone, DOX, DOX plus Mito-Q, and Mito-Q alone (Fig. 3). Throughout the duration of the experiment, electrocardiograms were obtained and myocardial tissues were isolated periodically from the four groups of animals and tested by ex vivo electron paramagnetic resonance (EPR) and histology.
Fig. 3.

DOX-induced cardiomyopathy in a rat model and the cardioprotective effect of Mito-Q. Echocardiography, EPR, and biochemical measurements were performed weekly. Reprinted from Biophysics Journal, 96, Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B, Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by Mito-Q, 1388–1398, Biophysical Society (2009), with permission from Elsevier.
6. DOX-induced cardiac dysfunction: Mitigation by Mito-Q
The cardiac function in DOX-treated rats was monitored by two-dimensional echocardiograpy. Two-dimensional strain echocardiography measures myocardial function [48,54]. Myocardial strain is a measure of intrinsic myocardial mechanics. The cardiac function in animals treated with DOX, Mito-Q, and DOX plus Mito-Q was measured every week. There was a progressive decline in heart function starting at eight weeks after DOX treatment. Fig. 4 shows the changes in myocardial strain after treatment of animals with DOX, DOX plus Mito-Q, and Mito-Q alone. As shown, DOX caused a significant reduction in global radial strain which, was reversed by co-administration of Mito-Q (Fig. 4).
Fig. 4.

Mito-Q inhibits DOX-induced cardiac dysfunction—two-dimensional strain echocardiography measurements. (A) End systolic two-dimensional B-mode images at mid- ventricular level. (B) Anatomical M-mode through the anterior and interior walls. (C) The radial strain from six equidistant segments of the left ventricle. Reprinted from Ultrasound in Medicine & Biology, 34, Migrino RQ, Aggarwal D, Konorev E, Brahmbhatt T, Bright M, Kalyanaraman B, Early detection of doxorubicin cardiomyopathy using two-dimensional strain echocardiography, 208–214, World Federation for Ultrasound in Medicine & Biology (2008), with permission from Elsevier.
7. Ex vivo EPR of heart tissues: DOX-induced changes in mitochondrial electron transport redox changes
The low-temperature EPR is a suitable experimental tool with which the status of the redox active component of heart tissues can be assessed [48,55]. Hearts were removed after 4, 8, 10, and 12 weeks of treatment protocols and frozen for EPR analysis. Fig. 5 shows the EPR spectra obtained from different groups at different treatment intervals. The low-temperature EPR spectra of control heart tissues show signals (observed at higher spectrometer gain) from paramagnetic iron sulfur centers from complexes I, II, and III. Most importantly, a strong signal at g = 6 (assigned to heme a3 of cytochrome c oxidase [CcO]) was observed in control heart tissues. As the duration of DOX treatment increased, the intensity of signal at g = 6 decreased, and at 12 weeks of treatment (when there was a significant decrease in myocardial function), the signal intensity was considerably decreased (Fig. 5). This suggests that DOX disrupts CcO in myocardial tissues. In the presence of DOX and Mito-Q, the signal intensity of heme a3 of CcO in restored to that of control heart tissues (Fig. 6). CcO catalyzes the electron transfer from cytochrome c to oxygen, forming water and enabling adenosine triphosphate (ATP) synthesis via proton pumping. Binding of DOX to CcO presumably uncouples the heme a3/CuB site that is involved in oxygen binding. It is plausible that Mito-Q inhibits the CcO/DOX interaction and restores the g = 6 signal responsible for heme a3 of CcO. Consistent with the EPR results, Mito-Q restored the loss of CcO activity induced by DOX, as illustrated in the model (Fig. 7).
Fig. 5.

Time-course EPR spectra of myocardial tissues. Low-temperature EPR of heart tissues isolated from rats treated with (A) DOX and (B) DOX and Mito-Q. Reprinted from Biophysics Journal, 96, Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B, Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by Mito-Q, 1388–1398, Biophysical Society (2009), with permission from Elsevier.
Fig. 6.

Time-course EPR spectral intensity (by monitoring the signal at g = 6.0). Reprinted from Biophysics Journal, 96, Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B, Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by Mito-Q, 1388–1398, Biophysical Society (2009), with permission from Elsevier.
Fig. 7.

DOX-induced inactivation of cytochrome c oxidase:proposed model. Reprinted from Biophysics Journal, 96, Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B, Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by Mito-Q, 1388–1398, Biophysical Society (2009), with permission from Elsevier.
Other studies [31] have shown that DOX could disrupt protein complexes between uncoupling protein 3 and cytochrome c oxidase via mitochondrial targeting of Bcl-2-like 19 kDa-interacting protein 3 (Bnip3). Inhibiting Bnip3 totally abrogated DOX cardiotoxicity. It is not known whether Mito-Q cardioprotection results from Bnip 3 inhibition.
8. Lack of evidence for increased oxidative marker in heart tissues of DOX-treated rat
EPR results showed that the signal intensity at g = 2.015 remained unchanged upon DOX and DOX plus Mito-Q treatments (Fig. 8). This signal is characteristic of oxidatively inactivated iron-sulfur centers of the enzyme aconitase. In the native form of aconitase, the 4Fe–4S center is EPR silent and ROS, such as O2•– or H2O2, react with the 4Fe–4S center and form the 3Fe–4S form of aconitase (inactivated aconitase) that is paramagnetic and EPR active. An increase in the 3Fe–4S center due to oxidative changes is associated with an increase in the g = 2.015 signal. Rotenone or antimycin A and other mitochondria-targeted agents that enhance superoxide formation in mitochondria increased the EPR signal intensity of the 3Fe–4S form of aconitase [55]. However, the 3Fe–4S form of aconitase remained unchanged in DOX-treated heart tissues at 12 weeks, suggesting a lack of evidence for increased oxidative damage being causally related to DOX-induced myocardial toxicity [48].
Fig. 8.

Lack of effect of Mito-Q on oxidative inactivation of aconitase. Reprinted from Biophysics Journal, 96, Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B, Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by Mito-Q, 1388–1398, Biophysical Society (2009), with permission from Elsevier.
9. Cardioprotection by Mito-Q is not related to ROS mechanism in the rat model of cardiomyopathy
Accumulation of mtDNA and respiratory chain defects were reported in chronic DOX cardiomyopathy [56]. Drugs capable of reversing or mitigating respiratory chain defects could decrease cardiomyopathy. DOX treatment caused a decrease in complex IV activity in heart mitochondria [57]. There was no significant decrease in complex IV activity in heart tissues from rats treated with Mito-Q alone; however, Mito-Q significantly restored the complex IV activity in DOX-treated rat heart mitochondrial tissues. Mito-Q clearly causes specific changes that affect the pathway by which DOX induced a decrease in complex IV activity. Mito-Q also inhibited DOX-mediated fibrosis and apoptosis (as measured by caspase-3 activity) in the cardiac tissues obtained from treated animals. Because there was no increase in the oxidative biomarker in DOX-treated and DOX plus Mito-Q treated rat heart tissues, we conclude that the cardioprotective mechanism of Mito-Q is not related to the ROS mechanism in the rat model of cardiomyopathy.
10. Cardioprotection by antioxidants, redox modulators, and iron chelators in cancer patients post DOX therapy: present status
Numerous investigators, including us, have shown that DOX treatment enhances dihydroethidium (DHE) or hydroethidine (HE)-derived red fluorescence; typically, there is a two-to three-fold increase in red fluorescence intensity in DOX-treated cells. Upon treatment with honokiol, iron chelators, or a host of different agents (typically described under the umbrella term “antioxidants”), the red fluorescence intensity decreased. These results suggest that there was increased oxidation of the redox dye, HE, to ethidium (E+), the two-electron oxidation product [55]. Increased apoptosis (release of cytochrome c) and/or increased peroxidatic activity in the presence of H2O2 could be responsible for oxidation of the dye. It is conceivable that other redox dyes (e.g., 2,7-dichlorodihydrofluorescein, dihydrorhodamine) could undergo a similar type of oxidation, giving rise to enhanced fluorescence of the oxidized dye [58]. Unless the products derived from HE are isolated and characterized, it is impossible to obtain any mechanistic details regarding the structure of the nature of oxidants responsible for oxidation of the dye [58].
Recently, a positron emission tomography tracer, 18F-DHMT (an 18F-labeled analog of dihydroethidium), was used to detect superoxide as an early index of cardiomyopathy in rodents [59]. Based on the similarity in reaction chemistry between ROS and hydroethidine and hydromethidine [60], it is impossible to claim that positron emission tomography imaging is due to superoxide-derived product (18F–2OH-ME+) and not to 18F-labeled methidium and 18F-labeled dimeric oxidation products [60]. Thus, any extrapolation of in vitro mechanistic interpretations to the animal model is not realistic, although only a few in vivo studies have used the chronic animal model of DOX cardiotoxicity in which cardioprotection by antioxidants, iron chelators, and redox modulating agents have been tested [48].
DXR (or ICRF-187) has been used in the clinic to mitigate DOX toxicity [41]. DXR was originally developed as an iron chelator, although its iron chelating efficacy and ability to inhibit ROS has been questioned [61]. DXR is a cell-permeable prodrug that is rapidly hydrolyzed to an EDTA-type iron-binding chelator [39]. DXR metabolites formed from hydrolysis did not, however, protect myocytes against DOX toxicity [39,62]. Recent studies have shown that DXR inhibits the catalytic activity of topoisomerase IIβ (TOP2β), so the cardioprotective mechanism of DXR was attributed to TOP2β inhibition rather than to iron chelation and ROS inhibition [61]. Topoisomerase is an enzyme present in all dividing cells and is involved in the mechanism of replication and transcription.
DOX treatment increased mitochondrial iron levels in cardiomyocytes and in mice hearts [63]. ATP-binding cassette (ABC) protein B8 (ABCB8 or ABCI), a member of the ABC protein family, was decreased following DOX treatment. ABCB8 presumably regulates mitochondrial iron export, and overexpression of ABCB8 decreased mitochondrial iron levels and reversed DOX-induced cardiac damage in cardiomyocytes and ABCB8 transgenic mice [63]. Deletion of ABCB8 in the heart exacerbated DOX cardiotoxicity. DXR treatment decreased mitochondrial iron levels and cardiac damage in DOX-treated myocytes and mice. However, administration of more potent iron chelators (e.g., desferrioxamine) had no effect on mitochondrial iron levels or cardiotoxicity in DOX-treated mice [63]. Although, the actual mechanism of cardioprotection afforded by DXR still remains enigmatic, it is plausible that ABCB8 and mitochondrial iron play an important role in DOX cardiotoxicity.
11. Implications of topoisomerase enzymes and not ROS mechanism in DOX cardiomyopathy
The current understanding is that DOX cytotoxicity in tumor cells is not related to ROS formation, and that it is related to the interaction between DOX and TOP2 levels in tumor cells [49,64,65]. Consistent with this proposal, published results suggest that supplementation with antioxidants or antioxidant enzymes in tumor cells does not affect DOX toxicity, ruling out the role of ROS as a mechanism responsible for DOX cytotoxicity in tumor cells [49].
The two forms of TOP2 enzymes are TOP2α and TOP2β. TOP2α is overexpressed in tumor cells but is undetectable in cardiomyocytes. It is well established that the ROS mechanism is not the major pathway by which DOX exerts cytotoxicity in tumor cells. DOX forms a ternary complex, TOP2α-DOX-DNA, that is responsible for DNA breakage and tumor cell death [49,64].
A recent study suggests that TOP2β is also the target of DOX-induced cardiotoxicity [61,66]. Adult cardiomyocytes predominantly express TOP2β but not TOP2α. The TOP2β-DOX-DNA ternary complex can induce DNA double-strand breaks in cardiomyocytes, resulting in cell death [65]. Depletion of TOP2β protected cardiomyocytes from DOX- induced double strand breaks. In the presence of TOP2β, DOX decreased the transcription of genes involved in mitochondrial biogenesis and mitochondrial function [49]. ROS levels were monitored using HE dye. As discussed previously, the use of redox dyes merely reports the two-electron oxidation of the dye; it does not measure ROS levels. Changes in TOP2β levels affect dye oxidation. Even these changes in dye oxidation were attributed to changes in mitochondrial transcriptome and not to redox cycling of DOX semiquinone [49]. A major conclusion of this study is that TOP2β is critical for stimulating DOX- induced cardiomyopathy and that cancer patients with higher levels [65] of TOP2β in cardiomyocytes are likely to be more susceptible to DOX cardiotoxicity [49]. Transgenic mice lacking TOP2β showed resistance to DOX toxicity in acute and chronic settings. TOP2β in cardiomyocytes as a major target in DOX cardiotoxicity is a provocative proposal that requires further testing in cancer patients.
In summary, the therapeutic effect of DOX against tumor cells is mediated through inhibition of Top2α and the cardiotoxic effect of DOX is mediated through inhibition of Top2β [67]. DXR belongs to a class of molecules, bis(2,6-dioxopiperazines), that are known to function as Top2β catalytic inhibitors. These compounds can antagonize the formation of Top2β-DNA complex, thereby inhibiting the formation of the Top2β-DOX-DNA complex and the DNA double-strand breaks in cardiomyocytes.
12. Alternate mechanisms by which mitochondria-targeted agents protect against DOX cardiac toxicity: Autophagy as a potential cardioprotective mechanism?
Autophagy is a well-regulated cellular trash removal process that occurs at the right place and right time [68]. Discovery of this novel phenomenon in cell biology led to a Nobel Prize [69,70]. In cancer biology, the role of autophagy is paradoxical, depending upon the timing during tumorigenesis and tumor progression [71]. The role of autophagy in DOX-induced cardiomyopathy is also contradictory with some studies suggesting that suppression of DOX-induced autophagy in cardiomyocytes is protective and other studies indicating that stimulation of autophagy affords cardioprotection [[72], [73], [74]]. Stimulating the autophagic process with Rapamycin, an mTOR inhibitor, before administering DOX attenuated DOX-induced cardiomyopathy [75,76]. In addition, upregulation of autophagy by other mechanisms (e.g., caloric restriction) in a rodent model afforded cardioprotection following DOX treatment [77]. However, pharmacologic inhibition of autophagy with 3-methyladenine (PI3 kinase inhibitor) following DOX treatment modulate cardiotoxicity [78]. Clearly, additional research specifically addressing the temporal effects of activating or inhibiting autophagy in DOX-induced mitochondrial function is needed to fully understand the short- and long-term implications of the autophagic process in DOX toxicity.
Several natural products, natural product derived-drugs, and FDA-approved drugs have shown protective effects in acute and chronic DOX-induced cardiotoxicity [79]. Metformin, one of the most widely prescribed antidiabetic drugs, mitigated DOX-induced toxicity. Metformin has a very high safety profile. Induction of AMP-activated protein kinase, autophagy, and inhibition of the mTOR pathway were attributed to playing a role in cardioprotection.
13. Enhancing mitochondrial biogenesis to strengthen mitochondria? An emerging cardioprotective strategy
Mitochondrial biogenesis (increasing the mitochondrial mass and mitochondrial function) in the myocardium is achieved by increasing the expression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and peroxisome proliferator-activated receptor gamma coactivator 1-beta (PGC-1β) in cardiomyocytes [80]. The burst of mitochondrial oxidative capacity is associated with the levels of PGC-1α expression. Both PGC-1α and PGC-1β coactivate several transcription factors (estrogen-related receptors, peroxisome proliferator- activated receptors, and nuclear respiratory factors) that induce the expression of a multitude of nuclear genes encoding most mitochondrial proteins involved in fatty acid oxidation, the tricarboxylic acid cycle, and the mitochondrial respiratory chain [81]. In addition to elevating the nuclear encoded genes, PGC-1α enhances the transcription of mitochondrial genes (transcription factor A, transcription factor binding motif, etc.) [82]. PGC-1α knockout mice developed cardiomyopathy, clearly implicating the cardioprotective role of PGC-1α [83].
Increasing the expression of PGC-1α in cardiomyocytes is thought to protect against drug-induced cardiomyopathy [84]. Exercise training (particularly short-term endurance training) that stimulated cardiac PGC-1α expression protected against DOX-induced cardiomyopathy [80,85]. Thus, upregulation of the PGC-1α signaling pathway decreased mitochondrial dysfunction in the heart [84].
14. Intervention with natural products: Honokiol inhibits DOX-induced mitochondrial damage
Honokiol is an active component extracted from the bark of the Magnolia officinalis that has been used to treat many inflammatory diseases [86,87]. Honokiol is also available as an over-the-counter supplement to improve mitochondrial biogenesis [88]. Honokiol supplementation protected the mouse heart against DOX-induced cardiomyopathy [89] and improved mitochondrial function. It was shown that honokiol- activated mitochondrial Sirtuin 3 (SIRT3) was attributed to a decrease in DOX-induced cardiotoxicity in mice [89]. Even in a tumor bearing mouse xenograft model, treatment with honokiol inhibited DOX-induced cardiotoxicity without compromising the chemotherapeutic ability of DOX [89]. SIRT3 protein was reported to improve mitochondrial respiration in H9c2 cardiomyocytes treated with DOX [90].
Because honokiol supplementation improves mitochondrial function via activation of SIRT3, and SIRT3 activation has been shown to afford cardioprotection [[89], [90], [91]], honokiol may be used prophylactically post-chemotherapy to prevent or mitigate cardiotoxicity in patients in remission.
15. L-glutamine supplementation affords cardioprotection
l-glutamine, an essential amino acid abundant in the bloodstream, afforded cardioprotection against DOX-induced cardiomyopathy in a rodent model [[92], [93], [94]]. Results indicate that glutamine supplementation diminished DOX-induced oxidative damage in cardiomyocytes via upregulation of glutathione [95]. l-glutamine is required for NAD+ synthesis, and oral administration of l-glutamine has been shown to increase the NAD redox ratio in sickle cells [96]. Both NAD+ and its reduced form, NADH, are needed to regulate the redox in cells. Oxidative damage is typically accompanied by a lower redox potential (NADH/NAD+). l-glutamine, under the commercial name Endari, is the first new therapeutic approved by the FDA in 50 years for treatment of sickle cell disease [96,97]. Increasing the NAD redox potential in sickle cells decreased their sensitivity to oxidative damage [96,97]. l-glutamine is also responsible for increasing glutathione and protein synthesis. l-glutamine enhances NAD by acting as a substrate for the glutaminase activity of NAD synthetase and using the ammonia released from glutamine catalyzed by the glutaminase activity. Repurposing l-glutamine as a prophylactic supplement may be considered to enhance the redox potential in cardiomyocytes following a patient's cessation of cytotoxic chemotherapy (e.g., DOX and chemotherapeutic drug cocktail) and radiation.
16. Current thinking on DOX cardiotoxicity: potential targets and cardioprotective strategies
Strong evidence supports the modulatory role of topoisomerase in DOX toxicity. Upregulation of the cellular stress response proteins mitigates DOX cardiotoxicity, and supplementation with agents enhancing mitochondrial proteins (PGC-1α and sirtuins) were shown to reverse DOX cardiotoxicity [98,99]. ROS generation was proposed to occur in a secondary mechanism via downregulation of topoisomerase β, PGC-1α, and manganese superoxide dismutase (MnSOD). This is shown in Fig. 9, which is a slight modification of the original scheme [100].
Fig. 9.

The current mechanistic model for DOX-induced cardiotoxicity.
17. Gut microbiota and DOX toxicity
An emerging area of great importance to cancer chemotherapy is the modulatory role of intestinal microbes in enhancing the efficacy of chemotherapeutic drugs and decreasing their toxic side effects [101]. One of the adverse effects of DOX is intestinal mucositis in addition to cardiotoxicity. Studies suggest that microbiota could be a therapeutic target as the composition of intestinal microbiome considerably affects the side effects of chemotherapy. Studies have identified the NADH dehydrogenase component of mitochondrial respiratory chain complex I as the major enzyme involved in the bacterial inactivation of DOX [101]. Studies also showed that the bacterial muramyl dipeptide prevented DOX-induced mucosal damage [102]. Selective microbial detoxification of DOX in noncancerous tissues could be a plausible therapeutic intervention strategy.
18. Conclusions
Clearly, systematic and detailed clinical trials using the FDA- approved drugs outlined here and elsewhere are crucial to understanding the cardiovascular complications in cancer survivors after cytotoxic chemotherapy/radiation therapy. Although, we have specifically addressed the cardiotoxic problems of DOX, cancer patients often are treated with a cocktail containing several cytotoxic antitumor drugs as well as radiation and other targeted-drug treatments. It is likely that such combination therapies will enhance cardiotoxicity in cancer survivors [103]. DOX reportedly has a “dose memory” that is prevalent long after the cessation of treatment due to defective mitochondrial respiratory chain caused by cumulative mitochondrial DNA lesions. It is possible that, like idiopathic cardiomyopathy that is often related to genetic predisposition, patients afflicted with a genetic disorder (DNA repair defects) are more susceptible to DOX-induced cardiomyopathy [104]. Therefore, the need to develop new strategies of intervention designed to enhance mitochondrial function of myocardium in cancer survivors is critical. A new beginning in interventional cardio-oncology (i.e., the cardiovascular care of cancer patients) is urgently needed [[105], [106], [107], [108], [109], [110]]. DXR, the only FDA-approved drug for treating DOX cardiotoxicity [39], may be used alone or more judiciously with other mitochondria-activating drugs (Coenzyme Q, honokiol, and the mitochondria-targeted analogs).
Funding
This work was supported by NIH/NCI grant R01 CA208648 and the Quadracci Endowment.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
I would like to thank my many collaborators and postdoctoral fellows for their scientific contributions, as well as Jane Thelaner and Lydia Washechek for helping me prepare this review.
References
- 1.Rosen M.R., Myerburg R.J., Francis D.P., Cole G.D., Marban E. Translating stem cell research to cardiac disease therapies: pitfalls and prospects for improvement. J. Am. Coll. Cardiol. 2014;64:922–937. doi: 10.1016/j.jacc.2014.06.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheng C.C., Amiri-Kordestani L., Palmby T., Force T., Hong C.C., Wu J.C., Croce K., Kim G., Moslehi J. 21st century cardio-oncology: identifying cardiac safety signals in the era of personalized medicine. JACC Basic Transl Sci. 2016;1:386–398. doi: 10.1016/j.jacbts.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardaway B.W. Adriamycin-associated cardiomyopathy: where are we now? updates in pathophysiology, dose recommendations, prognosis, and outcomes. Curr. Opin. Cardiol. 2019;34:289–295. doi: 10.1097/HCO.0000000000000617. [DOI] [PubMed] [Google Scholar]
- 4.Young R.C., Ozols R.F., Myers C.E. The anthracycline antineoplastic drugs. N. Engl. J. Med. 1981;305:139–153. doi: 10.1056/NEJM198107163050305. [DOI] [PubMed] [Google Scholar]
- 5.Blum R.H., Carter S.K. Adriamycin. A new anticancer drug with significant clinical activity. Ann. Intern. Med. 1974;80:249–259. doi: 10.7326/0003-4819-80-2-249. [DOI] [PubMed] [Google Scholar]
- 6.Bristow M.R., Billingham M.E., Mason J.W., Daniels J.R. Clinical spectrum of anthracycline antibiotic cardiotoxicity. Cancer Treat Rep. 1978;62:873–879. [PubMed] [Google Scholar]
- 7.M R.A., B R.S., G J.A. Adriamycin (NSC 123127) Cardiomyopathy: an overview with determination of risk factors. Cancer Chemother. Rep. 1975;6:198–201. [Google Scholar]
- 8.Singal P.K., Iliskovic N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 9.Doyle J.J., Neugut A.I., Jacobson J.S., Grann V.R., Hershman D.L. Chemotherapy and cardiotoxicity in older breast cancer patients: a population-based study. J. Clin. Oncol. 2005;23:8597–8605. doi: 10.1200/JCO.2005.02.5841. [DOI] [PubMed] [Google Scholar]
- 10.Sorensen K., Levitt G.A., Bull C., Dorup I., Sullivan I.D. Late anthracycline cardiotoxicity after childhood cancer: a prospective longitudinal study. Cancer. 2003;97:1991–1998. doi: 10.1002/cncr.11274. [DOI] [PubMed] [Google Scholar]
- 11.Iarussi D., Indolfi P., Casale F., Martino V., Di Tullio M.T., Calabro R. Anthracycline-induced cardiotoxicity in children with cancer: strategies for prevention and management. Paediatr Drugs. 2005;7:67–76. doi: 10.2165/00148581-200507020-00001. [DOI] [PubMed] [Google Scholar]
- 12.Lipshultz S.E., Lipsitz S.R., Sallan S.E., Dalton V.M., Mone S.M., Gelber R.D., Colan S.D. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2005;23:2629–2636. doi: 10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 13.Benson J.R., Jatoi I. vol 12. Future Oncol; 2016. pp. 893–896. (Highlights of the San Antonio Breast Cancer Symposium 2015: Part 1). [DOI] [PubMed] [Google Scholar]
- 14.Benson J.R., Jatoi I. vol 13. Future Oncol; 2017. pp. 1365–1369. (Highlights of the San Antonio Breast Cancer Symposium 2016: Part 2). [DOI] [PubMed] [Google Scholar]
- 15.Zhang J.C., Chen W.D., Alvarez J.B., Jia K., Shi L., Wang Q., Zou N., He K., Zhu H. Cancer immune checkpoint blockade therapy and its associated autoimmune cardiotoxicity. Acta Pharmacol. Sin. 2018;39:1693–1698. doi: 10.1038/s41401-018-0062-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handa K., Sato S. Stimulation of microsomal NADPH oxidation by quinone group-containing anticancer chemicals. Gan. 1976;67:523–528. [PubMed] [Google Scholar]
- 17.Bachur N.R., Gordon S.L., Gee M.V. Anthracycline antibiotic augmentation of microsomal electron transport and free radical formation. Mol. Pharmacol. 1977;13:901–910. [PubMed] [Google Scholar]
- 18.Svingen B.A., Powis G. Pulse radiolysis studies of antitumor quinones: radical lifetimes, reactivity with oxygen, and one-electron reduction potentials. Arch. Biochem. Biophys. 1981;209:119–126. doi: 10.1016/0003-9861(81)90263-0. [DOI] [PubMed] [Google Scholar]
- 19.Myers C.E., McGuire W.P., Liss R.H., Ifrim I., Grotzinger K., Young R.C. Adriamycin: the role of lipid peroxidation in cardiac toxicity and tumor response. Science. 1977;197:165–167. doi: 10.1126/science.877547. [DOI] [PubMed] [Google Scholar]
- 20.Myers C., Gianni L., Zweier J., Muindi J., Sinha B.K., Eliot H. Role of iron in adriamycin biochemistry. Fed. Proc. 1986;45:2792–2797. [PubMed] [Google Scholar]
- 21.Davies K.J., Doroshow J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986;261:3060–3067. [PubMed] [Google Scholar]
- 22.Minotti G., Cairo G., Monti E. Role of iron in anthracycline cardiotoxicity: new tunes for an old song? FASEB J. 1999;13:199–212. [PubMed] [Google Scholar]
- 23.Kalyanaraman B., Perez-Reyes E., Mason R.P. Spin-trapping and direct electron spin resonance investigations of the redox metabolism of quinone anticancer drugs. Biochim. Biophys. Acta. 1980;630:119–130. doi: 10.1016/0304-4165(80)90142-7. [DOI] [PubMed] [Google Scholar]
- 24.Goodman J., Hochstein P. Generation of free radicals and lipid peroxidation by redox cycling of adriamycin and daunomycin. Biochem. Biophys. Res. Commun. 1977;77:797–803. doi: 10.1016/s0006-291x(77)80048-x. [DOI] [PubMed] [Google Scholar]
- 25.Gutierrez P.L., Gee M.V., Bachur N.R. Kinetics of anthracycline antibiotic free radical formation and reductive glycosidase activity. Arch. Biochem. Biophys. 1983;223:68–75. doi: 10.1016/0003-9861(83)90572-6. [DOI] [PubMed] [Google Scholar]
- 26.Kalyanaraman B., Sealy R.C., Sinha B.K. An electron spin resonance study of the reduction of peroxides by anthracycline semiquinones. Biochim. Biophys. Acta. 1984;799:270–275. doi: 10.1016/0304-4165(84)90270-8. [DOI] [PubMed] [Google Scholar]
- 27.Bates D.A., Winterbourn C.C. Deoxyribose breakdown by the adriamycin semiquinone and H2O2: evidence for hydroxyl radical participation. FEBS Lett. 1982;145:137–142. doi: 10.1016/0014-5793(82)81222-2. [DOI] [PubMed] [Google Scholar]
- 28.Kalyanaraman B., Morehouse K.M., Mason R.P. An electron paramagnetic resonance study of the interactions between the adriamycin semiquinone, hydrogen peroxide, iron-chelators, and radical scavengers. Arch. Biochem. Biophys. 1991;286:164–170. doi: 10.1016/0003-9861(91)90023-c. [DOI] [PubMed] [Google Scholar]
- 29.Myers C.E., Gianni L., Simone C.B., Klecker R., Greene R. Oxidative destruction of erythrocyte ghost membranes catalyzed by the doxorubicin-iron complex. Biochemistry. 1982;21:1707–1712. doi: 10.1021/bi00537a001. [DOI] [PubMed] [Google Scholar]
- 30.Muindi J.R., Sinha B.K., Gianni L., Myers C.E. Hydroxyl radical production and DNA damage induced by anthracycline-iron complex. FEBS Lett. 1984;172:226–230. doi: 10.1016/0014-5793(84)81130-8. [DOI] [PubMed] [Google Scholar]
- 31.Dhingra R., Margulets V., Chowdhury S.R., Thliveris J., Jassal D., Fernyhough P., Dorn G.W., 2nd, Kirshenbaum L.A. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E5537–E5544. doi: 10.1073/pnas.1414665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang X., Wang H., Han D., Xie E., Yang X., Wei J., Gu S., Gao F., Zhu N., Yin X., Cheng Q., Zhang P., Dai W. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 2019;116:2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lebrecht D., Kokkori A., Ketelsen U.P., Setzer B., Walker U.A. Tissue-specific mtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J. Pathol. 2005;207:436–444. doi: 10.1002/path.1863. [DOI] [PubMed] [Google Scholar]
- 34.Yen H.C., Oberley T.D., Vichitbandha S., Ho Y.S., St Clair D.K. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J. Clin. Investig. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saad S.Y., Najjar T.A., Al-Rikabi A.C. The preventive role of deferoxamine against acute doxorubicin-induced cardiac, renal and hepatic toxicity in rats. Pharmacol. Res. 2001;43:211–218. doi: 10.1006/phrs.2000.0769. [DOI] [PubMed] [Google Scholar]
- 36.Kotamraju S., Konorev E.A., Joseph J., Kalyanaraman B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J. Biol. Chem. 2000;275:33585–33592. doi: 10.1074/jbc.M003890200. [DOI] [PubMed] [Google Scholar]
- 37.Swain S.M., Whaley F.S., Gerber M.C., Weisberg S., York M., Spicer D., Jones S.E., Wadler S., Desai A., Vogel C., Speyer J., Mittelman A., Reddy S. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J. Clin. Oncol. 1997;15:1318–1332. doi: 10.1200/JCO.1997.15.4.1318. [DOI] [PubMed] [Google Scholar]
- 38.Jirkovsky E., Jirkovska A., Bures J., Chladek J., Lencova O., Stariat J., Pokorna Z., Karabanovich G., Roh J., Brazdova P., Simunek T., Kovarikova P., Sterba M. Pharmacokinetics of the cardioprotective drug dexrazoxane and its active metabolite ADR-925 with focus on cardiomyocytes and the heart. J. Pharmacol. Exp. Ther. 2018;364:433–446. doi: 10.1124/jpet.117.244848. [DOI] [PubMed] [Google Scholar]
- 39.Hasinoff B.B., Herman E.H. Dexrazoxane: how it works in cardiac and tumor cells. Is it a prodrug or is it a drug? Cardiovasc. Toxicol. 2007;7:140–144. doi: 10.1007/s12012-007-0023-3. [DOI] [PubMed] [Google Scholar]
- 40.Hasinoff B.B., Hellmann K., Herman E.H., Ferrans V.J. Chemical, biological and clinical aspects of dexrazoxane and other bisdioxopiperazines. Curr. Med. Chem. 1998;5:1–28. [PubMed] [Google Scholar]
- 41.Lipshultz S.E., Cochran T.R., Franco V.I., Miller T.L. Treatment-related cardiotoxicity in survivors of childhood cancer. Nat. Rev. Clin. Oncol. 2013;10:697–710. doi: 10.1038/nrclinonc.2013.195. [DOI] [PubMed] [Google Scholar]
- 42.Lebrecht D., Geist A., Ketelsen U.P., Haberstroh J., Setzer B., Walker U.A. Dexrazoxane prevents doxorubicin-induced long-term cardiotoxicity and protects myocardial mitochondria from genetic and functional lesions in rats. Br. J. Pharmacol. 2007;151:771–778. doi: 10.1038/sj.bjp.0707294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harake D., Franco V.I., Henkel J.M., Miller T.L., Lipshultz S.E. Cardiotoxicity in childhood cancer survivors: strategies for prevention and management. Future Cardiol. 2012;8:647–670. doi: 10.2217/fca.12.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dickey J.S., Gonzalez Y., Aryal B., Mog S., Nakamura A.J., Redon C.E., Baxa U., Rosen E., Cheng G., Zielonka J., Parekh P., Mason K.P., Joseph J. Mito-tempol and dexrazoxane exhibit cardioprotective and chemotherapeutic effects through specific protein oxidation and autophagy in a syngeneic breast tumor preclinical model. PLoS One. 2013;8 doi: 10.1371/journal.pone.0070575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Konorev E.A., Kotamraju S., Zhao H., Kalivendi S., Joseph J., Kalyanaraman B. Paradoxical effects of metalloporphyrins on doxorubicin-induced apoptosis: scavenging of reactive oxygen species versus induction of heme oxygenase-1. Free Radic. Biol. Med. 2002;33:988. doi: 10.1016/s0891-5849(02)00989-9. [DOI] [PubMed] [Google Scholar]
- 46.Murphy M.P. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997;15:326–330. doi: 10.1016/S0167-7799(97)01068-8. [DOI] [PubMed] [Google Scholar]
- 47.Smith R.A., Porteous C.M., Gane A.M., Murphy M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. U. S. A. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chandran K., Aggarwal D., Migrino R.Q., Joseph J., McAllister D., Konorev E.A., Antholine W.E., Zielonka J., Srinivasan S., Avadhani N.G., Kalyanaraman B. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys. J. 2009;96:1388–1398. doi: 10.1016/j.bpj.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang S., Liu X., Bawa-Khalfe T., Lu L.S., Lyu Y.L., Liu L.F., Yeh E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 50.Pointon A.V., Walker T.M., Phillips K.M., Luo J., Riley J., Zhang S.D., Parry J.D., Lyon J.J., Marczylo E.L., Gant T.W. Doxorubicin in vivo rapidly alters expression and translation of myocardial electron transport chain genes, leads to ATP loss and caspase 3 activation. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghigo A., Li M., Hirsch E. New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim. Biophys. Acta. 2016;1863:1916–1925. doi: 10.1016/j.bbamcr.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 52.Sonowal H., Pal P., Shukla K., Saxena A., Srivastava S.K., Ramana K.V. Aldose reductase inhibitor, fidarestat prevents doxorubicin-induced endothelial cell death and dysfunction. Biochem. Pharmacol. 2018;150:181–190. doi: 10.1016/j.bcp.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwarz E.R., Pollick C., Dow J., Patterson M., Birnbaum Y., Kloner R.A. A small animal model of non-ischemic cardiomyopathy and its evaluation by transthoracic echocardiography. Cardiovasc. Res. 1998;39:216–223. doi: 10.1016/s0008-6363(98)00009-1. [DOI] [PubMed] [Google Scholar]
- 54.Migrino R.Q., Aggarwal D., Konorev E., Brahmbhatt T., Bright M., Kalyanaraman B. Early detection of doxorubicin cardiomyopathy using two-dimensional strain echocardiography. Ultrasound Med. Biol. 2008;34:208–214. doi: 10.1016/j.ultrasmedbio.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng G., Zielonka M., Dranka B., Kumar S.N., Myers C.R., Bennett B., Garces A.M., Dias Duarte Machado L.G., Thiebaut D., Ouari O., Hardy M., Zielonka J., Kalyanaraman B. Detection of mitochondria-generated reactive oxygen species in cells using multiple probes and methods: potentials, pitfalls, and the future. J. Biol. Chem. 2018;293:10363–10380. doi: 10.1074/jbc.RA118.003044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lebrecht D., Setzer B., Ketelsen U.P., Haberstroh J., Walker U.A. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation. 2003;108:2423–2429. doi: 10.1161/01.CIR.0000093196.59829.DF. [DOI] [PubMed] [Google Scholar]
- 57.Papadopoulou L.C., Tsiftsoglou A.S. Mitochondrial cytochrome c oxidase as a target site for daunomycin in K-562 cells and heart tissue. Cancer Res. 1993;53:1072–1078. [PubMed] [Google Scholar]
- 58.Kalyanaraman B., Cheng G., Hardy M., Ouari O., Bennett B., Zielonka J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biology. 2018;15:347–362. doi: 10.1016/j.redox.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boutagy N.E., Wu J., Cai Z., Zhang W., Booth C.J., Kyriakides T.C., Pfau D., Mulnix T., Liu Z., Miller E.J., Young L.H., Carson R.E., Huang Y. In vivo reactive oxygen species detection with a novel positron emission tomography tracer, (18)F-DHMT, allows for early detection of anthracycline-induced cardiotoxicity in rodents. JACC Basic Transl Sci. 2018;3:378–390. doi: 10.1016/j.jacbts.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalyanaraman B., Hardy M., Zielonka J. A critical review of methodologies to detect reactive oxygen and nitrogen species stimulated by NADPH oxidase enzymes: implications in pesticide toxicity. Curr Pharmacol Rep. 2016;2:193–201. doi: 10.1007/s40495-016-0063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bures J., Jirkovska A., Sestak V., Jansova H., Karabanovich G., Roh J., Sterba M., Simunek T., Kovarikova P. Investigation of novel dexrazoxane analogue JR-311 shows significant cardioprotective effects through topoisomerase IIbeta but not its iron chelating metabolite. Toxicology. 2017;392:1–10. doi: 10.1016/j.tox.2017.09.012. [DOI] [PubMed] [Google Scholar]
- 62.Hasinoff B.B., Schroeder P.E., Patel D. The metabolites of the cardioprotective drug dexrazoxane do not protect myocytes from doxorubicin-induced cytotoxicity. Mol. Pharmacol. 2003;64:670–678. doi: 10.1124/mol.64.3.670. [DOI] [PubMed] [Google Scholar]
- 63.Ichikawa Y., Ghanefar M., Bayeva M., Wu R., Khechaduri A., Naga Prasad S.V., Mutharasan R.K., Naik T.J., Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014;124:617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lyu Y.L., Kerrigan J.E., Lin C.P., Azarova A.M., Tsai Y.C., Ban Y., Liu L.F. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839–8846. doi: 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- 65.Vejpongsa P., Yeh E.T. Topoisomerase 2beta: a promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin. Pharmacol. Ther. 2014;95:45–52. doi: 10.1038/clpt.2013.201. [DOI] [PubMed] [Google Scholar]
- 66.Cui N., Wu F., Lu W.J., Bai R., Ke B., Liu T., Li L., Lan F., Cui M. Doxorubicin-induced cardiotoxicity is maturation dependent due to the shift from topoisomerase IIalpha to IIbeta in human stem cell derived cardiomyocytes. J. Cell Mol. Med. 2019;23:4627–4639. doi: 10.1111/jcmm.14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henriksen P.A. Anthracycline cardiotoxicity: an update on mechanisms, monitoring and prevention. Heart. 2018;104:971–977. doi: 10.1136/heartjnl-2017-312103. [DOI] [PubMed] [Google Scholar]
- 68.Rosenfeldt M.T., Ryan K.M. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32:955–963. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mohammadi D. 2016 Nobel prize in medicine goes to Japanese scientist. Lancet. 2016;388:1870. doi: 10.1016/S0140-6736(16)31797-4. [DOI] [PubMed] [Google Scholar]
- 70.Walton E.L. Food for thought: autophagy researcher wins 2016 Nobel prize in physiology or medicine. Biomed. J. 2017;40:1–4. doi: 10.1016/j.bj.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amaravadi R., Kimmelman A.C., White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913–1930. doi: 10.1101/gad.287524.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koleini N., Kardami E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget. 2017;8:46663–46680. doi: 10.18632/oncotarget.16944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li D.L., Wang Z.V., Ding G., Tan W., Luo X., Criollo A., Xie M., Jiang N., May H., Kyrychenko V., Schneider J.W., Gillette T.G., Hill J.A. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. 2016;133:1668–1687. doi: 10.1161/CIRCULATIONAHA.115.017443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abdullah C.S., Alam S., Aishwarya R., Miriyala S., Bhuiyan M.A.N., Panchatcharam M., Pattillo C.B., Orr A.W., Sadoshima J., Hill J.A., Bhuiyan M.S. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019;9:2002. doi: 10.1038/s41598-018-37862-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsutsumi Y., Numata S., Seo H., Ohashi H. Transaortic edge-to-edge mitral valve repair and left ventricular myectomy. Gen Thorac Cardiovasc Surg. 2013;61:223–225. doi: 10.1007/s11748-012-0131-2. [DOI] [PubMed] [Google Scholar]
- 76.Ding Y., Sun X., Xu X. TOR-autophagy signaling in adult zebrafish models of cardiomyopathy. Autophagy. 2012;8:142–143. doi: 10.4161/auto.8.1.18536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dutta D., Xu J., Dirain M.L., Leeuwenburgh C. Calorie restriction combined with resveratrol induces autophagy and protects 26-month-old rat hearts from doxorubicin-induced toxicity. Free Radic. Biol. Med. 2014;74:252–262. doi: 10.1016/j.freeradbiomed.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pizarro M., Troncoso R., Martinez G.J., Chiong M., Castro P.F., Lavandero S. Basal autophagy protects cardiomyocytes from doxorubicin-induced toxicity. Toxicology. 2016;370:41–48. doi: 10.1016/j.tox.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 79.Huang L., Zhang K., Guo Y., Huang F., Yang K., Chen L., Huang K., Zhang F., Long Q., Yang Q. Honokiol protects against doxorubicin cardiotoxicity via improving mitochondrial function in mouse hearts. Sci. Rep. 2017;7:11989. doi: 10.1038/s41598-017-12095-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Di W., Lv J., Jiang S., Lu C., Yang Z., Ma Z., Hu W., Yang Y., Xu B. PGC-1: the energetic regulator in cardiac metabolism. Curr. Issues Mol. Biol. 2018;28:29–46. doi: 10.21775/cimb.028.029. [DOI] [PubMed] [Google Scholar]
- 81.Austin S., St-Pierre J. PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012;125:4963–4971. doi: 10.1242/jcs.113662. [DOI] [PubMed] [Google Scholar]
- 82.Ventura-Clapier R., Garnier A., Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc. Res. 2008;79:208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 83.Karkkainen O., Tuomainen T., Mutikainen M., Lehtonen M., Ruas J.L., Hanhineva K., Tavi P. Heart specific PGC-1alpha deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc. Res. 2019;115:107–118. doi: 10.1093/cvr/cvy155. [DOI] [PubMed] [Google Scholar]
- 84.Ristow M. Oxidative metabolism in cancer growth. Curr. Opin. Clin. Nutr. Metab. Care. 2006;9:339–345. doi: 10.1097/01.mco.0000232892.43921.98. [DOI] [PubMed] [Google Scholar]
- 85.Kirkham A.A., Davis M.K. Exercise prevention of cardiovascular disease in breast cancer survivors. J Oncol. 2015;2015:917606. doi: 10.1155/2015/917606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Talarek S., Listos J., Barreca D., Tellone E., Sureda A., Nabavi S.F., Braidy N., Nabavi S.M. Neuroprotective effects of honokiol: from chemistry to medicine. Biofactors. 2017;43:760–769. doi: 10.1002/biof.1385. [DOI] [PubMed] [Google Scholar]
- 87.Munroe M.E., Arbiser J.L., Bishop G.A. Honokiol, a natural plant product, inhibits inflammatory signals and alleviates inflammatory arthritis. J. Immunol. 2007;179:753–763. doi: 10.4049/jimmunol.179.2.753. [DOI] [PubMed] [Google Scholar]
- 88.Wood Dos Santos T., Cristina Pereira Q., Teixeira L., Gambero A., J A.V., Lima Ribeiro M. Effects of polyphenols on thermogenesis and mitochondrial biogenesis. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19092757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pillai V.B., Kanwal A., Fang Y.H., Sharp W.W., Samant S., Arbiser J., Gupta M.P. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget. 2017;8:34082–34098. doi: 10.18632/oncotarget.16133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pillai V.B., Samant S., Sundaresan N.R., Raghuraman H., Kim G., Bonner M.Y., Arbiser J.L., Walker D.I., Jones D.P., Gius D., Gupta M.P. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015;6:6656. doi: 10.1038/ncomms7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Du Q., Zhu B., Zhai Q., Yu B. Sirt3 attenuates doxorubicin-induced cardiac hypertrophy and mitochondrial dysfunction via suppression of Bnip3. Am J Transl Res. 2017;9:3360–3373. [PMC free article] [PubMed] [Google Scholar]
- 92.Todorova V.K., Kaufmann Y., Hennings L., Klimberg V.S. Oral glutamine protects against acute doxorubicin-induced cardiotoxicity of tumor-bearing rats. J. Nutr. 2010;140:44–48. doi: 10.3945/jn.109.113415. [DOI] [PubMed] [Google Scholar]
- 93.Xue H., Ren W., Denkinger M., Schlotzer E., Wischmeyer P.E. Nutrition modulation of cardiotoxicity and anticancer efficacy related to doxorubicin chemotherapy by glutamine and omega-3 polyunsaturated fatty acids. JPEN - J. Parenter. Enter. Nutr. 2016;40:52–66. doi: 10.1177/0148607115581838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cao Y., Kennedy R., Klimberg V.S. Glutamine protects against doxorubicin-induced cardiotoxicity. J. Surg. Res. 1999;85:178–182. doi: 10.1006/jsre.1999.5677. [DOI] [PubMed] [Google Scholar]
- 95.Hudson C.D., Savadelis A., Nagaraj A.B., Joseph P., Avril S., DiFeo A., Avril N. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget. 2016;7:41637–41649. doi: 10.18632/oncotarget.9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Niihara Y., Miller S.T., Kanter J., Lanzkron S., Smith W.R., Hsu L.L., Gordeuk V.R., Viswanathan K., Sarnaik S., Osunkwo I., Guillaume E., Sadanandan S., Sieger L. A phase 3 trial of l-glutamine in sickle cell disease. N. Engl. J. Med. 2018;379:226–235. doi: 10.1056/NEJMoa1715971. [DOI] [PubMed] [Google Scholar]
- 97.Ortiz de Montellano P.R. A new step in the treatment of sickle cell DiseasePublished as part of the biochemistry series "biochemistry to bedside. Biochemistry. 2018;57:470–471. doi: 10.1021/acs.biochem.7b00785. [DOI] [PubMed] [Google Scholar]
- 98.Dolinsky V.W. The role of sirtuins in mitochondrial function and doxorubicin-induced cardiac dysfunction. Biol. Chem. 2017;398:955–974. doi: 10.1515/hsz-2016-0316. [DOI] [PubMed] [Google Scholar]
- 99.Coelho A.R., Martins T.R., Couto R., Deus C., Pereira C.V., Simoes R.F., Rizvanov A.A., Silva F., Cunha-Oliveira T., Oliveira P.J., Serafim T.L. Berberine-induced cardioprotection and Sirt3 modulation in doxorubicin-treated H9c2 cardiomyoblasts. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2017;1863:2904–2923. doi: 10.1016/j.bbadis.2017.07.030. [DOI] [PubMed] [Google Scholar]
- 100.Cowgill J.A., Francis S.A., Sawyer D.B. Anthracycline and peripartum cardiomyopathies. Circ. Res. 2019;124:1633–1646. doi: 10.1161/CIRCRESAHA.119.313577. [DOI] [PubMed] [Google Scholar]
- 101.Westman E.L., Canova M.J., Radhi I.J., Koteva K., Kireeva I., Waglechner N., Wright G.D. Bacterial inactivation of the anticancer drug doxorubicin. Chem. Biol. 2012;19:1255–1264. doi: 10.1016/j.chembiol.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 102.Ma W., Mao Q., Xia W., Dong G., Yu C., Jiang F. Gut microbiota shapes the efficiency of cancer therapy. Front. Microbiol. 2019;10:1050. doi: 10.3389/fmicb.2019.01050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farmakis D., Mantzourani M., Filippatos G. Anthracycline-induced cardiomyopathy: secrets and lies. Eur. J. Heart Fail. 2018;20:907–909. doi: 10.1002/ejhf.1172. [DOI] [PubMed] [Google Scholar]
- 104.Hantson P. Mechanisms of toxic cardiomyopathy. Clin. Toxicol. 2019;57:1–9. doi: 10.1080/15563650.2018.1497172. [DOI] [PubMed] [Google Scholar]
- 105.Bellinger A.M., Arteaga C.L., Force T., Humphreys B.D., Demetri G.D., Druker B.J., Moslehi J.J. Cardio-oncology: how new targeted cancer therapies and precision medicine can inform cardiovascular discovery. Circulation. 2015;132:2248–2258. doi: 10.1161/CIRCULATIONAHA.115.010484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pudil R. The future role of cardio-oncologists. Card. Fail. Rev. 2017;3:140–142. doi: 10.15420/cfr.2017:16:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vejpongsa P., Yeh E.T. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J. Am. Coll. Cardiol. 2014;64:938–945. doi: 10.1016/j.jacc.2014.06.1167. [DOI] [PubMed] [Google Scholar]
- 108.Lenneman C.G., Sawyer D.B. Cardio-oncology: an update on cardiotoxicity of cancer-related treatment. Circ. Res. 2016;118:1008–1020. doi: 10.1161/CIRCRESAHA.115.303633. [DOI] [PubMed] [Google Scholar]
- 109.Geisberg C.A., Sawyer D.B. Mechanisms of anthracycline cardiotoxicity and strategies to decrease cardiac damage. Curr. Hypertens. Rep. 2010;12:404–410. doi: 10.1007/s11906-010-0146-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Levis B.E., Binkley P.F., Shapiro C.L. Cardiotoxic effects of anthracycline-based therapy: what is the evidence and what are the potential harms? Lancet Oncol. 2017;18:e445–e456. doi: 10.1016/S1470-2045(17)30535-1. [DOI] [PubMed] [Google Scholar]