

Abstract
The efficacy of antigen-specific immunotherapy relies heavily on efficient antigen delivery to antigen-presenting cells and engagement of as many disease-relevant T cells as possible in various lymphoid tissues, which are challenging to achieve. Here, we compared two approaches to deliver mRNA encoding multiple epitopes targeting both CD4+ and CD8+ T cells: a lipid-based nanoparticle platform to target endogenous antigen-presenting cells in vivo versus ex vivo mRNA-electroporated dendritic cells. After intraperitoneal injection, the nanoparticle platform facilitated efficient entry of mRNA into various endogenous antigen-presenting cells, including lymph node stromal cells, and elicited robust T cell responses within a wider network of lymphoid tissues compared with dendritic cells. Following intravenous injection, mRNA-electroporated dendritic cells and the nanoparticle platform localized primarily in lung and spleen, respectively. When administered locally via an intradermal route, both platforms resulted in mRNA expression at the injection site and in robust T cell responses in draining lymph nodes. This study indicates that multiple epitopes, customizable for specific patient populations and encoded by mRNA, can be targeted to different lymphoid tissues based on delivery vehicle and route, and constitute the groundwork for future studies using mRNA to reprogram exogenous or endogenous APCs for immunotherapy.
Keywords: mRNA vaccine, autoimmune diabetes, precision medicine, beta cell antigens, neoepitopes, nanoparticle, nanomedicine, dendritic cell, stromal cell
Graphical Abstract
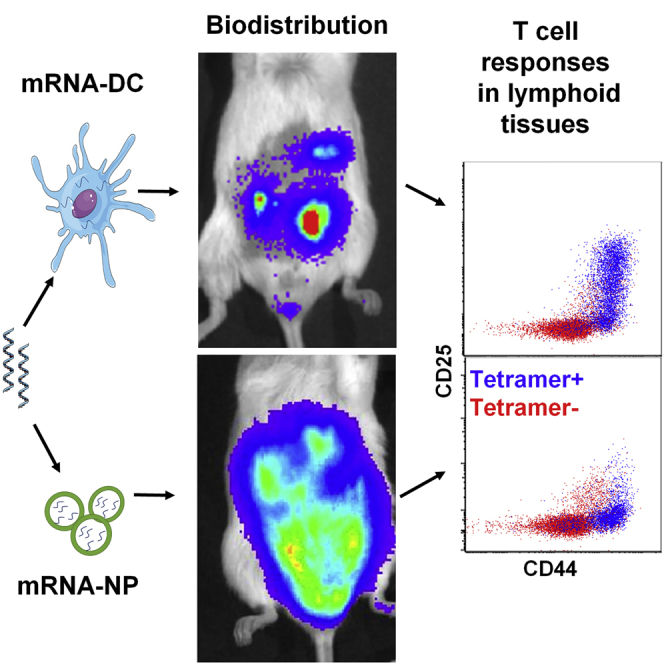
Introduction
Immunotherapies aim at manipulating immune responses to either overcome tolerance/exhaustion (in the case of cancer or chronic infections) or reestablish tolerance in the case of autoimmune diseases. In both cases, efficient engagement of antigen-specific T cells, preferably both CD4+ and CD8+ T cells, is essential to achieve effective and targeted clinical outcomes. Antigens may vary between patients or groups of patients, and are most conveniently produced by nucleic acid-based vectors, which are easy to design and manufacture. In cancer immunotherapy, such vectors have been delivered directly to patients in formulated forms1, 2, 3 or introduced into exogenous antigen-presenting cells (APCs) such as dendritic cells (DCs) for cell therapy.4, 5, 6 In several autoimmune diseases, DNA vaccines and DCs (with or without pulsed antigens) have also been evaluated clinically for tolerance induction;7, 8, 9, 10 however, mRNA has not yet been considered to express self-antigens in these settings.
Both plasmid DNA (pDNA) and mRNA enable the endogenous production of antigens in a more physiological form (post-translational modifications possible) and at a lower cost compared with antigens administered as recombinant proteins.1,11,12 However, mRNA has unique advantages over pDNA as vector, because it offers higher transfection efficiency (no nuclear entry required) allowing more effective delivery to quiescent cells in vivo, rapid and promoter-independent expression, as well as a relatively higher safety profile owing to lack of genomic integration.12, 13, 14 In recent years, substantial breakthroughs have been achieved to overcome the limitations of mRNA as a therapeutic, such as poor translation, inherent instability under physiological conditions, adverse immune reactions, and inefficient in vivo delivery. Modifications of the 5′ cap and poly(A), nucleoside substitutions, and codon optimization have all contributed to improved stability and dampened immunogenicity of mRNA,15, 16, 17, 18, 19 the latter being particularly crucial when considering mRNA for encoding self-antigens for tolerance. In addition, mRNA offers a versatile combinatorial platform to co-express antigens and immunomodulatory molecules to direct the immune response one way or another.20 However, efficient and safe in vivo delivery of mRNAs that bind and condense mRNA, protect it from degradation by the omnipresent RNases, and facilitate cellular uptake and endosomal escape into the cytosol without interfering with the cellular translational machinery is still challenging, yet key to the successful translation of mRNA therapeutics to the clinic.12,21
The mRNA construct in this study is based on a platform encoding multiple epitopes from different antigens and enabling effective presentation to both CD4+ and CD8+ T cells.22 A pertinent application of this platform is for the antigen-specific immunotherapy (ASIT) of type 1 diabetes (T1D), which is caused by diabetogenic CD4+ and CD8+ T cells that are reactive to multiple pancreatic β cell antigens and that eluded mechanisms of tolerance. ASITs are more targeted and safer than other immunosuppressive biologics tested, but have demonstrated limited clinical efficacy in T1D.23, 24, 25, 26 A gap in the field is that such ASITs have so far involved a single native antigen (in the form of recombinant protein, peptides, or pDNA-encoded protein) and lacked incorporation of neoepitopes.27, 28, 29 It is, however, becoming evident that neoepitopes play a key role in driving T1D and that islet-infiltrating T cells from T1D patients respond to diverse autoantigens,29,30 suggesting that the poor efficacy of ASITs may be linked to insufficient antigen coverage. The diversity of the T1D autoantigen targets is reflected in our platform with the combined incorporation of epitopes from multiple antigens along with unique neoepitopes/mimotopes. These constructs have already been tested as a DNA vaccine.31 This epitope-based platform can be applied to a variety of diseases, from cancer to autoimmune diseases, under conditions that potentiate or dampen specific immune responses, respectively. As far as autoimmune diseases are concerned, however, the use of antigen-encoding mRNA has not yet been reported. In this study, we have evaluated the delivery of mRNA-encoded epitopes using two systems, a lipid-based nanoparticle platform (mRNA-NP) versus ex vivo mRNA-electroporated dendritic cells (mRNA-DCs), with the goal to determine how T cell responses and their location differ. We show that the biodistribution of systemically injected mRNA-DCs is more restricted than mRNA-NPs, whereas mRNA-DCs may be better vehicles in the case of local injections. Interestingly, mRNA-NPs also target lymph node stromal cells (LNSCs), which constitute unique yet untapped populations of tolerogenic APCs for this particular application.32, 33, 34 These studies have important implications for the consideration of exogenous versus endogenous APCs to engage antigen-specific T cells.
Results
Preparation and Biophysical Characterization of mRNA-NPs
Naked mRNA is rapidly degraded by extracellular RNases and is also not efficiently internalized; thus, it relies on specific formulations that protect it and enhance its delivery to APCs.11,35, 36, 37 In our studies, we used jetMESSENGER, a preformed lipoplex made of ionizable mono-cationic lipids and co-helper phospholipids so far commercialized for in vitro transfection, and we tested this platform for in vivo delivery of mRNA encoding reporter genes or multiple epitopes (Figure 1A) to non-obese diabetic (NOD) mice, an animal model for T1D. We first evaluated the mRNA binding capacity of jetMESSENGER and determined the optimal mRNA/jetMESSENGER ratios for complex formation in mRNA buffer (supplied with jetMESSENGER). Formulation of different mRNAs with jetMESSENGER completely prevented their mobility in an agarose gel electrophoretic mobility shift assay (EMSA) at 1:2 mRNA/jetMESSENGER ratio (w/v) or lower, confirming the complexation of mRNA with little or no leaching (Figure 1B). Unbound mRNA was visible at higher mRNA/jetMESSENGER ratios (>1:2 [w/v]). We then measured the size and average surface charge of the nanoparticles made at the optimal 1:2 mRNA/jetMESSENGER ratio by dynamic light scattering. mRNA-NPs made with Firefly luciferase (Luc) mRNA had a mean particle size of 214 ± 6 nm (Figure 1C) with a narrow size distribution (polydispersity index of 0.124 ± 0.03). Furthermore, the zeta potential was +41.5 ± 3 (Figure 1D), indicating an excellent stability of the colloidal dispersions. Even though high positive zeta potential usually correlates with toxicity,38 we did not observe any significant toxicity that could be associated with this cationic surface charge of mRNA-NP at all doses tested in our subsequent in vitro transfections and in vivo administrations (data not shown).
Figure 1.

mRNA Construct and Biophysical Characterization of mRNA-NPs
(A) mRNA construct encoding major epitopes and mimotopes related to four different β cell antigens (insulin, ChgA, GAD65, and IGRP). It uses an endosome-targeting signal (ETS) to direct CD4 epitopes for processing and loading onto MHC class II. Some of the epitopes highlighted in color are recognized by T cell clones indicated underneath and used in this study. (B) EMSA assay showing the binding efficiency of mRNA and jetMESSENGER at different ratios for antigen (Ag), mCherry (MC), and Luc mRNAs (2 μg). (C and D) Biophysical characterization of mRNA-NPs containing Luc-mRNA by Zetasizer showing particle size distribution (C) and zeta potential (D). Each color represents a replicate.
mRNA-NPs Achieve High Transfection Efficiency in Various APCs
APCs in lymphoid tissues are ideal targets for ASIT due to their strategic position for interacting with circulating T cells. However, in autoimmune diseases such as T1D, APCs like DCs may be defective at maintaining tolerance,39 and targeting antigens to alternative APCs may prove beneficial. For example, non-hematopoietic APCs such as LNSCs have been shown to mediate tolerance by deleting or anergizing autoreactive T cells in mice.34,40, 41, 42 To assess the potential of mRNA-NPs to transfect these various cell types, we delivered mCherry- or EGFP-encoding mRNA both in vitro and in vivo. First, we compared the efficiency of mRNA-NP transfection with our mRNA electroporation protocol for bone marrow-derived DCs (BM-DCs). Efficient transfection (>90%) of BM-DCs was achieved by mRNA electroporation as compared with only 25% by mRNA-NPs after 24 h of culture (Figure S1A). Although DCs are more challenging to transfect with mRNA-NPs, they are also the most potent APCs, and this lower transfection efficiency is expected to be sufficient to induce antigen-specific T cell responses. We also tested mRNA-NPs on stromal cells such as DAPg7 fibroblasts43 and LNSC lines. DAPg7 cells, fibroblastic reticular cells (FRCs), and lymphatic endothelial cells (LECs) were efficiently transfected in vitro (>85% transfected) relative to BM-DCs (28%) (Figure 2A). We further tested mRNA-NPs on primary immune cells (splenocytes from NOD mice) cultured in vitro. Different APC subpopulations such as plasmacytoid DCs (pDCs), conventional resident DCs (cDC1), conventional migratory DCs (cDC2), B cells, and other immune cells (macrophages/monocytes/neutrophils [MMNs]) were analyzed for transfection efficiency by flow cytometry (FCM) (see gating strategy on Figure S1B). pDCs, cDC1, cDC2, and MMNs did all take up mRNA-NP and expressed the EGFP reporter after 48 h of in vitro culture (Figures S1C and S1D).
Figure 2.

mRNA-NP Transfection Efficiency in Various Cell Types
(A) In vitro transfection efficiency of mRNA-NPs in BM-DCs and stromal cell lines (LECs, FRCs, and DAPg7) after 48 h of culture using mCherry-encoding mRNA. The transfection efficiency of BM-DCs was significantly lower than any of the stromal cell lines (p ≤ 0.01). (B–E) In vivo transfection efficiency of mRNA-NPs in different cell types using reporter mRNA in PLNs 48 h after i.p. injection. Data shown in panels B and C are from a study using GFP mRNA and no tissue digestion, while those depicted in panels D and E are from a study using mCherry mRNA and collagenase digestion to retrieve all APC populations. Control mice were untreated mice whose untransfected tissues/cells were used to determine fluorescence background for each cell type. The data are shown as mean of three biological replicates ± SEM (B and D) or as representative plots (C and E). Significant differences in uptake and expression of mRNA indicated for several cell types are relative to LNECs (ECs) for the treated group (D).
To evaluate in vivo conditions of transfection with mRNA-NP, we injected EGFP mRNA-NPs intraperitoneally (i.p.) into NOD mice. After 2 days, five different lymphoid tissues (cervical, inguinal, pancreatic, and mesenteric lymph nodes [CLNs, ILNs, PLNs, and MLNs, respectively] and spleen) were collected, and the expression of EGFP in various cell types was quantified by FCM (same gating strategy; Figure S1B). We identified B220+CD11c+ pDCs (∼6%), B220−CD11c+ cDCs (∼4%), and BB220−CD11c−CD11b+ myeloid cells (∼5%) among mRNA-NP-transfected cells in PLNs (Figures 2B and 2C). Transfection of cDCs and pDCs was also observed to a lesser extent in spleen and MLNs located in the abdominal cavity, whereas there was no transfection detected in CLNs and ILNs outside the abdominal cavity (Figure S2A). To check whether LNSCs were efficiently transfected in vivo relative to DCs, we injected mCherry mRNA-NPs i.p. into NOD mice; lymphoid tissues were recovered 48 h later, and collagenase digested for release of stromal cells and analyzed for transfection by FCM. Lymph node endothelial cells (LNECs; CD45−CD31+) had the highest frequency of transfected cells (∼15%), followed by resident CD8α+ cDC1 (3%–4%) in the draining PLNs (Figures 2D and 2E), suggesting free drainage of mRNA-NPs to the local lymph nodes. Similarly, we also detected expression of EGFP in both CD45+CD11c+ cells (∼5%) and non-hematopoietic CD45− cells (∼8%) from PLNs after EGFP mRNA-NPs were injected i.p. into NOD mice (Figure S2B). Consistent with in vitro transfection results, our mRNA-NP delivery platform results in transfection of different APC populations in vivo, with a greater frequency of LNECs transfected as compared with DCs.
mRNA-NPs Achieve mRNA Delivery into a Broader Network of Lymphoid Tissues Than mRNA-DCs after i.p. Injection
The route of administration influences the amount of antigen produced and the location and duration of expression, and subsequently determines the efficacy of mRNA vaccine. In this respect, we further tested the in vivo application of the mRNA-NP delivery platform by studying its biodistribution relative to mRNA-DCs using bioluminescence imaging (BLI) after injection via four routes: i.p., intravenous (i.v.), intradermal (i.d.), and intranodal (i.n.). First, we injected either Luc-expressing mRNA-NPs at two doses (20 and 40 μg/mouse) or Luc-expressing mRNA-DCs at a dose of 6 μg mRNA/106 electroporated DCs/mouse via the i.p. route and performed BLI at 4 and 8 h. At both time points, we observed robust Luc signals around the abdominal and pelvic regions of mice treated by mRNA-NPs or mRNA-DCs (Figure 3). Analysis of lymphoid organs excised after the second full-body live imaging (8 h) revealed that i.p. injected mRNA-NPs localized primarily to PLNs, omentum, and spleen (Figure 3A), whereas mRNA-DCs mainly accumulated in PLNs and omentum (Figure 3B), consistent with our previous reports on DC homing.44 These results are in agreement with preferential in vivo uptake and expression of mRNA-NPs by professional APCs from PLNs and spleen after i.p. injection (Figure 2; Figure S2). Of note, the Luc signal from mRNA-NPs dropped between 4 and 8 h, whereas it remained relatively stable with mRNA-DCs, albeit with substantial variability, during the same period (Figures 3C and 3D). Some Luc signal was detected in nearby pancreatic tissue, but not consistently in all mice, and may be in part contributed by omental tissue associated with the pancreas. Luc expression from mRNA-NPs was also detected in the small intestine, stomach, liver, and kidney (Figure S3A), as well as in the epididymis in male mice (data not shown). Taken together, these results revealed that mRNA-NPs target a broader network of tissues compared with mRNA-DCs after i.p. injection.
Figure 3.

mRNA-NPs and mRNA-DCs Differ in Their In Vivo Biodistribution after i.p. Injection
(A and B) Luc expression 8 h after i.p. injection of Luc mRNA-NPs (A) or Luc mRNA-DCs (B) in live mice (top) and in excised lymphoid tissues (middle), and relative expression in each lymphoid tissue (bottom) at doses of 20 μg mRNA-NPs/mouse or 6 μg Luc-expressing mRNA/106 electroporated DCs. (C and D) Quantified Luc signal from Luc mRNA-NPs (C) or mRNA-DCs (D) at both 4- and 8-h time points displayed as total photons per second (based on region of interest). Mean ± SD from n = 3 mice.
mRNA-NPs Preferentially Deliver mRNA into Spleen, whereas mRNA-DCs Mainly Localize in Lung after i.v. Injection
Lipid nanoparticles (LNPs) are the most commonly used mRNA delivery tools, and they mainly target the liver when delivered i.v.11 To assess whether this is also true for our mRNA-NPs, we studied the biodistribution of mRNA-NPs and mRNA-DCs after i.v. injection into NOD mice. Most mRNA-DCs were mainly localized in lungs after 8 h, whereas mRNA-NPs accumulated in the spleen (Figures 4A–4D), illustrating a striking difference between these two delivery modalities when using this route. On the other hand, no Luc signal above background was detected in liver and other lymphoid tissues (Figure S3B; Figures 4A and 4B) in both delivery modalities. Although many DCs are apparently trapped in lung tissue, we expect that some of them eventually redistribute to the spleen and some lymph nodes at later time points.44 In contrast, mRNA-NPs more readily pass through the lungs and are readily taken up in the spleen, most likely by red pulp APCs.
Figure 4.

Localization of mRNA-NPs and mRNA-DCs after i.v. Injection
(A and B) Luc expression 4 h after i.v. injection of Luc-expressing mRNA-NPs (A) or mRNA-DCs (B) in live mice (top) and in excised lymphoid tissues 8 h after injection (bottom). (C and D) Quantification of Luc expression (total photons/s) from Luc-expressing mRNA-NPs (C) or mRNA-DCs (D) at 4- and 8-h time points (based on region of interest). Mean ± SD from n = 3 mice.
Local In Vivo Delivery of mRNA by mRNA-NPs and mRNA-DCs
Next, we investigated the fate of mRNA-NPs and mRNA-DCs inoculated into a tighter (i.d.) space. Following i.d. administration of mRNA-NP (two doses, 5 and 10 μg mRNA/mouse), Luc expression was observed only at the injection site, but not in the draining lymph nodes, suggesting that the dermis may be too dense for these particles to reach lymphatics and drain to lymph nodes in sufficient numbers for Luc signal to be detected (Figure 5A). By contrast, i.d. administration of mRNA-DCs (2 μg mRNA/106 DCs/mouse) resulted in Luc expression at both the injection site and the draining ILNs (Figure 5B). The reporter expression from i.d. injected mRNA-NPs and mRNA-DCs remained local, and in contrast with more systemic injections (i.v. and i.p.), we did not detect any signal in more distal tissues (data not shown). Moreover, and in contrast with systemic delivery, local injection of mRNA-NPs was not associated with any decrease in expression between the 4- and 8-h time points (Figures 5C and 5D), suggesting that this route may achieve more sustained protein expression. Additional studies will be needed to assess persistence of gene expression by mRNA-NPs beyond 8 h. It has been shown that i.d. administration of LNP-encapsulated mRNA leads to prolonged protein expression at the site of injection for up to 10 days.45 On the other hand, the inability of mRNA-NPs to drain to ILNs may be circumvented by direct i.n. injection, although it is technically more challenging. It offers the advantage of targeted antigen delivery to various APCs at the site of T cell activation, obviating the need for antigen drainage or DC migration to lymphoid tissues for antigen presentation. Such i.n. injection can be conducted in patients under direct ultrasonographic guidance and is being clinically evaluated for the delivery of protein antigens for T1D.26 Following a published protocol,46 we successfully achieved i.n. injection of mRNA-NPs into ILNs, evidenced by Luc expression in the nodes and not in the surrounding fat pad (Figure S3C).
Figure 5.

Localized Luc Expression from mRNA-NPs and mRNA-DCs after i.d. Injection
(A and B) Luc expression 4 h after i.d. injection of Luc-expressing mRNA-NPs at dose of 5 μg mRNA/mouse (A) or mRNA-DCs at dose of 2 μg mRNA/106 DCs/mouse (B) in live animal (top) and in excised lymphoid tissues (middle), and relative Luc expression in draining lymph nodes and injection site (bottom). (C and D) Quantification of Luc expression from Luc mRNA-NPs (C) or mRNA-DCs (D) at the site of injection (based on region of interest). Mean ± SD from n = 3 mice.
mRNA-NPs Stimulate Antigen-Specific T Cells in a Wider Network of Lymphoid Tissues Than mRNA-DCs upon i.p. or i.v. Vaccination
Encouraged by our transfection and biodistribution results from reporter genes, we investigated the immunological effect of our epitope-encoding mRNA construct (Figure 1A) delivered by both modalities to compare and evaluate their possible implications for mRNA-based immunotherapy. The construct expresses major epitopes and mimotopes covering four different β cell antigens (insulin, chromogranin A [ChgA], glutamate decarboxylase [GAD65], and islet-specific glucose-6-phosphatase catalytic subunit-related protein [IGRP]). We have previously shown that this mRNA construct can be effectively delivered to BM-DCs by mRNA electroporation and enables the stimulation of antigen-specific CD4+ and CD8+ T cells responses in vitro.22 We first tested mRNA-NPs for the delivery of our antigen mRNA construct in vitro. To that end, splenocytes from BDC2.5 mice, whose T cells are reactive to the p79 mimotope expressed by the construct, were transfected with antigen mRNA-NPs at the equivalent of 0.1, 1, and 10 μg of mRNA per 0.2 × 106 cells/well. Dose-dependent CD4+ T cell responses (measured by proliferation and upregulation of CD25) were seen in the presence of antigen mRNA-NPs (Figures S4A and S4B). Quantification of three secreted cytokines (interleukin-2 [IL-2], interferon-γ [IFN-γ], and IL-10) from the culture supernatant by ELISA also showed dose-dependent cytokine production (Figure S4C). Consistent with our previous data,22 these results also show that the target antigen/polypeptides were not only expressed from mRNA-NP formulation, but they were also effectively processed and presented to T cells by APCs and induced dose-dependent antigen-specific T cell responses in vitro. We then proceeded to validate the quality and amplitude of antigen-specific T cell responses to a number of expressed epitopes in the in vivo setting. We used an adoptive transfer model wherein donor CD4+CD25− and CD8+ T cells were isolated from T cell receptor (TCR)-transgenic BDC2.5 and NY8.3 mice, respectively, and transferred i.v. into congenic NOD.CD45.2 or Thy1.1 recipient mice (see gating strategy in Figure S5A). These T cells are specific to two of our expressed epitopes (Figure 1A).22 Following i.p. administration, mRNA-NPs elicited robust antigen-specific CD4+ and CD8+ T cell responses (as measured by proliferation and/or upregulation of CD25) within a range of lymphoid tissues such as PLNs, MLNs, spleen, and even as far as ILNs and CLNs (Figures 6A–6C; Figures S5B–S5D). By contrast, mRNA-DCs induced antigen-specific T cell responses in more localized lymphoid tissues (mainly in PLNs and to a limited extent in MLNs) after i.p. administration (Figures 6D–6F; Figures S5E–S5G). As expected, these TCR-transgenic T cells proliferated in PLNs in response to islet-derived endogenous antigens (independently from administration of antigen), but in contrast with those responding to our construct, they did not upregulate CD25 (Figures 6, S5, and S6), similar to delivery of this construct by DNA vaccine.31 We repeated this experiment using another CD4+ T clone from BDC12-4.1 mice that recognizes another epitope expressed from our construct (Figure 1A), and the results showed a T cell response profile similar to BDC2.5 T cells for both mRNA-NP and mRNA-DC approaches (Figures S6A–S6F). The magnitude of the BDC12-4.1 T cell response is reduced relative to BDC2.5 T cells, consistent with our previous work.22 We further confirmed these findings by measuring the response of endogenous antigen-specific T cells identified by MHC tetramer (see gating strategy in Figure S7A). Consistent with adoptive transfer experiments and biodistribution data using reporter mRNA, responses to i.p. injected mRNA-NPs were seen mainly in PLNs and spleen, but also in more distal sites such as MLNs and CLNs (Figures 7A–7C; Figures S7B and S7C), whereas responses to mRNA-DCs were primarily restricted to the PLNs, and to a limited extent to MLNs (Figures 7D and 7E; Figures S7B and S7D). Major differences between the two modalities were that mRNA-DCs generally elicited a stronger CD25 induction and no or limited response in the spleen relative to mRNA-NPs (Figures 6 and 7; Figures S6 and S7B–S7D). Despite a Luc signal predominant in spleen, i.v. injected mRNA-NPs induced robust antigen-specific CD4+ T cell responses in all lymphoid tissues tested (Figure S8). We also previously showed that mRNA-DCs induce T cell responses restricted to spleen, PLNs, MLNs, and lung-associated lymph nodes following i.v. injection.44 Taken together, these data show that mRNA-NPs achieve a broader antigen biodistribution than mRNA-DCs.
Figure 6.

mRNA-NPs Induce Antigen-Specific CD4+ T Cell Responses in a Wider Network of Lymphoid Tissues Compared with mRNA-DCs after i.p. Administration
(A–F) Responses of transferred BDC2.5 CD4+ T cells to mRNA-NPs at a dose of 10 μg mRNA/mouse (A–C) and mRNA-DCs at a dose of 2 μg/1 × 106 electroporated DCs/mouse (D–F) were analyzed in cervical (CLNs), inguinal (ILNs), pancreatic (PLNs), mesenteric lymph nodes (MLNs) and spleen. The results are depicted as representative dot plots (A and D), proliferation (percentage divided) (B and E), and CD25 upregulation (C and F). EGFP mRNA was used as control for both mRNA-NP and mRNA-DCs modalities. The bar graphs (B, C, E, and F) show the mean ± SEM from three biological replicates, and the significant differences indicated for several lymphoid tissues are relative to PLNs in the antigen-treated group.
Figure 7.

mRNA-NPs Target a Broader Network of Lymphoid Tissues Than mRNA-DCs for Antigen Presentation
(A–F) Endogenous antigen-specific CD4+ T cell responses to mRNA-NPs (A–C) and mRNA-DCs (D–F) in various lymphoid tissues. Mice were injected i.p. with mRNA-NPs (5 μg mRNA/mouse) or 1 × 106 mRNA-DCs (1 μg mRNA/mouse), whereby mRNAs express multiple epitopes (antigen) or mCherry (control), and lymphoid tissues were analyzed 3 days later. The percentage of CD44hi among p79-reactive CD4+ T cells indicating their engagement with antigen is shown in red on the dot plots (A and D). The bar graphs (B, C, E and F) show the mean ± SD from three biological replicates, and the significant differences indicated for several lymphoid tissues are relative to PLNs in the antigen-treated group.
mRNA-NPs and mRNA-DCs Induce Local Antigen-Specific T Cell Responses after i.d. Injection
Local antigen delivery can be a determinant factor for effective immunotherapy in some disease models where systemic delivery would otherwise result in unwanted side effects. Local antigen delivery (e.g., i.d. delivery) has also been shown to elicit significantly higher immune responses than other systemic routes of administration (e.g., i.v. route).47 Following i.d. vaccination of mice with mRNA-NPs or mRNA-DCs, we analyzed the response of adoptively transferred T cells in ILNs (draining lymph nodes), CLNs (internal negative control), and PLNs (internal positive control). Both modalities induced antigen-specific T cell responses characterized by upregulation of CD25 and proliferation in draining ILNs as compared with ILNs of mice vaccinated with control mRNA or with internal CLN controls (Figure 8). Although mRNA-NPs did not lead to measurable signal in draining ILNs by BLI imaging (Figure 5A), T cell responses similar to mRNA-DCs were observed (Figure 8), suggesting that antigens produced at the inoculation site eventually accessed the local LNs, either by draining on their own or by being transported by migratory DCs. There were no antigen-specific T cell responses induced by both modalities in CLNs and spleen after i.d. administration, confirming that the response remained local. As mentioned earlier, donor T cells proliferated in PLNs without upregulating CD25, and the proliferation was the same with and without antigen delivery, suggesting that locally delivered antigens likely did not reach the PLNs (Figure 8).
Figure 8.

Induction of Antigen-Specific T Cell Responses in Local Draining Lymphoid Tissues after i.d. Injection
(A–F) Responses of transferred BDC2.5 CD4+ T cells to mRNA-NPs at a dose of 5 μg mRNA/mouse (A–C) or mRNA-DCs at a dose of 0.6 μg/1 × 106 electroporated DCs/mouse (D–F) were analyzed 3 days after i.d. injection. The bar graphs (B, C, E, and F) show the mean ± SD from three to six biological replicates, and the significant differences indicated for several lymphoid tissues are relative to ILNs in the antigen-treated group.
Co-delivery of Antigens and Immune Modulator with mRNA-NPs and mRNA-DCs Modulates T Cell Responses
In order to achieve greater efficacy in ASIT, co-delivery of adjuvants (stimulatory or inhibitory cytokines/ligands) along with antigens may be necessary to modulate the immune response toward immunity or tolerance. Delivery systems based on mRNA make this particularly easy to do. In this case, we combined mRNA encoding epitopes and IL-27, and delivered them in vivo using mRNA-NPs or mRNA-DCs. Co-delivery of IL-27 mRNA enhanced the expression and secretion of the immunosuppressive cytokine IL-10 by responding T cells as compared with co-delivery of antigen with an equal amount of GFP mRNA (Figure S9), suggesting that immune modulation to enhance tolerance-promoting cytokines is feasible with these approaches.
Discussion
mRNA is an emerging versatile therapeutic class, but its inherent instability under physiological conditions and inefficient in vivo delivery have hindered the translation of mRNA-based therapy into clinics.11,48 The establishment of safe and effective in vivo delivery platforms that overcome these limitations is an important prerequisite to realize the full potential of mRNA therapeutics. In this study, we compared two different in vivo mRNA delivery modalities: mRNA-NP and mRNA-DC. First, we validated the delivery platforms using mRNA encoding reporter genes (EGFP, Luc, and mCherry mRNA) and applied them to delivery mRNA encoding for multiple epitopes and neoepitopes from several β cell antigens recognized in T1D. Our data demonstrate that the biodistribution of mRNA-DCs is more restricted than mRNA-NPs after i.p. or i.v. injection into NOD mice. Moreover, mRNA-NP uptake and expression occur predominantly in the draining lymph nodes by both professional and non-professional APCs (DCs and LNECs, respectively). Whereas mRNA-DCs involve an APC population that is rather homogeneous, well characterized, and can be manipulated ex vivo (e.g., for phenotype, function, or homing potential), mRNA-NPs target a wide variety of endogenous APCs such as DCs, macrophages, and LNECs in vivo. In this case, it is currently difficult to control which type of APC is preferentially transfected. Given the low amount of antigen mRNA needed to stimulate T cells, both modalities allow addition of accessory mRNA encoding immunomodulators to tweak the APC phenotype and T cell responses. For instance, co-delivery of antigen with IL-27 mRNA significantly enhanced expression and/or secretion of IL-10. The i.p. route, for both platforms, involved PLNs as the main lymph nodes where T cell engagement takes place, which is highly relevant in the case of T1D. However, the expression of CD25 by responding T cells hints to potential differences in the T cell phenotypes elicited by the two approaches.
Following i.v. injection, mRNA-NPs preferentially delivered mRNA into spleen, whereas mRNA-DCs mainly localized in lung. Kranz et al.49 have shown that positively charged LNPs resulted in predominant Luc expression in the lungs relative to the spleen, whereas a gradual decrease of the cationic lipid content shifted Luc expression from lungs to spleen. Others have shown that systemic delivery of cationic LNPs encapsulating mRNA predominantly targets hepatocytes.50,51 Differences in localization observed between previously reported LNPs and our mRNA-NPs may reflect a difference in time points used for analysis, in the physicochemical characteristics of the RNA formulations (e.g., size and surface charge), and in their compositions. Indeed, Fenton et al.52 recently demonstrated that LNP composition alone can be used to modulate the site of protein expression. Following i.v. dosing of LNPs, spleen or liver can be efficiently targeted based on the type of ionizable lipid (non-degradable or degradable) used in the composition of LNPs.52 Unlike other commonly used LNPs that are composed of an ionizable cationic lipid, cholesterol, lipid-linked polyethylene glycol, and naturally occurring phospholipids,11 jetMESSENGER has only two preassembled components: ionizable mono-cationic lipids and a co-helper phospholipid. Our data indicate that the measurement of antigen-specific T cell responses is more sensitive than imaging of reporter activity, and provide a more accurate assessment of biodistribution. However, both methods are complementary, because T cell responses may not provide a good readout for expression in non-lymphoid tissues.
In the case of local inoculation into tissues (e.g., i.d.), both modalities induced similar antigen-specific T cell responses, which were restricted to the local draining lymph nodes. However, mRNA-DCs may be better vehicles in the case of local injections (e.g., i.d.) owing to their migratory potential. Nonetheless, the mRNA-NP approach may be easier to translate clinically than the mRNA-DC approach, which requires complex manipulation processes and can be applied only in a personalized fashion.
Overall, the two platforms target similar or different tissues depending on the route of delivery, but mRNA-NPs have a broader biodistribution following systemic delivery, which may facilitate engagement of rare populations of antigen-specific T cells and possibly enhance the efficacy of ASIT. The demonstration of successful targeting of LNSCs with our mRNA-NP approach is novel and of significant interest because these non-professional APCs have tolerogenic potential,53,54 but have been largely unexplored in the context of ASIT for autoimmune diseases. More importantly, we recently reported that stromal cells in the PLNs of T1D patients (the same lymph nodes as those targeted by mRNA-NPs after i.p. in mice) had higher antigen presentation and tolerogenic potential than those in control individuals.55 Thus, the ability to target antigens to LNSCs with mRNA-NPs as shown here is therapeutically relevant. Future preclinical studies will provide more detailed phenotypic characterization of antigen-specific T cells induced by both modalities at different time points and will address whether such phenotypes are associated with successful tolerance induction. In summary, the present study describes the characteristics and therapeutic potential of two vehicles to deliver mRNA encoding tailored epitopes and to effectively engage antigen-specific T cells across a more diverse variety of epitopes. This precision medicine approach may help circumvent some of the limitations of current ASITs.
Materials and Methods
Mice
All mice used in this study were purchased from the Jackson Laboratory and bred in our barrier facility unless otherwise stated. NOD mice (Jax #001976) were used for the generation of BM-DCs, for assessing in vivo T cell responses using MHC tetramer staining, and for BLI. TCR-transgenic BDC2.5 mice (Jax #004460) and BDC12-4.1.TCRα knockout mice22 were used as donors for isolation of antigen-specific CD4+ T cells. Similarly, TCR-transgenic NY8.3 mice (Jax #005868) were used as donors for isolation of antigen-specific CD8+ T cells. Congenic mice (NOD.Thy1.1 [Jax #004483] or NOD.CD45.2 [Jax #014149]) were used as recipients for adoptive transfer of purified antigen-specific CD4+ and/or CD8+ T cells. Both male and female mice were used in those studies (one sex per experiment) at 8–12 weeks of age. All procedures on these mice were performed according to Columbia University Institutional Animal Care and Use Committee recommendations and approved protocols.
In-Vitro-Transcribed mRNA
All in-vitro-transcribed mRNAs used in these studies were obtained from TriLink Biotechnologies. Reporter-encoding mRNAs (EGFP, mCherry, Luc) were purchased off the shelf with CleanCap and 5-methoxyuridine. Other mRNAs were custom-made using codon optimization, anti-reverse capping analog (or CleanCap), and base substitution with 5-methylcytosine and pseudouridine. The antigen-encoding mRNA constructs (Figure 1A) express major epitopes and mimotopes covering four different β cell antigens (insulin, ChgA, GAD65, and IGRP).22 CD4 and CD8 epitopes are segregated as two polypeptides upon translation, with the CD4 epitopes targeted to MHC class II with the transferrin receptor endosome-targeting signal and CD8 epitopes processed through the MHC class I pathway, as previously described.22 IL-27 mRNA was made to express both Il27 and Ebi3 genes, encoding the cytokine’s two chains, on a single cistron with a P2A cleavage site in between.
Nanoparticle Preparation and Biophysical Characterization
To prepare mRNA-NPs, we used the new cationic lipid-based transfection reagent jetMESSENGER (Polyplus-transfection), which we tested for in vivo application for the first time. The validation of optimal mRNA/jetMESSENGER ratio was done with Luc, mCherry, and antigen-encoding mRNAs using EMSA. For this assay, 1 μg of mRNA was added to 0–4 μL jetMESSENGER in a final volume of 10 μL in mRNA buffer, which was provided along with jetMESSENGER. After thorough mixing by pipetting, the colloids were left at room temperature for 1 h prior to loading them onto a 1% agarose gel for electrophoresis. For other assays, mRNA was first diluted with mRNA buffer in a 1:100 w/v mRNA/buffer ratio, and mRNA-NP formulation was done according to the manufacturer’s instructions and EMSA validated optimal mRNA/jetMESSENGER ratio. Particle size and zeta potential of mRNA-NPs were measured on a Malvern Zetasizer ZSP in disposable cuvettes in triplicate at the Columbia Nano Initiative Facility (Columbia University). The average particle size and zeta potential values were determined at mRNA concentration of 10 μg/mL and reported as the mean ± SD.
In Vitro mRNA Transfection
Various APCs were used to test in vitro transfection. BM-DCs were generated in vitro from NOD mice at 8–12 weeks of age as described previously.22 In brief, tibia, femur, and pelvic bones were collected, and multiple rounds of crushing by mortar and pestle and washing with complete medium were performed until most bone marrow cells were recovered. Bone marrow cells were then subjected to separation using a 40% on 80% Percoll density gradient followed by red blood cell lysis, before being enriched for DC precursors by depleting T cells, B cells, and granulocytes using biotinylated antibodies to CD3, B220 and Gr-1 (BioLegend), and the Mouse Streptavidin RapidSpheres Isolation kit (STEMCELL Technologies). The cells were cultured with granulocyte macrophage colony stimulating factor (GM-CSF) and IL-4 (final concentrations of 10 ng/mL each) for 6 days before being used for assays. Stromal cell lines used for in vitro transfection were DAPg7 cells,22,43 as well as NOD FRCs and LECs that were immortalized in our lab by overexpression of human papilloma virus E6/E7 proteins. The cell lines were passaged at least two times before being used for transfection. Cells were plated in 96-well plates and cultured overnight in a humidified CO2 incubator at 37°C prior to transfection (10,000 cells/well for DCs and DAPg7; 3,000 cell/well for FRCs and LECs). mRNA-NP formulation was prepared using mCherry or EGFP mRNA at a dose of 0.1 μg mRNA in 20 μL mRNA buffer/well and incubated for 15 min at room temperature. The volume of transfection medium was adjusted to 50 μL using serum-free Opti-MEM medium (Thermo Fisher Scientific) per well in a 96-well plate. The culture medium was replaced with 50 μL of mRNA-NP suspensions in each well. After 4 h of culture, 50 μL of complete medium with 20% FBS was added to each well. After overnight culture, 100 μL complete medium was added to each well, and the cells were harvested 48 h post-transfection to determine transfection efficiency by FCM. For mRNA electroporation of DCs, 5 × 106 cells were added to a 4-mm cuvette in 200 μL of serum-free Opti-MEM medium and electroporated using the GenePulser XCell (Bio-Rad) with 325 V pulse and 10 ms time constant. Then, DCs were cultured at different time points in complete medium for mRNA expression and analyzed by FCM. For transfection of primary splenic APCs, splenocytes were isolated from NOD mice and cultured at 2 × 105 cells/well (U-bottom 96-well plates) in complete medium with or without 0.1 μg/well EGFP-encoding mRNA formulated as mRNA-NP. Cells were harvested at 24 h, washed twice, and stained with antibodies to CD45, B220, CD11c, CD11b, and CD8a (BioLegend) to differentiate APC subpopulations (Figure S1B), and were then analyzed on BD Fortessa for in vitro transfection efficiency.
In Vivo Cellular Uptake and Expression
For in vivo applications, DCs were injected immediately after electroporation. Similarly, mRNA-NP formulations were freshly prepared as described above using EGFP and mCherry mRNA, and injected i.p. into NOD mice at a dose of 20–30 μg mRNA per mouse. Various draining and control lymphoid tissues were excised 48 h after injection unless mentioned otherwise. Single-cell suspensions were prepared by triturating the tissues between the rough sides of two glass slides. For ex vivo analysis of in-vivo-transfected cells (using mCherry mRNA), tissues were digested in medium with 10% FCS plus 1 mg/mL collagenase IV (Worthington), 1 mg/mL Dispase (Invitrogen) and 100 μg/mL DNase I (Roche) at 37°C for 20 min with agitation to disperse aggregates and facilitate digestion to release stromal cells. Cells were filtered on a 70-μm cell strainer, washed twice, and stained with antibodies to CD45, B220, CD11c, CD11b, CD8a, Pdpn, and CD31 (BioLegend). Various APCs including DCs, macrophages, B cells, and CD45− stromal cells such as fibroblasts and endothelial cells were then analyzed on BD Fortessa for EGFP mRNA or LSRII for mCherry mRNA.
BLI Imaging
Luc-encoding mRNA-NPs were injected at a dose of 5–10 μg mRNA per mouse (i.d. and i.n.) or 20–40 μg mRNA per mouse (i.p. and i.v.). In order to compare the biodistribution of mRNA-DCs with mRNA-NPs, 30 μg Luc mRNA was introduced into 5 × 106 DCs by electroporation as described above and were directly injected into mice at a dose of 1 × 106 cells/mouse (i.d., i.p., or i.v.). Uptake, translation, and subsequent expression of Luc-mRNA in vivo were evaluated by BLI (IVIS® system; Caliper Life Sciences). Emitted photons from live anesthetized animals were quantified at 4- and 8-h time points after i.p. injection of sterile D-Luciferin substrate at 150 μg/g of body weight. Subsequently, lymphoid tissues such as CLNs, PLNs, MLNs, ILNs, and spleen, as well as other tissues (omentum, lungs, intestine, kidneys, etc.) were harvested for ex vivo tissue imaging to assess signal biodistribution. The images were analyzed with the IVIS® Living Image 4.0 software, and regions of interest were quantified as average radiance. All BLI experiments were performed at the Small Animal Imaging Core Facility (Columbia University).
T Cell Response Assays
In Vitro T Cell Responses
Splenocytes were isolated from NOD.BDC2.5 mice and were subjected to red blood cell lysis buffer. After being filtered through a 70-μm cell strainer and labeled with a violet cell proliferation dye eFluor 450 (VCPD) (10 μM for 15 min; eBioscience), the splenocytes were cultured at 2 × 105 cells with or without mRNA-NPs (titrated dose of 0.1, 1, and 10 μg antigen mRNA) per well for 3 days in a humidified CO2 incubator at 37°C. After supernatants were collected and kept at −20°C for cytokine analysis, the splenocytes were stained with CD4 and CD25 antibodies (BioLegend) to analyze T cell responses by FCM.
Adoptive Transfer Experiments
Spleen and pooled lymph nodes were collected from donor TCR-transgenic mice. Antigen-specific CD4+CD25− T cells were purified from BDC2.5 or BDC12-4.1.TCRα knockout mice using the EasySep Mouse CD4 T Cell Isolation Kit (STEMCELL Technologies) supplemented with biotinylated anti-CD25. Antigen-specific CD8+ T cells were purified from NY8.3 mice using the EasySep Mouse CD8 T Cell Isolation Kit. Cells were then labeled with VCPD and injected i.v. into NOD.Thy1.1 or NOD.CD45.2 congenic mice. On the same day, the recipient mice were injected i.d. or i.p. with either mRNA-DCs (1 × 106 electroporated DCs/mouse) or mRNA-NPs at 5 or 10 μg formulated mRNA/mouse, respectively, for each route. Because our electroporation protocol was optimized based on 30 μg total mRNA/cuvette, EGFP or mCherry mRNA was used as filler to adjust the total amount of mRNA per cuvette for mRNA-DCs. After 3 days, lymph nodes (ILNs, CLNs, PLNs, and MLNs separately) and spleen were isolated for single-cell suspension. The cells were stained with antibodies to CD4, CD8, CD25, CD44, as well as CD45.1 or Thy1.2 (CD90.2) (BioLegend), and T cell responses were analyzed by FCM on BD Fortessa.
In immunomodulation experiments, mice were immunized with mRNA-NPs containing 30 μg total mRNA (5 μg antigen mRNA and 25 μg IL-27 mRNA or GFP mRNA) or with 1 × 106 mRNA-DCs (electroporated with 3 μg antigen mRNA and 27 μg IL-27 mRNA or GFP mRNA per 5 × 106 cells).
Endogenous Antigen-Specific T Cell Responses
NOD mice were directly treated with either mRNA-NPs or mRNA-DCs, and after 3 days, the lymph nodes and spleen were collected as described above. In this case, lymph node or spleen cells were stained with MHC tetramers displaying the p79 “2.5” mimotope (I-Ag7/AAAAVRPLWVRMEAA) obtained from the NIH Tetramer Core Facility. The cells were then stained for different markers as described above.
Intracellular Cytokine Staining and Cytokine Secretion
Lymphocytes isolated from lymphoid tissues 2.5 days after vaccination were incubated in the presence of phorbol 12-myristate 13-acetate (25 ng/mL final; Sigma), ionomycin (1 μg/mL final; Sigma) for 4 (mRNA-DCs) or 48 h at half-dose (mRNA-NPs), and a mix of brefeldin A (1.5 μg/mL final; eBioscience) and monensin (1 μM final; BioLegend) during the last 4 h of culture. Restimulated cells were stained with Zombie Aqua dye and anti-CD4 (BioLegend), then fixed and permeabilized with BD Cytofix/Cytoperm buffer (BD Biosciences), and stained with anti-IL-10 (BioLegend) for quantification of IL-10 expression by FCM.
Supernatants, either from in-vitro-treated splenocytes or from cultured splenocytes obtained from treated mice, were collected after 3 days of culture and stored at −20°C. The concentrations of IL-2, IL-10, and/or IFN-γ were assessed by ELISA kits (BioLegend) using the manufacturer’s instructions.
Data Analysis
FCM data were analyzed using FCS Express 6 or FlowJo version 9.9.5. Data are shown as mean ± SD unless indicated otherwise. t tests were run on GraphPad Prism 7.05 for statistical significance, and the threshold was set to *p < 0.05, **p < 0.01, and *** p < 0.001.
Author Contributions
R.F.-F. designed and performed experiments, analyzed data, and wrote the manuscript. R.J.C. directed the research, designed experiments, and edited the manuscript. Both authors approved the manuscript.
Conflicts of Interest
The authors declare no competing interests. Columbia University was granted a patent protecting the constructs encoding multiple epitopes mentioned in these studies.
Acknowledgments
We thank Jorge Postigo for assistance with LNSC isolation, Valerie Kedinger and Patrick Erbacher at Polyplus-transfection for reagents and guidance, and Kris Thielemans (Vrije Universiteit Brussel) for kindly providing some in-vitro-transcribed mRNA for the very early stages of our studies. These studies were funded by NIH grant R21AI110812 to R.J.C. R.F.-F. was supported by a fellowship from the American Diabetes Association (grant 1-19-PMF-022). Research reported in this publication was performed using the CCTI Flow Cytometry Core, supported in part by the Office of the Director, NIH under awards S10RR027050 and S10OD020056, and by Diabetes Research Center grant P30DK063608. We thank the NIH Tetramer Core Facility, supported by contract HHSN272201300006C from the National Institute of Allergy and Infectious Diseases, for the MHC tetramer provided for these studies.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2019.10.015.
Supplemental Information
References
- 1.Riley R.S., June C.H., Langer R., Mitchell M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019;18:175–196. doi: 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guevara M.L., Persano S., Persano F. Lipid-Based Vectors for Therapeutic mRNA-Based Anti-Cancer Vaccines. Curr. Pharm. Des. 2019;25:1443–1454. doi: 10.2174/1381612825666190619150221. [DOI] [PubMed] [Google Scholar]
- 3.Hollingsworth R.E., Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7. doi: 10.1038/s41541-019-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garg A.D., Coulie P.G., Van den Eynde B.J., Agostinis P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017;38:577–593. doi: 10.1016/j.it.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Benteyn D., Heirman C., Bonehill A., Thielemans K., Breckpot K. mRNA-based dendritic cell vaccines. Expert Rev. Vaccines. 2015;14:161–176. doi: 10.1586/14760584.2014.957684. [DOI] [PubMed] [Google Scholar]
- 6.Dullaers M., Breckpot K., Van Meirvenne S., Bonehill A., Tuyaerts S., Michiels A., Straetman L., Heirman C., De Greef C., Van Der Bruggen P., Thielemans K. Side-by-side comparison of lentivirally transduced and mRNA-electroporated dendritic cells: implications for cancer immunotherapy protocols. Mol. Ther. 2004;10:768–779. doi: 10.1016/j.ymthe.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 7.Roep B.O., Solvason N., Gottlieb P.A., Abreu J.R.F., Harrison L.C., Eisenbarth G.S., Yu L., Leviten M., Hagopian W.A., Buse J.B., BHT-3021 Investigators Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8+ T cells in type 1 diabetes. Sci. Transl. Med. 2013;5:191ra82. doi: 10.1126/scitranslmed.3006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willekens B., Cools N. Beyond the Magic Bullet: Current Progress of Therapeutic Vaccination in Multiple Sclerosis. CNS Drugs. 2018;32:401–410. doi: 10.1007/s40263-018-0518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phillips B.E., Garciafigueroa Y., Trucco M., Giannoukakis N. Clinical Tolerogenic Dendritic Cells: Exploring Therapeutic Impact on Human Autoimmune Disease. Front. Immunol. 2017;8:1279. doi: 10.3389/fimmu.2017.01279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benham H., Nel H.J., Law S.C., Mehdi A.M., Street S., Ramnoruth N., Pahau H., Lee B.T., Ng J., Brunck M.E. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci. Transl. Med. 2015;7:290ra87. doi: 10.1126/scitranslmed.aaa9301. [DOI] [PubMed] [Google Scholar]
- 11.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowalski P.S., Rudra A., Miao L., Anderson D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019;27:710–728. doi: 10.1016/j.ymthe.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sullenger B.A., Nair S. From the RNA world to the clinic. Science. 2016;352:1417–1420. doi: 10.1126/science.aad8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeFrancesco L. The ‘anti-hype’ vaccine. Nat. Biotechnol. 2017;35:193–197. doi: 10.1038/nbt.3812. [DOI] [PubMed] [Google Scholar]
- 15.Youn H., Chung J.K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin. Biol. Ther. 2015;15:1337–1348. doi: 10.1517/14712598.2015.1057563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holtkamp S., Kreiter S., Selmi A., Simon P., Koslowski M., Huber C., Türeci O., Sahin U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108:4009–4017. doi: 10.1182/blood-2006-04-015024. [DOI] [PubMed] [Google Scholar]
- 17.Mauer J., Luo X., Blanjoie A., Jiao X., Grozhik A.V., Patil D.P., Linder B., Pickering B.F., Vasseur J.J., Chen Q. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature. 2017;541:371–375. doi: 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weissman D., Karikó K. mRNA: Fulfilling the Promise of Gene Therapy. Mol. Ther. 2015;23:1416–1417. doi: 10.1038/mt.2015.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thess A., Grund S., Mui B.L., Hope M.J., Baumhof P., Fotin-Mleczek M., Schlake T. Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015;23:1456–1464. doi: 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonehill A., Tuyaerts S., Van Nuffel A.M., Heirman C., Bos T.J., Fostier K., Neyns B., Thielemans K. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol. Ther. 2008;16:1170–1180. doi: 10.1038/mt.2008.77. [DOI] [PubMed] [Google Scholar]
- 21.Maruggi G., Zhang C., Li J., Ulmer J.B., Yu D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019;27:757–772. doi: 10.1016/j.ymthe.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dastagir S.R., Postigo-Fernandez J., Xu C., Stoeckle J.H., Firdessa-Fite R., Creusot R.J. Efficient Presentation of Multiple Endogenous Epitopes to Both CD4+ and CD8+ Diabetogenic T Cells for Tolerance. Mol. Ther. Methods Clin. Dev. 2016;4:27–38. doi: 10.1016/j.omtm.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atkinson M.A., Roep B.O., Posgai A., Wheeler D.C.S., Peakman M. The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol. 2019;7:52–64. doi: 10.1016/S2213-8587(18)30112-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serra P., Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat. Biotechnol. 2019;37:238–251. doi: 10.1038/s41587-019-0015-4. [DOI] [PubMed] [Google Scholar]
- 25.Roep B.O., Wheeler D.C.S., Peakman M. Antigen-based immune modulation therapy for type 1 diabetes: the era of precision medicine. Lancet Diabetes Endocrinol. 2019;7:65–74. doi: 10.1016/S2213-8587(18)30109-8. [DOI] [PubMed] [Google Scholar]
- 26.Ludvigsson J., Wahlberg J., Casas R. Intralymphatic Injection of Autoantigen in Type 1 Diabetes. N. Engl. J. Med. 2017;376:697–699. doi: 10.1056/NEJMc1616343. [DOI] [PubMed] [Google Scholar]
- 27.Cook D.P., Gysemans C., Mathieu C. Prospects of a type 1 diabetes vaccine. Expert Opin. Biol. Ther. 2017;17:403–406. doi: 10.1080/14712598.2017.1295035. [DOI] [PubMed] [Google Scholar]
- 28.Ludvigsson J. GAD65: a prospective vaccine for treating Type 1 diabetes? Expert Opin. Biol. Ther. 2017;17:1033–1043. doi: 10.1080/14712598.2017.1328495. [DOI] [PubMed] [Google Scholar]
- 29.James E.A., Pietropaolo M., Mamula M.J. Immune Recognition of β-Cells: Neoepitopes as Key Players in the Loss of Tolerance. Diabetes. 2018;67:1035–1042. doi: 10.2337/dbi17-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baekkeskov S., Hubbell J.A., Phelps E.A. Bioengineering strategies for inducing tolerance in autoimmune diabetes. Adv. Drug Deliv. Rev. 2017;114:256–265. doi: 10.1016/j.addr.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Postigo-Fernandez J., Creusot R.J. A multi-epitope DNA vaccine enables a broad engagement of diabetogenic T cells for tolerance in Type 1 diabetes. J. Autoimmun. 2019;98:13–23. doi: 10.1016/j.jaut.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fletcher A.L., Malhotra D., Turley S.J. Lymph node stroma broaden the peripheral tolerance paradigm. Trends Immunol. 2011;32:12–18. doi: 10.1016/j.it.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tewalt E.F., Cohen J.N., Rouhani S.J., Guidi C.J., Qiao H., Fahl S.P., Conaway M.R., Bender T.P., Tung K.S., Vella A.T. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood. 2012;120:4772–4782. doi: 10.1182/blood-2012-04-427013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ciré S., Da Rocha S., Ferrand M., Collins M.K., Galy A. In Vivo Gene Delivery to Lymph Node Stromal Cells Leads to Transgene-specific CD8+ T Cell Anergy in Mice. Mol. Ther. 2016;24:1965–1973. doi: 10.1038/mt.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fornaguera C., Guerra-Rebollo M., Ángel Lázaro M., Castells-Sala C., Meca-Cortés O., Ramos-Pérez V., Cascante A., Rubio N., Blanco J., Borrós S. mRNA Delivery System for Targeting Antigen-Presenting Cells In Vivo. Adv. Healthc. Mater. 2018;7:e1800335. doi: 10.1002/adhm.201800335. [DOI] [PubMed] [Google Scholar]
- 36.Van der Jeught K., De Koker S., Bialkowski L., Heirman C., Tjok Joe P., Perche F., Maenhout S., Bevers S., Broos K., Deswarte K. Dendritic Cell Targeting mRNA Lipopolyplexes Combine Strong Antitumor T-Cell Immunity with Improved Inflammatory Safety. ACS Nano. 2018;12:9815–9829. doi: 10.1021/acsnano.8b00966. [DOI] [PubMed] [Google Scholar]
- 37.Hunter Z., McCarthy D.P., Yap W.T., Harp C.T., Getts D.R., Shea L.D., Miller S.D. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano. 2014;8:2148–2160. doi: 10.1021/nn405033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomedicine. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Creusot R.J., Postigo-Fernandez J., Teteloshvili N. Altered Function of Antigen-Presenting Cells in Type 1 Diabetes: A Challenge for Antigen-Specific Immunotherapy? Diabetes. 2018;67:1481–1494. doi: 10.2337/db17-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fletcher A.L., Lukacs-Kornek V., Reynoso E.D., Pinner S.E., Bellemare-Pelletier A., Curry M.S., Collier A.R., Boyd R.L., Turley S.J. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J. Exp. Med. 2010;207:689–697. doi: 10.1084/jem.20092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cohen J.N., Guidi C.J., Tewalt E.F., Qiao H., Rouhani S.J., Ruddell A., Farr A.G., Tung K.S., Engelhard V.H. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J. Exp. Med. 2010;207:681–688. doi: 10.1084/jem.20092465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knoblich K., Cruz Migoni S., Siew S.M., Jinks E., Kaul B., Jeffery H.C., Baker A.T., Suliman M., Vrzalikova K., Mehenna H. The human lymph node microenvironment unilaterally regulates T-cell activation and differentiation. PLoS Biol. 2018;16:e2005046. doi: 10.1371/journal.pbio.2005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lombardi G., Arnold K., Uren J., Marelli-Berg F., Hargreaves R., Imami N., Weetman A., Lechler R. Antigen presentation by interferon-gamma-treated thyroid follicular cells inhibits interleukin-2 (IL-2) and supports IL-4 production by B7-dependent human T cells. Eur. J. Immunol. 1997;27:62–71. doi: 10.1002/eji.1830270110. [DOI] [PubMed] [Google Scholar]
- 44.Creusot R.J., Yaghoubi S.S., Chang P., Chia J., Contag C.H., Gambhir S.S., Fathman C.G. Lymphoid-tissue-specific homing of bone-marrow-derived dendritic cells. Blood. 2009;113:6638–6647. doi: 10.1182/blood-2009-02-204321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pardi N., Tuyishime S., Muramatsu H., Kariko K., Mui B.L., Tam Y.K., Madden T.D., Hope M.J., Weissman D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andorko J.I., Tostanoski L.H., Solano E., Mukhamedova M., Jewell C.M. Intra-lymph node injection of biodegradable polymer particles. J. Vis. Exp. 2014;83:e50984. doi: 10.3791/50984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niu L., Chu L.Y., Burton S.A., Hansen K.J., Panyam J. Intradermal delivery of vaccine nanoparticles using hollow microneedle array generates enhanced and balanced immune response. J. Control. Release. 2019;294:268–278. doi: 10.1016/j.jconrel.2018.12.026. [DOI] [PubMed] [Google Scholar]
- 48.Liu M.A. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines (Basel) 2019;7:e37. doi: 10.3390/vaccines7020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kranz L.M., Diken M., Haas H., Kreiter S., Loquai C., Reuter K.C., Meng M., Fritz D., Vascotto F., Hefesha H. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 50.Sahay G., Querbes W., Alabi C., Eltoukhy A., Sarkar S., Zurenko C., Karagiannis E., Love K., Chen D., Zoncu R. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013;31:653–658. doi: 10.1038/nbt.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang L., Berraondo P., Jericó D., Guey L.T., Sampedro A., Frassetto A., Benenato K.E., Burke K., Santamaría E., Alegre M. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018;24:1899–1909. doi: 10.1038/s41591-018-0199-z. [DOI] [PubMed] [Google Scholar]
- 52.Fenton O.S., Kauffman K.J., Kaczmarek J.C., McClellan R.L., Jhunjhunwala S., Tibbitt M.W., Zeng M.D., Appel E.A., Dorkin J.R., Mir F.F. Synthesis and Biological Evaluation of Ionizable Lipid Materials for the In Vivo Delivery of Messenger RNA to B Lymphocytes. Adv. Mater. 2017;29:1606944. doi: 10.1002/adma.201606944. [DOI] [PubMed] [Google Scholar]
- 53.Tostanoski L.H., Chiu Y.C., Gammon J.M., Simon T., Andorko J.I., Bromberg J.S., Jewell C.M. Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific. Cell Rep. 2016;16:2940–2952. doi: 10.1016/j.celrep.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.D’Rozario J., Roberts D., Suliman M., Knoblich K., Fletcher A. Leukocyte-Stromal Interactions Within Lymph Nodes. Adv. Exp. Med. Biol. 2018;1060:1–22. doi: 10.1007/978-3-319-78127-3_1. [DOI] [PubMed] [Google Scholar]
- 55.Postigo-Fernandez J., Farber D.L., Creusot R.J. Phenotypic alterations in pancreatic lymph node stromal cells from human donors with type 1 diabetes and NOD mice. Diabetologia. 2019;62:2040–2051. doi: 10.1007/s00125-019-04984-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.