

Abstract
Nanoparticles (NPs) are promising vehicles for drug delivery because of their potential to target specific tissues [1]. Although it is known that NP size plays a critical role in determining their biological activity, there are few quantitative studies of the role of NP size in determining biodistribution after systemic administration. Here, we engineered fluorescent, biodegradable poly(lactic-co-glycolic acid) (PLGA) NPs in a range of sizes (120 – 440 nm) utilizing a microfluidic platform and used these NPs to determine the effect of diameter on bulk tissue and cellular distribution after systemic administration. We demonstrate that small NPs (~120 nm) exhibit enhanced uptake in bulk lung and bone marrow, while larger NPs are sequestered in the liver and spleen. We also show that small NPs (~120 nm) access specific alveolar cell populations and hematopoietic stem and progenitor cells more readily than larger NPs. Our results suggest that size of PLGA NPs can be used to tune delivery to certain tissues and cell populations in vivo.
Keywords: Biodegradable nanoparticles, poly(lactic-co-glycolic acid) (PLGA), size, biodistribution, targeting, nanomedicine
Graphical Abstract
1. INTRODUCTION
Nanoparticles (NPs) hold great potential for controlled spatial and temporal delivery of a wide variety of therapeutic agents to disease-afflicted organs in vivo [2, 3]. Localizing agents to therapeutic target-sites can circumvent toxicity and reduce unnecessarily high system doses. However, achieving successful and tissue-specific delivery is difficult. NPs can be rapidly cleared from the bloodstream by the mononuclear phagocyte system (MPS), renal filtration, and endogenous enzymes [4, 5]. To overcome these processes and avoid non-specific uptake, NPs must be strategically engineered to reach their site of therapeutic action.
Altering NP size has been shown to control tissue-specific accumulation [6–8]. But the effectiveness of MPS uptake, renal filtration, and liver and spleen sequestration restricts the range of NP diameters that are able to access target-sites in substantial and impactful quantities. NPs with a hydrodynamic diameter greater than 200 nm, for example, are rapidly cleared from circulation and accumulate in the liver and spleen [4, 5, 7, 9]. This clearance is further promoted by the phagocytic action of Kupffer cells in the liver, which comprise 80 – 90% of the total body macrophage population [9]. Though critical to the body’s defense, these clearance processes limit the variety of NP sizes that can successfully reach their target sites in vivo.
While prior studies have evaluated the effect of NP size on in vivo biodistribution following intravenous (IV) administration [10–12], most have focused on inorganic NPs [10, 13]. Although they are readily available from a variety of commercial sources, and in a variety of sizes, these NPs cannot be easily engineered to deliver therapeutic cargo. Contrastingly, we and others have shown that biodegradable NPs made of poly(lactic-co-glycolic acid) (PLGA) can efficiently and safely deliver a variety of biological agents including chemotherapeutics [14–17], plasmid DNA (pDNA) [18–20], small-interfering RNAs (siRNAs) [21, 22], and peptide nucleic acids (PNAs) along with donor DNA molecules for genome modification [23–29]. Yet, the precise PLGA NP size that optimizes the delivery of these biological agents in vivo is not understood.
Control of NP size requires careful selection of the formulation method and control of relevant parameters. The most widely used methods to formulate polymeric NPs include double emulsion, nanoprecipitation, spray drying, and high-pressure homogenization [30–32]. Though effective, it is difficult to generate NPs within a narrow distribution of sizes with low polydispersity indices [30]. Double emulsion and spray drying typically result in NPs with a diameter greater than 300 nm and nanoprecipitation results in NPs with a diameter of 100 – 200 nm [30]. In addition, these methods are not easily scalable to meet the manufacturing demands of clinical trials. High-pressure homogenization can be scaled-up to meet large production demands, but factors including high volume of waste material, requirement of pre-emulsion due to phase separation, and manual handling of liquids can be limiting [31]. However, microfluidic systems—which were more recently developed—allow for control of NP size by varying polymer concentration and system flow rate [33]. One such microfluidic system is the benchtop NanoAssemblr™ from Precision NanoSystems Inc. Here, we used the NanoAssemblr™ to produce PLGA NPs of controlled size and low PDI [34].
In this report, we studied the effect of PLGA NP size on in vivo biodistribution after IV administration. We used two methods to quantify the effects of PLGA NP size in bulk tissue following systemic administration: an in vivo imaging system (IVIS) and flow cytometry. Also, using confocal microscopy, we visualized nanoparticle internalization and accumulation in cells. We demonstrate that, after IV administration, NPs approximately 120 nm in diameter access lung and bone marrow compartments in greater numbers than NPs of 160 nm in diameter or larger. This sharp threshold for size-dependent accumulation provides important guidance in the design of nanomaterials for drug delivery to the bone marrow and lung.
2. MATERIALS AND METHODS
2.1. Materials
Poly(D,L-lactide-co-glycolide; Mn = 10–15 kDa, LA:GA = 50:50) was purchased from PolySciTech (West Lafayette, IN). 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD) was ordered from Biotium (Fremont, CA). Acetonitrile (ACN) and dimethyl sulfoxide (DMSO) were obtained from J.T. Baker (Phillipsburg, NJ). 30 – 50 kDa poly(vinyl alcohol) (PVA) and Bovine Serum Albumin (BSA) were obtained from Sigma-Aldrich (St. Louis, MO). Amicon Ultra-15 filter tubes and heparin were obtained from MilliporeSigma (Burlington, MA). 378.3 g mol−1 D-(+)-trehalose dehydrate (trehalose) was purchased from MP Biomedicals (Irvine, CA). Slide-A-Lyzer MINI Dialysis Devices with 10K MWCO, Hoescht 33342, and CellTrace™ CFSE were purchased from Thermo Fisher (Waltham, MA). Tissue-Tek O.C.T. Compound was obtained from Sakura Finetek (Torrance, CA). DAKO Fluorescence Mounting Medium was purchased from Agilent Technologies (Santa Clara, CA). Chamber slides were purchased from Lab-Tek (Grand Rapids, MI). IsoThesia (Isoflurane) solution was obtained from Henry Schein Animal Health (Dublin, OH). 40 μm and 70 μm Sterile Cell Strainers were purchased from Fisher Scientific (Hampton, NH). RPMI 1640 Medium was purchased from Gibco (Gaithersberg, MD). Fetal Bovine Serum (FBS) was purchased from Atlanta Biologicals (Flowery Branch, GA). Collagenase, Type 2 was purchased from Worthington (Lakewood, NJ). ACK Lysing Buffer contained 154.95 mM ammonium chloride from JT Baker, 10 mM potassium bicarbonate from Sigma Aldrich, and 0.1 mM EDTA from Thermofisher. LIVE/DEAD Fixable Green Dead Cell Stain was ordered from Invitrogen. Antibody stains anti-mouse P2X7R, anti-mouse CD31, and anti-mouse F480 were purchased from BioLegend (San Diego, CA). EasySep Mouse CD117 (cKIT) Positive Selection Kit was obtained from StemCell Technologies (Vancouver, Canada).
2.2. Fabrication of NPs
PLGA NPs were produced using a benchtop NanoAssemblr™ instrument (Precision NanoSystems Inc., Vancouver, Canada). NPs with a range of sizes were formulated by dissolving PLGA at various concentrations in ACN. Fifteen mg of polymer was dissolved in 3 mL of ACN overnight to achieve NP-1, 20 mg of polymer was dissolved in 1 mL of ACN overnight to achieve NP-2, 30 mg of polymer was dissolved in 1 mL of ACN overnight to achieve NP-3, and 40 mg of polymer was dissolved in 1 mL of ACN overnight to achieve NP-4. To trace NPs in tissues, NPs were loaded with 0.5% DiD, a hydrophobic dye that has been used as a marker for NPs in prior studies [35, 36]. For dye-loaded NPs, 10 mg of DiD dissolved in 1 mL of dimethyl sulfoxide (DMSO) was added to the polymer solution at 0.5% wt:wt DiD:PLGA.
The organic phase comprised of the polymer/dye solution was injected into one port of the NanoAssemblr™ instrument. The aqueous phase consisting of 2% w:v PVA was simultaneously injected into the second port of the system to maintain a 1:1 aqueous:organic flow rate ratio. The total flow rate was maintained at 8 mL min−1. NP product was gathered in a 15 mL Falcon tube containing 2 mL of water, while separately disposing the initial 0.25 mL and the final volume of 0.05 mL of the NP solution. DI water was immediately added to bring the NPs solution to a volume of 15 mL and transferred to an Amicon Ultra-15 filter tube (100 K cutoff). NPs were washed at 4,000 g at 4°C for 45 min three times with DI water. Subsequently, the NPs were resuspended in 1 mg trehalose per 1 mg of NP, frozen at −80°C and then lyophilized. NPs were stored at −20°C after lyophilization until use.
2.3. In vitro characterization of NPs
The hydrodynamic diameter, polydispersity index (PDI), and surface charge (zeta-potential) of each NP formulation were measured at 0.05 mg mL−1 in water by dynamic light scattering (DLS) using a Zetasizer Nano-ZS (Malvern Instruments). A gold-palladium sputter coating was applied to NPs and morphologies were characterized by scanning electron microscopy (SEM) using a XL-30 scanning electron microscope (FEI/Philips).
To determine the loading of DiD in each NP formulation, 2 mg of NPs were resuspended by vortex and water bath sonication in 100 μL DMSO. Two-fold serial dilutions of the NP solutions were made into water and the concentration of DiD dye was quantified using a plate reader (ex/em 644/665 nm). Dye loading was calculated from a standard curve.
The release of DiD dye from PLGA NPs was analyzed using previously established methods [35, 36]. Briefly, 1 mg of NPs was resuspended by vortex and water bath sonication in 100 μL PBS, and then loaded into a Slide-A-Lyzer MINI Dialysis Device. The NPs were dialyzed in PBS at 37°C while shaking and the PBS solution was replaced at each pre-determined time point. For each time point, the nanoparticle solutions in the dialysis units were collected and the dye was quantified using a plate reader (ex/em 644/665).
2.4. Animal preparation
All procedures and experiments were performed in accordance with the guidelines and policies of the Yale Animal Resource Center (YARC) and approved by the Yale University Institutional Animal Care and Use Committee (IACUC). Male C57BL/6 mice (6 – 8 weeks old) obtained from Charles River Laboratories were used.
2.5. IVIS ex vivo biodistribution of NPs in whole organs
Two mg of fluorescent NPs were resuspended by vortex and water bath sonication in 1x dPBS to a concentration of 10 mg ml−1 and administered retro-orbitally to mice (n = 3). After 24 h, mice were sacrificed and perfused with heparinized saline. Tissues (brain, heart, lungs, liver, kidneys, spleen, pancreas, and bone marrow) were harvested for ex vivo imaging. Tissues were washed briefly in 1x PBS and imaged using a live imaging instrument (IVIS Spectrum, PerkinElmer) (ex/em 644/665 nm).
2.6. Cellular biodistribution of NPs
Flow cytometry was used to further assess and quantify NP biodistribution in vivo. Two mg of NPs were resuspended by vortex and water bath sonication in 1x dPBS to a concentration of 10 mg ml−1 and administered retro-orbitally to mice (n = 3). After 24 h, mice were sacrificed, perfused with heparinized saline, and tissues (brain, heart, lung, liver, kidney, spleen, pancreas, and bone marrow) were harvested. Tissues were homogenized to single cell suspensions in RPMI 1640 Medium through a 70 μm cell strainer. The resulting single cell suspensions were pelleted and resuspended in 200 μL of PBS containing 1% BSA (FACS buffer). The cells were stained with LIVE/DEAD Fixable Green Dead Cell Stain for 30 min at 4°C. After staining, cells were washed once with FACS buffer for 10 min and then resuspended in 200 μL of FACS buffer. Cell fluorescence was quantified using flow cytometry (Attune NxT, Invitrogen).
2.7. Fluorescence microscopy and NP foci analysis
Confocal microscopy was completed with an additional three animals for each treatment group, to measure cellular internalization of nanoparticles. Bone marrow cells were stained using CellTrace™ CFSE according to the manufacture’s protocol. 200,000 stained cells per 400 μL were seeded in chamber slides and fixed by adding an equal amount of 4% PFA in PBS to the media followed by a 15 min centrifugation step at 800 RCF in a swing bucket centrifuge. Subsequently, media/PFA mix was replaced with 4% PFA in PBS and samples were centrifuged again at 800 RCF for 15 min. DNA was stained with 2 μg/mL Hoechst 33342 in PBS for 15 min at room temperature. Lungs from at least three animals per condition were harvested, frozen in O.C.T. Compound, and sectioned. Ten μm sections were subsequently stained with 2 μg/mL Hoechst 33342 in PBS for 30 min at room temperature. After washing twice with PBS, bone marrow cells and lung samples were covered with coverslips using DAKO Fluorescence Mounting Medium. Images were analyzed with a Nikon Eclipse Ti fluorescence microscope with a Plan Apo 60X/1.40 Oil DIC h objective, a CSU-W1 confocal scanning unit with an iXon Ultra camera (Andor Technology), MLC 400B laser unit (Agilent Technologies), and NIS Elements 4.30 software (Nikon Corporation). Whole cell and nuclear NP foci were analyzed with the Focinator v2–31 software as previously described [37]. Images of the lung tissue section were quantified with the Stripenator software as previously described [37]. Representative images were generated using ImageJ.
2.8. NP uptake in type I alveolar epithelial cells, endothelial cells, and alveolar macrophage cells
Flow cytometry was used to assess NP distribution in specific lung cell populations. Mice (n = 3) were dosed with 2 mg of NP-1, NP-2, NP-3, and NP-4 at a concentration of 10 mg ml−1. After 24 hours, mice were sacrificed, perfused with heparinized saline, and lung tissue was harvested. The tissue was minced into several small pieces, incubated in 0.4% collagenase for 40 min at 37°C while shaking, and then homogenized through a 70 μm cell strainer. The resulting single cell suspensions were pelleted, resuspended in 2 mL of ACK Lysing Buffer, and incubated for 2 min at room temperature to lyse red blood cells and remove debris. To neutralize the ACK Lysing Buffer, 8 mL of RPMI 1640 containing 10% FBS was added to the solution. The cells were pelleted and resuspended in 200 μL of FACS buffer. The cells were stained with LIVE/DEAD Fixable Green Dead Cell Stain for 30 min at 4°C. After staining, cells were washed once with FACS buffer for 10 min and then incubated with anti-CD31 antibody, anti-F480 antibody, and anti-P2X7R antibody for 30 min. After staining, cells were washed once with FACS buffer for 10 min and then resuspended in 200 μL of FACS buffer. Cell fluorescence was quantified using flow cytometry.
2.9. NP uptake in hematopoietic stem and progenitor cells
Flow cytometry was used to assess NP distribution in bulk bone marrow and hematopoietic stem and progenitor cells. Mice (n = 3) were treated with 2 mg of NP-1, NP-2, NP-3, and NP-4 at a concentration of 10 mg ml−1. After 24 h, mice were sacrificed, perfused with heparinized saline, and femur and tibias harvested. Femurs and tibias were flushed with 5 mL 1x PBS [38]. Bone marrow cells were filtered through a 70 μm cell strainer and washed once with 1x PBS for 10 min. Hematopoietic stem and progenitor cells (HSPCs) were selected using the EasySep Mouse CD117 (cKIT) Positive Selection Kit (StemCell Technologies). CD117+ cells were stained with LIVE/DEAD Fixable Green Dead Cell Stain for 30 min at 4°C. After staining, CD117+ cells were washed once with FACS buffer for 10 min and then resuspended in 200 μL FACS buffer. Cell fluorescence was quantified using flow cytometry.
2.10. Data analysis
FlowJo v10.5.2 software was used to analyze flow cytometry data. GraphPad Prism 7 software was used for graphing and statistical analysis. Error bars represent standard error of the mean (SEM). Statistical significance was calculated by either a one-way ANOVA with a Bonferroni’s multiple comparisons test or an unpaired t-test (α = 0.05), which have been designated appropriately in each figure caption. Significance is represented on plots as ns p > 0.05, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
3. RESULTS
3.1. Formulation and characterization of NPs
To test whether the NanoAssemblr™ could be used to formulate DiD-loaded PLGA NPs of various sizes, several operational parameters—including flow rate ratio, stabilizer concentration, and PLGA concentration—were systematically varied and the size of resulting NPs was measured. Stable NPs with a hydrodynamic diameter less than 500 nm and PDI less than 0.25 were produced by maintaining an aqueous to organic phase flow rate ratio of 1:1 and surfactant concentration of 2% (Figure s1A–B). NPs with a range of sizes less than 500 nm were engineered by altering the concentration of PLGA dissolved in ACN (Figure s1C). Based on these results, we identified parameters for reproducible synthesis of 4 distinct NP populations: NP-1, NP-2, NP-3, and NP-4. The diameter of NP-1 was approximately 120 nm, NP-2 was 160 nm, NP-3 was 280 nm, and NP-4 was 440 nm (Figure 1B, Figure s2, Figure s3). The PDIs of all NP formulations were less than 0.25 (Figure 1B). These results were confirmed by SEM, which also demonstrated that NP morphology was spherical and fairly uniform for all NP formulations (Figure 1C–F). All NP formulations had a zeta-potential near −20 mV (Figure 1B). Further, all NPs demonstrated comparable DiD loading of 9 μg per mg of NP (Table s1), with less than 2% release over 24 h (Figure s4A–B).
Figure 1. Formulation and characterization of various sized DiD-loaded PLGA NPs.

(A) PLGA NPs are formulated by injecting ACN, PLGA, 0.5 wt% DiD and 2% PVA into the NanoAssemblr™. NPs are washed in water immediately following formulation. (B) Hydrodynamic diameter, PDI, and zeta-potential of NP formulations. Data is shown as mean ± SD (n = 3). SEM images of (C) NP-1 (D) NP-2 (E) NP-3 and (F) NP-4. Scale bar on images represents 1 μm.
3.2. IVIS biodistribution in whole organs depends on NP size
To test whether size of PLGA NPs influences whole tissue accumulation, DiD-loaded NPs were administered IV by retro-orbital injection. After 24 h, brain, heart, lung, liver, kidney, spleen, pancreas, and bone marrow tissue were excised and imaged using IVIS (Figure 2A). An average radiance (p/sec/cm2/sr) of DiD fluorescence was quantified using a tissue-specific region of interest (Figure 2B). Rigorous inter-organ statistical analyses were conducted to compare NP formulations to the untreated control and NP formulations to each other (Table s2). The average radiance in the brain and heart tissue was comparable to the untreated control for all NP formulations. In the lung, kidney, pancreas, and bone marrow, NP-1 demonstrated the greatest fluorescence accumulation while NP-4 demonstrated the lowest fluorescence accumulation. Further, the average radiance of NP-1 and NP-2 were significantly enhanced in the lung compared to the untreated control. The average radiance of NP-1, NP-2, and NP-3 were significantly enhanced in the kidney and bone marrow compared to the untreated control. The average radiance of NP-1 was significantly enhanced in the pancreas compared to the untreated control.
Figure 2. Whole organ ex vivo biodistribution of various sized PLGA NPs.

(A) IVIS imaging of organs 24 h post-injection. (B) Quantification of average radiance in organs. Data is shown as mean ± SEM (n = 3). Statistical significance was calculated using a one-way ANOVA test and significance is represented on graphs as *p ≤ 0.05. A more robust statistical analysis is shown in Table s2A.
Liver tissue showed the highest fluorescence accumulation of all tissues analyzed. While all NP formulations demonstrated significant accumulation in liver, NP-3 and NP-4 had the greatest average radiance in the liver. The average radiance in spleen tissue approached 5.0E0.8 p/sec/cm2/sr, with NP-1 having the lowest fluorescence accumulation and NP-3 having the greatest fluorescence accumulation.
3.3. In vivo biodistribution in bulk tissue depends on NP size
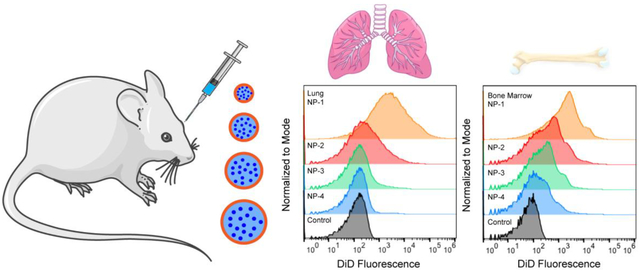
To further understand the effect of NP size on in vivo distribution, we used flow cytometry to assess cellular uptake. Twenty-four h after NP administration, tissues were excised and homogenized to form single cell suspensions. The mean fluorescence intensity of each NP formulation was quantified in all cells and normalized to the mean fluorescence intensity of the untreated control (nMFI) (Figure 3A). Rigorous inter-organ statistical analyses were conducted to compare NP formulations to the untreated control and NP formulations to one another (Table s3). The nMFI in brain, heart, and pancreas tissue after injection of all NP formulations was comparable to the untreated control. In the kidney, NP-1 demonstrated significantly enhanced uptake in comparison to the untreated control. The majority of the NPs accumulated in lung, liver, spleen, and bone marrow. In the liver, NP-4 had a significantly enhanced nMFI compared with the untreated control and all other NP formulations. In the spleen, injection of NP-1, NP-2, NP-3, and NP-4 formulations led to a significantly greater nMFI compared to the untreated control. In the lung, NP-1 had a significantly greater nMFI compared with the untreated control and all other NP formulations (Figure 3B–C). Similarly, in bone marrow, NP-1 had a significantly greater nMFI compared with all the untreated control and all other NP formulations (Figure 3D–E).
Figure 3. In vivo biodistribution of various sized PLGA NPs in bulk tissue.

(A) nMFI of DiD expression in all tissues quantified by flow cytometry. (B) Representative flow cytometry histograms showing DiD fluorescence in bulk lung tissue and (C) nMFI of DiD expression. (D) Representative flow cytometry histograms showing DiD fluorescence in bulk bone marrow and (E) nMFI of DiD expression. Data is shown as mean ± SEM (n = 3). Statistical significance was calculated using a one-way ANOVA test and significance is represented on graphs as *p ≤ 0.05, ***p ≤ 0.001, and ****p ≤ 0.0001. A more robust statistical analysis is shown in Table s3A.
3.4. Nanoparticle internalization in bulk bone marrow and lung cells
To further confirm that cells internalize NPs, we performed confocal microscopy in bone marrow and lung tissue. Twenty-four h after injection, bulk bone marrow cells were harvested, fixed, and stained with Hoechst 33342 for imaging and quantification. Visually, NPs seemed to overlap with the blue Hoechst staining of the cellular DNA. Further, NP-1 demonstrated enhanced internalization and accumulation within bone marrow cells after administration (Figure 4B) when compared to NP-2 (Figure 4C). When quantified, NP-1 had an average fluorescent intensity of ~5000, a nearly two-fold (and statistically significant) increase over NP-2 (Figure 4D).
Figure 4. Confocal imaging of bulk bone marrow and lung tissue from animals administered with NP-1 and NP-2.

Representative images of bone marrow 24 h post-administration for (A) untreated control, (B) NP-1, and (C) NP-2, which are quantified in (D) as fluorescent intensity. Scale bars on images equal 10 μm. Representative images of lung tissue 24 h post-administration for (E) untreated control, (F) NP-1, and (G) NP-2, which are quantified in (H) as NP intensity. Scale bars on images equal 50 μm. Data is shown as mean ± SEM (n = 3). Statistical significance was calculated using an unpaired t-test and significance is represented on graphs as *p ≤ 0.05 and **p ≤ 0.01.
To visualize NP internalization in lung tissue, tissues were harvested, frozen in O.C.T., and sectioned 24 h after injection. NP-1 demonstrated greater internalization and accumulation within lung cells after administration (Figure 4F) when compared to NP-2 (Figure 4G). When this was quantified, NP-1 had an average NP intensity of ~4500, nearly two-fold greater than NP-2, which was significantly different (Figure 4H).
3.5. Flow cytometry reveals enhanced uptake of NP-1 in type I alveolar epithelial and alveolar macrophage cells
Since NP-1 exhibited extensive uptake in lung tissue, we wanted to determine the specific lung cell populations in which NPs accumulated. Twenty-four h after injection, lung tissue was harvested, digested, and processed into a single cell suspension for flow cytometry. Type I alveolar epithelial cells (AEC I) were identified by staining with antibodies to P2X7R+ and were found to represent 14.4% of the overall lung cell population (Figure 5A). Alveolar macrophages were identified by staining with antibodies to F480+ and were found to represent 17.7% of the overall lung cell population (Figure 5B). Endothelial cells were identified by staining with antibodies to CD31+ and were found to represent 13.3% of the overall lung cell population (Figure 5C). At 24 h, NP-1, NP-2, NP-3, and NP-4 treated mice resulted in an increase in DiD fluorescence in AEC I (Figure 5D), alveolar macrophages (Figure 5E), and endothelial cells (Figure 5F) compared to the untreated controls. The nMFI for NP-1 treated mice was significantly greater than NP-2, NP-3, and NP-4 treated mice in AEC I (Figure 5G). In AEC I, the nMFI was not significantly different between NP-2 and NP-3 treated mice, however the nMFI was significantly different between NP-2 and NP-4 treated mice. A significant increase in nMFI was observed for NP-1 treated mice in comparison with all other NP formulations in alveolar macrophages (Figure 5H). NP-2, NP-3, and NP-4 treated mice had comparable nMFI values in alveolar macrophages. In endothelial cells, NP-1 and NP-3 treated mice did not have significantly different nMFIs, however both NP-1 and NP-3 presented a significant increase in nMFI over NP-2 and NP-4 treated and control mice (Figure 5I).
Figure 5. Cell-specific association of PLGA NPs in lung tissue.

Gating for (A) alveolar epithelial type I cells (P2X7R+), (B) alveolar macrophages (F480+), and (C) endothelial cells (CD31+). Black contour plots are unstained controls. Representative histogram showing DiD fluorescence in (D) P2X7R+, (E) F480+, and (F) CD31+ cell populations isolated from lung tissue 24 h post-injection with NP-1, NP-2, NP-3, and NP-4. nMFI of (G) P2X7R+ DiD+, (H) F480+ DiD+, and (I) CD31+ DiD+ cell populations at 24 h. Data is shown as mean ± SEM (n = 3). Statistical significance was calculated using a one-way ANOVA test and significance is represented on graphs as ns p > 0.05, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001. A more robust statistical analysis is shown in Table s4A.
For all NP formulations, the percentage of DiD+ AEC I (Figure s5A, Table s6A) and DiD+ alveolar macrophages (Figure s5B, Table s6A) was significantly greater than the untreated control. The difference in the percentage of DiD+ endothelial cells between NP-1 and all other NP formulations were statistically significant, whereas the difference in the percentage of DiD+ endothelial cells between NP-2 and NP-3 were not statistically significant (Figure s5C, Table s6A). All NP formulations had a significantly greater percentage of DiD+ endothelial cells than NP-4.
3.6. Flow cytometry exhibits enhanced uptake of NP-1 in isolated hematopoietic stem and progenitor cells
NP-1 demonstrated significant uptake in bulk bone marrow, so we investigated the accumulation of NP-1 in HSPCs. Twenty-four h post-injection, bulk bone marrow was harvested and processed using the EasySep Mouse CD117 (cKIT) Positive Selection Kit. The majority (83.6%) of positively selected cells were confirmed as HPSCs (CD117+) by flow cytometry (Figure 6A). NP-1, NP-2, NP-3, and NP-4 treated mice demonstrated an increase in DiD fluorescence in HSPCs compared to untreated controls (Figure 6B). The nMFI in HPSC populations for NP-1 treated mice was significantly greater than NP-2, NP-3, and NP-4 treated mice (Figure 6C). There was no significant difference in the nMFI of NP-2, NP-3, and NP-4 treated mice (Figure 6C).
Figure 6. Cell-specific association of PLGA NPs in hematopoietic stem and progenitor cells.

(A) Gating for CD117+ bone marrow cells (HSPCs). Black contour plot is unprocessed bulk bone marrow. (B) Representative histogram showing DiD fluorescence in HPSCs 24 h post-injection with NP-1, NP-2, NP-3, and NP-4. (C) nMFI of HSPCs at 24 h. Data is shown as mean ± SEM (n = 3). Statistical significance was calculated using a one-way ANOVA test and significance is represented on graphs as ns p > 0.05 and **p ≤ 0.01. A more robust statistical analysis is shown in Table s5A.
For all NP formulations, the percentage of DiD+ HSPCs (Figure s6, Table s7) was significantly greater than the untreated control. The difference in the percentage of DiD+ endothelial cells between NP-1 and all other NP formulations were statistically significant, whereas the difference in the percentage of DiD+ HSPCs between NP-2, NP-3, and NP-4 were not statistically significant (Figure s6, Table s7).
4. DISCUSSION
The principal advantage of NPs as drug carriers is their small size, which allows them to traverse biological barriers, enter various tissues, and associate with specific cell populations. Size, therefore, is one of the main parameters that define the effectiveness of NPs for preferential delivery to desired cell populations. However, only a few studies of polymer NPs have carefully examined the effect of size on biodistribution [39–45]. Rather, the influence of NP size on distribution in cells and tissues has been more thoroughly explored using inorganic NPs, given their ease of manufacturing in controlled size fractions [10, 46]. Size-dependent biodistribution was observed in a study of PEGylated gold NPs [46]: small PEGylated gold NPs (20 nm) demonstrated significantly greater accumulation compared with NPs (80 nm) in A341 tumor-xenografted mice. Outside of the tumor, NPs (20 nm) had prolonged blood circulation and decreased uptake by the liver and spleen, while larger NPs (80 nm) were taken up more readily by the liver and spleen [46].
It is not obvious that results obtained with inorganic NPs will be translatable to polymer NPs, which have advantages over inorganic NPs with regard to drug encapsulation and release. PLGA NPs, for example, have successfully delivered a variety of therapeutic agents, including chemotherapy drugs, pDNA, siRNA, and PNAs [14–29]. However, the effect of PLGA NP size on localization in tissues and cells after injection is still poorly understood. Here, we report the synthesis of fluorescent, PLGA NPs of various sizes, using the NanoAssemblr™ microfluidic device. The NanoAssemblr™ allows for the control of several NP characteristics including size by parameters such as flow rate ratio, total flow rate, formulation volume, surfactant concentration, and polymer concentration [34]. We optimized each parameter to formulate NPs with a PDI less than 0.25 (Figure s1A–C). Specifically, similar to other studies [47], we observed that decreasing the concentration of PLGA in the organic phase decreased the diameter of the NPs (Figure 1B). Together, these data show that we are able to synthesize NPs with specific average sizes using a scalable microfluidic approach.
We hypothesized that IV injection of PLGA NPs of different sizes would result in altered in vivo biodistribution. To test this hypothesis, size-differentiated DiD-loaded NPs were injected retro-orbitally into mice. Twenty-four h after injection, we studied the accumulation of NPs in various tissues, selecting this window of time to ensure that majority of the NPs were removed from circulation [48]. Here, we used three different methods to evaluate accumulation of NPs in organs: IVIS imaging, flow cytometry, and confocal microscopy. While IVIS imaging allows for rapid assessment of biodistribution, this method is unable to resolve cellular uptake. Therefore, signals detected by IVIS may result from interstitial accumulation, rather than cell-specific uptake. Although flow cytometry requires additional processing steps prior to analysis, we have found that this technique more accurately predicts uptake by therapeutically relevant cell populations at the specified time point [27, 49–51]. Finally, confocal microscopy confirmed that NPs were internalized and accumulated in bone marrow and lung tissue.
Using the aforementioned techniques, we found that NP size greatly affected organ and cellular distribution. In the brain and heart, we observed no significant accumulation at any NP size. In the pancreas and kidney, small NPs accumulated in the tissue to some extent, but few of those NPs were associated with cells strongly enough for us to observe by flow cytometry (Figure 3A). In the spleen and liver, the largest NPs have the highest levels of accumulation. In contrast, in the lung and bone marrow, the smallest NPs have the highest levels of accumulation, including accumulation within cell types that are of major interest in delivery of new therapies for lung and blood disorders.
For all NP formulations, the level of fluorescence detected in the heart and brain were insignificant. The low level of detection in the heart makes sense: biodistribution studies have demonstrated that polymer NPs > 100 nm do not have significant uptake in heart tissue [39, 41]. The low level of fluorescence observed in the brain also makes sense; many earlier studies have shown that passage from the systemic circulation to the brain through the blood-brain barrier (BBB) for unmodified PLGA NPs is low (< 1%) [52–54]. Although there are reports of accumulation of certain NPs—particularly those that are decorated with certain surface ligands—into the brain, it is invariably a small fraction (~1–2%) of the total injected dose [55].
Using IVIS, the three smallest preparations (NP-1, NP-2, and NP-3) led to small, but significant, accumulation of fluorescence in the kidney. Using flow cytometry, however, only the smallest formulation (NP-1) resulted in a significant fluorescence signal. The discrepancy observed between these two methods is likely due to accumulation of NPs in the interstitial space, which would be detected by IVIS. However, preparation of tissues for flow cytometry requires a series of digestion, homogenization, and wash steps to create a single cell suspension. These additional processing steps would result in loss of any NPs residing in the interstitial space, while preserving NPs that have been taken up by cells.
Similarly, we observed a discrepancy between IVIS and flow cytometry when analyzing the pancreas. By IVIS significant NP-1 accumulation was detected but was not observed by flow cytometry. These results suggest that NP-1 particles were able to accumulate in the interstitium but were not internalized by cells as measured by flow cytometry.
In contrast to these other tissues, both IVIS and flow cytometry revealed higher accumulation of the largest NPs in the spleen and liver. In the spleen, IVIS quantification and flow cytometry showed significant DiD fluorescence levels for the three largest NP formulations (NP-2, NP-3, and NP-4). These results align with previous studies, which have shown that NPs with a diameter greater than 200 nm are rapidly removed from circulation and sequestered in the spleen [4, 7, 9].
It is well known that NPs with a diameter greater than 200 nm are rapidly cleared from the blood by the liver [4, 7, 9]. Further, inside the liver sinusoidal capillaries, Kupffer cells are responsible for the clearance of particulates, including NPs [9]. In previous work, using PLGA NPs that were similar in size to NP-3, we showed that injected NPs are internalized by 98% of Kupffer cells, 89% of liver sinusoidal endothelial cells, 56% of hepatic stellate cells, and 7% of hepatocytes [49]. Here, we demonstrate that the liver has the greatest level of NP-associated fluorescence by IVIS and flow cytometry. This high level of fluorescence is likely mediated by substantial NP uptake in Kupffer cells.
Our smallest formulation, NP-1, demonstrated significantly enhanced uptake in the lung (Figure 3C, Figure 4F). These results align with previous findings, which have shown that after IV injection, small NPs more readily accumulate in the lung when compared to larger NPs [41]. However, these prior studies looked at bulk biodistribution, using high performance liquid chromatography (HPLC) of tissue extracts, which are unable to assess biodistribution at a cellular level [41]. Therefore, it is unclear from this prior work whether NPs are gaining entry into parenchymal cells or simply accumulating in the pulmonary interstitial or vasculature spaces. Here, we used flow cytometry to determine whether NPs associated with therapeutically relevant cell populations, including AEC I, alveolar macrophages, and endothelial cells. Our smallest formulation, NP-1, demonstrated the highest level of DiD fluorescence by flow cytometry in AEC I and alveolar macrophages. These results suggest that NPs that are sufficiently small are able to successfully escape from the systemic circulation, through lung endothelial fenestrations, and into AEC I more readily than NPs with larger diameters. Interestingly, alveolar macrophages and epithelial cells have been shown to express the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-dependent chloride channel, and may contribute to the hyperinflammatory immune response observed in patients diagnosed with cystic fibrosis (CF) [56]. This suggests that therapeutic cargo can be optimally delivered to these cells in the lung using NPs approximately 120 nm in diameter, or perhaps even smaller.
Similar to the lung, the smallest NPs (NP-1) demonstrated significantly greater uptake in bone marrow compared to larger NPs (Figure 4A–D). Further, we used flow cytometry to study whether NPs accessed HSPCs, a therapeutically pertinent cell population in the bone marrow. In HSPCs, NP-1 demonstrated the highest level of fluorescent uptake by flow cytometry, suggesting that small NPs escape from circulation, through bone marrow fenestrations, and into HSPCs more easily than larger NPs at the time point evaluated. Although the discontinuous endothelial fenestrations of the bone marrow have not been studied in detail, prior studies have shown that PLGA NPs with diameters of 150 – 300 nm accumulate in the bone marrow [25, 57]. In fact, our previous work used PLGA NPs approximately 300 nm in diameter to deliver PNA/donor DNA combinations to the bone marrow to edit HSPCs in a mouse model of β-thalassemia [28, 29]. While we have demonstrated successful gene modification using these particles (similar in size to NP-3), we hypothesize that a smaller formulation similar in size to NP-1 may improve delivery of PNA/donor DNA to bone marrow.
It is evident from our results that IVIS quantification and flow cytometry do not always provide the same information. Here, IVIS served as a preliminary screening method to detect NP fluorescence in intact organs, while flow cytometry was used to measure fluorescence associated with individual cells. While useful as a general screening tool, IVIS imaging does not differentiate among NPs remaining in the vasculature, distributed in the interstitial space, or associated with cells. Flow cytometry, on the other hand, accurately provides information on cellular distribution, but limits understanding of broad NP distribution in whole organs or associated vasculature. Therefore, when paired together with confocal microscopy, these complementary methods can provide a comprehensive understanding of NP biodistribution, and their localization in cells or interstitial spaces after 24 h. Given the dynamic nature of NP uptake, future studies should look at additional time points to further assess NP accumulation in tissues over time.
5. CONCLUSION
We have demonstrated that the NanoAssemblr™, a scalable microfluidic platform, can be used to engineer size-differentiated PLGA NPs. After IV injection, small (~120 nm) PLGA NPs demonstrate significantly greater uptake in the lung and bone marrow compared with larger NPs. Further, these small, 120 nm NPs associated with AEC I and alveolar macrophages in the lung, and HPSCs in the bone marrow more readily than larger NPs. Thus NPs of similar size to NP-1 may be more effective for improved delivery of agents to therapeutically pertinent tissues, by avoiding sequestration in the liver and spleen, and by crossing tissue barriers to reach relevant cellular targets. Our study demonstrates the potential for optimizing biodegradable NP size to passively target tissues and specific sub-cellular populations.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Young-Eun Seo, Alexander Josowitz, Sofia Griff, Lorenzo Arvanitis, and Zoe Moscato for technical assistance. We also thank Zhenting Jiang for technical assistance with SEM. We are grateful to Amy Kauffman for helpful discussions. This work was supported by the NIGMS Medical Scientist Training Program T32GM07205 (to E.Q.), an institutional training grant 5T32GM007223-43 (to E.Q.), a grant from NIH (UG3 HL147352), and a sponsored research agreement from Trucode Gene Repair.
Footnotes
CONFLICT OF INTEREST
P.M.G., W.M.S. and E.Q. are consultants for Trucode Gene Repair, Inc. P.M.G. and W.M.S. also have equity interests in Trucode Gene Repair, Inc.
REFERENCES
- 1.Patra JK, et al. , Nano based drug delivery systems: recent developments and future prospects. Journal of Nanobiotechnology, 2018. 16(1): p. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JH and Yeo Y, Controlled drug release from pharmaceutical nanocarriers. Chemical Engineering Science, 2015. 125: p. 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salata OV, Applications of nanoparticles in biology and medicine. Journal of Nanobiotechnology, 2004. 2(1): p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albanese A, Tang PS, and Chan WCW, The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems 2012. 14(1): p. 1–16. [DOI] [PubMed] [Google Scholar]
- 5.Owens DE and Peppas NA, Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. International Journal of Pharmaceutics, 2006. 307(1): p. 93–102. [DOI] [PubMed] [Google Scholar]
- 6.Lundy DJ, et al. , Distribution of Systemically Administered Nanoparticles Reveals a Size-Dependent Effect Immediately following Cardiac Ischaemia-Reperfusion Injury. Scientific Reports, 2016. 6: p. 25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoshyar N, et al. , The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine (London, England), 2016. 11(6): p. 673–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips MA, Gran ML, and Peppas NA, Targeted nanodelivery of drugs and diagnostics. Nano Today, 2010. 5(2): p. 143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertrand N and Leroux JC, The journey of a drug-carrier in the body: an anatomo-physiological perspective. J Control Release, 2012. 161(2): p. 152–63. [DOI] [PubMed] [Google Scholar]
- 10.De Jong WH, et al. , Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials, 2008. 29(12): p. 1912–1919. [DOI] [PubMed] [Google Scholar]
- 11.Pérez-Campaña C, et al. , Biodistribution of Different Sized Nanoparticles Assessed by Positron Emission Tomography: A General Strategy for Direct Activation of Metal Oxide Particles. ACS Nano, 2013. 7(4): p. 3498–3505. [DOI] [PubMed] [Google Scholar]
- 12.Liao W-Y, et al. , Comprehensive characterizations of nanoparticle biodistribution following systemic injection in mice. Nanoscale, 2013. 5(22): p. 11079–11086. [DOI] [PubMed] [Google Scholar]
- 13.Huang J, et al. , Effects of Nanoparticle Size on Cellular Uptake and Liver MRI with Polyvinylpyrrolidone-Coated Iron Oxide Nanoparticles. ACS Nano, 2010. 4(12): p. 7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawyer AJ, et al. , Convection-enhanced delivery of camptothecin-loaded polymer nanoparticles for treatment of intracranial tumors. Drug Delivery and Translational Research, 2011. 1(1): p. 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malinovskaya Y, et al. , Delivery of doxorubicin-loaded PLGA nanoparticles into U87 human glioblastoma cells. International Journal of Pharmaceutics, 2017. 524(1): p. 77–90. [DOI] [PubMed] [Google Scholar]
- 16.Bowerman CJ, et al. , Docetaxel-Loaded PLGA Nanoparticles Improve Efficacy in Taxane-Resistant Triple-Negative Breast Cancer. Nano Letters, 2017. 17(1): p. 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Householder KT, et al. , Intravenous delivery of camptothecin-loaded PLGA nanoparticles for the treatment of intracranial glioma. International Journal of Pharmaceutics, 2015. 479(2): p. 374–380. [DOI] [PubMed] [Google Scholar]
- 18.Blum JS and Saltzman WM, High loading efficiency and tunable release of plasmid DNA encapsulated in submicron particles fabricated from PLGA conjugated with poly-L-lysine. Journal of Controlled Release, 2008. 129(1): p. 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao K, et al. , Preparation and Efficacy of Newcastle Disease Virus DNA Vaccine Encapsulated in PLGA Nanoparticles. PLOS ONE, 2013. 8(12): p. e82648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santos DM, et al. , PLGA nanoparticles loaded with KMP-11 stimulate innate immunity and induce the killing of Leishmania. Nanomedicine: Nanotechnology, Biology and Medicine, 2013. 9(7): p. 985–995. [DOI] [PubMed] [Google Scholar]
- 21.Woodrow KA, et al. , Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nature Materials, 2009. 8: p. 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cun D, et al. , Preparation and characterization of poly(dl-lactide-co-glycolide) nanoparticles for siRNA delivery. International Journal of Pharmaceutics, 2010. 390(1): p. 70–75. [DOI] [PubMed] [Google Scholar]
- 23.McNeer NA, et al. , Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nature communications, 2015. 6: p. 6952–6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNeer NA, et al. , Nanoparticles Deliver Triplex-forming PNAs for Site-specific Genomic Recombination in CD34+ Human Hematopoietic Progenitors. Molecular Therapy, 2011. 19(1): p. 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNeer NA, et al. , Systemic delivery of triplex-forming PNA and donor DNA by nanoparticles mediates site-specific genome editing of human hematopoietic cells in vivo. Gene Therapy, 2012. 20: p. 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schleifman EB, et al. , Site-specific Genome Editing in PBMCs With PLGA Nanoparticle-delivered PNAs Confers HIV-1 Resistance in Humanized Mice. Molecular therapy. Nucleic acids, 2013. 2(11): p. e135–e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fields RJ, et al. , Modified poly(lactic-co-glycolic acid) nanoparticles for enhanced cellular uptake and gene editing in the lung. Advanced healthcare materials, 2015. 4(3): p. 361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bahal R, et al. , In vivo correction of anaemia in β-thalassemic mice by γPNAmediated gene editing with nanoparticle delivery. Nature Communications, 2016. 7: p. 13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ricciardi AS, et al. , In utero nanoparticle delivery for site-specific genome editing. Nature Communications, 2018. 9(1): p. 2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang W and Zhang C, Tuning the Size of Poly(lactic-co-glycolic Acid) (PLGA) Nanoparticles Fabricated by Nanoprecipitation. Biotechnol J, 2018. 13(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Operti MC, et al. , A comparative assessment of continuous production techniques to generate sub-micron size PLGA particles. International Journal of Pharmaceutics, 2018. 550(1): p. 140–148. [DOI] [PubMed] [Google Scholar]
- 32.Dong Y and Feng S-S, Poly(d,l-lactide-co-glycolide) (PLGA) nanoparticles prepared by high pressure homogenization for paclitaxel chemotherapy. International Journal of Pharmaceutics, 2007. 342(1): p. 208–214. [DOI] [PubMed] [Google Scholar]
- 33.Kucuk I and Edirisinghe M, Microfluidic preparation of polymer nanospheres. Journal of nanoparticle research : an interdisciplinary forum for nanoscale science and technology, 2014. 16(12): p. 2626–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morikawa Y, et al. , The Use of an Efficient Microfluidic Mixing System for Generating Stabilized Polymeric Nanoparticles for Controlled Drug Release. Biological and Pharmaceutical Bulletin, 2018. 41(6): p. 899–907. [DOI] [PubMed] [Google Scholar]
- 35.Deng Y, et al. , The effect of hyperbranched polyglycerol coatings on drug delivery using degradable polymer nanoparticles. Biomaterials, 2014. 35(24): p. 6595–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu C-MJ, et al. , Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proceedings of the National Academy of Sciences, 2011. 108(27): p. 10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oeck S, et al. , High-throughput Evaluation of Protein Migration and Localization after Laser Micro-Irradiation. Scientific reports, 2019. 9(1): p. 3148–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madaan A, et al. , A stepwise procedure for isolation of murine bone marrow and generation of dendritic cells. 2014, 2014. [Google Scholar]
- 39.Cruz LJ, et al. , Effect of PLGA NP size on efficiency to target traumatic brain injury. Journal of Controlled Release, 2016. 223: p. 31–41. [DOI] [PubMed] [Google Scholar]
- 40.Yadav K, et al. , Effect of Size on the Biodistribution and Blood Clearance of Etoposide-Loaded PLGA Nanoparticles. Vol. 65 2011. 131–9. [PubMed] [Google Scholar]
- 41.Kulkarni SA and Feng S-S, Effects of Particle Size and Surface Modification on Cellular Uptake and Biodistribution of Polymeric Nanoparticles for Drug Delivery. Pharmaceutical Research, 2013. 30(10): p. 2512–2522. [DOI] [PubMed] [Google Scholar]
- 42.He C, et al. , Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials, 2010. 31(13): p. 3657–3666. [DOI] [PubMed] [Google Scholar]
- 43.Vila A, et al. , PLA-PEG particles as nasal protein carriers: the influence of the particle size. International Journal of Pharmaceutics, 2005. 292(1): p. 43–52. [DOI] [PubMed] [Google Scholar]
- 44.Liu M, et al. , Pharmacokinetics and biodistribution of surface modification polymeric nanoparticles. Archives of Pharmacal Research, 2008. 31(4): p. 547–554. [DOI] [PubMed] [Google Scholar]
- 45.Caster JM, et al. , Effect of particle size on the biodistribution, toxicity, and efficacy of drug-loaded polymeric nanoparticles in chemoradiotherapy. Nanomedicine: Nanotechnology, Biology and Medicine, 2017. 13(5): p. 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang G, et al. , Influence of anchoring ligands and particle size on the colloidal stability and in vivo biodistribution of polyethylene glycol-coated gold nanoparticles in tumor-xenografted mice. Biomaterials, 2009. 30(10): p. 1928–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abstiens K and Goepferich AM, Microfluidic manufacturing improves polydispersity of multicomponent polymeric nanoparticles. Journal of Drug Delivery Science and Technology, 2019. 49: p. 433–439. [Google Scholar]
- 48.Panagi Z, et al. , Effect of dose on the biodistribution and pharmacokinetics of PLGA and PLGA-mPEG nanoparticles. International journal of pharmaceutics, 2001. 221: p. 143–52. [DOI] [PubMed] [Google Scholar]
- 49.Park J-K, et al. , Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomedicine : nanotechnology, biology, and medicine, 2016. 12(5): p. 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cui J, et al. , Poly(amine-co-ester) nanoparticles for effective Nogo-B knockdown in the liver. Journal of Controlled Release, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fields RJ, et al. , Surface modified poly(β amino ester)-containing nanoparticles for plasmid DNA delivery. Journal of Controlled Release, 2012. 164(1): p. 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J and Sabliov C, PLA/PLGA nanoparticles for delivery of drugs across the blood-brain barrier, in Nanotechnology Reviews 2013. p. 241. [Google Scholar]
- 53.Lu W, et al. , Brain delivery property and accelerated blood clearance of cationic albumin conjugated pegylated nanoparticle. Journal of Controlled Release, 2007. 118(1): p. 38–53. [DOI] [PubMed] [Google Scholar]
- 54.Hu K, et al. , Lactoferrin conjugated PEG-PLGA nanoparticles for brain delivery: Preparation, characterization and efficacy in Parkinson’s disease. International Journal of Pharmaceutics, 2011. 415(1): p. 273–283. [DOI] [PubMed] [Google Scholar]
- 55.Saucier-Sawyer JK, et al. , Systemic delivery of blood–brain barrier-targeted polymeric nanoparticles enhances delivery to brain tissue. Journal of Drug Targeting, 2015. 23(7–8): p. 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di A, et al. , CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nature Cell Biology, 2006. 8: p. 933. [DOI] [PubMed] [Google Scholar]
- 57.Swami A, et al. , Engineered nanomedicine for myeloma and bone microenvironment targeting. Proceedings of the National Academy of Sciences, 2014. 111(28): p. 10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.