

Abstract
mTOR inhibition extends life span in multiple organisms. In mice, when metformin treatment (Met) is added to the mTOR inhibitor rapamycin (Rap), median and maximal life span is extended to a greater degree than with Rap or Met alone. Treatments that extend life span often maintain proteostasis. However, it is less clear how individual tissues, such as skeletal muscle, maintain proteostasis with life span–extending treatments. In C2C12 myotubes, we used deuterium oxide (D2O) to directly measure two primary determinants of proteostasis, protein synthesis, and degradation rates, with Rap or Met+Rap treatments. We accounted for the independent effects of cell growth and loss, and isolated the contribution of autophagy and mitochondrial fission to obtain a comprehensive assessment of protein turnover. Compared with control, both Rap and Met+Rap treatments lowered mitochondrial protein synthesis rates (p < .001) and slowed cellular proliferation (p < .01). These changes resulted in greater activation of mechanisms promoting proteostasis for Rap, but not Met+Rap. Compared with control, both Rap and Met+Rap slowed protein breakdown. Autophagy and mitochondrial fission differentially influenced the proteostatic effects of Rap and Met+Rap in C2C12 myotubes. In conclusion, we demonstrate that Met+Rap did not increase protein turnover and that these treatments do not seem to promote proteostasis through increased autophagy.
Keywords: Protein turnover, Healthspan, Autophagy, C2C12 myotubes
A decline in mitochondrial function is associated with the aging process, in part, due to an accumulation of damaged proteins in the mitochondrial reticulum. Interventions that can prevent the accumulation of damage, perhaps through increased protein turnover, may, therefore, slow the aging process. There is some belief that treatments that slow aging also slow protein synthesis (1). The prevailing notion is that treatments that slow aging also increase protein breakdown through activation of autophagy (2). Decreases in protein synthesis with increased protein breakdown would create an unsustainable cellular condition that results in a progressive net loss of protein. Our previous data from long-lived murine models show that, when cell proliferation is accounted for, turnover of mitochondrial proteins is increased (3–7). By simultaneously assessing rates of protein synthesis and rates of cellular proliferation, we find that treatments that slow aging often reduce cellular proliferation. Therefore, what is commonly thought to be a decrease in the rate of protein synthesis is predominately associated with decreased cellular proliferation. When protein synthesis is increased relative to DNA synthesis, we interpret this increased ratio of protein to DNA synthesis as indicative of an energetic trade-off to maintain protein homeostasis (proteostasis) at the cost of cellular growth (7).
Cellular proliferation is an energetically costly process as proliferation includes the duplication of both the proteome and the genome (8). Increased allocation of energetic resources to proteostasis is particularly important for post-mitotic tissues, such as skeletal muscle, that cannot rely on proliferation to remove excessive intracellular damage (9). In many treatments that slow aging, such as calorie restriction, there are energetic constraints that trigger an increase in 5′ AMP-activated protein kinase (AMPK) signaling and a decrease in mechanistic target of rapamycin complex 1 (mTORC1) signaling (10,11). We have shown that these signaling changes induce reallocation of cellular resources from proliferation/growth to somatic maintenance (ie, proteostasis (4,6).
Rapamycin (Rap) inhibits mTORC1, reducing global protein translation and cellular growth by preventing cell cycle progression (12). Metformin (Met) increases activation of AMPK, potentially through moderate inhibition of the mitochondrial complex I (13), although the precise mechanism is not confirmed. Chronic administration of Rap increases median and maximal mouse life span (14), whereas chronic Met treatment increases life span in most (15–17), but not all interventions (18). The combination of metformin and rapamycin (Met+Rap) activates AMPK and decreases mTORC1 signaling, events that could increase allocation of energetic resources to maintaining proteostasis.
Treatment with Met+Rap further extends median and maximal life span compared with Rap alone (18). Our previous study shows that chronic Rap treatment in mice reduces cellular proliferation in multiple tissues, but does not decrease mitochondrial protein synthesis proportionately, suggesting an increased energetic allocation to mitochondrial protein turnover at the expense of cellular proliferation (4). To our knowledge, no studies have examined the effects of Met+Rap compared with Rap treatment on protein turnover, cellular proliferation, or AMPK and mTORC1 signaling.
Both Rap and Met independently stimulate autophagic flux (19,20). Autophagy is an intracellular degradation process that recycles intracellular components, including organelles (21). It is thought that autophagy is required for the life span–extending effects of calorie restriction (22,23), and contributes to cellular homeostasis through degradation of dysfunctional mitochondria (24). Mitochondria-specific autophagy (mitophagy) contributes to mitochondrial proteostasis by isolating damaged portions of the organelle through mitochondrial fission (25) for degradation and recycling (26). Maintenance of a functional mitochondrial reticulum may contribute to proper cellular function across the life span (27,28). To date, however, direct assessments of the effects of Rap or Met+Rap treatments on autophagic flux or mitochondrial fission are not available. Furthermore, measuring autophagy in vivo is often limited to static assessments of autophagic structures. We chose a skeletal muscle cell line because skeletal muscle accounts for ~40% of the body, and age-related loss of skeletal muscle mass has detrimental effects on function, health, and overall independence. Further, autophagy and mitochondrial dynamics are integral processes for maintaining skeletal muscle function.
No direct comparisons of the effects of Rap and Met+Rap treatments on protein turnover and cellular proliferation have been made. It is also unclear if, and to what extent, autophagy contributes to the effects of Rap and Met+Rap on protein turnover. Therefore, the purpose of this investigation was to (i) assess protein synthesis and degradation and cellular proliferation and degradation during treatment with Rap or Met+Rap and (ii) determine how autophagy or mitochondrial fission contribute to protein turnover and cellular proliferation during treatment with Rap or Met+Rap. In the current study, we used murine C2C12 myotubes to examine the effects of Rap and Met+Rap treatments on protein and cellular turnover during pharmacological inhibition of autophagy and mitochondrial fission using bafilomycin-A1 and Mdivi. We hypothesized that Rap and Met+Rap treatments would decrease the rates of protein synthesis and breakdown but that both treatments would decrease rates of cellular proliferation to a greater extent than the decrease in protein turnover, leading to an increased energetic allocation toward protein turnover. We also hypothesized that autophagy contributes to the proteostatic effects of Rap and Met+Rap treatments.
Methods
Experimental Overview and Reagents
All experiments conducted were with a commercially available cell line, and no ethical approval was needed. C2C12 myoblasts were obtained from American Type Culture Collection (ATCC; Manassas, VA) and cultured in growth medium (Dulbecco’s modified Eagle medium + 10% fetal bovine serum + 1% penicillin/streptomycin) in a humidified 37°C, 5% CO2 incubator. Myoblasts were plated on 100-mm culture plates, grown to approximately 90% confluence, and then differentiated into myotubes by culturing in differentiation medium (Dulbecco’s modified Eagle medium containing 2% horse serum and 1% penicillin/streptomycin) for 5 days. All treatments for these experiments were diluted in growth medium containing 10% deuterium oxide (D2O).
Protein and DNA Synthesis Experiments
To measure protein and DNA synthesis in C2C12 myotubes, treatments were prepared in growth medium as listed previously and supplemented with 10% sterile D2O. Cells were treated in triplicate for each experimental condition at all time points. To assess protein and DNA synthesis, cultured myotubes were treated for 4 and 24 hours with Rap (5 nM), or Met+Rap (2 mM + 5 nM), medium alone (Con), or medium with vehicle (dimethyl sulfoxide [DMSO]) and harvested at 4 and 24 hours in triplicate. These concentrations were determined to be the lowest effective doses to inhibit mTOR (Rap) and to activate AMPK (Met). Each treatment was applied in growth medium enriched with 10% D2O to measure the incorporation of deuterium into alanine (protein synthesis) or deoxyribose (DNA synthesis) as described previously. To assess the contribution of autophagy to protein synthesis rates during Rap or Met+Rap treatments, bafilomycin-A1 (Baf) was used to pre- and co-treat myotubes. Four hours prior to the start of the stable isotope labeling, plates that were to be treated with Rap, Met+Rap, or Baf were pretreated with 100 nM Baf. At the end of the pretreatment, cells were rinsed twice with sterile phosphate-buffered saline, and treatments were applied with Baf cotreatment (B-Rap, B-Met+Rap, Baf) in growth medium enriched with 10% D2O. Control and DMSO treatments did not include Baf at any point during the experiment. The experiment to assess the contributions of mitochondrial fission to protein synthesis was completed as the autophagy experiment, replacing bafilomycin pre- and co-treatment with Mdivi-1 pre- and co-treatment.
Protein and DNA Breakdown Experiments
To measure protein and DNA breakdown in myotubes, myoblasts were plated onto 100-mm plates. Differentiation media for the breakdown experiments consisted of Dulbecco’s modified Eagle medium, 2% horse serum, antibiotics, and the media was enriched to 15% with sterile D2O to pre-label the cellular proteins and DNA. Differentiation media with D2O was replaced every 48 hours during differentiation over the course of 5 days. At the end of the differentiation period, cells were rinsed twice with sterile phosphate-buffered saline, and then treatments in growth media without D2O were applied. A set of cells were harvested at the time of switching medium from 15% D2O enriched to unenriched to determine the starting protein and DNA enrichment for the experiments (ie, 0-hour time point). Each treatment was applied in growth medium without D2O to facilitate assessment of loss of deuterium-labeled alanine and deoxyribose as proteins and DNA were degraded. To prevent recycling of tracer from rapidly degraded proteins, medium was removed after 1 hour of treatment, cells were rinsed with sterile phosphate-buffered saline, and replenished with fresh D2O-free medium. Myotubes were treated for 4 or 24 hours with Rap, Met+Rap, Con medium, and DMSO medium in triplicate. Treatment concentrations for Rap, Met+Rap, DMSO, Con, Baf, and Mdivi-1 were the same as in the synthesis experiments. For the autophagy and mitochondrial fission inhibition experiments, cells were pre- and co-treated with Baf or Mdivi-1 as described above.
Preparation of Analytes for Mass Spectrometric Analyses
For the assessment of subcellular protein synthesis or breakdown, we used differential centrifugation techniques to isolate mitochondrial protein and cytoplasmic protein-enriched protein fractions as previously described (29). The pentafluorobenzyl-N,N-di(pentafluorobenzyl) derivative of alanine was analyzed on an Agilent 7890A GC coupled with an Agilent 5975C MS as previously described (30). To assess cellular DNA synthesis or breakdown, we prepared the pentafluorobenzyl-hydroxylamine hydrochloride derivative of extracted DNA and analyzed on an Agilent 7890A GC coupled with an Agilent 5975C MS as previously described (4,29).
An important consideration for the D2O labeling studies used in the current investigation is how the deuterium label is incorporated into DNA. Deuterium from D2O is incorporated into deoxynucleosides (dN) during de novo dN synthesis creating a pool of enriched dN that is used to synthesize DNA (31). Importantly, D2O does not label dN that arise from the dN salvage pathway, which is the primary source of dN for DNA repair. Therefore, our measurements are indicative of de novo DNA synthesis and not DNA repair (31).
Calculation of Protein:DNA Ratios
To account for changes in protein synthesis and breakdown independent of proliferation and cell loss, we compare protein synthesis and breakdown rates to DNA synthesis and breakdown rates (protein:DNA). From this calculation, we interpret an increased ratio as greater turnover of the proteome at the expense of cell turnover. The media enrichment for each plate was used to calculate the precursor enrichment (alanine and deoxyribose, respectively) for protein and DNA synthesis by using the appropriate mass isotopomer distribution analysis adjustment (32). Protein and DNA fractional synthesis was calculated by dividing the fraction of newly synthesized proteins or DNA at the time of cell harvest by the duration, in hours, of the treatment period. Mitochondrial and cytoplasmic protein:DNA ratios were calculated by dividing the respective protein fractional synthesis rates by the corresponding DNA fractional synthesis rate from the same plate. Fractional breakdown rates for both protein fractions and DNA were calculated based on the fractional decrease from the starting enrichment (0 hour) for each treatment over time. The protein:DNA breakdown ratio was calculated by dividing fractional breakdown rates for protein and DNA.
Western blotting
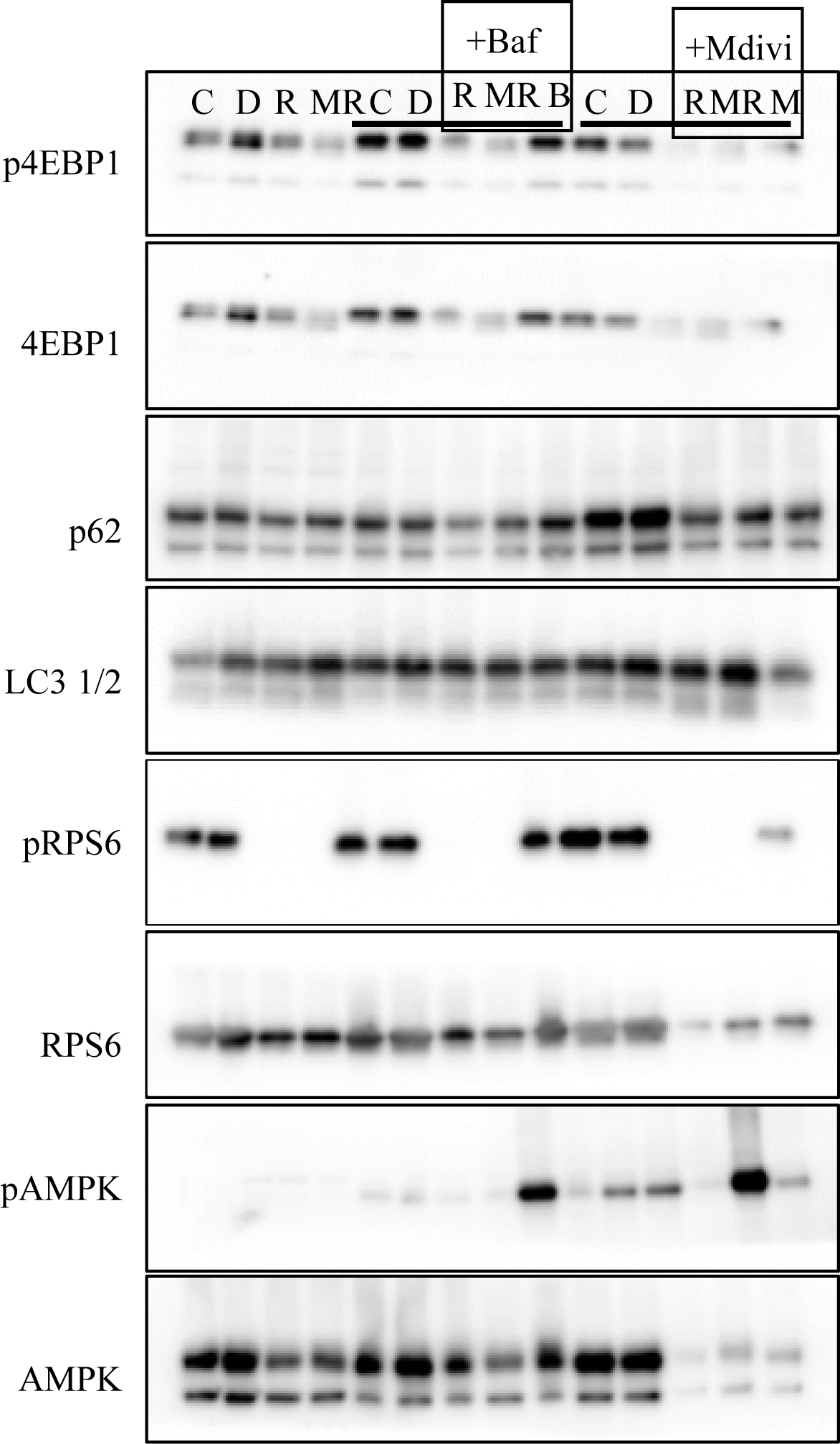
For the assessment of protein content, an aliquot of cytoplasmic-enriched proteins from each sample was run on a 4%–20% gel for phosphorylated and total RPS6, 4EBP-1, and AMPK as well as p62, and microtubule-associated protein 1A/1B-light chain 3 (LC3) lipidation. Both p62 and LC3 are well-established markers of autophagic flux. Accumulation of p62 indicates reduced autophagy flux, and the ratio of the LC3-phosphatidylethanolamine conjugate (LC3-II) expressed relative to the cytosolic form of LC3 (LC3-I) provides an indication of autophagic activity. Approximately 11 µg of protein per sample was loaded, and membranes were stained with 0.1% amido-black (Sigma–Aldrich) and total protein staining quantified to control for protein loading and transfer quality. Primary antibodies were diluted 1:1,000 and secondary antibodies 1:5,000. All antibodies were obtained from Cell Signaling Technologies (Danvers, MA). Protein content was assessed using densitometry.
Statistics
Statistical analyses were performed using Prism v8 (GraphPad Software, Inc. La Jolla, CA). One-way analysis of variance tests with Tukey’s multiple comparisons test were used to determine the effect of treatment for each experimental outcome. All treatments for the following experiments were diluted in DMSO. Thus, we performed nonparametric t-tests for all nonvehicle control (Con) samples compared with DMSO samples. There were no significant differences between Con and DMSO for protein synthesis, DNA synthesis, protein:DNA, protein breakdown, or DNA breakdown values in any experiment. Significance for these experiments was set a priori at p < .05 with additional notations for p < .01, p < .001, and p < .0001 where appropriate. Data are presented as mean ± SEM.
Results
Protein Synthesis, DNA Synthesis, and Protein:DNA
There were no differences between Rap and Met+Rap treatment for mitochondrial protein synthesis rates compared with vehicle control (DMSO) after 4 hours (Supplementary Figure S1). Rap lowered (p < .0001) mitochondrial protein synthesis rates compared with DMSO after 24 hours, whereas Met+Rap treatment significantly lowered mitochondrial protein synthesis rates (p < .0001) compared with Rap and DMSO at 24 hours (Figure 1). In addition, Met+Rap lowered cytoplasmic protein synthesis rates after 4 hours of treatment compared with DMSO (p < .01) and Rap (p < .01; Supplementary Figure S1). After 24 hours, Met+Rap lowered cytoplasmic protein synthesis rates compared with DMSO (p < .001) and Rap (p < .01), and Rap was not different from DMSO (p = .084; Figure 1). Rap and Met+Rap did not alter DNA synthesis rates after 4 hours of treatment compared with DMSO. Rap (p < .001) and Met+Rap (p < .001) significantly lowered DNA synthesis rates compared to DMSO after 24 hours (Figure 1).
Figure 1.

Effects of 24-h Rap and Met+Rap treatments on protein turnover, DNA turnover, and protein:DNA. In the top half of the figure, the effects of DMSO, Rap, and Met+Rap on cellular synthetic processes are reported. Data from the 24-h time point for mitochondrial protein synthesis rates, DNA synthesis rates, mitochondrial protein:DNA synthesis, cytoplasmic protein synthesis rates, and cytoplasmic protein:DNA synthesis are presented above the horizontal bar. On the bottom half of the figure, we present the data from the breakdown experiments including mitochondrial protein breakdown rates, DNA breakdown rates, mitochondrial protein:DNA breakdown, cytoplasmic protein breakdown rates, and cytoplasmic protein:DNA breakdown. *p < .05 compared with DMSO, **p < .01 compared with DMSO, ***p < .001 compared with DMSO, and ****p < .0001 compared with control. #p < .05 compared with corresponding Rap treatment.
Rap and Met+Rap did not significantly alter mitochondrial or cytoplasmic protein:DNA after 4 hours compared with DMSO (Supplementary Figure S1). However, after 24 hours, mitochondrial protein:DNA was significantly higher in Rap compared with DMSO (p < .01) and Met+Rap treatments (p < .05; Figure 1). After 24 hours, cytoplasmic protein:DNA synthesis was significantly higher in Rap compared with DMSO (p < .01) and Met+Rap (p < .01; Figure 1).
Protein Breakdown, DNA Breakdown, and Protein:DNA
Rap and Met+Rap did not significantly alter mitochondrial protein degradation rates after 4 hours of treatment (Supplementary Figure S1). However, after 24 hours, Rap and Met+Rap treatments significantly reduced mitochondrial protein breakdown rates compared with DMSO (p < .001; Figure 1). Met+Rap reduced mitochondrial protein breakdown rate compared with Rap (p < .01) at 24 hours (Figure 1). Rap and Met+Rap significantly reduced cytoplasmic protein breakdown rates compared to DMSO (p < .001, and p < .01, respectively) after 4 hours of treatment (Supplementary Figure S1). After 24 hours of treatment, Rap reduced cytoplasmic protein breakdown rates compared with DMSO (p < .001), whereas Met+Rap reduced cytoplasmic protein breakdown rates compared with DMSO (p < .0001) and Rap (p < .01; Figure 1). Neither Rap nor Met+Rap changed DNA breakdown rates compared with DMSO at either time point (Figure 1). As a result, there were no significant changes in mitochondrial or cytoplasmic protein:DNA measurements during the breakdown experiments (Figure 1).
Contributions of Autophagy and Mitochondrial Fission to Protein Breakdown
Bafilomycin-A1, a vacuolar ATPase inhibitor, did not alter mitochondrial protein degradation rates compared with DMSO after 4 hours (Supplementary Figure S2). However, Baf significantly lowered (p < .001) mitochondrial protein breakdown rate compared with DMSO after 24 hours (~29%; Figure 2). In the cytoplasmic fraction, protein breakdown rates were higher in Baf compared with DMSO after 4 hours, perhaps due to compensatory activation of the proteasome system, as discussed below (p < .01; Supplementary Figure S2). However, after 24 hours, Baf lowered cytoplasmic protein breakdown rates compared with DMSO (p < .05; Figure 2).
Figure 2.

Contributions of autophagy and mitochondrial fission to Rap and Met + Rap effects on protein synthesis, DNA synthesis, and protein:DNA. The top portion of the figure reports data from synthesis experiments during autophagic inhibition with Baf. This includes 24-h mitochondrial protein synthesis rates, cellular DNA synthesis rates, mitochondrial protein:DNA synthesis, cytoplasmic protein synthesis rates, and cytoplasmic protein:DNA synthesis. The bottom portion of the figure presents the data from synthesis experiments during Mdivi treatment including mitochondrial protein synthesis rates, cellular DNA synthesis rates, mitochondrial protein:DNA synthesis, cytoplasmic protein synthesis rates, and cytoplasmic protein:DNA synthesis. *p < .05 compared with DMSO, **p < .01 compared with DMSO, ***p < .001 compared with DMSO, and ****p < .0001 compared with control. #p < .05 compared with corresponding Rap treatment. @p < .05 compared with corresponding Met+Rap treatment.
Mitochondrial protein degradation rates were not altered by treatment with the small molecule inhibitor of mitochondrial fission Mdivi-1 (10 µM) after 4 or 24 hours compared with DMSO (Figure 2 and Supplementary Figure S2). Similarly, treatment with Mdivi-1 did not alter cytoplasmic protein breakdown rates compared with DMSO at 4 or 24 hours (Figure 2 and Supplementary Figure S2).
Impact of Bafilomycin or Mdivi on Protein:DNA
Bafilomycin and rapamycin cotreatment (B-Rap) did not change mitochondrial protein:DNA synthesis compared with rapamycin alone or DMSO after 24 hours (Figure 3). However, cytoplasmic protein:DNA was significantly higher in B-Rap compared with Rap (p < .001) and DMSO (p < .0001) after 24 hours. Mitochondrial and cytoplasmic protein:DNA breakdown ratios were not different between any treatment at 24 hours. Baf treatment did not alter mitochondrial or cytoplasmic protein:DNA at 4 hours.
Figure 3.

Inhibition of autophagic flux does not influence mitochondrial protein:DNA, but inhibition of mitochondrial fission does. The data in the current figure provide comparisons across all three synthesis experiments (top four panels) and all three breakdown experiments (bottom four panels). (A) Twenty-four-hour mitochondrial protein synthesis rates, (B) 24-h cytoplasmic protein synthesis rates, (C) 24-h mitochondrial protein:DNA synthesis, (D) 24-h cytoplasmic protein:DNA synthesis, (E) 24-h mitochondrial protein breakdown rates, (F) 24-h cytoplasmic protein breakdown rates, (G) 24-h mitochondrial protein:DNA breakdown, and (H) 24-h cytoplasmic protein:DNA breakdown. *p < .05 compared with DMSO, **p < .01 compared with DMSO, ***p < .001 compared with DMSO, and ****p < .0001 compared with control. #p < .05 compared with corresponding Rap treatment. @p < .05 compared with corresponding Met+Rap treatment. @@p < .01 compared with corresponding Met+Rap treatment.
Mdivi-1 altered the protein:DNA ratio for cytoplasmic-enriched fraction. Specifically, Mdivi in combination with Met+Rap (M-Met+Rap) significantly lowered cytoplasmic protein:DNA compared with Rap alone (p < .05), as well as M-Rap (p < .05). Mdivi-1 treatment alone did not change the mitochondrial protein:DNA synthesis ratio. M-Rap mitochondrial protein:DNA was significantly lower (p < .05) compared with Rap alone. M-Met+Rap mitochondrial protein:DNA was also significantly lower than Rap or Met+Rap alone (p < .001, and 0.05, respectively). However, when protein:DNA ratios were calculated from the fractional breakdown rates, there were no significant differences between any treatments and control.
Protein Signaling
All Western blotting data are presented as summary graphs (Figure 4), and full immunoblot images are presented in Supplementary Figure S3. Rap and Met+Rap completely abolish phosphorylated RPS6 in all experiments (p < .0001; Figure 4). AMPK phosphorylation was only detectable in Met+Rap-treated cells, including during Baf or Mdivi cotreatment (p < .0001; Figure 4). The ratio of LC32:1 was not increased during Rap or Met+Rap treatments.
Figure 4.

Protein signaling associated with life span-extending treatments. Quantified immunoblot values from the Rap and Met+Rap only experiment (A–D), the autophagy inhibition experiment (E–H), and the mitochondrial division inhibition experiment (I–L). (A) phosphorylated:total AMPK, (B) phosphorylated:total RPS6, (C) p62 protein content, (D) LC32:1 protein content, (E) phosphorylated:total AMPK, (F) phosphorylated:total RPS6, (G) p62 protein content, (H) LC32:1 protein content, (I) phosphorylated:total AMPK, (J) phosphorylated:total RPS6, (K) p62 protein content, (L) LC32:1 protein content. *p < .05 compared with DMSO, **p < .01 compared with DMSO, ***p < .001 compared with DMSO, and ****p < .0001 compared with control. ##p < .01 compared with corresponding Rap treatment. @@p < .01 compared with corresponding Met+Rap treatment.
Discussion
The primary objective of this investigation was to directly compare the effects of Rap and Met+Rap treatments on mitochondrial and cytoplasmic protein turnover, cellular proliferation, and protein:DNA ratio in cultured skeletal myotubes. We demonstrated that both Rap and Met+Rap treatments lowered protein synthesis and breakdown and concomitantly lowered cellular proliferation. In addition, we found that acute treatment with Rap in vitro increased mitochondrial protein:DNA synthesis compared with control, similar to our previous findings in heart, liver, and skeletal muscle from Rap-treated mice (4). Contrary to our hypothesis, however, Met+Rap treatment did not alter protein:DNA synthesis or breakdown compared with control during assessments of protein synthesis or breakdown, despite slower cellular proliferation and breakdown. These are the first data to demonstrate that Rap and Met+Rap treatments differ in their effects on mitochondrial and cytoplasmic protein:DNA synthesis.
The relationship between protein synthesis and cellular proliferation can provide insight into energetic resource allocation for protein turnover (7). Specifically, cellular proliferation is an energetically costly process, primarily due to the energy required for replication of the proteome (33). However, when cells do not replicate (eg, are post-mitotic), protein turnover is still required to maintain a functional proteome. Examining the relationship between protein synthesis and DNA synthesis helps to inform whether protein synthesis is allocated to cellular proliferation or to protein turnover (7). We found that both Rap and Met+Rap treatments concomitantly lower protein synthesis and DNA synthesis rates. However, mitochondrial protein:DNA synthesis was greater in Rap-treated cells compared with control, suggesting that Rap increased the maintenance or turnover of mitochondrial proteins. These findings are in line with our previous data in Rap-treated mice (4). These data are the first experimental evidence indicating that Met+Rap treatment does not alter mitochondrial protein:DNA synthesis. A potential explanation for the differing effects of Rap and Met+Rap treatments on mitochondrial protein turnover in the current investigation is that Met treatment in vivo is thought to primarily affect hepatic glucoregulation, not skeletal muscle (34). Despite no effect of acute Met+Rap treatment on mitochondrial protein:DNA synthesis or breakdown in cultured skeletal myotubes, chronic administration may be required for to affect skeletal muscle protein turnover as well as the life span–extending effect.
Rap treatment has been reported to increase protein breakdown through an increase in autophagy (35). Our experiments allowed us to distinguish the effects of Rap on autophagy as a component of total breakdown, which presumably includes proteasomal breakdown. We found that both Rap and Met+Rap treatments slowed protein degradation compared to control. When autophagy was inhibited with Baf treatment, there was no additional decrease in mitochondrial protein breakdown with either treatment, but cytosolic protein breakdown in Met+Rap slowed. These data indicate that in vitro only Met+Rap treatment activated autophagy of cytosolic proteins to an appreciable extent. However, when protein:DNA breakdown was compared, there were no changes with any treatment indicating that differences in breakdown might solely be attributed to changes in cell loss.
In addition to protein breakdown, we also sought to understand how changes in autophagic flux affected protein synthesis during Rap and Met+Rap treatments. A priori we expected that Rap would activate autophagy and that this may in turn influence protein synthesis as a proteostatic mechanism. However, as indicated above, our treatments did not increase autophagy. Similarly, Rap and Met+Rap treatments did not alter mitochondrial protein:DNA synthesis, supporting that changes seen in vitro are likely due to changes in the cell cycle (36). In contrast to the lack of change in mitochondrial protein:DNA synthesis, there was a major effect of autophagic inhibition on cytoplasmic protein:DNA synthesis. Cytoplasmic, but not mitochondrial, proteins have been identified as major substrates for autophagic flux in quiescent fibroblasts (37), potentially explaining the significant increase in cytoplasmic protein:DNA synthesis during autophagic inhibition (Figure 3). In sum, our data support that Rap and Met+Rap have differing effects on cytosolic protein turnover, while having minimal influence on mitochondrial protein turnover. In addition, many, but not all, of the apparent changes in protein synthesis in vitro can be explained by changes in cell cycling.
Finally, we sought to examine the contribution of mitochondrial fission, an event preceding mitophagy, to protein turnover during Rap and Met+Rap treatments. Inhibiting mitochondrial fission during Rap or Met+Rap treatments lowered mitochondrial protein:DNA synthesis, but did not influence mitochondrial protein breakdown, suggesting that inhibiting mitochondrial fission decreases mitochondrial density through reduced mitochondrial protein accretion. Burman and colleagues recently reported that inhibiting Drp1-dependent mitochondrial fission increased the rate of mitophagy through a Parkin-dependent mechanism (38). Therefore, inhibiting mitochondrial fission during Rap and Met+Rap treatments may reduce mitochondrial protein:DNA synthesis by increasing autophagic degradation of mitochondria, but reduce the selective mitophagy of protein aggregates, as both Rap and Met+Rap treatments lower mitochondrial protein synthesis rates. Together, these findings highlight a key role for mitochondrial remodeling and potentially selective mitophagy contributing to the effects of Rap and Met+Rap treatments on mitochondrial protein turnover.
There are two potential limitations of the current study. Our calculation of protein:DNA assumes that the source of DNA synthesis is nuclear, and that it is indicative of a homogenous pool of protein and DNA. Regarding our first assumption, the ratio of mtDNA to nuclear DNA is approximately 2,000:1 in C2C12 myotubes (39). However, the number of base pairs, which is what is analyzed on GC-MS, is 16,700 for mtDNA and 3,300,000,000 for nuclear DNA. Therefore, mtDNA contributes 1% of the total number of base pairs. In addition, the turnover of mtDNA in vivo is on the order of 1 month (40), although this is not known in C2C12 myotubes in vitro. If we assume that only a fraction of mtDNA is new in our 24-hour labeling period, it would represent an even smaller fraction of the total DNA. Finally, even if mtDNA is 100% new in the labeling period, the maximal 1% it could account for is far less than the 10%–15% new DNA we measure over the 24-hour labeling period. Therefore, we are confident that mtDNA is a small, if any, contributor to the measured DNA synthesis rates. Regarding our second assumption, it is possible that a small pool of cells are replicating, while protein synthesis is distributed throughout all cells. Tracer approaches assume that there is a single homogenous pool, which we believe to be safe under these conditions. In our experiment, the ratio of fractional synthesis rates is on the order of approximately 1.5:1 to 4:1, indicating to us that the changes in proliferation are probably a primary factor in determining changes in protein synthesis. If we measured ratios that were skewed further in the direction of protein (eg, 100:1 or 1,000:1), we would be more concerned about the influence of a nonhomogenous DNA pool distinct from the homogenous protein pool. Further studies should confirm these assumptions.
Together, our data reveal that there are differential effects of the life span–extending treatments Met+Rap and Rap on cellular resource allocation toward protein turnover at the expense of proliferation. In addition, Rap and Met+Rap treatments seemingly reduced mitochondrial protein turnover, but only Rap treatment increases mitochondrial protein:DNA synthesis. Our findings provide evidence that, in cultured skeletal myotubes, Rap and Met+Rap have different effects on protein turnover. In addition, it appears that autophagy was not a significant contributor to changes in mitochondrial protein turnover with these life span–extending treatments. Future investigations should further consider the independent roles of protein turnover and cell turnover when considering mechanisms of life-span extension with Rap and Met+Rap.
Funding
This work was supported by the National Institute on Aging at the National Institutes of Health (R01 AG042569 to K.L.H. and B.F.M.).
Conflict of Interest
None reported.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank the members of the Translational Research on Aging and Chronic Disease Laboratory at Colorado State University for valuable discussion of these data and technical assistance.
References
- 1. Hipkiss AR. On why decreasing protein synthesis can increase lifespan. Mech Ageing Dev. 2007;128:412–414. doi: 10.1016/j.mad.2007.03.002 [DOI] [PubMed] [Google Scholar]
- 2. Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015;125:85–93. doi: 10.1172/JCI73946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hamilton KL, Miller BF. Mitochondrial proteostasis as a shared characteristic of slowed aging: the importance of considering cell proliferation. J Physiol. 2017;22:1350. doi: 10.1113/JP274335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drake JC, Peelor FF 3rd, Biela LM, et al. Assessment of mitochondrial biogenesis and mTORC1 signaling during chronic rapamycin feeding in male and female mice. J Gerontol A Biol Sci Med Sci. 2013;68:1493–1501. doi: 10.1093/gerona/glt047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Drake JC, Bruns DR, Peelor FF 3rd, et al. Long-lived crowded-litter mice have an age-dependent increase in protein synthesis to DNA synthesis ratio and mTORC1 substrate phosphorylation. Am J Physiol Endocrinol Metab. 2014;307:E813–E821. doi: 10.1152/ajpendo.00256.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drake JC, Bruns DR, Peelor FF 3rd, et al. Long-lived Snell dwarf mice display increased proteostatic mechanisms that are not dependent on decreased mTORC1 activity. Aging Cell. 2015;14:474–482. doi: 10.1111/acel.12329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miller BF, Drake JC, Naylor B, Price JC, Hamilton KL. The measurement of protein synthesis for assessing proteostasis in studies of slowed aging. Ageing Res Rev. 2014;18:106–111. doi: 10.1016/j.arr.2014.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731 [DOI] [PubMed] [Google Scholar]
- 9. Murach KA, Englund DA, Dupont-Versteegden EE, McCarthy JJ, Peterson CA. Myonuclear domain flexibility challenges rigid assumptions on satellite cell contribution to skeletal muscle fiber hypertrophy. Front Physiol. 2018;9:635. doi: 10.3389/fphys.2018.00635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miller BF, Robinson MM, Reuland DJ, et al. Calorie restriction does not increase short-term or long-term protein synthesis. J Gerontol A Biol Sci Med Sci. 2013;68:530–538. doi: 10.1093/gerona/gls219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Civitarese AE, Carling S, Heilbronn LK, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. doi: 10.1371/journal.pmed.0040076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cuyàs E, Corominas-Faja B, Joven J, Menendez JA. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. Methods Mol Biol. 2014;1170:113–144. doi: 10.1007/978-1-4939-0888-2_7 [DOI] [PubMed] [Google Scholar]
- 13. Hur KY, Lee MS. New mechanisms of metformin action: focusing on mitochondria and the gut. J Diabetes Investig. 2015;6:600–609. doi: 10.1111/jdi.12328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miller RA, Harrison DE, Astle CM, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66:191–201. doi: 10.1093/gerona/glq178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martin-Montalvo A, Mercken EM, Mitchell SJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anisimov VN, Piskunova TS, Popovich IG, et al. Gender differences in metformin effect on aging, life span and spontaneous tumorigenesis in 129/Sv mice. Aging. 2010;2:945–958. doi: 10.18632/aging.100245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Haes W, Frooninckx L, Van Assche R, et al. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc Natl Acad Sci USA. 2014;111:E2501–E2509. doi: 10.1073/pnas.1321776111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Strong R, Miller RA, Antebi A, et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell. 2016;15:872–884. doi: 10.1111/acel.12496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci USA. 2015;112:15790–15797. doi: 10.1073/pnas.1521919112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teng AC, Miyake T, Yokoe S, et al. Metformin increases degradation of phospholamban via autophagy in cardiomyocytes. Proc Natl Acad Sci USA. 2015;112:7165–7170. doi: 10.1073/pnas.1508815112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol. 2011;76:397–402. doi: 10.1101/sqb.2011.76.011023 [DOI] [PubMed] [Google Scholar]
- 22. Minina EA, Sanchez-Vera V, Moschou PN, et al. Autophagy mediates caloric restriction-induced lifespan extension in Arabidopsis. Aging Cell. 2013;12:327–329. doi: 10.1111/acel.12048 [DOI] [PubMed] [Google Scholar]
- 23. Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–599. [DOI] [PubMed] [Google Scholar]
- 24. Palikaras K, Tavernarakis N. Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol. 2014;56:182–188. doi: 10.1016/j.exger.2014.01.021 [DOI] [PubMed] [Google Scholar]
- 25. Diot A, Morten K, Poulton J. Mitophagy plays a central role in mitochondrial ageing. Mamm Genome. 2016;27:381–395. doi: 10.1007/s00335-016-9651-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drake JC, Yan Z. Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing. J Physiol. 2017;595:6391–6399. doi: 10.1113/JP274337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Konopka AR, Sreekumaran Nair K. Mitochondrial and skeletal muscle health with advancing age. Mol Cell Endocrinol. 2013;379:19–29. doi: 10.1016/j.mce.2013.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Musci RV, Hamilton KL, Miller BF. Targeting mitochondrial function and proteostasis to mitigate dynapenia. Eur J Appl Physiol. 2018;118:1–9. doi: 10.1007/s00421-017-3730-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bruns DR, Ehrlicher SE, Khademi S, et al. Differential effects of vitamin C or Protandim on skeletal muscle adaptation to exercise. J Appl Physiol. 2018;509:565. doi: 10.1152/japplphysiol.00277.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miller BF, Ehrlicher SE, Drake JC, et al. Assessment of protein synthesis in highly aerobic canine species at the onset and during exercise training. J Appl Physiol. 2015;118:811–817. doi: 10.1152/japplphysiol.00982.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Busch R, Neese RA, Awada M, Hayes GM, Hellerstein MK. Measurement of cell proliferation by heavy water labeling. Nat Protoc. 2007;2:3045–3057. doi: 10.1038/nprot.2007.420 [DOI] [PubMed] [Google Scholar]
- 32. Hellerstein MK, Neese RA. Mass isotopomer distribution analysis: a technique for measuring biosynthesis and turnover of polymers. Am J Physiol. 1992;263(5 Pt 1):E988–E1001. doi: 10.1152/ajpendo.1992.263.5.E988 [DOI] [PubMed] [Google Scholar]
- 33. Lynch M, Marinov GK. The bioenergetic costs of a gene. Proc Natl Acad Sci USA. 2015;112:15690–15695. doi: 10.1073/pnas.1514974112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–1585. doi: 10.1007/s00125-017-4342-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ko JH, Yoon SO, Lee HJ, Oh JY. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFκB pathways in autophagy- and p62-dependent manners. Oncotarget. 2017;8:40817–40831. doi: 10.18632/oncotarget.17256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohkuma S, Shimizu S, Noto M, Sai Y, Kinoshita K, Tamura H. Inhibition of cell growth by bafilomycin A1, a selective inhibitor of vacuolar H(+)-ATPase. In Vitro Cell Dev Biol Anim. 1993;29A:862–866. doi:10.1007/BF02631364 [DOI] [PubMed] [Google Scholar]
- 37. Zhang T, Shen S, Qu J, Ghaemmaghami S. Global analysis of cellular protein flux quantifies the selectivity of basal autophagy. Cell Rep. 2016;14:2426–2439. doi: 10.1016/j.celrep.2016.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burman JL, Pickles S, Wang C, et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol. 2017;216:3231–3247. doi: 10.1083/jcb.201612106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frangini M, Franzolin E, Chemello F, et al. Synthesis of mitochondrial DNA precursors during myogenesis, an analysis in purified C2C12 myotubes. J Biol Chem. 2013;288:5624–5635. doi: 10.1074/jbc.M112.441147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poovathingal SK, Gruber J, Lakshmanan L, Halliwell B, Gunawan R. Is mitochondrial DNA turnover slower than commonly assumed? Biogerontology. 2012;13:557–564. doi: 10.1007/s10522-012-9390-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.