

Abstract
The response of eosinophils to respiratory virus has emerged as an important link between pulmonary infection and allergic asthmatic exacerbations. Eosinophils activate innate immune responses through toll-like receptor (TLR) signaling. Here, using mouse and human eosinophils and mice lacking the prolyl isomerase Pin1 selectively in eosinophils, we show that Pin1 is indispensable for eosinophilopoiesis in the bone marrow and mature cell function in the presence of TLR7 activation. Unbiased in vivo analysis of mouse models of allergic airway inflammation revealed that TLR7 activation in KO mice resulted in systemic loss of eosinophils, reduced IFN production, and an inability to clear respiratory viruses. Consistent with this finding, bone marrow mouse eosinophil progenitors lacking Pin1 showed markedly reduced cell proliferation and survival after TLR7 activation. Mechanistically, unlike WT cells, Pin1 null mouse eosinophils were defective in the activation of the ER stress-induced unfolded-protein response (UPR). We observed significant reductions in the expression of UPR components and target genes, aberrant TLR7 cleavage and trafficking and reduced granule protein production in KO eosinophils. Our data strongly suggest that Pin1 is required for bone marrow eosinophil generation and function during concurrent allergen challenge and viral infection.
NTRODUCTION
Eosinophils (Eos) are frequently the major component of airway inflammation in acute allergic asthma. They promote pulmonary pathology by driving goblet cell hyperplasia and mucus overproduction, facilitate pulmonary inflammation and contribute to chronic airway remodeling. Exacerbations of asthma with eosinophilic inflammation are often associated with respiratory viral infection, especially in children (1). The anti-viral defense includes the local release of IFNα and β from mononuclear cells, epithelium and Eos (2, 3), suggesting allergic inflammation can have beneficial consequences. The balance between inflammation and infection along with other potential risk factors (e.g. genetic predisposition, allergic sensitization, bronchial anatomy) likely determine the pathologic trajectory of disease.
Allergen-induced pulmonary eosinophilia is preceded by increased Eos differentiation in bone marrow. Eos differentiate from hematopoietic stem cells (HSC) under the control of multiple cytokines including IL-5 which induces the terminal differentiation of Eos progenitors (EoP) from common myeloid progenitors (CMP) and stimulates the exit of mature cells from bone marrow (4). Over several days early in differentiation, Eos actively synthesize highly basic granule proteins including EDN, EPX and MBP. After production, these proteins traffic through the secretory pathway, temporarily inducing ER stress and the unfolded protein response (UPR) (5, 6). Xbp1 is a central component of three evolutionarily conserved UPR pathways located in the ER lumen that are expressed in cells with a secretory phenotype (7). In response to excess and/or unfolded proteins, Xbp1 mRNA is spliced and translated, leading to the transcription of genes that, in aggregate, suppress ER protein influx, catabolize misfolded proteins, and improve protein folding. Consistent with the importance of this response, Eos are completely eliminated in the bone marrow of Xbp1 KO mice (6).
UPR can also be activated by external stimuli such as bacterial and viral infection via TLR7/9 mediated signaling (8). Agonists of TLR7/9 are currently in human clinical trials and have been shown to reduce pulmonary eosinophilia and acute airway hyper-responsiveness (AHR) although the mechanisms underlying these effects are unclear (9). Eos express TLR7 and 9 suggesting these agonists may act directly on these cells. TLR signaling has recently been linked to the prolyl isomerase, Pin1 (10, 11). Pin1 is a ubiquitously expressed, cis-trans peptidyl-prolyl isomerase with substrate specificity for phosphorylated Ser-Pro or Thr-Pro peptide bonds. Pin1 regulates eukaryotic cell-cycle progression as well as a variety of other signaling pathways (12). In Eos, Pin1 facilitated IL-5/GM-CSF pro-survival signaling, enhanced cytokine expression through the stabilization of coding mRNAs and was necessary for cytokinesis toward EBI2 ligands released from asthmatic lung (13–17). Conversely, systemic genetic ablation or chemical inhibition of Pin1 significantly attenuated pulmonary Eos accumulation and airway remodeling in rodent models of asthma (16, 17), akin to the effects seen after TLR7 agonists. In response to dsRNA, Pin1 bound IRF3 to trigger its ubiquitination and subsequent degradation in cancer cells (10) while Pin1 loss led to enhanced IRF3-dependent IFN-β production and subsequent reduction of virus replication. In primary DCs, activation of TLR7 or TLR9 (which sense ssRNA and dsDNA, respectively) induced Pin1 binding to IRAK1, leading to IRF7 activation, IFN-α/β production and viral clearance (11). Thus, Pin1 plays an important role at multiple levels in the regulation of Eos function, TLR signaling and anti-viral immunity which likely depends on cell type.
In order to further characterize the biological function of Pin1 in Eos, we generated mice with floxP sites flanking exon 2. Breeding with eoCre mice led to selective deletion of Pin1 in bone marrow EoP and the entire Eos lineage. We show that under basal conditions, the loss of Pin1 had modest effects on Eos differentiation and function. However, under stress induced by TLR7 activation, Pin1 null Eos showed abnormal maturation, attenuated UPR, reduced granularity, shortened life span, reduced IFNs production, and inability to produce anti-viral interferons. Biochemically, Pin1 interacted with IRAK4 and Eos lacking Pin1 showed aberrant TLR7 cleavage and localization while physiologically consequences included an inability to clear pulmonary viruses. Our data demonstrate an unexpected interplay between TLR7 signaling and UPR and indicates that Pin1 modulates these processes during eosinophil development.
METHODS
Reagents:
Anti-TLR7 (against N-terminal for WB), anti-ATF4 and anti-Xbp1 were from Abcam. Anti-TLR7 (against C-terminal for WB) was purchased from Cell Signaling. Anti-TLR7 (against N-terminal) from Invitrogen, anti-TLR7 (against C-terminal) and anti-TLR7 (against C-terminal for IF) from Noves Biologicals. Anti-Pin1 and anti-ATF6 from Santa Cruz Biotechnology. SYBR Green PCR Master Mix was from Bio-Rad. R848, OVA and anti-β-actin were from Sigma. PCR primers were purchased from IDT, Inc. Protease Inhibitor Mixture was from Calbiochem. The DyLight 800/680 secondary antibodies and IMJECT Alum were purchased from Thermo Scientific. TaqMan Universial PCR Master Mix (for Rhinovirus) and TaqMan Gene Expression Assay (for SeV) were from Applied Biosystem. Sendai virus was from ATCC. Mouse TLR1–9 Agonist kit was from InvivoGen. Murine IL-5, mSCF and mFLT3L were from PeproTech. EDN (RNase2) ELISA kit, MBP (Prg2) ELISA kit and ECP ELISA kit were from MyBioSource. Anti-PRG2, anti-ECP and anti-EDN were from Biorbyt. LEGENDplx Mouse Proinflammatory Chemokine Panel was from Biolegend.
Mice
Pin1fl/fl mice was created in the lab on a pure C57Bl6 background. Exon2 is flanked by two loxP sites. EoCre mice (C57Bl6) were obtained from Dr. James J. Lee (Mayo Clinic). Animal care was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Our protocol was approved by the Committee on the Ethics of Animal Experiments of the University of Texas Southwestern Medical Center (Permit Number: 2011–0139). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering. Mixed gender (balanced in numbers) were used for all experiments unless otherwise indicated.
Induction of allergic airway inflammation and virus infection
Mice, 10–13 weeks of age, were sensitized (day 0 and 14) by intraperitoneal injection of OVA (20 μg) combined with adjuvant (Alum). After 1 week (day 21) mice were challenged with aerosolized OVA (1% wt/vol solution in PBS) or 1× PBS for 20 minutes for 4 consecutive days. On day 18, mice received an intravenous or intratracheal injection of R848 or vehicle (1× PBS). R848 stock was prepared in DMSO (2.4 mg/ml) and diluted with 1× PBS to obtain a dose of 100 μg/mouse. 50 μl of Sendai virus (SeV) (2500 CEI/mouse) were given by intratracheal injection. Control injections contained 50 μl of 1× PBS. One or four days after last aerosol OVA challenge, BAL fluid and cells were collected. Lungs were fixed with 4% of buffered formalin by filling to total lung capacity by gravity or the right lung was excised and snap frozen in liquid nitrogen and crushed with a mortar and pestle for RNA and protein analysis. Paraffin sections of the right lung were prepared and stained with HE.
Eos differentiation from bone marrow
Eos were differentiated from bone marrow cells as described previously (18). Briefly, bone marrow were collected from femurs by flushing out with RPMI-1640 medium. Red blood cells were lysed and remained cells were cultured at 106/ml in RPMI-1640 with 20% FBS, 100 IU/mL penicillin and 10 μg/mL streptomycin, 2 mM glutamine, 25 mM HEPES and 1× non-essential amino acids and 1 mM sodium pyruvate and 50 μM β-mercaptoethanol and supplemented with 100 ng/ml SCF and 100 ng/mL FLT3L from day 0 to day 4. On day 4, the media was replaced with fresh media containing 10 ng/ml IL-5 only. Every other days from this point forward, 50% of the media was replaced with fresh media containing IL-5. For the experiment, dead cells were removed with Dead Cell Removal Kit (Miltenyi Biotec), and only preparations with >96% viable cell were used for these studies.
Human eosinophil preparation
Peripheral blood was obtained by venipuncture from healthy or mildly atopic donors. Eosinophils from the blood were purified as described (19). Only populations >96% eosinophils were used for the studies. Eosinophils were cultured at a density of 1 × 106cells/ml in RPMI-1640 medium and 10% FBS. All participants have a clinical record at the University of Wisconsin Hospital and written informed consent was obtained according to an approved protocol of the University of Wisconsin Hospital Institutional Review Board. The review board also specifically approved this study before initiation. The viability of cells was determined after each cell isolation and cell lysates/RNA were generated if the viability was >96% at day 0 and >80% at day 4 after incubation with 200 pM IL-5.
Reverse transcription and real-time PCR
RNA was extracted with TriReagent. cDNA Quantitative PCR was performed with a SYBR PCR master mix with the primers shown. An ABI 7500 thermocycler (Applied Biosystems, Foster City, CA) was used for 45 cycles of PCR. ΔCT calculates the differences between target CT values and the normalizer (housekeeping gene) for each sample: ΔCT = CT (target) − CT (normalizer). The comparative ΔΔCT calculates the differences between each sample ΔCT value and the baseline ΔCT. The comparative expression level (fold changes) was obtained transforming the logarithmic values to absolute values using 2−ΔΔCT. All data from untreated control cells was normalized to fold change “1” or 100%.
Immunostaining and immunoblots
For the analysis of TLR7 localization to the endosome, cells on cytospun slides were permeabilized, blocked, and incubated with appropriate dilutions of primary antibodies followed by FITC-conjugated secondary antibodies and DAPI. Images were collected by fluorescent microscopy. At least 6 fields/slide and 3 mice/group were analyzed. For immunoprecipitation and immunoblots, cell lysates were prepared in nonidet P-40 buffer. For immunoprecipitation, cell lysates were cleared with normal IgG and protein G sepharose beads before incubation with Pin1 antibody (R&D). Proteins were transferred onto PVDF membranes and probed with primary and secondary antibodies. Protein bands were detected and quantified using LI-COR® Odyssey®Imaging System.
Recombinant TAT prot eins
TAT-WW and its mutant (W34A) were chemically synthesized and purified by the UT-Southwestern Protein Core. The cDNA encoding the full-length of Pin1 were cloned in-frame into pHis-TAT. Proteins were expressed in E. coli and were purified on a Ni2+ chelate column (QIAGEN) as described by the manufacturer. The TAT-linked proteins were more than 90% pure, based on Coomassie blue staining of SDS gels.
Pin1 Activity Assay
Activity was measured as described previously (20) with slight modifications.
Statistics
All p values were calculated by one-way ANOVA using GraphPad Prism. Data are represented as mean ± SD. p< 0.05 was considered significant.
RESULTS
Pin1 knockout alters allergic airway inflammation and BM Eos production in response to TLR7 activation.
Conditional mice were created by the insertion of loxP sites flanking exon 2 of Pin1 (Fig. 1A) with recombination eliminating the isomerase active site. Eos-specific deletion was accomplished by breeding Pin1f/f mice with eoCre mice where Cre recombinase is driven by the eosinophil specific EPX promoter and expressed at the EoP stage (21). As expected, Cre-mediated exon 2 deletion occurred in Eos during bone marrow differentiation in vitro (Fig. 1B) as well as in vivo in mature Eos in the blood or spleen under basal conditions, or in the lung after allergen challenge (Fig. 1B, Fig 1S). Based on flow cytometry analysis, all other immune cells (T and B cells, macrophages, dendritic cells) expressed Pin1 normally (not shown).
Figure 1. Pin1 KO mice exhibit systemic reduction of Eos numbers after TLR7 activation in allergic airway inflammation.
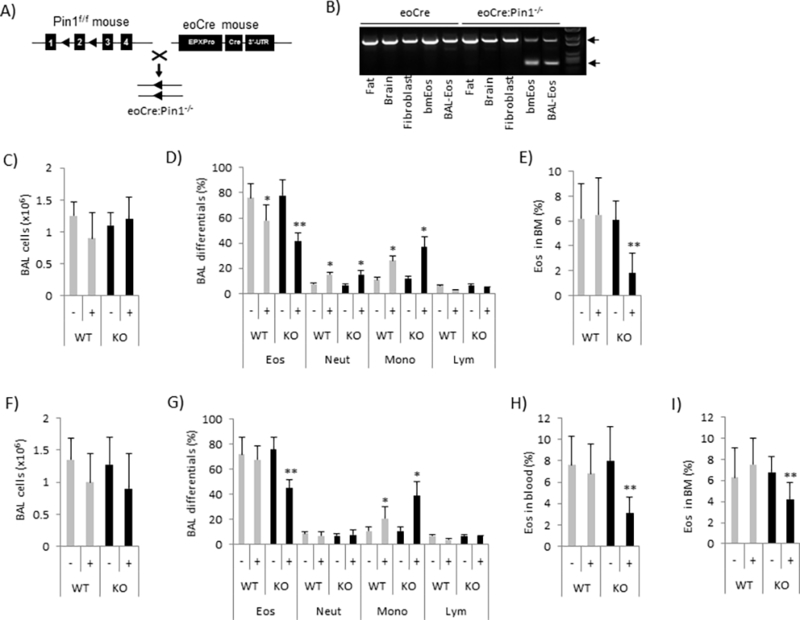
(A) Pure strain C57BL/6 Pin1f/f mice were crossed with eoCre mice to delete exon 2 of Pin1. (B) Cre-mediated exon 2 deletion occurs only in Eos. Top arrow points to WT while bottom arrow points to recombined band. (C-E) The total BAL cell counts (C), BAL differential cell counts (D) and percentage of Eos in BM one day after last OVA-challenge, +/− 100 μg R848/mouse (intratracheally). (F-I) The total BAL cells counts (F), BAL differential cell counts (G), and percentages of Eos in blood (H) and bone marrow (I) four days after last OVA-challenge, +/− 100 μg R848/mouse. Data are expressed as mean±SD. * (between treatments in same genotype) and ** (between genotypes after treatment) denote p<0.05 by one-way ANOVA with 10–15 mice each group.
To explore how selective deletion of Pin1 in Eos affects allergic disease, we sensitized and challenged adult Pin1−/−:eoCre (KO) or parental Pin1f/f (WT) mice with OVA. Before challenge, some mice were treated with a single intra-tracheal (IT) dose of R848 (Fig. 2S), a potent, cell-permeable TLR7 agonist (22). Mice were harvested one day or four days after the last OVA challenge. Irrespective of R848, total BAL inflammatory cell counts were similar between genotypes with Eos predominant (Fig. 1C, 1D and Fig 3S). Consistent with the previous observations (23), R848 treated WT mice showed dose dependent shifts in BAL inflammatory infiltrates highlighted by reductions in Eos (57% from 76%) and lymphocytes (3% from 6%) but increased neutrophils (15% from 8%) and monocytes (26% from 11%) (Fig. 1C, 1D and Fig. 3S).
However, in the presence of R848, BAL Eos counts were further significantly reduced in KO compared to WT controls 1 day after the completion of allergen challenge (Fig. 1D). Eos were the only BAL inflammatory cells negatively affected by Pin1 deficiency after R848 (Fig. 1D). Surprisingly, reduced Eos counts were also seen in the peripheral blood and most notably, in the bone marrow of KO mice (Fig. 1E). Similar data were obtained with mice harvested 4 days after the last OVA challenge or if R848 was administered intravenously rather than IT (Fig. 1F–1I) (24, 25). WT and KO BAL and blood Eos showed similar viability based on annexin V (not shown) and caspase 3 staining (not shown), similar expression of cell-surface IL5RA (not shown) and of BAL chemokines (eotaxin, RANTES and MCP-1) (Fig. 4S, A and B). Interestingly, the B-lymphocyte chemoattractant (CXCL13) and LPS-induced CXC chemokine which are not predominantly produced by Eos were decreased in KO BALF, implying an indirect effect of selective Pin1 KO on other cells. Similar lung histology was observed after TLR7 agonists in WT and KO mice (not shown). Taken together, these data suggest that Eos reductions in KO animals are possibly due to reduced or defective proliferation or enhanced Eos death in the BM after TLR7 activation, thereby implicating a role of Pin1 in Eos stress responses.
Knockout mice show reduced respiratory virus clearance, granule release and anti-viral interferons.
Next, we asked whether Pin1 loss influenced Eos effector function during stress. We tested this by infecting WT and KO mice with Sendai virus after the last aerosol challenge and measuring residual virus in BAL by Taq-Man qPCR. Viral infections are frequent causes of asthma exacerbations in humans (1) and promote airway Eos antiviral responses that include degranulation, the release of ROS and anti-viral cytokines (26–28). Of note, viral clearance was significantly attenuated (~8 fold) in KO mice compared to WT (Fig. 2A). We probed for mechanism by measuring MBP, EDN and EPX which are the major granule proteins released by activated Eos in response to infection (29). In the absence of R848, granule protein release was equivalent in WT and KO mice (Fig. 2B–2E). While R848 treatment triggered significantly greater release of Eos granule proteins (MBP and EDN) into the airway irrespective of genotype, BAL EDN was significantly lower in KO mice compared to WT even after normalization by absolute Eos number (Fig. 2C, 2E). These results suggest that KO Eos have abnormal granular contents, contain fewer granules and/or have reduced protein/granule.
Figure 2. Pin1 KO mice exhibit reduced granular proteins and virus clearance in the lung.
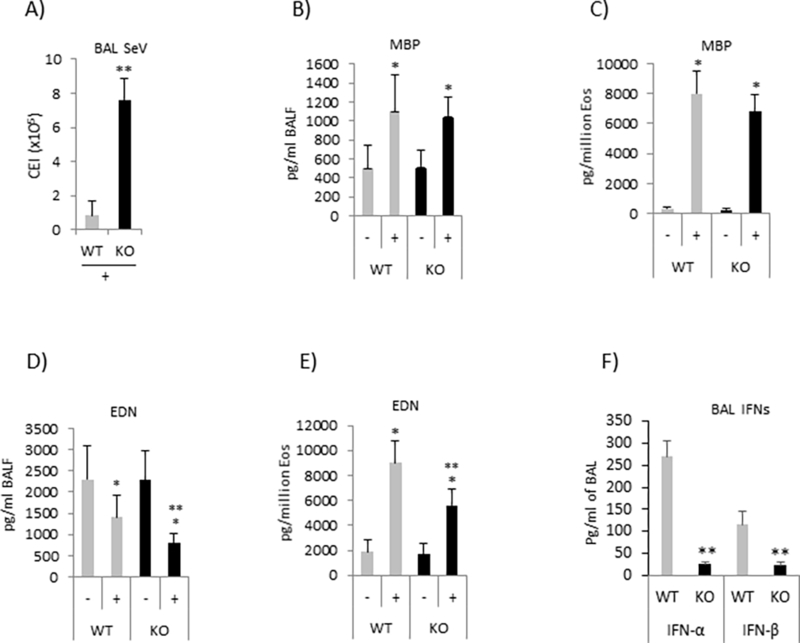
(A) Sendai virus (SeV) (2500 CEI/mouse) were injected intratracheally on the day of last OVA challenge, +/−100 μg R848/mouse. Four days later the virus titers in BAL fluid were determined by qPCR using TaqMan probes. (B-E) The protein levels of MBP and EDN in BAL fluid were measured by ELISA, and normalized by volume of fluid (B and D) or absolute Eos counts (C and E). (F) IFNs in BAL fluid were measured by ELISA and normalized by volume of fluid. Data are expressed as mean±SD. * (between treatments in same genotype) and ** (between genotypes after treatment) denotes p<0.05 by one-way ANOVA with 10–15 mice each group.
We also measured the release of reactive oxygen species (ROS) after infection. When normalized with absolute Eos counts or by BAL volume, there was no significant difference between WT and KO (not shown). However, anti-viral IFNs (α and β) in the BAL were highly significantly reduced in KO mice treated with R848 (Fig. 2F), suggesting the change was due to reduced production by infiltrating Eos. In order to clarify the mechanism, we inhibited Pin1 in primary human peripheral blood Eos (PBE) with TAT-WW or TAT-W34A after priming cells with IL-5. IL-5 treated PBE are a commonly used surrogate for BAL Eos (29). The TAT tag permits rapid and complete transduction of cells and in this case, delivers a highly specific, dominant negative Pin1 inhibitor peptide (WW) or an inactive control peptide that differs by one amino acid (W34A). When pre-treated with Pin1 inhibitor but not control TAT-peptide, PBE were unable to upregulate anti-viral IFN mRNAs after exposure to R848 or ssRNA (Fig. 3A and 3B). The acute Pin1 blockade with TAT-WW or R848 treatment did not affect cell viability within 24 h compared to untreated or TAT-W34A treated controls (data not shown) but TAT-WW moderately reduced the viability of activated Eos after 3 days (15). Similar results were observed with murine, BM-derived Pin1 KO Eos (Fig. 3C and 3D). These results indicate that Pin1 is required for TLR7 signaling that culminates in anti-viral cytokine gene expression.
Figure 3. Pin1 is required for IFN responses by mature Eos after TLR7 activation.

(A-B) Purified human peripheral blood Eos (PBE) were primed with IL-5 (10 pM) before Pin1 inhibitor peptide (TAT-WW) (200 nM) or its mutant control (W34A) for 4 h followed by R848 treatment (30 μg/ml) for 18 h. * denotes p<0.05 between WW and W34A treatments. (C-D) Mature mouse Eos differentiated in vitro from bone marrow were treated with R848 for 18 h before qPCR for IFN expression. * denotes p<0.05 between genotypes after treatment. (E) Human PBE were treated with R848 for 10 min. PPIase activity of Pin1 in cell lysates was measured as described in Methods. * denotes p<0.05 between indicated treatments. Data are expressed as mean±SD and from 3–4 independent experiments.
As Pin1 is typically inactive in circulating PBE (14, 15), we asked if Pin1 was itself downstream of TLR7 signaling. PBE were pretreated with TAT-WW or TAT-W34A prior to R848 for 10 minutes. Lysates showed significantly higher Pin1 isomerase activity after R848 that could be suppressed by TAT-WW but not by control TAT-W34A (Fig. 3E) consistent with previous observations in peripheral blood mononuclear cells (11). These data show that Pin1 is relatively inactive in resting cells but is strongly upregulated by TLR7 signaling which is essential for a variety of anti-viral responses in both circulating as well as tissue based Eos.
Pin1 null Eos undergo apoptosis in bone marrow after TLR7 activation.
As shown earlier, KO mice displayed systemic reductions in circulating and BM Eos after stress induced by TLR7 agonists (Fig. 1E, 1H and 1I). Those Eos that reached the inflamed airways showed defective anti-viral responses, presumably due to reduced release of granule protein and/or anti-viral interferons. These data suggest that Pin1 is not only essential for TLR7 signaling in mature Eos (Fig. 3), but also plays an important role in Eos differentiation in bone marrow. This is consistent with prior reports demonstrating Eos lineage ablation in the BM of Xbp1 KO mice (6).
In order to identify the underlying mechanism, we cultured WT and Pin1 null BM and exposed them to TLR7 agonists during Eos differentiation. BM cultures can be driven nearly exclusively toward Eos in the presence of stem cell factors and IL-5. In the absence of R848 or ssRNA, Eos developed normally in both genotypes as assessed by the appearance of cell surface markers (IL-5RA, Siglec-F and CCR3), viability, yield and morphology (Fig. 4A–4B and data not shown). However, when TLR7 agonists (R848 or ssRNA) were added to the culture on day 8 before the appearance of granules, KO Eos underwent accelerated programmed cell death starting on day 14 (Fig. 4C and data not shown) while WT Eos remained healthy (>90% viability). Cell death was entirely reversed in KO cells if reconstituted with TAT-Pin1 on day 12 (Fig. 4D). There was no change in differentiation marker expression (IL-5RA, Siglec-F and CCR3) on KO cells compared to WT cells (not shown). Accelerated apoptosis was also observed in WT TLR7 agonized BM cultures after treatment with Pin1 inhibitor peptide (TAT-WW) (Fig. 4E). The KO BM cultures also experienced reduced cell division detectable on day 14 (Fig. 4F). When R848 (or ssRNA) was added to BM cultures on day 14 or beyond, KO cells showed normal viability (Fig. 4G). Moreover, accelerated cell death was exclusively triggered by TLR7 (R848 or ssRNA) but not by agonists that engage other TLRs (Fig. 4H) or cytokines (IL-4, IL-8, IL-10, IL-12, IL-1β, TNF-α, eotaxin, and TGF-β) (not shown). Interestingly, WT cells, but not KO were sensitive to TLR5 agonists but the molecular mechanism remains to be explored (Fig. 4H)
Figure 4. Eos lacking Pin1 undergo apoptosis during differentiation after TLR7 activation.
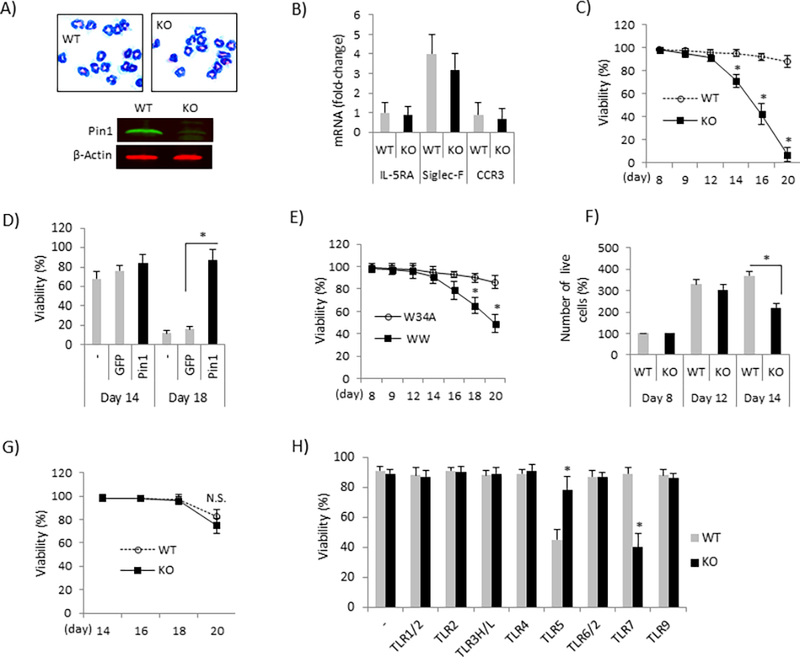
(A) WT and KO Eos were differentiated (days in vitro 14) from bone marrow in vitro and stained with Wright-Giemsa (upper) or immunoblotted (bottom) with antibodies shown. (B) qPCR for differentiation markers in KO and WT Eos. (C) WT and KO Eos were differentiated as in (A) but R848 (30 μg/ml) was added to the culture every other day starting from day 8. Cell viabilities were examined on the days indicated. * denotes p<0.05 between treatments in same day. (D) KO Eos were differentiated as in (A) but TAT-Pin1 protein (TAT-GFP as control) and R848 were added to the culture every other day starting from day 8. Cell viability was measured on day 14 and 18. (E) WT Eos were differentiated as in (A) but TAT-WW (200 nM) or its mutant, inactive control (TAT-W34A) and R848 were added to the culture every other day starting from day 8. Cell viability was determined at time points shown. * denotes p<0.05 between treatments in same day. (F) WT and KO Eos were differentiated from bone marrow as in (A). R848 was added every other day from day 8 and the number of live cells was determined by Trypan blue on the days indicated. (G) Mature WT and KO Eos (day 14) were incubated with R848 and cell viability was determined at time points indicated. (H) Eos were differentiated as in (A) but TLR agonists were added to the culture every other day starting from day 8. Cell viability was examined on day 16. * denotes p<0.05 between genotypes. Data were expressed as mean±SD and from 3–5 independent experiments.
Pin1 null Eos show defective UPR signaling.
Next, we asked how TLR7 signaling at day 8 of culture induced Eos death in the absence of Pin1. This time point coincides with the production, trans-Golgi ER trafficking and packaging of highly basic, secretory granular proteins EDN, MBP and EPX (6). These events induce UPR, leading to the suppression of translation, and the upregulation of Xbp1s and IRF dependent cytokine gene expression (6, 7). TLR7, in addition to activating a pro-inflammatory and anti-viral cytokine response via MyD88-IRAK1/4-TRAF3/6 (11), can also activate UPR via IRE1-Xbp1s-ATF4/6 (6). If UPR is compromised, ER stress triggers Eos lineage apoptosis as seen in Xbp1 KO mice (6). Therefore, we analyzed UPR signaling in differentiating KO and WT Eos agonized with TLR7 agonists on day 8. qPCR analysis on day 12 showed significant reductions in the expression of critical UPR genes in KO cells compared to WT (Fig. 5A and 5B). Immunoblots revealed reduced Xbp1s production and ATF6α cleavage in KO Eos while PERK phosphorylation was unaffected (Fig. 5C and data not shown). Pin1 inhibition with TAT-WW peptide added at day 8–12 of differentiating WT Eos also reduced Xbp1 expression and ATF6a cleavage (Fig. 5D and data not shown). Conversely, KO cells treated with TAT-Pin1 showed restoration of Xbp1s mRNA even in the absence of IL5 (Fig. 5E). In aggregate, these results confirm that Pin1 loss also prevents the TLR7 signaling in immature Eos as measured by reduced UPR and cell survival. Interestingly, terminally differentiated Eos (day 14) lost nearly all expression of Xbp1 and ATF6a (Fig. 5C), coincident with completion of granule production. Similarly, RNAseq and proteomic analysis detected only trace amounts of Xbp1 and ATF6α in mature, human peripheral blood Eos (30, 31).
Figure 5. Defective UPR and granule formation in Pin1 KO Eos.
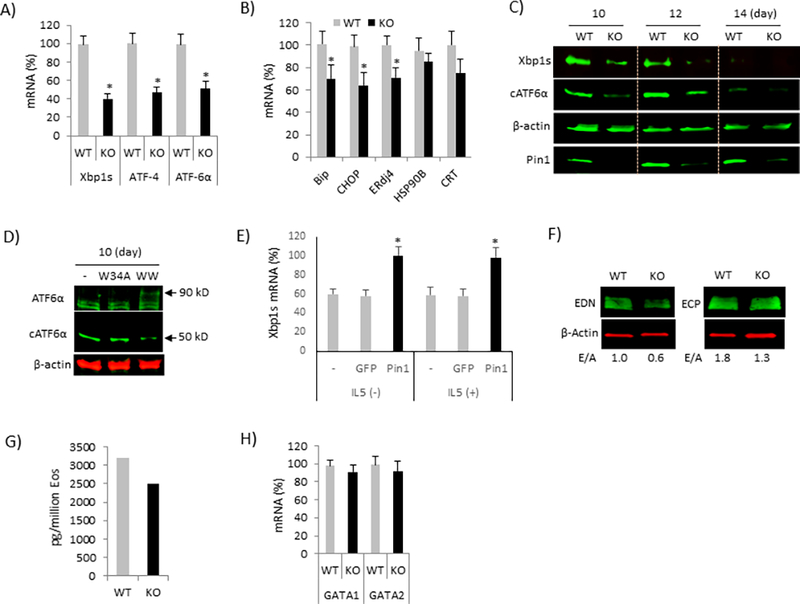
(A-B) BM Eos (day 8) were treated with R848 (30 μg/ml) every other day prior to analysis for UPR (A) and its target genes (B) on day 12 by qPCR. (C) Cell were treated as in (A) for indicated times prior to immunoblot of cell lysates with the antibodies shown. (D) WT Eos were treated with dominant negative TAT-WW and its mutant control (TAT-W34A) in the presence of R848 and immunoblotted on day 10. (E) KO Eos were differentiated as in (A) and TAT-Pin1 or TAT-GFP and R848 were added to the culture every other day starting from day 8 in the presence or absence of IL-5. The levels of Xbp1s mRNA were measured on day 14. * denotes p<0.05 between GFP and Pin1. (F) WT and KO Eos (day 16) treated with R848 were immunoblotted with the antibodies shown. E/A: EDN or ECP to β-Actin ratio. (G) The level of EDN protein in cell lysate (F) was measured by ELISA. (H) Cells were treated as in (A) prior to analysis for GATA1 and GATA2 mRNA on day 12 by qPCR. * in (A-B) denotes p<0.05 between genotypes. Data are expressed as mean±SD and from 3–4 independent experiments.
In the absence of UPR, differentiating Eos displayed defective maturation and granule protein production, leading to reduced cell survival (6). Therefore, we analyzed the granule protein content in Pin1 KO Eos by immunoblot of whole cells and indirectly, the intracellular granule number by autofluorescence. Mature KO Eos contained reduced EDN and ECP (Fig. 5F and 5G) if treated with R848 on day 8. Transcriptional analyses of granule proteins (data not shown) and master Eos lineage-commitment factors GATA1 and GATA2 (Fig. 5H) were unchanged by Pin1 deficiency. These data suggest that Pin1 is required for post-translational granule protein processing and maturation in differentiating Eos and that failure of this program leads to cell death.
Pin1 null Eos are defective in TLR7 processing, activation and signaling
Pin1-null DC and lymphocytes (11) and mature Eos (Fig. 3A–3D) showed reduced TLR7-mediated, anti-viral IFN responses. Thus, we sought to determine how Pin1 loss impaired normal TLR7 signaling in differentiating and mature Eos. After interacting with ligand in endosomes, TLR7/9 is usually cleaved as a prelude to recruiting MyD88, IRAK1/4 and TRAF leading to subsequent downstream signaling (32, 33). As expected, WT BM Eos cultures treated with R848 on day 8 - day 10, showed loss of full-length TLR7 (110 kD) with appearance of 60 kD (Fig. 6A) (32) and 53 kD products (not shown) that were recognized by C-terminal and N-terminal specific antibodies, respectively. Similar cleavage was also seen in IL5-activated human PBE after TLR7 engagement with ligand (not shown). However, in KO Eos, the proteolytic process was markedly attenuated (Fig. 6A) despite multiple R848 exposures (Fig. 6A, ++ lane). Deficiencies in the chaperone UNC93B can prevent TLR7 cleavage in DCs (34). However, we were unable to detect any differences in its expression between genotypes irrespective of R848 treatment (data not shown).
Figure 6. Defective TLR7 processing and signaling in Pin1 KO Eos.
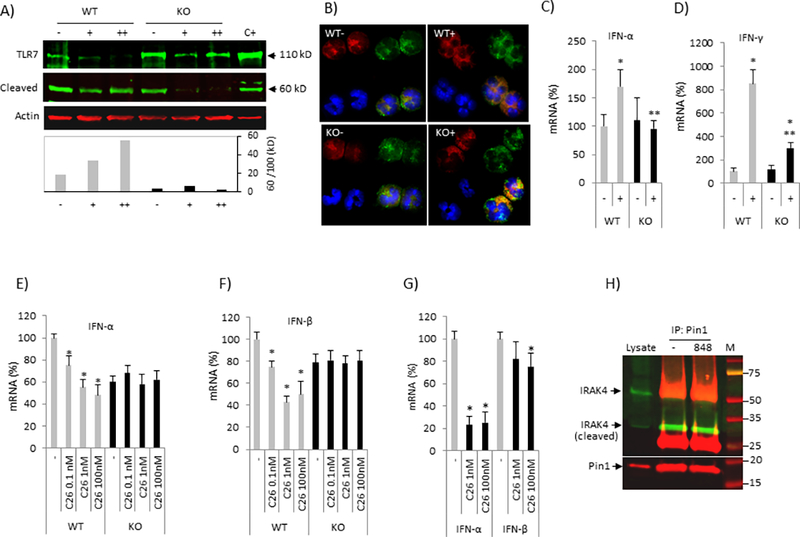
(A) BM Eos (day 8) were treated with or without R848 prior to immunoblot of whole cell lysates on day 12. (+): R848 added on day 8, (++): R848 added on day 8 and 10. Bottom: Cleaved TLR7 (60 kD) was normalized to full-length TLR7 (110 kD). (B) BM EOS (day 8) were treated with or without R848 on day 8 and 10 prior to immunostaining on day 12 with anti-TLR7 (red), anti-EEA1 (endosome marker, green), DAPI (nuclei, blue) and green-red merged (yellow). (C-D) BM Eos (day 10) were treated with R848 for 18 h prior to analysis for the IFN-α and IFN-γ mRNA by qPCR. * and ** denote p<0.05 between treatments in same genotype and between genotypes after treatment, respectively. (E-F) BM Eos (day 10) were treated with IRAK4 inhibitor (C26) and R848 for 18 h prior to analysis for IFN-α (E) and IFN-β (F) mRNA by qPCR. * denotes p<0.05 between non-treatment (−) and treatment (C26) in same genotype. (G) Human PBE were primed with IL-5 before IRAK4 inhibitor (C26) for 4 h followed by R848 treatment for 18 h. * denotes p<0.05 between (−) and (C26). (H) Human PBE were treated with R848 for 10 min. Cell were lysed in NP-40 buffer and the supernatant was used for immunoprecipitation (IP) with anti-Pin1 followed by immunoblot with the antibodies shown (see Method). 10% of supernatant (without IP) was loaded as input (lysate). Data were expressed as mean±SD and from 3–4 independent experiments.
In macrophages and DCs, proteolytic cleavage of TLR7/9 in the endolysosomes was a prerequisite for their activation (35, 36). Chimeric TLR9 redirected to the plasma membrane was resistant to cleavage and nonfunctional (36). Therefore, we analyzed TLR7 localization in WT and KO Eos treated with R848. Confocal analysis of day-12 WT cells using an anti-C-terminal Ab showed predominantly endosomal full length and cleaved TLR7 irrespective of R848 (Fig. 6B, WT panels) (32). R848 treated WT cells showed increased, punctuate C-terminal fragment staining, consistent with cleavage (Fig. 6B, WT+ panel). Untreated KO cells exhibited similar post-endosomal distribution as resting WT Eos but with less total TLR7 signal (Fig.6B, KO-). However, treatment with R848 caused markedly increased receptor retention in the endosomal compartments (Fig. 6B, KO+). Trafficking abnormalities were associated with signaling defects as R848-induced types I and II IFN responses were significantly reduced in KO compared to WT cells (Fig. 6C and 6D). Similar attenuation in cytokine responses and Pin1 isomerase activity was observed in WT cells pretreated with the highly specific IRAK4 inhibitor C26 (Fig. 6E, 6F and data not shown), demonstrating IRAK4 is downstream of TLR7 but upstream of IFN gene expression in Eos. Mature, human PBE stimulated with R848 showed a similar reduction in anti-viral IFN expression after IRAK4 inhibition (Fig. 6G) which was not seen in KO cells (Fig. 6E and 6F).
After TLR engagement, MyD88, IRAK1 and 4 form the so-called myddosome (37). IRAK4-mediated phosphorylation activates IRAK1 and causes dissociation of both IRAK1 and 4 from MyD88. Phospho-IRAK1 then associates with TRAF6 before being ubiquitinylated and degraded by the proteasome. As Pin1 KO, inhibition and IRAK4 blockade equivalently reduced IFN upregulation after R848, we asked if these proteins interacted in mature PBE. Indeed, Pin1 was reproducibly pulled down with cleaved IRAK4 (32 kD) (recognized by anti-C-terminal antibody) (Fig. 6H) but not with full length IRAK4. Activation (10 min - 4 h) of TLR7 with R848 did not affect the level of IRAK4 (protein or mRNA) nor the interaction of Pin1 with cleaved IRAK4. These results suggest that Pin1 regulates TLR7 signaling through both a modulation of TLR7 processing and localization and the function of myddosome complex.
DISCUSSION
In this study, we used eoCre mice to selectively delete Pin1 in eosinophil-lineage committed cells, permitting us to better define the role of Pin1 in these granulocytes. Unexpectedly, we found an absolute requirement for Pin1 in Eos differentiation, survival and IFNs expression in the context of TLR7 activation both in vivo and in vitro. In the absence of Pin1, stressed BM Eos exhibited reductions in UPR gene expression, resulting in incomplete post-translational maturation and packaging of granule proteins. These failures likely led to significant reductions in cell survival. In vitro, reconstitution of KO cells with Pin1 gene completely rescued cells from apoptosis and restored UPR. Mechanistically, Pin1 null cells neither cleaved nor localized TLR7 appropriately, providing a biochemical rationale for the aberrant signaling. Pin1 interacted with IRAK4, a critical component in TLR7 signaling regulating IFNs and proinflammatory cytokine gene expression. Using in vivo rodent models of allergic disease, we showed that KO mice exhibit reduced pulmonary Eos accumulation and granule protein release and attenuated ability to clear viral infection. Our data present new evidence that Pin1 plays an important role in eosinophil differentiation and function.
Cytokines (e.g. IL-5, IFN-γ and IL-17) (38, 40), transcription factors (e.g. GATA1, C/EBP and PU.1) (41) and signaling molecules (SHP2 and Xbp1) (42, 6) play critical roles in EoP formation and terminal Eos differentiation in bone marrow. However, there is limited information regarding granulocyte differentiation in the setting of infection. While the transcription factors play instructive roles in the differentiation of EoPs from CMPs, IL-5 enhances EoP differentiation and proliferation and strongly supports mature Eos survival, expansion and maturation (38). The proinflammatory cytokines (IFN-γ and IL-17) (39, 40) and protein tyrosine phosphatase SHP2 (42) coordinately regulate EoP formation through Erk-MAPK and GATA1 dependent pathways (43, 44). In the absence of stress, Pin1 null EoP differentiated normally. However, when TLR7 was agonized under conditions mimicking viral infection, a common and oft-times inciting event in patients with asthma, Pin1 null Eos were unable to successfully engage UPR or anti-viral responses, leading to both cell death and an inability to clear virus.
GATA1 is essential for the expression of granule proteins (41) whereas Xbp1 is required for subsequent protein folding and maturation. In series, both proteins are required for successful, terminal Eos differentiation (6). In the absence of Pin1, Xbp1 was down-regulated along with other UPR related genes (Fig. 5A–5D), explaining the observed reduction in intracellular granule proteins and accelerated cell death. The regulation of UPR and Xbp1 expression supports an unappreciated role by Pin1 in ER homeostasis in Eos specifically and possibly secretory cells more generally. The mechanism for these effects remains enigmatic. We did not observe changes in the activity (protein level, phosphorylation, endoribonuclease activity) (data not shown) of IRE1α, the splicing of Xbp1 mRNA (not shown) or the phosphorylation of PERK (not shown). IP/IB failed to reveal direct protein-protein interactions between Pin1 and UPR components (IRE1α, eIF2α, ATF4, ATF6α or Xbp1), unlike in tumor cells where Pin1 bound to Xbp1 and promoted Xbp1-induced cell proliferation and transformation (45). Alternatively, the regulation of UPR genes by Pin1 may involve a TLR7 induced, autocrine mediator such as IL-4. Differentiating B cells exposed to IL-4 upregulated Xbp1 mRNA through a STAT6 dependent signal (46). IL-4 also induced other UPR components including the ER chaperone GRP78 and the transcription factor CHOP (46). Thus, a cytokine driven, feed-forward mechanism may be involved. In support, Pin1 interacts with IRAK1 (11) and IRAK4 (Fig. 6H) which was essential for TLR7/9 mediated, type I IFNs responses (11) (Fig. 3A–3D) and participates in IL-18 and IL-33 signaling (47, 48). While TLR2/4 signaling induces Xbp1 mRNA splicing, IL-6 and IFN-β are also induced in macrophages (49). Therefore, Pin1 acts downstream of TLR signaling to induce both transcription factor (Xbp1) and UPR responses involved in innate immune response and cell differentiation.
TLR7 is preferentially confined to intracellular compartments such as ER, endosomes and lysosomes, rather than to the cell surface (32). In unstimulated cells, TLR7 localizes in the ER, and upon activation, is rapidly transported to the endosome for proteolytic cleavage (32). Rarely, cleavage occurs earlier in proximal biosynthetic compartments such as ER and Golgi (50). After cleavage, the C-terminal fragment (60 kD) is competent for signaling and indispensable for recruiting the MyD88 complex while the N-terminal fragment can act as a dominant negative and inhibit C-terminal function (51). In Eos, R848 induced TLR7 cleavage (Fig. 6A) but in the absence of Pin1, excess full length protein accumulated in the endosome (Fig. 6B). Presumably, reduced C-terminal production inhibits signaling and impairs UPR during pathogen infection. While activated TLR9 was retained in the endosomes of pDCs (52), downstream anti-viral IFNs were still produced, suggesting proteolytic and topologic decisions are cell and receptors specific. While UNC93B1 has been implicated in the regulation of TLR3/7/9 transport and cleavage (53, 54), Pin1 KO Eos showed no alterations in UNC93B1 expression (data not shown). Other possibilities include Pin1 dependent regulation of intracellular proteases involved in TLR processing including the acidic-pH-dependent cathepsins B/K (55), furin (50) and AEP (56, 57), all of which are expressed by Eos.
A growing body of experimental data and ongoing clinical trials highlights the important role for and potential of TLR7 in asthma. Among its functions, this receptor activates Eos and promotes viral clearance, a common accelerant of asthma recurrence (58, 59). However, it remains unclear how Eos contribute to viral eradication. The levels of granule proteins, IRF7, IFN-β and NOS-2 were positively correlated with the degree of airway eosinophilia and negatively correlated with viral load in the lung (58). Purified Eos released EDN and NO after exposure to supernatants from infected epithelial cells (58), consistent with the hypothesis that the highly abundant granule proteins are anti-viral (30, 31), In vivo TLR7 signaling synergized with allergen challenge to increase the release of Eos granule proteins (Fig. 2B - 2E) as well as the expression of anti-viral IFNs (Fig. 2F) which were attenuated by the absence of Pin1. In vitro incubation of human PBE with R848 significantly induced pro- and anti-inflammatory cytokines such as TNF-α, IFNs and IL-1α and β (Fig. 3A–3D and data not shown), which was also dependent on Pin1 through a TLR7-IRAK4 dependent pathway (Fig. 6E-6G). These data strongly suggest that TLR7-mediated, anti-viral innate immune responses are Pin1 dependent and adequate to eliminate virus.
In summary, our data provide new insights into how Pin1 contributes to the anti-viral response by pulmonary Eos. Given that R848 is a potential inhibitor of type 2 cell-driven inflammatory responses without giving tissue damage and generating autoimmunity or immune incompetence (25, 60), a strategy toward targeting Pin1 and TLR7 signaling in both immature and mature cells may be a new option to treat eosinophilic disorders.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health grant P01HL088594.
REFERENCES
- 1.Jartti T and Gern JE. 2017. Role of viral infections in the development and exacerbation of asthma in children. J. Allergy Clin. Immunol. 140: 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lorente F, Laffond E, Moreno E and Dávila I. 2001. [Viral infection and asthma: immunologic mechanisms]. Allergol. Immunopathol (Madr). 29: 126–133. [DOI] [PubMed] [Google Scholar]
- 3.Domachowske JB, Dyer KD, Bonville CA, and Rosenberg HF. 1998. Recombinant human eosinophil-derived neurotoxin/RNase 2 functions as an effective antiviral agent against respiratory syncytial virus. J. Infect. Dis. 177: 1458–1464. [DOI] [PubMed] [Google Scholar]
- 4.Denburg JA, Woolley M, Leber B, Linden M & O’Byrne P. 1994. Basophil and eosinophil differentiation in allergic reactions. J. Allergy Clin. Immunol. 94: 1135–1141. [DOI] [PubMed] [Google Scholar]
- 5.Bainton DF and Farquhar MG. 1970. Segregation and packaging of granule enzymes in eosinophilic leukocytes. J. Cell Biol. 45: 54–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bettigole SE, Lis R, Adoro S, Lee AH, Spencer LA, Weller PF, Glimcher LH. 2015. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol. 16: 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwakoshi NN, Lee AH & Glimcher LH. 2003. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 194: 29–38. [DOI] [PubMed] [Google Scholar]
- 8.Celli J and Tsolis RM. 2015. Bacteria, the endoplasmic reticulum and the unfolded protein response: friends or foes? Nat. Rev. Microbiol. 13: 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drake MG, Kaufman EH, Fryer AD & Jacoby DB. The therapeutic potential of Toll-like receptor 7 stimulation in asthma. Inflamm. Allergy Drug Targets 2012; 11: 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, Akira S, Yamamoto N, Lu KP and Yamaoka S. 2006. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol. 7: 598–605. [DOI] [PubMed] [Google Scholar]
- 11.Tun-Kyi A, Finn G, Greenwood A, Nowak M, Lee TH, Asara JM, Tsokos GC, Fitzgerald K, Israel E, Li X, Exley M, Nicholson LK, Lu KP 2011. Essential role for the prolyl isomerase Pin1 in Toll-like receptor signaling and type I interferon-mediated immunity. Nat Immunol. 12: 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu KP and Zhou XZ. 2007. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8: 904–916. [DOI] [PubMed] [Google Scholar]
- 13.Shen ZJ, Esnault S, Schinzel A, Borner C and Malter JS. 2009. The peptidyl-prolyl isomerase Pin1 facilitates cytokine-induced survival of eosinophils by suppressing Bax activation. Nat. Immunol. 10: 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen ZJ, Hu J, Kashi VP, Kelly EA, Denlinger LC, Lutchman K, McDonald JG, Jarjour NN and Malter JS. 2017. Epstein-Barr Virus-induced Gene 2 Mediates Allergen-induced Leukocyte Migration into Airways. Am J Respir Crit Care Med. 195:1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen ZJ, Esnault S and Malter JS. 2005. The peptidyl-prolyl isomerase Pin1 regulates the stability of granulocyte-macrophage colony-stimulating factor mRNA in activated eosinophils. Nat. Immunol. 6 : 1280–1287. [DOI] [PubMed] [Google Scholar]
- 16.Shen ZJ, Esnault S, Rosenthal LA, Szakaly RJ, Sorkness RL, Westmark PR, Sandor M and Malter JS. 2008. Pin1 regulates TGF-beta1 production by activated human and murine eosinophils and contributes to allergic lung fibrosis. J Clin Invest. 118: 479–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esnault S, Rosenthal LA, Shen ZJ, Sedgwick JB, Szakaly RJ, Sorkness RL and Malter JS. 2007. A critical role for Pin1 in allergic pulmonary eosinophilia in rats. J Allergy Clin Immunol. 120:1082–1088. [DOI] [PubMed] [Google Scholar]
- 18.Dyer KD, Mose JM, Czapiga M, Siegel SJ, Percopo CM and Rosenberg HF. 2008. Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow. J. Immunol. 181: 4004–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansel TT, De Vries IJ, Iff T, Rihs S, Wandzilak M, Betz S, Blaser K and Walker C. 1991. An improved immunomagnetic procedure for the isolation of highly purified human blood eosinophils. J Immunol Methods. 145:105–110 [DOI] [PubMed] [Google Scholar]
- 20.Hennig L, Christner C, Kipping M, Schelbert B, Rücknagel KP, Grabley S, Küllertz G, and Fischer G. 1998. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry. 37: 5953–5960. [DOI] [PubMed] [Google Scholar]
- 21.Doyle AD, Jacobsen EA, Ochkur SI, Willetts L, Shim K, Neely J, Kloeber J, Lesuer WE, Pero RS, Lacy P, Moqbel R, Lee NA, and Lee JJ. 2013. Homologous recombination into the eosinophil peroxidase locus generates a strain of mice expressing Cre recombinase exclusively in eosinophils. J Leukoc Biol. 94:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smits EL, Cools N, Lion E, Van Camp K, Ponsaerts P, Berneman ZN, and Van Tendeloo VF. 2010. The Toll-like receptor 7/8 agonist resiquimod greatly increases the immunostimulatory capacity of human acute myeloid leukemia cells. Cancer Immunol Immunother. 59: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jirmo AC, Daluege K, Happle C, Albrecht M, Dittrich AM, Busse M, Habener A, Skuljec J, and Hansen G. 2016. IL-27 Is Essential for Suppression of Experimental Allergic Asthma by the TLR7/8 Agonist R848 (Resiquimod). J Immunol. 197: 4219–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koga-Yamakawa E, Dovedi SJ, Murata M, Matsui H, Leishman AJ, Bell J, Ferguson D, Heaton SP, Oki T, Tomizawa H, Bahl A, Takaku H, Wilkinson RW, and Harada H. 2013. Intratracheal and oral administration of SM-276001: a selective TLR7 agonist, leads to antitumor efficacy in primary and metastatic models of cancer. Int J Cancer. 132: 580–590. [DOI] [PubMed] [Google Scholar]
- 25.Drake MG, Kaufman EH, Fryer AD, and D.B. 2012. Jacoby. The therapeutic potential of Toll- like receptor 7 stimulation in asthma. Inflamm Allergy Drug Targets. 11: 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Percopo CM, Dyer KD, Ochkur SI, Luo JL, Fischer ER, Lee JJ, Lee NA NA, and J. B. 2014. Domachowske, Rosenberg HF. Activated mouse eosinophils protect against lethal respiratory virus infection. Blood. 123: 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Z, Xu X, Leng X, He M, Wang J, Cheng S, and Wu H. 2017. Roles of reactive oxygen species in cell signaling pathways and immune responses to viral infections. Arch Virol.162: 603–610. [DOI] [PubMed] [Google Scholar]
- 28.Message SD and Johnston SL. 2001. The immunology of virus infection in asthma. Eur. Respir. J. 18: 1013–1025. [DOI] [PubMed] [Google Scholar]
- 29.Devos R, Plaetinck G, Cornelis S, Guisez Y, Van der Heyden J, and Tavernier J. 1995. Interleukin-5 and its receptor: a drug target for eosinophilia associated with chronic allergic disease. J. Leukoc. Biol. 57: 813–819. [DOI] [PubMed] [Google Scholar]
- 30.Shen ZJ, Hu J, Esnault S, Dozmorov I, and Malter JS. 2015. RNA Seq profiling reveals a novel expression pattern of TGF-β target genes in human blood eosinophils. Immunol. Lett. 167: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkerson EM, Johansson MW, Hebert AS, Westphall MS, Mathur SK, Jarjour NN, Schwantes EA, Mosher DF, and Coon JJ. 2016. The Peripheral Blood Eosinophil Proteome. J Proteome Res. 15:1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ewald SE and Barton GM. 2011. Nucleic acid sensing Toll-like receptors in autoimmunity. Curr. Opin. Immunol. 23: 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, and Barton GM. 2008. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 456: 658–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YM, Brinkmann MM, Paquet ME, and Ploegh HL. 2008. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 452: 234–238. [DOI] [PubMed] [Google Scholar]
- 35.Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, and Ploegh HL. 2008. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol. 9: 1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barton GM, Kagan JC, and Medzhitov R. 2006. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat. Immunol. 7: 49–56. [DOI] [PubMed] [Google Scholar]
- 37.Kawasaki T and Kawai T. 2014. Toll-like receptor signaling pathways. Front. Immunol. 5: 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dent LA, Strath M, Mellor AL and Sanderson CJ. 1990. Eosinophilia in transgenic mice expressing interleukin 5. J. Exp. Med. 172: 1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Bruin AM, Buitenhuis M, van der Sluijs KF, van Gisbergen KP, Boon L, and Nolte MA. 2010. Eosinophil differentiation in the bone marrow is inhibited by T cell-derived IFN-gamma. Blood. 116: 2559–2569. [DOI] [PubMed] [Google Scholar]
- 40.Tian BP, Hua W, Xia LX, Jin Y, Lan F, Lee JJ, Lee NA, Li W, Ying SM, Chen ZH, and Shen HH. 2015. Exogenous interleukin-17A inhibits eosinophil differentiation and alleviates allergic airway inflammation. Am J Respir Cell Mol Biol. 52: 459–470. [DOI] [PubMed] [Google Scholar]
- 41.Fulkerson PC 2017. Transcription Factors in Eosinophil Development and As Therapeutic Targets. Front. Med. (Lausanne). 4: 115 Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xia LX, Hua W, Jin Y, Tian BP, Qiu ZW, Zhang C, Che LQ, Zhou HB, Wu YF, Huang HQ, Lan F, Ke YH, Lee JJ, Li W, Ying SM, Chen ZH, and Shen HH. 2016. Eosinophil differentiation in the bone marrow is promoted by protein tyrosine phosphatase SHP2. Cell Death Dis 7: e2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pazdrak K, Schreiber D, Forsythe P, Justement L and Alam R. 1995. The intracellular signal transduction mechanism of interleukin 5 in eosinophils: the involvement of lyn tyrosine kinase and the Ras-Raf-1-MEK-microtubule-associated protein kinase pathway. J. Exp. Med. 181: 1827–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pazdrak K, Adachi T and Alam R. 1997. Src homology 2 protein tyrosine phosphatase (SHPTP2)/Src homology 2 phosphatase 2 (SHP2) tyrosine phosphatase is a positive regulator of the interleukin 5 receptor signal transduction pathways leading to the prolongation of eosinophil survival. J. Exp. Med. 186: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chae U, Park SJ, Kim B, Wei S, Min JS, Lee JH, Park SH, Lee AH, Lu KP, Lee DS, and Min SH. 2016. Critical role of XBP1 in cancer signalling is regulated by PIN1. Biochem J. 473: 2603–2610. [DOI] [PubMed] [Google Scholar]
- 46.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, and Glimcher LH. 2003. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4: 321–329. [DOI] [PubMed] [Google Scholar]
- 47.Ali MR, Rahman MA, Wu Y, Han T, Peng X, Mackey MA, Wang D, Shin HJ, Chen ZG, Xiao H, Wu R, Tang Y, Shin DM, and El-Sayed MA. 2017. Efficacy, long-term toxicity, and mechanistic studies of gold nanorods photothermal therapy of cancer in xenograft mice. Proc Natl Acad Sci U S A. 114: E3110–E3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nechama M, Kwon J, Wei S, Kyi AT, Welner RS, Ben-Dov IZ, Arredouani MS, Asara JM, Chen CH, Tsai CY, Nelson KF, Kobayashi KS, Israel E, Zhou XZ, Nicholson LK, and Lu KP. 2018. The IL-33-PIN1-IRAK-M axis is critical for type 2 immunity in IL-33-induced allergic airway inflammation. Nat Commun. 9: 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinon F, Chen X, Lee AH and Glimcher LH. 2010. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11: 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hipp MM, Shepherd D, Gileadi U, Aichinger MC, Kessler BM, Edelmann MJ, Essalmani R, Seidah NG, Reis e Sousa C, and Cerundolo V. 2013. Processing of human toll-like receptor 7 by furin-like proprotein convertases is required for its accumulation and activity in endosomes. Immunity. 39: 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinha SS, Cameron J, Brooks JC and C.A. 2016. Leifersd. Complex Negative Regulation of TLR9 by Multiple Proteolytic Cleavage Events. J. Immunol. 197: 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, and Taniguchi T. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 434:1035–1040. [DOI] [PubMed] [Google Scholar]
- 53.Kim YM, Brinkmann MM, Paquet ME, and Ploegh HL. 2008. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 452: 234–238. [DOI] [PubMed] [Google Scholar]
- 54.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, Shamel L, Herskovits AA, Portnoy DA, Cooke M, Tarantino LM, Wiltshire T, Steinberg BE, Grinstein S, and Beutler B. 2006. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 7:156–164. [DOI] [PubMed] [Google Scholar]
- 55.Matsumoto F, Saitoh S, Fukui R, Kobayashi T, Tanimura N, Konno K, Kusumoto Y, Akashi-Takamura S, and Miyake K. 2008. Cathepsins are required for Toll-like receptor 9 responses. Biochem Biophys Res Commun. 367: 693–699. [DOI] [PubMed] [Google Scholar]
- 56.Sepulveda FE, Maschalidi S, Colisson R, Heslop L, Ghirelli C, Sakka E, Lennon-Duménil AM, Amigorena S, Cabanie L, and Manoury B. 2009. Critical role for asparagine endopeptidase in endocytic Toll-like receptor signaling in dendritic cells. Immunity. 31: 737–748. [DOI] [PubMed] [Google Scholar]
- 57.Maschalidi S, Hässler S, Blanc F, Sepulveda FE, Tohme M, Chignard M, van Endert P, Si-Tahar M, Descamps D, and Manoury B. 2012. Asparagine endopeptidase controls anti-influenza virus immune responses through TLR7 activation. PLoS Pathog. 8: e1002841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Phipps S, Lam CE, Mahalingam S, Newhouse M, Ramirez R, Rosenberg H. f., Foster PS, and K. I. 2007. Matthaei. Eosinophils contribute to innate antiviral immunity and promote clearance of respiratory syncytial virus. Blood. 110:1578–1586. [DOI] [PubMed] [Google Scholar]
- 59.Drake MG, Bivins-Smith ER, Proskocil BJ, Nie Z, Scott GD, Lee JJ, Lee NA, Fryer AD, and Jacoby DB. 2016. Human and Mouse Eosinophils Have Antiviral Activity against Parainfluenza Virus. Am J Respir Cell Mol Biol. 55: 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gunzer M, Riemann H, Basoglu Y, Hillmer A, Weishaupt C, Balkow S, Benninghoff B, Ernst B, Steinert M, Scholzen T, Sunderkötter C, and Grabbe S. 2005. Systemic administration of a TLR7 ligand leads to transient immune incompetence due to peripheral-blood leukocyte depletion. Blood. 106: 2424–2432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.