

Abstract
Our previous study demonstrated that the tyrosine kinase receptor inhibitor sunitinib induces acquired drug resistance in endothelial cells. The present study explored the role of lysosomal sequestration of sunitinib in the acquisition of drug resistance in human microcapillary endothelial HMEC-1 cells. Resistance was induced by escalating concentrations of sunitinib and a shift in IC50 from 12.8 to >20 µM was detected. The results of time-lapse fluorescence microscopy illustrated an instantaneous emergence of fluorescent vesicles in living cells once sunitinib was added. Most of these vesicles emerged in the juxtanuclear area, and exhibited the characteristics of growing autophagosomes and lysosomes. The vesicles were identified as autophagosomes and lysosomes because they co-located with the lysosomal tracers Lyso-ER and Lyso-NIR, and the protein markers lysosomal-associated membrane protein 1 (LAMP-1) and microtubule-associated protein 1A/1B-light chain 3 (LC3). The results of western blotting demonstrated that sunitinib induced upregulation of LAMP-1 and LC3-II, and downregulation of sequestosome 1/p62, indicating the activation of autophagy. Bafilomycin A1, which suppresses lysosomal acidification, completely blocked sunitinib sequestration; however, chloroquine, which blocks lysosomal fusion with autophagosomes, exhibited no effect. Notably, bafilomycin A1 and chloroquine significantly counterbalanced HMEC-1 drug-resistance. These results provided evidence for autophagy-flux-associated sunitinib lysosomal sequestration in endothelial cells, leading to isolation of the drug from the cytoplasm; a key process involved in the development of drug resistance during antiangiogenic therapy. These data supported the notion that inhibiting autophagy may be a potential strategy to prevent drug sequestration and resistance to antiangiogenic therapy.
Keywords: endothelial cells, tyrosine kinase receptor inhibitor, drug resistance, lysosomal drug sequestration, autophagy
Introduction
Endothelial cells of blood vessels are in direct contact with circulating components; therefore, they are inevitably exposed to the highest concentrations of drugs during drug treatment. This is well illustrated by the blood-brain barrier, where endothelial cells overexpressing the ABC transporter are a major mechanism underlying drug resistance (1–4). Previous studies demonstrated that acquired multiple drug resistance can be induced in endothelial cells, and that this resistance affects the efficacy of anticancer treatment in vitro and in vivo (1–4). Notably, resistance in endothelial cells can be induced by antiangiogenic drugs targeting tyrosine kinase receptors, including sunitinib (2,3).
Various factors involved in the process of tumor angiogenesis and tumor growth are angiogenesis-dependent (5). Therefore therapeutic agents targeting tumor endothelial cells have been developed for anticancer treatment, including compounds such as sunitinib, which target angiogenic growth factor receptors, predominantly tyrosine kinase receptors (6,7). These inhibitors were initially expected not to induce clinical resistance; however, after >10 years of clinical use, resistance to antiangiogenic therapies has been widely reported (7–9). Several mechanisms have now been described, including alternative angiogenic pathways, selective pressure of hypoxia, cancer stem cells, recruitment of vascular progenitors and modulators, and tumor dormancy (9–11).
It is known that lysosomes sequester lipophilic amine drugs through a non-enzymatic and non-transporter-mediated process (12–14). As a hydrophobic (logP, 2.93), weak base (pKa, 9.04) molecule, sunitinib has been reported to accumulate in acidic lysosomes (15,16). The extent of lysosomal drug sequestration depends on the pH gradient between the acidic luminal pH of the lysosome and that of the cytoplasm. Consequently, drugs are sequestered away from their intracellular target sites (17). Notably, certain multidrug resistance transporters of the ABC superfamily, such as ATP binding cassette subfamily B member 1 (ABCB1; also known as P-gp), are highly expressed on the lysosomal membrane, and further accelerate ATP-dependent lysosomal drug sequestration (16–18). A previous in vivo study detected higher intratumoral concentrations of sunitinib than those found in plasma, further supporting the clinical relevance of sunitinib lysosome sequestration (19).
Studies on tumor cells have indicated that lysosome sequestration of sunitinib may induce autophagy-associated resistance in these cells (20–23). Lysosomes are spherical membrane-bound organelle vesicles with a lumen pH of 4.5–5.0; these vesicles contain a panel of hydrolytic enzymes that enable biomolecular hydrolysis. Lysosomes are involved in various cellular processes, including secretion, plasma membrane repair, cell signaling and energy metabolism (24,25). Autophagosome biogenesis and lysosome activity are essential for autophagy, which enables the breakdown and recycling of cellular components. Autophagy is constitutively expressed in all mammalian cells, and the overall mechanisms initiating and terminating autophagy are at present being extensively investigated (24–26). Currently, the role of autophagic drug flux in sunitinib lysosomal sequestration in cancer cells is well demonstrated and autophagy activation is considered a key step in drug sequestration (17). The modulation of autophagy is expected to be an effective strategy for developing novel anticancer drugs (27,28).
Our previous study demonstrated the induction of multiple drug resistance in human microcapillary endothelial HMEC-1 cells following exposure to sunitinib, with upregulated P-gp expression (2). The present study investigated the occurrence of lysosome sequestration of sunitinib in endothelial cells and explored the relevant mechanisms.
Materials and methods
Materials
Anti-microtubule-associated protein 1A/1B-light chain 3 (LC3; cat. no. 0231-100/LC3-5F10) was obtained from Enzo Life Sciences, Inc.; anti-lysosomal-associated membrane protein 1 (LAMP-1; H4A3; cat. no. sc-20011) was purchased from Santa Cruz Biotechnology, Inc.; anti-sequestosome 1 (SQSTM1)/p62 (cat. no. 5114) was obtained from Cell Signaling Technology, Inc.; horseradish peroxidase-labeled anti-rabbit/mouse IgG antibodies (cat. no. 31460/31430) were obtained from Invitrogen; Thermo Fisher Scientific, Inc.; anti-β-actin (cat. no. A5316) was purchased from Sigma-Aldrich; Merck KGaA; goat anti-mouse IgG and goat anti-rabbit IgG coupled to Alexa Fluor 594 (cat. no. A-11005/A-11012) were also obtained from Invitrogen; Thermo Fisher Scientific, Inc. Bafilomycin A1 (BAF; 10 nM) and chloroquine (CQ; 20 µM) were purchased from Sigma-Aldrich; Merck KGaA; sunitinib was from Selleck Chemicals; and Lyso-ER, Lyso-NIR and DAPI were purchased from Abcam.
Cell culture and drug resistance induction
The HMEC-1 cell line (ATCC® CRL-3243™) was cultured in MCDB-131 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 2 mM L-glutamine, 100 µg/ml streptomycin, 100 U/ml penicillin and 15% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), Sunitinib-resistant HMEC-1 cells were obtained in two ways: i) By continuously exposing HMEC-1 cells to escalating concentrations of sunitinib, from 0.01 to 16 µM, over a period of >8 weeks. ii) By continuously exposing HMEC-1 cells to escalating concentrations of sunitinib, from 0.01 to 6 µM, then maintaining cells in culture with 6 µM sunitinib over 8–12 weeks (HMEC6 µM). All cells were digested with trypsin-EDTA once or twice a week and were cultured at 37°C in a 100% humidified atmosphere at 5% CO2. No mutagenic agents were used in the establishment of these sunitinib-resistant HMEC-1 cells.
Subcellular colocalization studies
The HMEC-1 cells were incubated with or without sunitinib 6 µM for 24 h, and then incubated with Lyso-ER or Lyso-NIR (Abcam) for 30 min at 37°C. Subsequently, those cells were fixed with 2% para-formaldehyde for 10 min at room temperature without light. Images of the fixed cells were obtained under a fluorescence microscope (Carl Zeiss AG).
Immunofluorescence
HMEC-1 and HMEC6 µM cells were seeded on glass coverslips (5×104 cells) in 24-well dishes with or without sunitinib 6 µM at 37°C. After 24 or 48 h, the cells were washed at 37°C, fixed at room temperature for 20 min with 2% paraformaldehyde, and permeabilized with Tris-buffered saline (TBS) containing 0.1% Triton X-100 (Sigma-Aldrich; Merck KGaA) for 30 min at room temperature prior to being exposed to anti-LC3 (1:200) and anti-LAMP-1 (1:50) antibodies and incubated overnight at 4°C. The cells were washed three times with TBS and were then incubated in the dark for 1 h at room temperature with a 1:1,000 dilution of anti-mouse or anti-rabbit Alexa Fluor 594-labeled secondary antibody (Invitrogen; Thermo Fisher Scientific, Inc.). The cells were mounted using DAPI (Abcam). Fluorescence images were collected under a Microscopy Imaging system (Carl Zeiss AG) and examined. Sunitinib absorbance is in the range of 340–480 nm and the maximum absorption is 429 nm. Sunitinib exhibits strong green fluorescence with a maximum of 540 nm.
Flow cytometry
The HMEC-1 cells were incubated with or without sunitinib (6 µM) for 24 or 48 h, and the HMEC6 µM cells were treated with sunitinib (6 µM) for 2 or 3 months at 37°C. Then, cultured cells were incubated with or without LysoTracker Red DND-99 for 15 min at 37°C (cat. no. L7528; Invitrogen; Thermo Fisher Scientific, Inc.), then all those cells were trypsinized and washed with PBS. After fixing in 2% paraformaldehyde for 20 min at room temperature, ~106 cells were resuspended in PBS and analyzed using a fluorescence-activated cell sorter (Canton II flow cytometer FACscan; BD Biosciences), and data were analyzed using FlowJo v7.6.1 and BD FACSDiva v4.1 (BD Biosciences).
Western blot analysis
The HMEC-1 cells were treated with or without sunitinib 6 µM for 24 h, or with the autophagy inducer rapamycin (Sigma-Aldrich; Merck KGaA; cat. no. 553210; 5 µM) for 8 h, or Hanks' solution (Sigma-Aldrich; Merck KGaA; cat. no. H6648) for 24 h at 37°C. All cells were collected and incubated for 30 min in radioimmunoprecipitation assay lysis buffer (Sigma-Aldrich; Merck KGaA) on ice, centrifuged at 15,000 × g for 5 min and the total proteins in the supernatant were recovered. The amount of extracted protein was determined using the RC-DC™ Protein Assay kit (Bio-Rad Laboratories, Inc.). The proteins (12.5 µg in 50 µl), in the presence of Laemmli 1X buffer (diluted from Laemmli 2X buffer; cat. no. 161-0737; Bio-Rad Laboratories, Inc.) and β-mercaptoethanol, were denatured at 100°C for 5 min and were then loaded onto an SDS-PAGE gel (4-20% Mini-Protean® TGX Stain-Free™; Bio-Rad Laboratories, Inc.). The proteins were transferred to a PVDF membrane (Trans-Blot® Turbo™ Transfer kit, Mini Format, 0.2 µm PVDF; Bio-Rad Laboratories, Inc.), using the Turbo™ Transfer system (Trans-Blot® Turbo™ Transfer system; Bio-Rad Laboratories, Inc.). The membrane was blocked with 5% skimmed milk diluted in PBS-0.1% Tween (PBS-T) for 1 h at room temperature and the membrane was then incubated with primary anti-LC3 (1:1,000), anti-SQSTM1/p62 (1:1,000), anti-LAMP1 (1:200) or anti-β-actin (1:5,000) antibodies in 5% bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA) at 4°C overnight. The membrane was then washed three times with PBS-0.1% Tween-20 for 5 min and incubated with secondary antibodies. Horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibodies were diluted 1:3,500 in 5% BSA and were incubated with the membrane for 1 h at room temperature. After three washes, HRP-ECL reagents (Santa Cruz Biotechnology, Inc.) were added to the membrane and chemiluminescence was revealed using a Fuji LAS-3000 system (Fujifilm). The ratios of blot density signal of specific protein bands to the control band were determined using ImageJ v1.51 software (National Institutes of Health).
Cell proliferation and cell viability assays
HMEC-1 or HMEC6 µM cells (5×104 cells/well) were placed in the presence of sunitinib (0, 3, 6, 12 or 24 µM) for 72 h, or under treatment with 10 nM BAF or 20 µM CQ, with or without sunitinib (6 µM) for 48 h, in a 24-well plate at 37°C. Then, the cells in each well were washed twice with PBS, detached with 200 µl trypsin and added to 800 µl medium. Cells were counted using KOVA Slide II (cat. no. 87118; Kova International, Inc.) or an automated cell-counter (Z2; Beckman Coulter, Inc.). The cell number in each well was divided by the initial seeded cell number to obtain the fold increase in cell number. The cell number of the treated groups was divided by the cell number of the appropriate control group to obtain the cell survival rate (%). The replication time was the average duration for a cell division.
Time-lapse imaging
HMEC-1 cells (2×104/500 µl) with sunitinib (6 µM) for 72 h or HMEC6 µM cells (2×104/500 µl) without sunitinib (6 µM) for 72 h were incubated in 4-well Hi-Q4 dishes (Biovalley) at 37°C in an incubator with 100% humidified atmosphere at 5% CO2. The images were recorded in real time using the BioStation IM-Q Time Lapse Imaging system (Nikon Corporation). Following the addition of sunitinib, culture plates were immediately put into the culture chamber of the time-lapse imaging system. The video recording usually began after a lapse of 15 min, which was required for the choice of image fields under microscopy.
Statistical analysis
Data are presented as the mean ± SD or mean ± SEM of more than three independent experiments carried out in triplicate. Results were statistically analyzed by one-way ANOVA using GraphPad Prism 6 (GraphPad Software, Inc.). P<0.05 was considered to indicate a statistically significant difference.
Results
Influx of sunitinib in HMEC-1 cells, and its inhibitory and toxic effect
The present study investigated the impact of different concentrations of sunitinib on the in vitro culture of HMEC-1 cells. Sunitinib was added to HMEC-1 cells for 24, 48 and 72 h, and the cells were then trypsinized, collected and counted under microscopy. The results demonstrated that sunitinib inhibited cell division at low concentrations, and induced cell death at higher concentrations (Fig. 1A). There were very few floating dead cells observed at concentrations <12 µM, but much more at higher concentrations, and few survived following treatment with 24 µM sunitinib. Below 12 µM, cell proliferation was inhibited; however, cell numbers still increased, indicating that cell division was still occurring. The IC50 of sunitinib in normal HMEC-1 cells was 12.8±0.76 µM (Fig. 1B). This indicated that sunitinib concentrations <12 µM were compatible with the survival of HMEC-1 cells. During the experiments, it was demonstrated that 6 µM was a good compromise dose, since it satisfied three essential conditions for further experiments. It enabled the maintenance of cell culture in the long term; it provided a clear view of the development of drug resistance; and it enabled clear observation of sequestration of sunitinib using fluorescence microscopy. Therefore, 6 µM concentration was selected for further exploration.
Figure 1.

Sunitinib suppresses HMEC-1 cells growth and penetrates HMEC-1 cells with the formation of fluorescent perinuclear vesicles. (A) Inhibitory effect of sunitinib at the indicated concentrations during different time periods. HMEC-1 cells were treated with 3, 6, 12 and 24 µM sunitinib for 24, 48 and 72 h. The cells were then washed, trypsinized and counted. Data are presented as the mean ± SEM. (B) Cell proliferation rate of HMEC-1 cells following treatment with sunitinib for 72 h compared with the control cells (100%). The inset graph shows the percentage of survived cells compared with the untreated control cells under the indicated sunitinib concentrations. Data are presented as the mean ± SEM. (C) Typical microscope image of HMEC-1 cells following treatment with 6 µM sunitinib, exhibiting visible fluorescent granules (magnification, ×200). (D) Typical fluorescence microscope image of HMEC-1 cells following treatment with 6 µM sunitinib, exhibiting fluorescent granules (magnification, ×100). (E) Amplified picture of a typical image of 6 µM sunitinib-treated HMEC-1 cells under fluorescence microscopy (magnification, ×400). (F) Evaluation of intracellular sunitinib quantity in HMEC-1 cells. The cells ware incubated with 6 µM sunitinib for 30 min, and 24 and 48 h, washed and immediately analyzed using flow cytometry. (G) Video images of living HMEC-1 cells following the addition 6 µM sunitinib. After the addition of sunitinib, culture plates were immediately placed into the culture chamber of a time-lapse imaging system. After image zone selection, video recording usually began 15 min after the addition of the drug. The images were taken every 12 min and images at the indicated time points are shown (magnification, ×40). Sunit, sunitinib.
Under light microscopy, the organelles of normal flat HMEC-1 cells were not clearly discernible. However, since solubilized sunitinib displays a bright yellowish color, it was possible to visualize sunitinib accumulation at sufficient concentrations >1 µM. The cells were stained yellow after overnight treatment and a large number of bright yellow vesicles could be seen within the cells (Fig. 1C). These bright vesicles were concentrated around the nucleus, with enhanced refraction.
Under fluorescence microscopy, green fluorescent sunitinib was enclosed within intracellular granules (Fig. 1D). With higher amplification, the green fluorescent granules were seen clearly around the cell nucleus (Fig. 1E). Subsequently, flow cytometry was conducted to follow the dynamic accumulation of 6 µM sunitinib in living cells. The results revealed that 30 min after adding sunitinib, the cells displayed clear green fluorescence. Much stronger fluorescence was obtained after 24 h and it appeared to approach its maximum level at 48 h (Fig. 1F).
To more intuitively and dynamically observe the formation of fluorescent sunitinib granules, a time-lapse imaging system coupled with fluorescence microscopy was used to record normal living HMEC-1 cells for 72 h following 6 µM sunitinib treatment (Fig. 1G and Video S1). The video revealed that sunitinib influx into the cells was immediate and extremely efficient. In <20 min, the intracellular fluorescence level was seen to rise within the cytoplasm and bright fluorescent granules began to emerge. The majority of the florescent granules emerged in the juxtanuclear area of the cells and a few of the granules emerged in the cytoplasm and then moved centripetally toward the nucleus. During the first 24 h, these moving granules grew in size and quickly clustered together. Subsequently, they either moved to one cell pole or continually rotated around the cell nucleus. An increase in the intensity of the drug fluorescence was observed during the first 24 h and the brightness then gradually stabilized. The spatial distribution of these vesicles fully complied with the characteristic positioning of lysosomes in the perinuclear pool near the microtubule-organizing center. Sunitinib is a weak base known to induce cytosol alkalinization, which could also facilitate the return of lysosomes to their central location (29).
To further verify these results, two lysosomal tracers, Lyso-ER and Lyso-NIR, were added to the sunitinib-treated cells and the cells were examined. The results confirmed that the lysosomal tracers and sunitinib markedly overlapped in HMEC-1 cells (Fig. 2).
Figure 2.
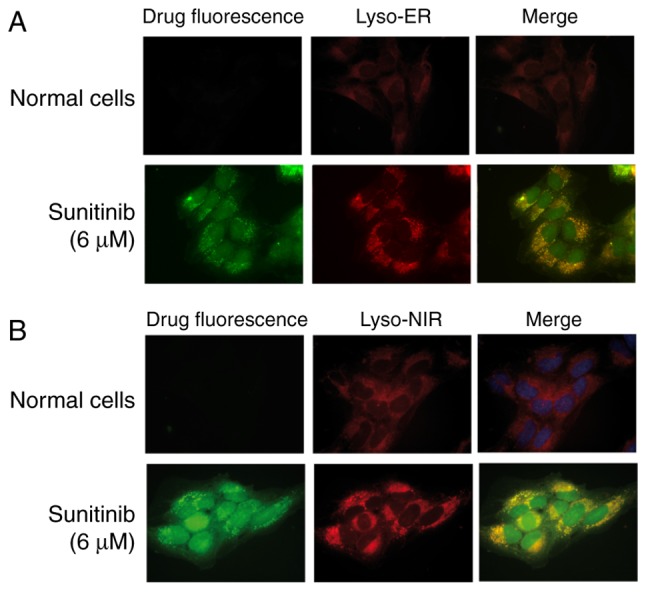
Colocalization of lysosome tracers and sunitinib fluorescence. (A) HMEC-1 cells were treated with 6 µM sunitinib for 24 h. The cells were washed, and Lyso-ER was added to the cells and left for 30 min. Washed cells were then fixed and underwent fluorescence microscopy. Lyso-ER exhibited red fluorescence (magnification, ×100). (B) HMEC-1 cells were treated with 6 µM sunitinib for 24 h. The cells were washed, and Lyso-NIR was added to the cells and left for 30 min. Washed cells were then fixed and underwent fluorescence microscopy. DAPI was added to stain the nuclei. Lyso-NIR exhibited red fluorescence (magnification, ×100).
Induction of drug resistance and activation of autophagy by sunitinib in HMEC-1 cells
HMEC-1 cells were exposed to escalating doses of sunitinib, up to 24 µM, to induce drug resistance. The results revealed that the time required for cell division progressively increased, with more floating dead cells observed when the concentrations reached >12 µM. To avoid cell death, cells were incubated with a fixed concentration of 6 µM sunitinib for 3 months to induce drug resistance. Notably, the cell division cycle was gradually prolonged (Fig. 3A). After 3 months, the IC50 value of sunitinib in 6 µM sunitinib-induced HMEC-1 cells had progressively shifted to ~25 µM compared with 12.8 µM for normal HMEC-1 cells (Fig. 3B and C). These cells were maintained in long-term culture with 6 µM sunitinib for subsequent experiments (HMEC6 µM). These data indicated that the induction of sunitinib resistance in HMEC-1 cells was dose- and time-dependent.
Figure 3.

Induction of drug resistance in HMEC-1 cells and sunitinib-induced activation of autophagy. (A) Replication times of cultured HMEC-1 cells in the presence of indicated concentrations of sunitinib. Data were calculated from more than two repetitions. (B) Evolution of the IC50 of sunitinib in HMEC-1 cells after treatment with 6 µM sunitinib for different time periods. (C) Plotted survival rates of HMEC-1 cells and sunitinib-resistant HMEC6 µM cells. Normal HMEC-1 cells and sunitinib-resistant HMEC6 µM cells were exposed to the indicated concentrations of sunitinib for 72 h, and the cells were trypsinized and counted. Non-treated cells were set as 100%. (A-C) Curves are derived from the means of more than three experiments. (D) LysoTracker DND-99 density of HMEC-1 cells following incubation with 6 µM sunitinib for different time periods. Cells were treated with sunitinib for 24 or 48 h and LysoTracker DND-99 was added followed by flow cytometric analysis. (E) LysoTracker DND-99 density of sunitinib-treated HMEC-1 cells and HMEC6 µM cells. The cells were washed five times and then incubated with LysoTracker DND-99. Fluorescence was measured with the control cells without LysoTracker DND-99 staining using flow cytometry. (F-H) Western blot analysis using anti-LAMP-1, anti-LC3 and anti-p62 antibodies. HMEC-1 and HMEC6 µM cells were exposed to 6 µM sunitinib, or autophagy inducer 5 µM rapamycin, or Hanks' media for 24 h. Subsequently, the cells were collected and extracted proteins were used for western blotting. The experiments were repeated at least three times. *P<0.05 vs. control HMEC-1 cells. (I) Colocalization of LC3 protein labeling with green sunitinib fluorescence. HMEC-1 and HMEC6 µM cells were treated with 6 µM sunitinib for 24 h. Subsequently, the cells were stained with anti-LC3 (red fluorescence) (magnification, ×100). DAPI was added to stain the nuclei. (J) Colocalization of LAMP-1 protein labeling with green sunitinib fluorescence. HMEC-1 and HMEC6 µM cells were treated with 6 µM sunitinib for 24 h. Subsequently, the cells were stained with anti-LAMP-1 (red fluorescence) (magnification, ×100). DAPI was added to stain the nuclei. LAMP-1, lysosomal-associated membrane protein 1; LC3, microtubule-associated protein 1A/1B-light chain 3; SQSTM1, sequestosome 1; Suni, sunitinib.
The aforementioned results strongly indicated that the detected lysosomes were the sunitinib-sequestering vesicles, and LysoTracker DND-99 was used to quantify the volumetric change in lysosomes following exposure to sunitinib. The results demonstrated that together with the increase in the intensity of sunitinib autofluorescence (Fig. 1F), the intensity of LysoTracker DND-99 in the sunitinib-treated cells was increased in parallel compared to the untreated control cells, indicating the expansion of lysosomes and their fusion with autophagosomes (Fig. 3D). Lysosomal mass was compared between HMEC-1 and HMEC6 µM cells. The HMEC6 µM cells exhibited moderately higher DND-99 fluorescence, suggesting a limited expansion of the lysosomal mass following long term exposure to sunitinib (Fig. 3E).
This study investigated the activation of autophagy in response to sunitinib treatment using autophagy molecular markers (30). The results of western blot analysis revealed that the protein expression levels of LAMP-1 were significantly increased after 24 h of sunitinib treatment, and a further increase was observed in HMEC6 µM cells (Fig. 3F) (31). In addition, the ratio of LC3-II/LC3-I was significantly increased in these cells, which indicated the active formation of autophagosomes (Fig. 3G). Furthermore, the significant downregulation of SQSTM1/p62 protein in these cells confirmed the ongoing process of active autophagy (Fig. 3H). These findings suggested that autophagy was activated in response to sunitinib treatment. Immunolabelling experiments with sunitinib-treated cells exhibited colocalization of anti-LC3-II (Fig. 3I) and a similar highly overlapped colocalization of anti-LAMP-1 (Fig. 3J) with the fluorescent sunitinib granules. Therefore, it was concluded that sunitinib was sequestered in autophagosomes and lysosomes in these cells.
Counterbalancing drug resistance by inhibiting lysosomal function
The aforementioned results indicated that drug resistance was associated with sunitinib sequestration in lysosomes. It is reasonable to postulate that the blockage of lysosomal sequestration would abolish drug resistance. Therefore, the effect and the efficiency of two autophagy inhibitors, the H+-ATPase inhibitor BAF, which blocks acidification of lysosomes, and CQ, which suppresses the fusion of autophagosomes with lysosomes, was investigated (32,33).
Fluorescence microscopy revealed that LAMP-1-labeled lysosomes were not disorganized, but were conserved in BAF- and CQ-treated cells (Fig. 4A and B). However, in the BAF-treated cells, green sunitinib granules disappeared, indicating the cessation of sunitinib sequestration in the LC3-labeled autophagosomes and lysosomes (Fig. 4A and B). Notably, the green sunitinib granules in CQ-treated cells had not disappeared or diminished (Fig. 4A and B). This observation was verified with lysosome tracers (Lyso-ER and Lyso-NIR) and the results confirmed the presence of fluorescent sunitinib granules in the CQ-treated HMEC-1 cells (Fig. 4C and D). These findings indicated the dependence of suitinib sequestration on intra-lysosomal pH.
Figure 4.

Reversal of resistance to sunitinib in HMEC-1 cells by autophagy inhibitors. (A-D) HMEC-1 cells were treated with 6 µM sunitinib for 24 h, either alone or in the presence of 10 nM BAF or 20 µM CQ for 4 h. DAPI was added to stain the nuclei. (A) Cells were then stained with anti-LAMP and observed under fluorescence microscopy. (B) Treated and non-treated HMEC-1 cells underwent anti-LC3 staining and were observed under fluorescence microscopy. (C) Treated or non-treated HMEC-1 cells underwent Lyso-ER staining for 30 min and were observed under fluorescence microscopy. (D) Treated or non-treated HMEC-1 cells underwent Lyso-NIR staining for 30 min and were (A-D) Magnification, ×63. (E) Survival rates of HMEC-1 cells following treatment with sunitinib with or without BAF. HMEC-1 cells were incubated with or without 6 µM sunitinib or 10 nM BAF for 48 h. Cells were trypsinized and counted. *P<0.05. (F) Survival rates for HMEC-1 cells following treatment with sunitinib with or without CQ. HMEC-1 cells were incubated with or without 6 µM sunitinib or 20 µM CQ for 48 h. Cells were trypsinized and counted. *P<0.05. (G) Survival rates for HMEC6 µM cells following treatment with sunitinib with or without BAF or CQ. HMEC6 µM cells were incubated with or without 6 µM sunitinib, 10 nM BAF or 20 µM CQ for 48 h. Cells were trypsinized and counted. *P<0.05. The results in e, f and g are presented as the mean ± SEM. (H) Steady decrease in sunitinib fluorescent density in cell vesicles following the withdrawal of sunitinib from culture media. HMEC6 µM cells were washed and cultured in normal culture medium. The cells were immediately placed into the culture chamber of a time-lapse imaging system. After image zone selection, image recording usually began after 15 min. The images were taken every 12 min and the images at indicated time points are shown. The upper row shows optic microscope images, the middle row shows green fluorescent microscope images and the lower row shows merged photos (magnification, ×40). BAF, bafilomycin A1; CQ, chloroquine; LAMP-1, lysosomal-associated membrane protein 1; LC3, microtubule-associated protein 1A/1B-light chain 3; Suni, sunitinib.
Cell resistance to sunitinib was evaluated following the addition of BAF and CQ. The addition of BAF or CQ significantly sensitized HMEC-1 cells to sunitinib and increased the toxic effect of sunitinib (Fig. 4E and F; P<0.05). The cytotoxicity experiments were repeated with HMEC6 µM cells, which have a higher resistance capacity. The results were similar; both autophagy inhibitors significantly increased the sensitivity of HMEC6 µM cells to sunitinib (Fig. 4G). These findings suggested that inhibition of lysosomal autophagy function counterbalanced HMEC-1 cell resistance to sunitinib.
Reversible resistance to Sunitinib
To analyze the reversibility of this drug resistance, HMEC6 µM cells were cultured normally after the withdrawal of sunitinib from the culture media, and their IC50 values were regularly checked. It was revealed that the cells had regained their sensitivity to sunitinib after 2 weeks of culture without sunitinib, with an IC50 equivalent to normal HMEC-1 (data not shown). The video-recorded images with the time-lapse imaging system coupled with fluorescence microscopy revealed that the fluorescence density of sunitinib granules in recovered HMEC6 µM cells had decreased by approximately half in the first 12 h and continued to recede. This indicated that once the extracellular concentration of sunitinib had decreased, the efflux of sunitinib sequestered in lysosomes began immediately. The decrease in fluorescence resulted from both decreases in fluorescent vesicle volume and reduction in the number of fluorescent vesicles (Fig. 4H).
Discussion
Our previous studies reported the induction of drug resistance by doxorubicin and sunitinib in endothelial cells in vitro and in vivo (1–3). Previous studies have demonstrated lysosomal sequestration in renal and colorectal cancer cells, which is an important mechanism implicated in the development of sunitinib resistance (15,17). These results prompted the study of sunitinib lysosomal sequestration in endothelial HMEC-1 cells and its relationship with multiple drug resistance.
Since sunitinib is an autofluorescent molecule in culture medium, a time-lapse imaging system was used to monitor the influx of 6 µM sunitinib into HMEC-1 cells. This concentration was chosen because it was sufficient to induce drug resistance and avoided highly injurious toxicity in long-term culture. When viewing the video of time-lapse fluorescence microscopy, an instant influx was systematically noted as sunitinib was added to the culture medium. This suggests a very effective diffusion of sunitinib across the cell membrane. A rapid emergence of green fluorescent granules was detected a few minutes later, particularly in the juxtanuclear zone where the lysosomes were concentrated. The sharp contrast of the bright fluorescence of the vesicles compared to that of the cytosol background strongly indicated that drug accumulation in the vesicles was driven by an active transport of sunitinib from the cytosol to the vesicles, most probably by ABC family molecules highly expressed in endothelial cells (1–3,18). The video images also revealed that intracellular fluorescent particles were in permanent movement, and they either rotated around the nucleus or concentrated markedly at one cell pole. These features were indeed significant for lysosomes because normal cytosol acidification should cause dispersal of the perinuclear lysosome population, whereas sunitinib-induced alkalinization could return them to their central location (29,34).
The formation of autophagosomes and their fusion with pre-existing lysosomes is a well-known natural process of autophagic flux. Western blotting with anti-LAMP-1, anti-LC3 and anti-p62 antibodies was used to evaluate autophagy activation. The results revealed that sunitinib treatment for 24 h activated autophagy, as determined by a significant increase in LC3-II/I ratio and LAMP-1 expression, and a decrease in p62. Experiments with HMEC6 µM cells exhibited similar results, indicating that autophagy remained active (30). These results may explain the increase in size and numbers of green fluorescent granules in the video following sunitinib treatment. Immunostaining of sunitinib-treated HMEC-1 and HMEC6 µM cells with anti-LAMP-1 and anti-LC3 antibodies revealed colocalization of these two autophagy-lysosome markers with sunitinib green fluorescence. In addition, the staining of the cells with two fluorescent lysosome tracers, Lyso-ER and Lyso-NIR, revealed colocalization with sunitinib green fluorescence. Flow cytometry using LysoTracker DND-99 staining indicated an expansion of vacuole compartments under sunitinib treatment. These results confirmed that the vesicles in the juxtanuclear zones enclosing sunitinib and isolating the drug from the cytosol were indeed lysosomes/autophagosomes. However, further work is required to clarify the mechanisms underlying sunitinib-associated autophagic flux in endothelial cells.
This study also revealed that the development of sunitinib resistance in HMEC-1 cells was dose- and time-dependent, and resistance remained as long as the sunitinib was present in the culture medium. HMEC-1 cells tolerated sunitinib well until the concentration reached >12 µM. This implied that therapeutic doses of sunitinib may not massively damage the endothelium, because in vivo concentrations were estimated to be <10 µM (6,19). The decrease in the proliferation rate of HMEC-1 cells at low concentrations may be attributed to the blockage of endothelial cell growth factor receptors; however, it is also reasonable to suppose that the interruption of lysosome hydrolytic activity by sunitinib-induced basification also affects HMEC-1 metabolism and proliferation. This study evaluated the effect of two autophagy inhibitors on sunitinib-induced drug resistance. The results of fluorescence microscopy revealed that the H+-ATPase inhibitor BAF halted sunitinib sequestration, although the LAMP-1-positive lysosomal structure was still intact. Another autophagy inhibitor, CQ, is known to inhibit autophagosome-lysosome fusion and to induce an accumulation of autophagosomes (33). The present results demonstrated that CQ did not affect the sequestration of sunitinib in lysosomes, which is consistent with a previous result (33). Therefore, the difference in the observed effect could be adequately explained by differences in mechanism, in particular because sunitinib sequestration is highly dependent on the H+-ATPase-associated acidic environment, but much less dependent on the fusion of autophagosomes with lysosomes. Likewise, although both inhibitors suppressed cell resistance to sunitinib, BAF appeared more efficient than CQ. These results are in agreement with other reports using BAF and CQ in vitro (21-23,27). Overall, these data support the notions that inhibition of autophagic flux sensitizes cells to sunitinib and other drugs, and that lysosomal drug sequestration is one of the important underlying mechanisms (12–15,20–23,35). Notably, the anti-autophagy mechanism of CQ has not yet been fully understood, whereas its pharmaceutical potential has attracted much attention because it affects tumor cells and endothelial cells through complex mechanisms, including overcoming drug resistance (27,35–37).
Finally, this study evaluated the reversibility of HMEC6 µM resistance. The results revealed that resistance gradually disappeared after sunitinib had been removed from the culture media for ~2 weeks. This is in line with the 3-day observations in the video, which illustrate a slow decrease in sunitinib fluorescence during culture.
Antiangiogenic sunitinib was designed to target endothelial cells (4,38). Lysosomal sequestration of drugs, and its associated toxicity and drug resistance in endothelial cells, are of particular importance. Endothelial cells are exposed to the highest peak levels of sunitinib (10 µM, t1⁄2>40 h) following absorption by the digestive system and its penetration into the blood circulation (6,19). Therefore, the present results obtained with 6 µM sunitinib are relevant for clinical dosage, and sunitinib lysosomal sequestration can reasonably be supposed to occur in patients. These results raised several clinically relevant questions. For example, sunitinib neutralizes the acidic environment of lysosomes, which can lead to the impairment or cessation of normal intracellular metabolism, but at present little is known about the significance of a defective lysosome system of this sort in a clinical setting. Furthermore, the overall clinical interpretation of lysosomal sequestration of sunitinib also appears complex. It could be considered beneficial if it prevents the massive destruction of the endothelium by reducing toxicity, or on the contrary, it could be considered harmful if it reduces therapeutic efficacy by causing drug resistance. Further study is potentially required to clarify and understand this delicate balance, in order to improve daily clinical practice by better management of the dosage of sunitinib treatment, particularly in patients with cardiovascular comorbidity (39,40).
Sequestration-associated drug resistance is not restricted to sunitinib, as many drug compounds share similar physico-chemical properties (12–14). The present study indicated that the sensitizing effect of H+-ATPase blockers, such as BAF, appears to be a more attractive means for the development of novel drugs reversing sunitinib resistance. It is clear that tumors are able to develop drug resistance through adaptive mechanisms under chemotherapy. In particular, the upregulation of ABC transporter molecules simultaneously contributes to the development of drug resistance in endothelial cells and tumor cells (1–3). The blocking of ABCB1 and ATP binding cassette subfamily G member 2 by elacridar has demonstrated therapeutic efficacy (23). Notably, each anticancer drug has specific targets, specific physicochemical properties and distinct mechanisms, although they are often used interchangeably in clinical settings. Clinically, drug resistance is closely associated with drug efficacy and drug toxicity (7,8,11,39). In addition, there is growing interest in reviewing the role of stromal cells, including in blood vessels, in the development of resistance to anticancer drugs (9). The present study may be helpful in providing a better understanding of the pharmacological mechanisms of anticancer drugs, and may improve the clinical management of drug resistance and toxicity during therapy.
Supplementary Data
Acknowledgments
Not applicable.
Abbreviations
- CQ
chloroquine
- BAF
bafilomycin A1
- LAMP-1
lysosomal-associated membrane protein 1
- LC3
microtubule-associated protein 1A/1B-light chain 3
- SQSTM1
sequestosome 1
Funding
This study was supported by the China Scholarship Council. LH is supported by the Science and Technology Department of Guizhou Province (grant nos. J-2015-2088 and Qian-P-Ren-2017-5611), the Guiyang Municipal Science and Technology Bureau (grant nos. 20141001-62 and Zhukehetong-2017-5-1) and the National Natural Science Foundation of China (grant nos. 81660451, 31860325 and 31460312).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
SW, LH, ZH, HoL and HeL designed the study. SW, RS, MBC, PZ and CH participated in the experiments. MDB, AJ and GB participated in the analysis of the data. SW, LH and HL prepared the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Authors' information
Currently, RS works at the Department of Hematology of Ruijin Hospital School of Medicine, Shanghai Jiao Tong University. PZ works at North China University of Water Resources and Electric Power. CH works at The First Affiliated Hospital of Guizhou Medical University.
References
- 1.Huang L, Perrault C, Coelho-Martins J, Hu C, Dulong C, Varna M, Liu J, Jin J, Soria C, Cazin L, et al. Induction of acquired drug resistance in endothelial cells and its involvement in anticancer therapy. J Hematol Oncol. 2013;6:49. doi: 10.1186/1756-8722-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang L, Hu C, Di Benedetto M, Varin R, Liu J, Wang L, Vannier JP, Jin J, Janin A, Lu H, Li H. Induction of multiple drug resistance in HMEC-1 endothelial cells after long-term exposure to sunitinib. Onco Targets Ther. 2014;7:2249–2255. doi: 10.2147/OTT.S67251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang L, Hu C, Di Benedetto M, Varin R, Liu J, Jin J, Wang L, Vannier JP, Janin A, Lu H, Li H. Cross-drug resistance to sunitinib induced by doxorubicin in endothelial cells. Oncol Lett. 2015;9:1287–1292. doi: 10.3892/ol.2014.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naito H, Wakabayashi T, Kidoya H, Muramatsu F, Takara K, Eino D, Yamane K, Iba T, Takakura N. Endothelial side population cells contribute to tumor angiogenesis and antiangiogenic drug resistance. Cancer Res. 2016;76:3200–3210. doi: 10.1158/0008-5472.CAN-15-2998. [DOI] [PubMed] [Google Scholar]
- 5.Weis SM, Cheresh DA. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat Med. 2011;17:1359–1370. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 6.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmaco-dynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- 7.Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: Current status and future directions. Lancet. 2016;388:518–529. doi: 10.1016/S0140-6736(15)01088-0. [DOI] [PubMed] [Google Scholar]
- 8.Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S. Drug resistance in cancer: An overview. Cancers (Basel) 2014;6:1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huijbers EJ, van Beijnum JR, Thijssen VL, Sabrkhany S, Nowak-Sliwinska P, Griffioen AW. Role of the tumor stroma in resistance to anti-angiogenic therapy. Drug Resist Updat. 2016;25:26–37. doi: 10.1016/j.drup.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Gu Y, Lu H, Boisson-Vidal C, Li H, Bousquet G, Janin A, Di Benedetto M. Resistance to anti-angiogenic therapy: A clinical and scientific current issue. Med Sci (Paris) 2016;32:370–377. doi: 10.1051/medsci/20163204015. In French. [DOI] [PubMed] [Google Scholar]
- 11.Nunes T, Hamdan D, Leboeuf C, El Bouchtaoui M, Gapihan G, Nguyen TT, Meles S, Angeli E, Ratajczak P, Lu H, et al. Targeting cancer stem cells to overcome chemoresistance. Int J Mol Sci. 2018;19:E4036. doi: 10.3390/ijms19124036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacIntyre AC, Cutler DJ. The potential role of lysosomes in tissue distribution of weak bases. Biopharm Drug Dispos. 1988;9:513–526. doi: 10.1002/bod.2510090602. [DOI] [PubMed] [Google Scholar]
- 13.Duvvuri M, Krise JP. Intracellular drug sequestration events associated with the emergence of multidrug resistance: A mechanistic review. Front Biosci. 2005;10:1499–1509. doi: 10.2741/1634. [DOI] [PubMed] [Google Scholar]
- 14.Kazmi F, Hensley T, Pope C, Funk RS, Loewen GJ, Buckley DB, Parkinson A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells) Drug Metab Dispos. 2013;41:897–905. doi: 10.1124/dmd.112.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gotink KJ, Broxterman HJ, Labots M, de Haas RR, Dekker H, Honeywell RJ, Rudek MA, Beerepoot LV, Musters RJ, Jansen G, et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin Cancer Res. 2011;17:7337–7346. doi: 10.1158/1078-0432.CCR-11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamagishi T, Sahni S, Sharp DM, Arvind A, Jansson PJ, Richardson DR. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J Biol Chem. 2013;288:31761–31771. doi: 10.1074/jbc.M113.514091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhitomirsky B, Assaraf YG. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget. 2015;6:1143–1156. doi: 10.18632/oncotarget.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapuy B, Koch R, Radunski U, Corsham S, Cheong N, Inagaki N, Ban N, Wenzel D, Reinhardt D, Zapf A, et al. Intracellular ABC transporter A3 confers multidrug resistance in leukemia cells by lysosomal drug sequestration. Leukemia. 2008;22:1576–1586. doi: 10.1038/leu.2008.103. [DOI] [PubMed] [Google Scholar]
- 19.Gotink KJ, Broxterman HJ, Honeywell RJ, Dekker H, de Haas RR, Miles KM, Adelaiye R, Griffioen AW, Peters GJ, Pili R, Verheul HM. Acquired tumor cell resistance to sunitinib causes resistance in a HT-29 human colon cancer xenograft mouse model without affecting sunitinib biodistribution or the tumor microvasculature. Oncoscience. 2014;1:844–853. doi: 10.18632/oncoscience.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santoni M, Amantini C, Morelli MB, Liberati S, Farfariello V, Nabissi M, Bonfili L, Eleuteri AM, Mozzicafreddo M, Burattini L, et al. Pazopanib and sunitinib trigger autophagic and non-autophagic death of bladder tumour cells. Br J Cancer. 2013;109:1040–1050. doi: 10.1038/bjc.2013.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeda T, Ishii KA, Saito Y, Miura M, Otagiri A, Kawakami Y, Shimano H, Hara H, Takekoshi K. Inhibition of autophagy enhances sunitinib-induced cytotoxicity in rat pheochromocytoma PC12 cells. J Pharmacol Sci. 2013;121:67–73. doi: 10.1254/jphs.12158FP. [DOI] [PubMed] [Google Scholar]
- 22.Abdel-Aziz AK, Shouman S, El-Demerdash E, Elgendy M, Abdel-Naim AB. Chloroquine synergizes sunitinib cytotoxicity via modulating autophagic, apoptotic and angiogenic machineries. Chem Biol Interact. 2014;217:28–40. doi: 10.1016/j.cbi.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 23.Giuliano S, Cormerais Y, Dufies M, Grépin R, Colosetti P, Belaid A, Parola J, Martin A, Lacas-Gervais S, Mazure NM, et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy. 2015;11:1891–1904. doi: 10.1080/15548627.2015.1085742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 25.Turco E, Martens S. Insights into autophagosome biogenesis from in vitro reconstitutions. J Struct Biol. 2016;196:29–36. doi: 10.1016/j.jsb.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antonioli M, Di Rienzo M, Piacentini M, Fimia GM. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. 2017;42:28–41. doi: 10.1016/j.tibs.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: From pathophysiology to treatment. J Hematol Oncol. 2017;10:67. doi: 10.1186/s13045-017-0436-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pu J, Guardia CM, Keren-Kaplan T, Bonifacino JS. Mechanisms and functions of lysosome positioning. J Cell Sci. 2016;129:4329–4339. doi: 10.1242/jcs.196287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017;18:E1865. doi: 10.3390/ijms18091865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27:495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Gagliardi S, Rees M, Farina C. Chemistry and structure activity relationships of bafilomycin A1, a potent and selective inhibitor of the vacuolar H+-ATPase. Curr Med Chem. 1999;6:1197–1212. [PubMed] [Google Scholar]
- 33.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, Coppes RP, Engedal N, Mari M, Reggiori F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heuser J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol. 1989;108:855–864. doi: 10.1083/jcb.108.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–1836. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol Med. 2013;19:428–446. doi: 10.1016/j.molmed.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell. 2014;26:190–206. doi: 10.1016/j.ccr.2014.06.025. [DOI] [PubMed] [Google Scholar]
- 38.Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J, Vandenbeldt K, Qian CN, Teh BT. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 2010;70:1053–1062. doi: 10.1158/0008-5472.CAN-09-3722. [DOI] [PubMed] [Google Scholar]
- 39.Moslehi JJ. Cardiovascular toxic effects of targeted cancer therapies. N Engl J Med. 2016;375:1457–1467. doi: 10.1056/NEJMra1100265. [DOI] [PubMed] [Google Scholar]
- 40.Bretagne M, Boudou-Rouquette P, Huillard O, Thomas-Schoemann A, Chahwakilian A, Orvoen G, Arrondeau J, Tlemsani C, Cessot A, Cabanes L, et al. Tyrosine kinase inhibiting the VEGF pathway and elderly people: Tolerance, pre-treatment assessment and side effects management. Bull Cancer. 2016;103:259–272. doi: 10.1016/j.bulcan.2015.10.020. In French. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.