

Abstract
Background
Acute lymphoblastic leukemia (ALL) is the most common cancer in children and can arise in B or T lymphoid lineages. Although risk loci have been identified for B-ALL, the inherited basis of T-ALL is mostly unknown, with a particular paucity of genome-wide investigation of susceptibility variants in large patient cohorts.
Methods
We performed a genome-wide association study (GWAS) in 1191 children with T-ALL and 12 178 controls, with independent replication using 117 cases and 5518 controls. The associations were tested using an additive logistic regression model. Top risk variants were tested for effects on enhancer activity using luciferase assay. All statistical tests were two sided.
Results
A novel risk locus in the USP7 gene (rs74010351, odds ratio [OR] = 1.44, 95% confidence interval [CI] = 1.27 to 1.65, P = 4.51 × 10–8) reached genome-wide significance in the discovery cohort, with independent validation (OR = 1.51, 95% CI = 1.03 to 2.22, P = .04). The USP7 risk allele was overrepresented in individuals of African descent, thus contributing to the higher incidence of T-ALL in this race/ethnic group. Genetic changes in USP7 (germline variants or somatic mutations) were observed in 56.4% of T-ALL with TAL1 overexpression, statistically significantly higher than in any other subtypes. Functional analyses suggested this T-ALL risk allele is located in a putative cis-regulatory DNA element with negative effects on USP7 transcription. Finally, comprehensive comparison of 14 susceptibility loci in T- vs B-ALL pointed to distinctive etiology of these leukemias.
Conclusions
These findings indicate strong associations between inherited genetic variation and T-ALL susceptibility in children and shed new light on the molecular etiology of ALL, particularly commonalities and differences in the biology of the two major subtypes (B- vs T-ALL).
Acute lymphoblastic leukemia (ALL) is the most common malignancy in children and can arise in B or T lymphoid lineages (1). Comprising 15% of childhood and 25% of adult ALL, T-ALL is associated with more aggressive-presenting features (older age and higher initial leukocyte count) and historically inferior survival compared to B-ALL (2). Also notably, there is an overrepresentation of African Americans in children with T-ALL compared with those with B-ALL (3,4), and the basis for this racial disparity remains unknown. Molecular profiling studies show that T-ALL has a unique landscape of somatic genomic alterations largely absent in B-ALL (5–7). For example, a major class of oncogenes in T-ALL are transcription factors that are aberrantly expressed as a result of chromosomal rearrangement or enhancer mutations, TAL1, TAL2, LMO1, LMO2, MYC, and so forth (7). In fact, gene expression-based molecular subtypes of T-ALL also largely fall in line with major genomic abnormalities involving one of these transcription factors (5). Therefore, the etiology of B- vs T-ALL is likely distinctive, with separate molecular processes invoked during leukemogenesis.
Because B-ALL is much more common than T-ALL, genome-wide association studies (GWAS) of ALL susceptibility thus far have primarily focused on the former with a number of risk loci identified, including genes involved with hematopoiesis (eg, ARID5B, IKZF1, CEBPE, GATA3, and BMI1) and tumor suppressor pathways (eg, CDKN2A) (8–15). Some studies also examined the effects of these risk variants in small cohorts of T-ALL: for example, CDKN2A risk allele appears to influence both B- and T-ALL (16). However, there is a paucity of comprehensive investigation of susceptibility variants of T-ALL at a genome-wide level, and the inherited basis of this ALL subtype is poorly understood.
In this study, we performed a discovery GWAS in 1191 children with T-ALL and 12 178 controls to systematically identify novel genetic factors for T-ALL susceptibility in children, evaluate their associations with ALL clinical features and somatic genomics, and explore functional effects of these risk alleles.
Methods
Subjects and Samples
In the discovery GWAS, childhood T-ALL cases (n = 1191) were from the Children’s Oncology Group (COG) AALL0434 (NCT00408005) (17) and St. Jude Total XVI (NCT00549848) (18) clinical trials for newly diagnosed ALL. Non-ALL controls (n = 12 178) were unrelated subjects from the Health and Retirement Study (HRS; dbGaP: phs000428). The replication series included 117 children with T-ALL enrolled on the St. Jude Total Therapy XIIIA, XIIIB, and XV ALL protocols (NCI-T93-0101D and NCT00137111) (12) and 5518 non-ALL controls from the Multi-Ethnic Study of Atherosclerosis (MESA) study (dbGAP phs000209.v9) (11). Demographic and clinical features of T-ALL cases included in this study are summarized in Supplementary Table 1 (available online). To estimate effects of known risk variants on susceptibility to B-ALL, we included childhood B-ALL cases (n = 1824) enrolled in COG P9904/9905/9906 (NCT00005585/NCT00005596/NCT00005603) (19) with the MESA cohort as non-ALL controls. Genetic ancestry (European, African, East Asian, and Native American) was determined by using ADMIXTURE (version 1.3.0) (20), with the sum of these four ancestries being 100% for any given subject. European American (EA), African American (AA), and Asian were defined as having more than 95% European genetic ancestry, more than 70% African ancestry, and more than 90% Asian ancestry, respectively. Hispanics were individuals for whom Native American ancestry was more than 10% and greater than African ancestry, as previously described (12). To further characterize population structure within T-ALL cases and controls in the GWAS, we also performed principal components analysis (PCA) by applying EIGENSTRAT (21) to all samples and single-nucleotide polymorphisms (SNPs) that passed quality control above. The top three principal components (PCs) explained 6.8% of total genetic variation: PC1 with 6.2%, PC2 with 0.5%, and PC3 with 0.1%, corresponding to African, Asian, and Native American genetic ancestry, respectively. This study was approved by the respective institutional review boards and informed consent was obtained from parents, guardians, or patients, as appropriate.
Genotyping, Imputation, and Quality Control
SNP genotyping was performed in germline DNA using the Infinium Omni2.5Exome BeadChip from Illumina (San Diego, CA; COG AALL0434) (17), the Affymetrix Human SNP Array 6.0 (St. Jude Total XVI, COG P9904/9905) (12), the Affymetrix GeneChip Human Mapping 500 K Array (St. Jude Total XIIIB/XV and COG P9906) (11,12), or the Axiom Genome-Wide KP UCSF Array (St. Jude Total XIIIA). For non-ALL controls, the HRS and the MESA cohorts were genotyped using the Illumina Omni2.5 and Affymetrix SNP 6.0 arrays, respectively. Genotype calls were determined as described previously (12). The sample quality control procedures were performed on the basis of SNP call rate and minor allele frequency (Supplementary Figure 3, available online). Individuals with one of the following features were excluded: discordant sex; genotype failure rate no less than .05; heterozygosity rate no less than 5 SD from the mean; heterozygosity rate no less than 3 SD from the mean and genotype failure rate no less than .03; identity-by-descent score greater than .185 and lower call rate than other individuals.
Using a large reference panel of human haplotypes from the Haplotype Reference Consortium (HRC r1.1 2016) (22) in Michigan Imputation Server (22,23) with ShapeIT (v2.r790) (24) as the phasing tool, we imputed additional SNPs genome-wide for all qualified cases and controls. The SNPs used in the GWAS were filtered on the basis of imputation quality, allele frequency, call rate, and deviation from Hardy-Weinberg equilibrium (HWE). Specifically, SNPs with one of the following features were excluded: imputation quality metric R2 less than .3 (indicating inadequate accuracy of the imputed genotype); minor allele frequency in cases and controls less than .01; HWE P less than 5.00 × 10–4 in cases and controls classified as EA; and differences in allele frequency in two non-ALL cohorts (HRS vs MESA) with P less than .001 by logistic regression test adjusting genetic ancestry.
Because original genotyping was performed using different SNP arrays for different cohorts, we also compared the imputation results between cohorts to rule out any bias arising from differences in array platforms. To this end, we imputed SNPs, calculated the allele frequency for each SNP in each cohort genotyped on a different array, and determined the correlation coefficient in allele frequency of these 30 000 imputed SNPs between any two arrays (eg, Illumina Omni2.5 Exome vs Affymetrix SNP 6.0; Affymetrix SNP 6.0 vs Affymetrix Axiom UCSF Array; and Illumina Omni2.5 Exome vs Affymetrix Axiom UCSF Array). We have conducted this analysis separately for T-ALL cases and controls and observed an average of 0.98 correlation across genotyping platforms (Supplementary Figure 4, available online). We therefore believe that differences in genotyping chemistry and array platform had minimal impact on imputation and were unlikely to influence our GWAS.
Genome-Wide Association and Statistical Analyses
In the discovery GWAS, the association between each SNP and ALL susceptibility was tested by comparing the genotype frequencies between ALL cases and non-ALL controls with a logistic regression model in PLINK (version 1.90) (25), in which genetic ancestry (European, African, and Native American) was included as continuous covariates to control for population stratification. Under the additive model, allelic dosages were used to associate with T-ALL status. Quantile-quantile plots indicated minimal inflation at the tail of the distribution (λ = 1.07, Supplementary Figure 5, available online), but hits from the discovery GWAS were subjected to replication to rule out potential spurious findings arising from population stratification. In the replication studies, we tested the USP7 variants using the additive logistic regression model with genetic ancestry included as covariates. Regional plots were illustrated through LocusZoom (26) with the structure of linkage equilibrium at USP7 locus based on 1000 Genomes Nov 2014 EUR (hg19). We also tested the USP7 SNP genotype for association with T-ALL in the discovery cohort with PC1, PC2, and PC3 as covariate, and the results were largely identical to analyses in which genetic ancestries were used as covariates for population structure (Supplementary Table 2, available online).
The differences in relative luciferase activity between DNA sequences with different USP7 genotypes were tested by using Student t test. The association of USP7 status with T-ALL subgroups was evaluated by Fisher exact test; the correlation between USP7 genotype and genetic ancestry was examined by general regression test; SNP genotype associations with patient sex, age at diagnosis (≥ or <10 years of age) were evaluated by logistic regression test, adjusting for genetic ancestry. Somatic lesions in leukemia blasts were ascertained as described previously (5), and T-ALL subtypes were defined by integrated analyses on the basis of leukemia gene expression profile as well as somatic genomic abnormalities (5). R (version 3.3.3) statistical software was used for all analyses unless indicated otherwise. All statistical tests were two sided and P < .05 was used as the cut point for statistical significance unless indicated otherwise.
Luciferase Reporter Assay for USP7 Variants
The sequences flanking GWAS top hits were cloned into pGL4.23 [luc2/miniP] luciferase vector (Promega; Fitchburg, WI). Site directed mutagenesis was performed to generate plasmids containing each risk allele (QuikChange II Site-Directed Mutagenesis Kit, Agilent Technologies, Santa Clara, CA). Detailed information including primers and cloned regions can be found in Supplementary Table 3 (available online). For luciferase assay, 5 × 106 Jurkat cells cultured in 12-well plates were transiently transfected with 6 µg of pGL4.23 construct (luciferase gene with USP7 intronic sequences spanning GWAS hit SNPs for each allele) and 1 µg of pGL-TK (Renilla luciferase) using Lonza Cell Line Nucleofector Kit V (Lonza; Basel, Switzerland). Firefly luciferase activity was measured 24 hours post-transfection and normalized to Renilla luciferase activity. Relative luciferase activity indicates the ratio over the value from pGL4.23 vector alone. All experiments were performed in triplicate and repeated three times for rs74010351.
Results
We imputed SNP genotypes genome-wide, and after quality control, evaluated the association between genotype and T-ALL for 7 967 910 variants using an additive logistic regression model. Genetic ancestry was adjusted to control for population structure. Two loci reached genome-wide significance (ie, P < 5.00 × 10–8) (Figure 1 and Supplementary Table 1, available online): CDKN2A at 9q21.3 and USP7 at 16p13.3. The CDKN2A variants (eg, rs2188127, odds ratio [OR] = 0.65, 95% confidence interval [CI] = 0.55 to 0.76, P = 5.00 × 10–8) were previously linked to B-ALL susceptibility with similar effects on T-ALL (16). Encoding human ubiquitin-specific protease 7, USP7 is somatically mutated in approximately 12% of children and young adults with T-ALL (5,27). A cluster of USP7 variants (eg, rs74010351, OR = 1.44, 95% CI = 1.27 to 1.65, P = 4.51 × 10–8) (Table 1 and Figure 2A) in its intronic region showed genome-wide statistically significant associations. In an independent replication cohort of 117 T-ALL patients from the St. Jude Total Therapy XIIIA, XIIIB, and XV cohorts and 5518 non-ALL controls from the MESA cohort, the association was confirmed for the same USP7 variant (OR = 1.51, 95% CI = 1.03 to 2.22, P = .04) (Table 1). Multivariable analyses in the discovery cohort conditioning on this SNP did not provide any evidence for additional variants with independent association at this locus (Supplementary Figure 1, available online), although our sample size is limited to evaluate the full spectrum of causal variants in USP7.
Figure 1.

Genome-wide association study of childhood T-ALL. The association between genotype and ALL was evaluated by using a logistic regression model for 7 967 910 genotyped/imputed single-nucleotide polymorphisms (SNPs) in 1191 T-ALL cases and 12 178 unrelated non-ALL controls. P values (−log10P) were plotted against the respective chromosomal position of each SNP. Gene symbols are indicated for two loci achieving genome-wide significance (two-sided P < 5 × 10–8, dashed black horizontal line): CDKN2A (9p21) and USP7 (16p13.3). T-ALL = T-cell acute lymphoblastic leukemia.
Table 1.
Association of USP7 variants with T-ALL susceptibility in the discovery GWAS and replication cohorts*
| rs74010351 (USP7) |
European American RAF |
African American RAF |
All ethnicities |
|||
|---|---|---|---|---|---|---|
| P† | OR‡ (95% CI) | |||||
| Case | Control | Case | Control | |||
|
Discovery GWAS cohort (n = 1191 cases and 12 178 controls) |
0.10 | 0.07 | 0.26 | 0.19 | 4.51 × 10−8 | 1.44 (1.27 to1.65) |
|
Replication cohort (n = 117 cases and 5518 controls) |
0.11 | 0.06 | 0.23 | 0.17 | 0.04 | 1.51 (1.03 to 2.22) |
Ethnicity was defined by single-nucleotide polymorphism genotype-based European, African, East Asian, and Native American genetic ancestry. CI = confidence interval; GWAS = genome-wide association study; OR = odds ratio; RAF = risk allele frequency; T-ALL = T-cell acute lymphoblastic leukemia.
P values were calculated by a two-sided logistic regression test in PLINK.
Individuals who do not carry the risk allele were considered as the reference group.
Figure 2.

Functional annotation and activity screen of regulatory DNA at the USP7 locus. A) Functional annotation of regulatory DNA at the USP7 locus. Genomic positions and scale for the human genome assembly February 2009 (GRCh37/hg19) are shown on the top. Genome-wide statistically significant T-ALL risk variants in USP7 (P < 5 × 10–8) are marked in the middle panel, and the log-transformed P values (for association with T-ALL) are shown in the bed graph. The gene structure, ChIP-seq signals for histone modifications (ie, H3K4me3, H3K27ac, and H3K79me2) in T-ALL cell lines (ie, DND41 and Jurkat) (28), and ATAC-seq signals of hematopoietic cells (29) are also included. B) Functional activity screen of cis-elements with different genotypes/haplotypes by luciferase reporter assay in Jurkat cells. Jurkat cells were transiently transfected with pGL4.23 [luc2/minP] construct (luciferase gene with USP7 intronic sequences spanning GWAS hit SNPs for each allele) and pGL-TK (Renilla luciferase). Firefly luciferase activity was measured 24 hours post-transfection and normalized to Renilla luciferase activity. Relative luciferase activity indicates the ratio over the value from pGL4.23 vector alone. All experiments were performed in triplicate, and repeated three times for rs74010351. Statistical significance was evaluated by using two-sided Student's t test. Error bars indicate SD. SNP = single-nucleotide polymorphisms; T-ALL = T-cell acute lymphoblastic leukemia; WT = wild-type.
To explore functional effects of risk variants at the USP7 locus, we first examined lineage-specific chromatin accessibility inferred from the ATAC-seq of human hematopoietic cells and also histone modification marks in human T-ALL cell lines (ie, DND41 and Jurkat) (28–30). Of the eight USP7 SNPs that rose to genome-wide significance, four clustered close to the USP7 transcription start site (ie, rs61426394, rs74010349, rs59591814, and rs74010351), and this region is marked by strong promoter-active histone modifications H3K4me3 and H3K27ac in both T-ALL cell lines and also overlapped with multiple open chromatin segments within this region (Figure 2A and Supplementary Figure 2, available online). In fact, the rs74010351 variant aligned exactly within an open chromatin signal specific to T and B lymphocytes (Figure 2A and Supplementary Figure 2, available online). In contrast, the other four genome-wide statistically significant USP7 variants (ie, rs113177770, rs75172776, rs7202585, and rs16964691) were located in the intronic region with minimal overlap with regulatory elements identified from ATAC-seq or by histone marks. Taken together, we thus postulated that the association of the USP7 risk variant with T-ALL susceptibility might be mediated by its effects on USP7 expression. We performed luciferase assays to test the effects of each of the eight USP7 risk alleles on transcription regulation. As shown in Figure 2B, the rs74010351 variant exhibited the most statistically significant difference in reporter gene transcription between the reference and risk alleles in this assay. The substitution of the wild-type allele A with the risk allele G at this SNP resulted in a 2.9-fold decrease in transcription activity (P = .03) (Figure 2B), in line with the predicted regulatory DNA function of this segment.
USP7 is a ubiquitin-specific protease in the deubiquitinating enzyme family, and loss-of-function somatic mutations in USP7 are observed recurrently in pediatric and young adult T-ALL (∼12%) (5). In a subset of 190 T-ALL in our cohort with both somatic and germline genomic data available (5), we comprehensively evaluated the relationship between USP7 SNP genotype (rs74010351) and leukemia genomic abnormalities. The prevalence of the USP7 risk allele varied substantially across T-ALL molecular subtypes (defined by leukemia gene expression profile and somatic genomic lesions [5]), with a higher frequency in cases with TAL1 or HOXA deregulation and lower in cases with TLX1 or TLX3 aberrations (Figure 3A). Interestingly, within the TAL1 subtype, germline and somatic USP7 variations were almost mutually exclusive (P = .03) (Figure 3A). In fact, the frequency of either type of USP7 variation approached 56.4% in TAL1 cases, statistically significantly higher than any other T-ALL subtypes (P = 9.25 × 10–5) (Figure 3A). We next evaluated the relationship between germline risk genotype of USP7 and clinical features of T-ALL in the COG AALL0434 cohort, which represented an unselected childhood T-ALL population from North America across risk and demographic groups (n = 1092). We found that the USP7 risk allele G at rs74010351 was associated with higher levels of African ancestry (P = .002) (Figure 3B) and older age at diagnosis (P = .007) (Figure 3C), but was not related to sex.
Figure 3.

Relationship of USP7 genotype and/or somatic mutation with T-ALL subgroups and clinical features. A) This analysis was restricted to 190 T-ALL children in the COG AALL0434 cohort with both germline and somatic genomic data available. Proportion of T-ALL patients with USP7 risk genotype at rs74010351 and/or USP7 somatic mutation was calculated within each T-ALL subgroup with at least 10 cases (TLX3, TLX1, LMO2/LYL1, HOXA, TAL1, and T-other) (5). The association of USP7 status with T-ALL subgroups was evaluated by Fisher exact test. B and C) The analysis was restricted to patients with T-ALL in the COG AALL0434, representing a largely unselected and nationwide patient population. The correlation between USP7 genotype and genetic ancestry was tested by generalized linear regression test. The association of USP7 genotype at rs74010351 with age at diagnosis was evaluated by logistic regression test (ie, age < 10y vs age ≥ 10y) adjusting for genetic ancestry. All statistical tests were two sided. T-ALL = T-cell acute lymphoblastic leukemia.
Finally, we compared the effects of all known ALL risk variants on the susceptibility to T-ALL (1191 cases and 12 178 controls from discovery GWAS) vs to B-ALL (1824 cases from the COG P9900 cohort and 5518 controls). Of 14 risk loci, eight (ie, IKZF1, TP63, SP4, GATA3, LHPP, ELK3, CEBPE, and 2q22.3) were statistically significant only for B-ALL risk and one was specific for T-ALL (USP7). Risk variants at CDKN2A/B, ARID5B, IKZF3, PIP4K2A, and 8q24.21 were statistically significantly associated with susceptibility both to B- and T-ALL, of which 8q24.21 and PIP4K2A showed comparable effect sizes across lineages and ARID5B had greater effects on B-ALL (Figure 4).
Figure 4.
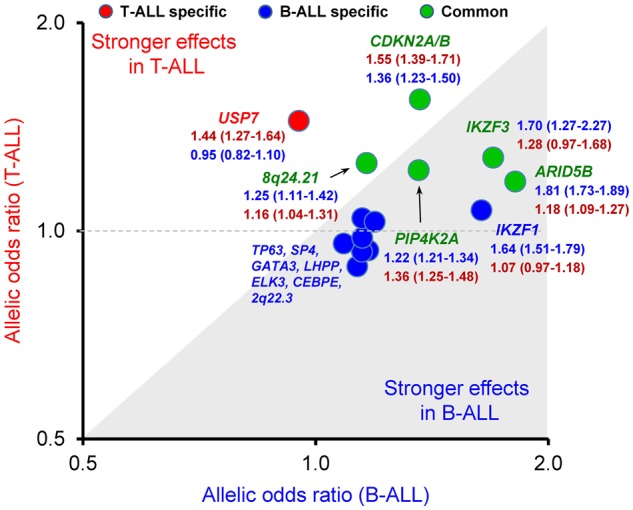
Similarities and differences in genetic predisposition to ALL between B and T lineage. The association between genotype and ALL was evaluated by using a logistic regression model for GWAS hits reported previously in B-ALL (ie, ARID5B [rs10821936], IKZF1 [rs11978267], CDKN2A/B [rs11978267], IKZF3 [rs17607816], PIP4K2A [rs7088318], 8q24.21 [rs28665337], TP63 [rs17505102], SP4 [rs2390536], GATA3 [rs3824662], LHPP [rs35837782], ELK3 [rs4762284], CEPBE [rs2239633], and 2q22.3 [rs17481869]) or newly identified in T-ALL (ie, USP7 [rs74010351]) in 1191 T-ALL cases (COG AALL0434 and St. Jude Total Therapy XVI protocols) vs 12 178 unrelated non-ALL controls (HRS), and 1824 B-ALL cases (COG P9904/9905/9906) vs 5518 unrelated non-ALL controls (MESA). The locus specifically statistically significant (P < .05) in T-ALL is marked in red, with B-ALL marked in blue, and common marked in green. ALL = acute lymphoblastic leukemia; COG = Children’s Oncology Group; HRS = Health and Retire Study; MESA = Multi-Ethnic Study of Atherosclerosis.
Discussion
In summary, we have performed the first T-ALL susceptibility GWAS and identified USP7 as a novel risk locus for this type of leukemia. These findings provide new insight into the molecular etiology of ALL in children, particularly some commonalities and differences in the biology of the two major subtypes (B- vs T-ALL). The higher prevalence of the USP7 risk allele in Africans points to a genetic basis for the increased incidence of T-ALL in African Americans, an observation hitherto without a plausible explanation. Future comprehensive functional investigations are warranted to pinpoint the causal variants and to fully understand the contribution of USP7 to ALL pathogenesis.
As a deubiquitinase, USP7 modulates the stability of a multitude of human transcription factors, tumor suppressors (eg, TP53, FOXO4, and PTEN), and epigenetic regulators (eg, DNMT1 and PRC1), and has been implicated in several solid malignancies (31,32). In leukemia, USP7 somatic mutations are exclusively restricted to T-ALL and almost always frameshift or nonsense changes that result in the truncation of the catalytic domains, suggesting that the loss of USP7 function contributes to leukemogenesis in this context. The co-segregation of USP7 mutations with TAL1 deregulation in leukemia cells also points to potential cooperating effects to promote leukemia transformation. Remarkably, the relationship between germline USP7 risk allele and ALL mirrors that of somatic mutations in this gene: uniquely associated with T-ALL susceptibility with little effect on B-ALL, and overrepresentation in the TAL1 subtype of T-ALL. Because germline and somatic USP7 variations co-occur only minimally in T-ALL, we stipulate they both impair USP7 function although somatic mutations are likely to have much more severe effects. Therefore, these results argue that USP7 is a tumor suppressor in T-ALL. However, a recent report showed that USP7 directly regulates NOTCH1 stability and its intact activity might be important for the oncogenic effects of NOTCH1 complex, suggesting a leukemogenic effect of USP7 (33). In fact, small molecule inhibitor of USP7 potently inhibited T-ALL cell growth (33). Future mechanistic studies are warranted to reconcile these seemingly conflicting observations and to understand the exact mechanism by which USP7 influences the pathogenesis of T-ALL.
Given the differences in the landscape of somatic genomic abnormalities in T- vs B-ALL, it is not completely surprising that germline variants also have lineage-specific effects on ALL susceptibility. For example, IKZF1 is a key modulator of B-cell development with germline variants that directly result in severe immune deficiency and familial B-ALL (34). Consistently, common variants in IKZF1 have a much stronger effect in B-ALL than in T-ALL, which is also true for leukemia risk variants in a number of other transcription factor risk genes (eg, GATA3, ARID5B, and CEBPE). In contrast, ALL risk genes in the tumor suppressor class are more likely to have lineage-independent effects on leukemia susceptibility (eg, CDKN2A/B and MYC [signified by variants in a cis distal enhancer region 8q24.21]). However, it should be noted that the sample size for the T-ALL susceptibility association analysis is statistically significantly smaller than B-ALL, which plausibly could have contributed to the difference in effect sizes of the risk variants.
There are a number of limitations of this study. For example, the sample size of the discovery GWAS is still relatively small compared with similar studies of B-ALL, and therefore risk variants with modest effects on T-ALL susceptibility would be unlikely to emerge above the genome-wide significance threshold in this study. Furthermore, there is considerable molecular heterogeneity within T-ALL, and therefore risk variants with subtype-specific effects could have been missed in our GWAS. These limitation can be overcome in future GWAS of larger cohorts of T-ALL.
In conclusion, we have identified a novel T-ALL risk gene USP7, and our results suggest that loss-of-function variants at this locus (either somatic or germline) promote T-ALL leukemogenesis. These data shed new light on the etiology of this subtype of childhood ALL but also beg for future mechanistic studies to characterize the molecular processes linking USP7 to normal and malignant hematopoiesis in the T lineage.
Funding
This work was partly supported by National Institutes of Health grants P50 GM115279, CA156449, CA21765, CA36401, CA98543, CA114766, CA98413, CA140729, CA176063, CA180886, CA196173, CA180899, GM92666, and HHSN261200800001E, and the American Lebanese Syrian Associated Charities.
Notes
Affiliations of authors: Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN (MQ, XZ, WY, YG, CS, YL, WEE, MVR, JJY); Children’s Hospital and Institutes of Biomedical Sciences, Fudan University, Shanghai, China (MQ, XZ); Department of Biostatistics, College of Medicine, Public Health and Health Professions, University of Florida, Gainesville, FL (MD); Institute for Genomic Medicine, Nationwide Children’s Hospital, and Departments of Pathology and Pediatrics, Ohio State University, Columbus, OH (JMG-F); Department of Laboratory Medicine, Precision Medicine Center, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, China (HX, SZ); Department of Global Pediatric Medicine, St. Jude Children’s Research Hospital, Memphis, TN (SJ); Hematological Malignancies Program, St. Jude Children’s Research Hospital, Memphis, TN (SJ, C-HP, WEE, MVR, JJY); Cancer Science Institute of Singapore, National University of Singapore, Singapore (TS); Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore (TS); Children’s Minnesota Research Institute, Children’s Minnesota, Minneapolis, MN (SSW); Children’s Hematology and Oncology, Carilion Clinic, Roanoke, VA (KPD); Division of Pediatric Hematology and Oncology, New York University Langone Health, New York, NY (EAR, WLC); Division of Pediatric Hematology and Oncology, University of Texas Southwestern Medical Center, Dallas, TX (NJW); Texas Children’s Cancer and Hematology Centers, Baylor College of Medicine, Houston, TX (KRR); Immunogen, Inc, Waltham, MA (PAZ-M); Department of Laboratory Medicine and Division of Hematopathology, University of Washington, Seattle, WA (BW); Department of Oncology, St. Jude Children’s Research Hospital, Memphis, TN (C-HP, JJY); Department of Pediatrics and Center for Childhood Cancer Research, Children’s Hospital of Philadelphia and the Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA (SPH); Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN (CGM); Department of Pediatrics, Benioff Children’s Hospital and the Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, CA (MLL).
MLL is the UCSF Benioff Chair of Children’s Health and Deborah and Arthur Ablin Endowed Chair in Pediatric Molecular Oncology. EAR is a KiDS of New York University Foundation Professor at New York University Langone Health. SPH is the Jeffrey E. Perelman Distinguished Chair in the Department of Pediatrics at the Children’s Hospital of Philadelphia. The other authors have no conflicts of interest to declare.
JJY is the principal investigator of this study, has full access to all the study data, and takes responsibility for the integrity of the data and the accuracy of the data analysis. MQ and WY performed data analysis; MQ and JJY wrote the manuscript; XZ, MD, CS, YG, JMG-F, YL, HX, SZ, SJ, TS, SSW, KPD, EAR, WLC, NJW, KRR, PAZ-M, and BW contributed reagents, materials, and/or data; MQ, WY, C-HP, WEE, SPH, XZ, CGM, MVR, MLL, and JJY interpreted the data and the research findings. All the co-authors reviewed the manuscript.
The study sponsors were not directly involved in the design of the study, the collection, analysis, and interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript. The authors thank the patients and parents who participated in the clinical protocols included in this study and the clinicians and research staff at participating institutions.
Supplementary Material
Reference
- 1. Linabery AM, Ross JA.. Trends in childhood cancer incidence in the U.S. (1992-2004). Cancer. 2008;112(2):416–432. [DOI] [PubMed] [Google Scholar]
- 2. Hunger SP, Mullighan CG.. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373(16):1541–1552. [DOI] [PubMed] [Google Scholar]
- 3. Pui CH, Sandlund JT, Pei D, et al. Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA. 2003;290(15):2001–2007. [DOI] [PubMed] [Google Scholar]
- 4. Pui CH, Behm FG, Singh B, et al. Heterogeneity of presenting features and their relation to treatment outcome in 120 children with T-cell acute lymphoblastic leukemia. Blood. 1990;75(1):174–179. [PubMed] [Google Scholar]
- 5. Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Girardi T, Vicente C, Cools J, et al. The genetics and molecular biology of T-ALL. Blood. 2017;129(9):1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belver L, Ferrando A.. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016;16(8):494–507. [DOI] [PubMed] [Google Scholar]
- 8. Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trevino LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Migliorini G, Fiege B, Hosking FJ, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013;122(19):3298–3307. [DOI] [PubMed] [Google Scholar]
- 11. Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45(12):1494–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu H, Yang W, Perez-Andreu V, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105(10):733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moriyama T, Relling MV, Yang JJ.. Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood. 2015;125(26):3988–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vijayakrishnan J, Studd J, Broderick P, et al. Genome-wide association study identifies susceptibility loci for B-cell childhood acute lymphoblastic leukemia. Nat Commun. 2018;9(1):1340.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wiemels JL, Walsh KM, de Smith AJ, et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat Commun. 2018;9(1):286.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sherborne AL, Hosking FJ, Prasad RB, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010;42(6):492–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Winter SS, Dunsmore KP, Devidas M, et al. Improved survival for children and young adults with T-lineage acute lymphoblastic leukemia: results from the Children's Oncology Group AALL0434 methotrexate randomization. J Clin Oncol. 2018;36(29):2926–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fernandez CA, Smith C, Yang W, et al. HLA-DRB1*07: 01 is associated with a higher risk of asparaginase allergies. Blood. 2014;124(8):1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang JJ, Cheng C, Devidas M, et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet. 2011;43(3):237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexander DH, Novembre J, Lange K.. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Patterson N, Price AL, Reich D.. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64 976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Das S, Forer L, Schönherr S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delaneau O, Marchini J, Zagury JF.. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;9(2):179–181. [DOI] [PubMed] [Google Scholar]
- 25. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li M, Chen D, Shiloh A, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416(6881):648–653. [DOI] [PubMed] [Google Scholar]
- 28. Mansour MR, Abraham BJ, Anders L, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346(6215):1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Corces MR, Buenrostro JD, Wu B, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48(10):1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanda T, Lawton LN, Barrasa MI, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nicholson B, Suresh Kumar KG.. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60(1–2):61–68. [DOI] [PubMed] [Google Scholar]
- 32. Salmena L, Pandolfi PP.. Changing venues for tumour suppression: balancing destruction and localization by monoubiquitylation. Nat Rev Cancer. 2007;7(6):409–413. [DOI] [PubMed] [Google Scholar]
- 33. Jin Q, Martinez CA, Arcipowski KM, et al. USP7 cooperates with NOTCH1 to drive the Oncogenic Transcriptional Program in T-Cell leukemia. Clin Cancer Res. 2019;25(1):222–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Churchman ML, Qian M, Te Kronnie G, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell. 2018; 33(5):937–948. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.