

SUMMARY
CRISPR-Cas9 systems provide powerful tools for genome editing. However, optimal employment of this technology will require control of Cas9 activity so that the timing, tissue specificity, and accuracy of editing may be precisely modulated. Anti-CRISPR proteins, which are small, naturally occurring inhibitors of CRISPR-Cas systems, are well suited for this purpose. A number of anti-CRISPR proteins have been shown to potently inhibit subgroups of CRISPR-Cas9 systems, but their maximal inhibitory activity is generally restricted to specific Cas9 homologs. Since Cas9 homologs vary in important properties, differing Cas9s may be optimal for particular genome-editing applications. To facilitate the practical exploitation of multiple Cas9 homologs, here we identify one anti-CRISPR, called AcrIIA5, that potently inhibits nine diverse type II-A and type II-C Cas9 homologs, including those currently used for genome editing. We show that the activity of AcrIIA5 results in partial in vivo cleavage of a single-guide RNA (sgRNA), suggesting that its mechanism involves RNA interaction.
Graphical Abstract
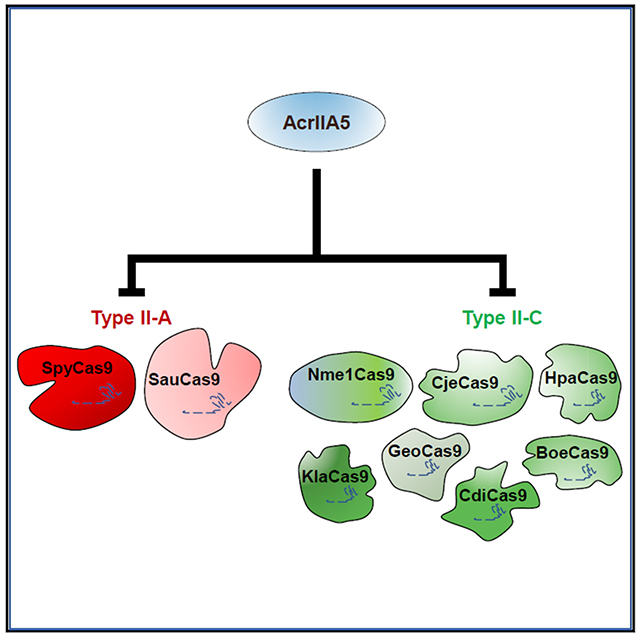
In Brief
Garcia et al. show that anti-CRISPR protein AcrIIA5 strongly inhibits all of the CRISPR-Cas9 homologs that are commonly used for genome editing. They show that it functions effectively in bacterial and mammalian cells. This anti-CRISPR will be useful for a wide variety of biotechnological applications.
INTRODUCTION
CRISPR-Cas9 systems combine a single effector protein, Cas9, with a single-guide RNA (sgRNA) molecule to target specific DNA sequences for precise genome manipulation. Their ability to program these systems to target any desired DNA sequence has led to their widespread usage for creating genomic knockouts and knockins, editing single bases, and gene activation and silencing (Doudna and Charpentier, 2014; Hess et al., 2017; Komor et al., 2017). However, there are concerns about the ability to safely and effectively control this technology, particularly in the case of applications like gene drives (Baltimore et al., 2015; Gantz and Bier, 2015; Hammond et al., 2016).
One mechanism by which CRISPR-Cas9 activity can be controlled is through the use of small, naturally occurring protein inhibitors known as anti-CRISPRs (Borges et al., 2017; Pawluk et al., 2018). These proteins have been shown to function as off switches for CRISPR-Cas9 genome editing in human cells (Lee et al., 2018; Pawluk et al., 2016; Rauch et al., 2017; Shin et al., 2017). They have also been used to control gene activation (CRISPRa) and gene interference (CRISPRi) in yeast and mammalian cells (Nakamura et al., 2019) and to decrease the toxicity of CRISPR-Cas9 delivered by an adenovirus vector to human stem cells (Li et al., 2018). Since the methods of in vivo delivery for CRISPR-Cas9, which include viral vectors and nano-particles, do not have high tissue specificity, it is crucial to avoid editing in non-targeted tissues, which would increase the risk of unwanted side effects (Cox et al., 2015). Recently, a Cas9-ON switch based on microRNA-dependent expression of an anti-CRISPR protein was used to control gene editing in a cell-specific manner (Hoffmann et al., 2019), including in the tissues of adult mice in vivo (Lee et al., 2019). These applications of anti-CRISPRs are varied, and their potential for further development is enormous.
While many different Cas9 proteins exist in nature, only a few are commonly used for genome engineering applications. These include the type II-A Cas9 proteins derived from Streptococcus pyogenes (SpyCas9) and Staphylococcus aureus (SauCas9) (Colella et al., 2017; Ran et al., 2015) and the type II-C Cas9 proteins from Neisseria meningitidis (Nme1Cas9) and Campylobacter jejuni (CjeCas9) (Ibraheim et al., 2018; Kim et al., 2017; Lee et al., 2016; Mir et al., 2018b; Zhang et al., 2015). These Cas9 homologs vary in features such as protospacer adjacent motif (PAM) specificity, size, and off-target activity, which makes each more or less advantageous for particular genome-editing applications. Anti-CRISPRs that target some of these Cas9 proteins have been identified (Harrington et al., 2017; Hynes et al., 2017; Pawluk et al., 2016; Rauch et al., 2017), but none of these efficiently inhibit all of them. The identification of a well-characterized, universal anti-CRISPR protein that could function to control Cas9 activity in a variety of different applications—including genome editing, gene drives, and CRISPRi/CRISPRa—would have broad utility and could hasten the development of these technologies. Thus, the goal of this work was to identify an anti-CRISPR with broad and potent activity.
In this study, we investigated the spectra of inhibition of a variety of previously described anti-CRISPRs that showed activity against type II-A (Hynes et al., 2018, 2017; Rauch et al., 2017; Uribe et al., 2019) and type II-C (Mir et al., 2018b; Pawluk et al., 2016) CRISPR-Cas9 systems using an efficient E. coli phage-based assay system. We discovered that the previously identifed anti-CRISPR, AcrIIA5 from Streptococcus thermophilus phage D4276 (Hynes et al., 2017), has the broadest Cas9 inhibitory activity described to date, inhibiting all of the Cas9 proteins commonly used in genome-editing applications.
RESULTS
AcrIIA5 Inhibits a Broad Spectrum of Cas9 Proteins
A key initial goal of this work was to develop a system to identify anti-CRISPRs with the broadest possible spectrum of activity for use in Cas9-based technologies. To quantitatively compare the specificity profiles of a large number of anti-CRISPR proteins, we expanded upon a previously described phage-targeting assay in which Cas9 from Geobacillus stearothermophilus (GeoCas9) was engineered to prevent infection by E. coli phage Mu (Harrington et al., 2017). In this assay, GeoCas9 was co-expressed with an sgRNA that targets phage Mu and prevents its replication by cleaving its genome. In the current work, we expressed a diverse group of Cas9 homologs (Figures 1A and 1B) in E. coli, each engineered to target phage Mu. These Cas9 homologs include those commonly used in genome-editing applications, including SpyCas9, SauCas9, CjeCas9, and Nme1Cas9. We also chose six additional Cas9 homologs distributed across the phylogeny of Cas9s occurring in bacteria (Figure 1A). All three subtypes (II-A, II-B, and II-C) were represented among these Cas9s, which range in pairwise sequence identity from 19% to 66% and utilize a variety of PAM sequences (Figure 1B).
Figure 1. AcrIIA5 Displays a Broad Spectrum of Activity against Cas9 Proteins.

(A) Phylogenetic tree of a non-redundant dataset of Cas9 proteins with <90% sequence identity. Cas9 homologs tested for AcrIIA5 inhibitory activity are indicated at the ends of branches on the tree. Clades are colored by Cas9 subtype.
(B) Summary of Cas9 proteins used in the phage Mu targeting assays. The length in amino acids, subtype classification, PAM sequence, and all-versus-all pairwise sequence identity are shown.
(C) Representative E. coli phage Mu plaque assays for each of the Cas9 systems tested. Ten-fold serial dilutions of phage Mu lysate were plated on lawns expressing the anti-CRISPR noted above the columns. Representative pictures of at least three biological replicates are shown.
(D) The inhibitory activity of all tested anti-CRISPRs against diverse Cas9 homologs is represented. The darkness of the cell in the table indicates the degree of inhibition of theCas9 homolog by the indicated anti-CRISPR, with the darkest cell representing >106-fold inhibition of the Cas9 system (i.e., plaquing efficiency of phage Mu increases >106-fold in the presence of the anti-CRISPR). The lightest-shaded cells indicate that the given anti-CRISPR displayed no inhibition of the Cas9 homolog. This figure represents data obtained through at least three biological replicates of each assay.
We tested 10 previously identified anti-CRISPRs in the phage Mu targeting assay, including four that were shown to inhibit type II-A and five that inhibit type II-C CRISPR-Cas systems. As seen in Figure 1C, the targeted cleavage activity of each of these Cas9 proteins reduced the plaquing efficiency of phage Mu by at least 105-fold compared to strains expressing the same Cas9 proteins with non-targeting sgRNA (Figure 1C). The co-expression of anti-CRISPRs completely reversed the Cas9-mediated reduction of plaquing efficiency in some cases (Figure 1C). However, in other cases, anti-CRISPR co-expression caused no increase or only a partial increase in plaquing efficiency. The level of phage Mu plaquing in the presence of a particular Cas9/anti-CRISPR combination provides a quantitative measure of the effectiveness of the anti-CRISPR in inhibiting a given Cas9 homolog. Some anti-CRISPRs, such as AcrIIA4, are very specific, inhibiting only one or a few CRISPR-Cas9 systems, while others, such as AcrIIC1, strongly inhibited many different Cas9s (Figure 1D). Overall, the results in Figure 1D show that the strength of anti-CRISPRs may vary over many orders of magnitude, and the specificity profile of each anti-CRISPR is unique.
In contrast to all of the other anti-CRISPRs tested, AcrIIA5 was able to completely inhibit every type II-A and II-C Cas9 tested, failing to block only the type II-B Cas9 from Francisella novicida (Figures 1C and 1D). AcrIIA5 was the only anti-CRISPR able to block the highly divergent CdiCas9, emphasizing its unusually broad activity. A previous in vitro study noted the ability of AcrIIA5 to inhibit CjeCas9 and a homolog of AcrIIA5 to inhibit Nme1Cas9 (Marshall et al., 2018). The uniquely broad specificity of AcrIIA5 inspired us to further investigate its properties.
AcrIIA5 Inhibits Genome Editing Mediated by Type II-A and Type II-C CRISPR Systems in Mammalian Cells
Although AcrIIA5 was previously shown to inhibit genome editing mediated by SpyCas9 and Streptococcus thermophilus Cas9 (St1Cas9) in mammalian cells (Hynes et al., 2017), its activity against other Cas9 proteins in genome-editing applications had not been tested. To determine if AcrIIA5 could inhibit genome editing mediated by the four Cas9 homologs commonly used for genome-editing purposes in mammalian cells, we transiently co-transfected mouse Neuro-2a (N2a) (Figure 2A) or human HEK293T (Figure 2B) cells with plasmids expressing anti-CRISPR proteins, Cas9s and their respective sgRNAs designed to target specific genomic sites. Tracking of indels by decomposition (TIDE) analyses (Brinkman et al., 2014) revealed that AcrIIA5 inhibited the activities of SpyCas9, Nme1Cas9, SauCas9, and CjeCas9. These results were confirmed using a previously described T7 endonuclease I (T7E1) assay (Figure 2C) (Pawluk et al., 2016). We further probed the ability of AcrIIA5 to inhibit Cas9 homologs using a variation of the traffic light reporter (TLR) system (Certo et al., 2011), which contains an artificial locus harboring Cas9 target sites. In this assay, an out-of-frame mCherry gene is targeted for Cas9 editing, resulting in a subset of indels that restore the proper reading frame for mCherry, thereby generating a fluorescent signal. Co-transfection of Cas9 homologs and their respective sgRNAs targeting the TLR locus resulted in cells with mCherry expression ranging from 5% to 20%, depending on the Cas9 used for editing. Expression of AcrIIA5 by transient transfection reduced the editing at the TLR locus by all of the Cas9 homologs tested (Figure 2D). Collectively, these results show that AcrIIA5 efficiently inhibits the in vivo genome-editing activity of four diverse Cas9 proteins in both bacterial and mammalian cells. Furthermore, AcrIIA5 inhibits genome editing with similar potency to previously utilized anti-CRISPRs.
Figure 2. AcrIIA5 Inhibits Genome Editing Mediated by Nme1Cas9, SpyCas9, CjeCas9, and SauCas9 in Mammalian Cells.

(A) Co-transfection of Cas9 orthologs and their respective sgRNA expression plasmids show inhibition of genome editing by AcrIIA5 in mammalian cells. AcrIICI and AcrIIA4 are used as positive controls for SauCas9 and SpyCas9 inhibition, respectively. Genome editing was quantified using TIDE analysis (Brinkman et al., 2014) in mouse cells.
(B) Co-transfection of Cas9 orthologs and their respective sgRNA expression plasmids targeting either the ARHGEF9 (SpyCas9 and Nme1Cas9) or AAVS1 locus (CjeCas9) show inhibition of genome editing by AcrIIA5 in a human HEK293T cell line. Editing was quantified by TIDE analysis (Brinkman et al., 2014). AcrIIC1 and ArIIA4 were used as positive controls for type II-C (Nme1Cas9 and CjeCas9) and type II-A (SpyCas9) inhibition, respectively.
(C) Genome editing in the cell lines used in (A) and (B) were analyzed by T7E1 experiments. The image is representative of at least three replicates.
(D) Co-transfection of Cas9 orthologs, their respective sgRNAs, and AcrIIA5 expression plasmids in a cell line stably expressing TLR-MCV1, a variation of the traffic light reporter (TLR) system (Certo et al., 2011). TLR-MCV1 contains an artificial locus harboring target sites for a wide range of Cas9 orthologs. Upon double-strand break induction by a Cas9 ortholog, a subset of non-homologous end-joining (NHEJ) repair events generate indels that place mCherry in frame. The percentage (%) of mCherry cells was used as an estimate of genome-editing efficiency. Anti-CRISPRs used as controls were AcrIIC1 for type II-C Cas9 homologs (Nme1Cas9 and CjeCas9) and AcrIIA4 for type II-A SpyCas9.
In (A), (B), and (D), the values and error bars represent the mean ± the SD of three independent biological replicates.
AcrIIA5 Activity Prevents DNA Binding and Leads to sgRNA Cleavage
To investigate how AcrIIA5 inhibits Cas9 activity, we developed a luminescence-based bioassay in which we targeted the catalytically inactive dSpyCas9 (Gilbert et al., 2014) to a constitutively expressed artificial promoter that drives expression of the luxCDABE luminescence genes in E. coli (Figure 3A). sgRNA-targeted binding of dSpyCas9 to the promoter of the luxCDABE operon repressed transcription, and no luminescence was detected (Figure 3B). Expression of AcrIIA5 relieved this repression, leading to an increase in luminescence and showing that DNA binding was inhibited. Similarly, expression of AcrIIA4, which was previously shown to inhibit SpyCas9 DNA binding (Dong et al., 2017; Shin et al., 2017; Yang and Patel, 2017), also led to an increase in luminescence. By contrast, expression of AcrIIC1, which does not inhibit SpyCas9 (Harrington et al., 2017; Pawluk et al., 2016), showed no increase in luminescence, as expected. These results demonstrate that AcrIIA5 blocks binding of dSpyCas9 to target DNA and impedes its function as a transcriptional repressor.
Figure 3. AcrIIA5 Prevents DNA Binding and Leads to sgRNA Cleavage.

(A) Overview of the luminescence-based bioassay. Mutations in the catalytic domains of SpyCas9 yield a dead variant (dSpyCas9) that binds but does not cleave DNA. When dSpyCas9 is programmed to target the promoter controlling the lux expression, binding of dSpyCas9 to the promoter blocks transcription. Anti-CRISPR proteins can block the binding of dSpyCas9 to the target DNA, restoring transcription and expression of the lux cassette.
(B) The luminescence signal displayed by cells expressing dSpyCas9 targeting a promoter driving lux expression in the presence of the indicated anti-CRISPR is shown. Data represent the mean and SD of luminescence measurements for three replicates.
(C) His6-Nme1Cas9 was co-expressed and co-purified with pCDF-1b (no anti-CRISPR [-]), AcrIIC1, or AcrIIA5. Ribonucleoprotein complexes were analyzed by SDS-PAGE gel (top) and polyacrylamide/Urea gel (bottom). The name of the anti-CRISPR co-expressed with Nme1Cas9 is indicated at the top of the SDS-PAGE gel.
(D) DNA cleavage mediated by Nme1Cas9 co-expressed with AcrIIA5. A plasmid containing the target protospacer was linearized and incubated with the indicated Nme1Cas9/anti-CRISPR combination at 37°C. Samples were taken at the indicated time points and analyzed by DNA gel electrophoresis.
(E) A schematic of the Nme1Cas9 sgRNA with bound target protospacer DNA is shown. The positions of RNA cleavage detected by sequencing in the sgRNA bound to Nme1Cas9 co-expressed with AcrIIA5 are indicated. The sgRNA secondary structure shown is predicted from other Cas9/sgRNA structures. The image is representative of at least three replicates.
(F) Activity of AcrIIA5 against Cas9 bound to modified crRNA and tracrRNA. A ribonucleoprotein complex composed of SpyCas9 and modified or unmodified crRNA/tracrRNA was electroporated into HEK293T cells (No Acr) or HEK293T cell line stably expressing AcrIIA5. Efficiency of genome editing at the genomic VEGFA target site was measured by TIDE analysis (Brinkman et al., 2014). The bar graph represents the data plotted as mean of three replicates with SD.
After co-expression of His6-tagged Nme1Cas9 and AcrIIA5, AcrIIA5 did not co-elute with Nme1Cas9, while a control, AcrIIC1, did co-elute (Figure 3C). Nevertheless, Nme1Cas9 expressed in the presence of AcrIIA5 was unable to cleave DNA in vitro (Figure 3D). Thus, co-expression of AcrIIA5 with Nme1Cas9 caused a loss of activity even though the anti-CRISPR did not form a stable complex with Cas9. Electrophoretic examination of the sgRNA bound to Nme1Cas9 purified in the presence of AcrIIA5 surprisingly showed that a sizable proportion was smaller compared to the sgRNA bound to Nme1Cas9 expressed without AcrIIA5 or with AcrIIC1 (Figures 3C, S1A, and S1B). The full-length and cleaved sgRNA molecules seen in these gels were excised, reverse transcribed into DNA, and sequenced. We found that a portion of the Nme1Cas9 co-expressed with AcrIIA5 was bound to full-length sgRNA that was indistinguishable from that of Nme1Cas9 controls, but it was also frequently bound to truncated forms (Figure S1B). These truncations mapped to stem-loop 1 and stem-loop 2 of the sgRNA (Figures 3E and S1C). It was recently shown that Nme1Cas9 can mediate RNA cleavage that is catalyzed by the Cas9 HNH endonuclease domain (Rousseau et al., 2018). However, the formation of the truncated sgRNA molecules seen here was not mediated by either of the nuclease domains of Nme1Cas9 (Figure S1D).
To investigate the relationship between AcrIIA5 activity and CRISPR RNA(crRNA) in a genome-editing application in mammalian cells, we tested its ability to inhibit editing efficiency in the presence of chemically modified crRNA/trans-activating crRNA (tracrRNA) molecules that were previously described (Mir et al., 2018a). In a cell line that stably expresses AcrIIA5, editing efficiency was completely abrogated when an unmodified guide (C0:T0) was used (see STAR Methods for details of the modified RNA molecules) but was only partially compromised (~3-fold) when an RNase-resistant heavily modified guide (C20:T2) was used (Figure 3F). The protection from AcrIIA5 activity provided by chemical modification of the crRNA/tracrRNA is consistent with this anti-CRISPR interacting with RNA. A caveat to this experiment is that the unmodified RNA mediated genome editing much less efficiently (~5-fold) than the modified RNA. However, given that TIDE methodology can detect editing levels of ~1%–2% (Brinkman et al., 2014), and editing by Cas9 bound to unmodified RNA was undetectable, we estimate that AcrIIA5 reduced editing mediated by unmodified RNA by at least 15-fold, while that mediated by modified RNA was only reduced by ~3-fold.
sgRNA Cleavage Is a Conserved Feature of AcrIIA5 Homologs
Despite extensive efforts, AcrIIA5 could not be purified in a soluble and active form on its own. In an effort to circumvent this problem, we also cloned and expressed five additional AcrIIA5 family members (Figures S2A and S2B), which ranged from 87% to 48% in sequence identity at the amino acid level from the homolog from S. thermophilus phage D4276 that we characterized here. Although each of these homologs robustly inhibited all Cas9s tested (Figure S2C) and were also well expressed in E. coli (Figure S2D), none could be purified in a soluble and active form. Thus, we were unable to carry out the detailed in vitro experiments necessary to further elucidate the AcrIIA5 inhibitory mechanism.
To address the question of whether sgRNA cleavage is a consistent feature of the AcrIIA5 family, we purified Nme1Cas9 co-expressed with each of the five AcrIIA5 homologs described above. Notably, the sgRNA co-purifying in these Nme1Cas9 preparations displayed similar levels of partial cleavage in every case (Figure S2E). To establish that sgRNA cleavage was not due to a unique feature of Nme1Cas9, we also co-expressed the AcrIIA5 homologs with SpyCas9 and purified the resulting Cas9/sgRNA complexes. The sgRNA molecules associated with SpyCas9 were also cleaved in the presence of each of the AcrIIA5 homologs (Figure S2F).
Finally, we constructed three AcrIIA5 mutants bearing amino acid substitutions at residues potentially involved in catalysis (Figure S2A). Two of these mutants displayed no inhibitory activity against all tested Cas9 homologs (Figures S3A and S3B). Interestingly, the fourth mutant (H66N70H73) was fully active against SpyCas9 but was unable to inhibit the other Cas9 homologs. This indicates that there may be distinct regions of AcrIIA5 responsible for binding to different Cas9 homologs. Purified Nme1Cas9 that was co-expressed with the inactive mutants was not associated with cleaved sgRNA, supporting the connection between AcrIIA5 inhibition of Cas9 and sgRNA cleavage (Figure S3C).
DISCUSSION
AcrIIA5 is a remarkably broad specificity anti-CRISPR that functions through a unique mechanism. The co-expression of AcrIIA5 with Nme1Cas9 results in the truncation of the sgRNA from the 3′ end. This sgRNA truncation was seen consistently in six different AcrIIA5 family members when co-expressed with Nme1Cas9 or SpyCas9. The enigmatic feature of this sgRNA truncation is that the relative amounts of truncated products vary in different experiments, as do the apparent numbers of truncated products (i.e., the number of higher-mobility bands seen in the gels) (Figures 3C, S1B, S1D, S2E, S2F, and S3C). In addition, there is always some sgRNA remaining bound to Cas9 co-expressed with AcrIIA5 that displays the mobility of full-length sgRNA in denaturing polyacrylamide/Urea gels, and sequencing confirmed that this sgRNA is indistinguishable from that bound to Nme1Cas9 in the absence of AcrIIA5 (Figure S1C). We conclude that sgRNA cleavage alone cannot account for the potent inhibitory activity of AcrIIA5. Rather, the action of AcrIIA5 may partially dislodge the sgRNA from Cas9, leaving it prone to digestion by intracellular RNases. The portion of the sgRNA that we observe to be digested, stem-loops 1 and 2, are the more exposed parts of the sgRNA in the Cas9/sgRNA complex. We speculate that this AcrIIA5-induced effect on the sgRNA interaction with Cas9 could be due to an irreversible conformational change or a post-translational modification. An anti-CRISPR has been described that inhibits Cas12a by acetylating a key residue in the DNA-binding interface (Dong et al., 2019).
We cannot rule out that AcrIIA5 itself does have nuclease activity, as there are many examples of small ribonucleases associated with toxin-antitoxin systems that specifically digest mRNA, tRNA, or rRNA (Masuda and Inouye, 2017), and anti-CRISPR AcrVA1 is a specific crRNA nuclease (Knott et al., 2019; Zhang et al., 2019). However, we were unable to detect any resemblance between AcrIIA5 and ribonucleases by performing extensive Position-Specific Iterative Basic Local Alignment Search Tool (PSI-BLAST) (Altschul et al., 1997) searches or through analysis using HHpred (Söding et al., 2005). Furthermore, no structural similarity to ribonucleases was predicted by either the Iterative Threading ASSEmbly Refinement (I-TASSE) (Yang and Zhang, 2015) or Phyre (Kelley et al., 2015) protein structure prediction servers. Some role for sgRNA in the activity of AcrIIA5 is supported by the relative insensitivity of modified sgRNA-bound Cas9 to inhibition by this anti-CRISPR. Chemical modifications may provide protection from cleavage by AcrIIA5, prevent RNA recognition by AcrIIA5, or protect sgRNA from intracellular RNases when it is dislocated from Cas9 by AcrIIA5 activity.
In summary, we have characterized AcrIIA5, a potent inhibitor of all type II-A and type II-C Cas9 homologs tested. This anti-CRISPR functions in a distinct manner from other characterized Cas9 inhibitors. As the number of described anti-CRISPRs has rapidly increased in recent years, the contemplated biotechnological uses for them have also grown. Since different Cas9 homologs possess distinct properties (e.g., small size, thermotolerance, and distinct PAM specificities) that make them more suited for particular applications, the identification of a single anti-CRISPR that can potently inhibit the broadest possible range of Cas9 homologs is of great value.
STAR★METHODS
CONTACT FOR REAGENT AND MATERIALS AVAILABILYITY
Please direct any requests for further information or reagents generated in this study to the lead contact, Alan R. Davidson (alan.davidson@utoronto.ca). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Escherichia coli BB101 (DE3)
E. coli BB101 (DE3) cells were used for protein expression for in vivo phage targeting experiments, in vitro studies, and RNP expression and purification. Cells were cultured in Luria broth (LB) at 37°C. To ensure plasmid maintenance, LB media was supplemented with chloramphenicol (34 μg/mL) and streptomycin (34 μg/mL) for the co-expression of the plasmid expressing Cas9s and the Anti-CRISPR proteins for the phage targeting experiments. For RNP expression and purification in the presence of Anti-CRISPR proteins, ampicillin (100 μg/mL) and streptomycin (34 μg/mL) were used for plasmid maintenance.
Phage
E. coli Mu phage was propagated at 30°C. Mu phage production was induced by increasing the temperature to 45°C for 15 minutes and transferred to 37°C until cell lysis occurred. Mu phage was stored at 4°C.
Cell lines
HEK293T and Traffic Light Reporter (TLR) cell line (TLR-MCV1, unpublished) were cultured in Dulbecco’s modified Eagle’s minimum essential medium (DMEM; Life Technologies) supplemented with 10% (vol/vol) FBS (Sigma) and 1% Penicillin/Streptomycin.
Neuro-2a cells were cultured at 37°C, 5% CO2in DMEM + 10% FBS + 1% Penicillin/Streptomycin.
METHOD DETAILS
Phylogenetic analysis of Cas9 proteins
The Neisseria meningitidis Cas9 sequence from strain 8013 (accession number PHP22510) was used to query the NCBI complete bacterial genome database (four psiBLAST iterations, max target sequences 30,000). Protein sequences corresponding to unique accession numbers (e-value < 0.1) were collected. A non-redundant set of proteins was compiled by filtering out proteins with > 90% sequence identity. The resulting sequences were aligned using MAFFT (Katoh et al., 2002) and Clustal Omega (Sievers et al., 2011) to generate multiple sequence alignment and tree files (Newick format). Phylogenetic trees were visualized using FigTree 1.4.3 (http://tree.bio.ed.ac.uk/software/).
Plasmid construction
DNA sequences encoding anti-CRISPR genes were cloned into pCDF-1b (Novagen) for in vivo phage Mu targeting experiments, luminescence assays, and co-expression in E. coli. Mutations were introduced into the AcrIIA5 open reading frame contained in the pCDF-1b-derived plasmid by site-directed mutagenesis. For each mutation, two 40-bp complementary primers containing desired mutations were designed. The PCR reaction was conducted using Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific), followed by Dpnl digestion. The resulting DNA product was used to transform E.coli DH5α cells. Plasmids were isolated from streptomycin resistant colonies and all mutations were verified by sequencing.
The plasmids expressing GeoCas9 and HpaCas9 used for phage targeting assays were previously described (Harrington et al., 2017; Lee et al., 2018). The non-targeting plasmids expressing Cas9 orthologs were constructed from pGeoCas9-sgRNA (Harrington et al., 2017), replacing the Cas9 coding sequence and the sgRNA scaffolds (listed in Table S1). The Cas9 homologs from Kiloniella laminariae and Brackiella oedipodis, which have not been previously described, were cloned and expressed in the same manner as the other Cas9 proteins. sgRNA and PAM sequences were determined using the same approach as used previously by our group (Lee et al., 2018). The detailed characterization of these Cas9 homologs will be described in a future publication.
The sgRNA scaffolds were ordered as gblocks (IDT) and were cloned into the plasmid using a Gibson Assembly reaction (NEB). A unique BsaI restriction site was included in the non-targeting plasmids to clone DNA fragments encoding a crRNA that targets phage Mu or the J23119 promoter (http://parts.igem.org/Part:BBa_J23119). These DNA fragments were generated by phosphorylation and annealing of ssDNA oligos (Eurofins Genomics).
The AcrIIA5 mammalian expression vectors were generated by cloning a codon-optimized anti-CRISPR sequence into pCDest2 (pEJS1004-pCDest-ACRIIA5-FLAG-NLS) or a lentiviral vector (pEJS1005-pLenti-AcrIIA5-FLAG/NLS-HygR) using Hifi-Assembly (NEB). The plasmid used for the mouse cell line genome editing experiments was previously described for AcrIIC1 (Pawluk et al., 2016; Addgene #85679). The same plasmid was used for expression of AcrIIA4 and AcrIIA5. Addgene plasmids were used for the SpyCas9-sgRNA (#62988) and SauCas9-sgRNA (#61591) experiments.
Expression plasmid 6x-His-tagged SpyCas9-sgRNA was constructed from 6x-His-tagged Nme1Cas9-sgRNA in pMCSG7 (Pawluk et al., 2016), replacing the Cas9 coding sequence and the sgRNA scaffolds. Both plasmids were used for the co-expression and co-purification experiments.
In vivo phage Mu plaquing assays
E. coli BB101 cells were co-transformed with plasmids expressing Cas9-sgRNA combinations targeting phage Mu and a pCDF-1b plasmid expressing the different anti-CRISPR proteins. Cells containing both plasmids were sub-cultured in LB supplemented with chloramphenicol and streptomycin and grown for two hours, at which point anti-CRISPR expression was induced with 0.01mM IPTG for 3h. Cells were then mixed with soft LB-agar and top-plated on LB supplemented with both antibiotics and 200 ng/mL aTc, 0.2% arabinose, and 10 mM MgSO4. Serial dilutions of phage Mu were spotted on top and the plates were incubated overnight at 37°C. Experiments were performed in triplicate. To confirm anti-CRISPR expression in E. coli, 500 μL of cells after IPTG induction were collected by centrifugation, resuspended in 100 μL SDS loading buffer and analyzed by SDS-PAGE on a 15% Tris-Tricine gel, followed by Coomassie Blue staining.
Cell culture, transfection, and stable cell line construction
Cells were cultured in Dulbecco’s modified Eagle’s minimum essential medium (DMEM; Life Technologies) supplemented with 10% (vol/vol) FBS (Sigma) and 1% P/S (Life Technologies). Plasmids transfection was performed using Polyfect reagent as described (Pawluk et al., 2016). Transient transfection of 100 ng Cas9, 100 ng sgRNA, and either 200 ng or 300 ng of anti-CRISPR were used for HEK293T and a Traffic Light Reporter (TLR) cell line (TLR-MCV1, unpublished), respectively. For the “NoAcr” conditions 200 ng or 300 ng of a stuffer plasmid was included in the transfection to keep the total amount of DNA constant. Lentiviral transduction was performed as described (Ma et al., 2016). Briefly, viruses were produced and collected by transfecting HEK293T (ATCC) with the lentiviral vector plasmid, pEJS1005-pLenti-c.o.AcrIIA5-FLAG-NLS-HygR that expresses AcrIIA5 (driven by the EF1-a promoter) and packaging helper plasmids (VSV-G and ΔR8.2). HEK293T and HEK293T-TLR-MCV1 target cells were transduced with viruses and then selected with hygromycin, resulting in HEK293T-AcrIIA5 and HEK293T-TLR-MCV1-AcrIIA5 cell lines.
Neuro-2a cells [cultured at 37°C, 5% CO2 in DMEM + 10% FBS+ 1% Penicillin/Streptomycin (GIBCO)] were transiently transfected with 250 ng of either SpyCas9/sgRNA targeting the NPC1 locus or SauCas9/sgRNA targeting the NPC1 locus, and 250 ng of anti-CRISPR expressing plasmid.
Electroporation of mammalian cells
The HEK293T cells or HEK293T-AcrIIA5 cells were electroporated using the Neon transfection system (ThermoFisher) with an in vitro-formed ribonucleoprotein complex of SpyCas9, crRNAs (C0 or C20), and tracrRNAs (T0 or T2) that were synthesized as described previously (Mir et al., 2018a). Briefly, 80 pmol SpyCas9, 100 pmol crRNA, and 100 pmol tracrRNA were incubated in Buffer R at room temperature for 30 minutes and electroporated into 100,000 cells.
Flow cytometry
The mCherry-positive TLR-MCV1 cells were analyzed on a MACSQuant® VYB from Miltenyi Biotec using a yellow laser with a 561 nm excitation and emission 615/20 nm filter. FlowJo® v10.4.1. was used for gating single cells based on FSC-A and FSC-H after removal of debris. The percentage of cells expressing mCherry was used to estimate the Cas9-mediated editing efficiency.
Indel analysis by T7E1 and TIDE
Genomic DNA from cells was harvested using DNeasy Blood and Tissue kit (QIAGEN) according to the manufacturer protocol. PCR was used to amplify the locus surrounding the targeted site [DreamTaq Green PCR Master Mix 2X (Thermo Scientific)]. The PCR reactions were subsequently used for T7E1 assay as previously described (Pawluk et al., 2016). The amplicons were also sequenced by Sanger sequencing using the forward amplification primer. TIDE decomposition software (https://tide.deskgen.com/) was used to assess editing percentage for all transfections (Brinkman et al., 2014).
Luminescence assay
DNA encoding a crRNA targeting the constitutive promoter region of J23119 was cloned into the BsaI site of the pCRISPathBrick plasmid (Cress et al., 2015). This plasmid was co-transformed into E. coli BL21 cells with pCM-str, a plasmid in which the J23119 artificial promoter drives constitutive expression of the luxCDABE operon from Photorhabdus luminescens (Winson et al., 1998). These cells were then co-transformed with a pCDF-1b plasmid expressing the anti-CRISPR proteins and a protospacer targeting the J23119 promoter. Cells containing the three plasmids were grown in LB supplemented with kanamycin, chloramphenicol and streptomycin until they reached an optical density at 600 nm (OD600) of 0.6. The cultures were then diluted to an OD600 of 0.1 in LB containing 200 ng/mL aTc, 0.2% arabinose and 0.01 mM IPTG, and 100 μl was dispensed into a 96-well plate. The plate was incubated with shaking at 37°C using a Synergy H1 reader controlled by Gen5 2.09 software (BioTek Instruments Inc.), and the OD600 and luminescence was monitored for 24 hours.
Co-expression and co-purification of Nme1Cas9/sgRNA and anti-CRISPR
E. coli BB101 cells were co-transformed with 6x-His-tagged Nme1Cas9/sgRNA in pMCSG7 (Pawluk et al., 2016) or 6x-His-tagged SpyCas9/sgRNA in pMCSG7 and a pCDF-1b vector encoding untagged anti-CRISPR protein. Cells were grown in LB at 37°C to an OD of 0.8. Protein expression was induced by the addition of 1mM IPTG, and the cells were incubated for an additional 3 hours at 37°C. Cells were collected by centrifugation, resuspended in binding buffer [50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 5% glycerol, 20 mM imidazole], and lysed by sonication. Clarified lysates were incubated with Ni-NTA agarose (QIAGEN) for 30 minutes at 4°C, washed with binding buffer supplemented with 30 mM imidazole, and bound protein was eluted with binding buffer supplemented with 300 mM imidazole. The purified ribonucleoprotein complexes were analyzed by SDS-PAGE using a 15% Tris-Tricine gel, and the proteins were visualized using Coomassie stain. The co-purifying sgRNA was examined using a denaturing 12.5% polyacrylamide/Urea gel and visualized by SYBR™ Gold (ThermoFisher Scientific) staining.
For size exclusion chromatography experiments, a Superdex 200 10/300 column was used for Nme1Cas9-sgRNA and Nme1Cas9sgRNA purified from cells expressing the AcrIIA5. Fractions were analyzed on a 15% PAGE gel and stained with Coomassie Blue. The co-purifying sgRNA was examined using a denaturing 12.5% polyacrylamide/Urea gel and visualized by SYBR™ Gold (ThermoFisher Scientific) staining.
In vitro DNA cleavage assays
DNA cleavage reactions were conducted in Cleavage Buffer [75 mM NaCl, 20 mM Tris (pH 7.5), 5 mM MgCl2, 1 mM TCEP] at 37°C. Cas9/sgRNA complexes purified from cells expressing anti-CRISPR proteins were added to the reactions at a concentration of 500 nM. Linear DNA substrates generated by restriction digestion were used at a concentration of 20 nM. Samples removed at various time points were quenched by the addition of EDTA to a final concentration of 10 nM. Cleavage products were analyzed on a 1.25% agarose gel stained with RedSafe (FroggaBio).
RNA cloning and sequencing
sgRNA bound to affinity purified Nme1Cas9 in the presence or absence of AcrIIA5 or in the presence of AcrIIC1 were electrophoresed on a denaturing 12.5% polyacrylamide/Urea gel and visualized by SYBR™ Gold (ThermoFisher Scientific) staining. Bands corresponding to full-length sgRNA were excised for each sample and bands with higher mobility than full-length sgRNA were excised from the sample of Nme1Cas9 purified from cells grown in the presence of AcrIIA5. The gel slices were soaked in 250 μL of DNA Gel Elution Buffer (New England Biolabs) supplemented with 1:100 SUPERase· In RNase Inhibitor (ThermoFisher Scientific) and rotated overnight at 4°C. The eluate was filtered through a Nanosep® MF 0.45 μm column (Pall Laboratory, ODM45C35). The RNA was ethanol precipitated and reconstituted in ultrapure water. Libraries were prepared with the NEBNext Small RNA Library Prep Set for Illumina (New England Biolabs) following the protocol provided by the manufacturer. 100 ng of immunoprecipitated RNA was used as starting material. The resulting DNA library was visualized using 8% PAGE and bands corresponding to the sgRNA fragments were excised. The DNA was eluted from the excised bands by rotating overnight in DNA Gel Elution buffer at room temperature. The eluate was filtered through a Nanosep® MF 0.45 μm column and the DNA was ethanol precipitated and resuspended in ultrapure water. DNA fragments were then ligated to the TOPO Blunt vector (ThermoFisher Scientific), DNA was purified from single colonies and inserts were sequenced using the M13F or M13R primers.
Assays with chemically modified sgRNA molecules
To perform assays with chemically modified sgRNA molecules, we generated a stable human HEK293T cell line that expresses AcrIIA5 through lentiviral vector transduction and analyzed the editing efficiency of a SpyCas9/crRNA/tracrRNA ribonucleoprotein (RNP) complex at a well-validated site within the VEGFA gene (Mir et al., 2018a). The “unmodified” dual crRNA/tracrRNA guide RNA called C0:T0 has three phosphorothioates at each end of both the crRNA (C0) and the tracrRNA (T0) to help protect against cellular exonucleases. The guide RNA referred to as “modified,” called C20:T2, is heavily modified: the C20 crRNA has a mix of 2′-O-methyl and 2′-fluoro residues, and only six unmodified ribose moieties, each of which is adjacent to a phosphorothioate modification and is RNase-resistant. The 67-nucleotide T2 tracrRNA is ~82% modified, with a mix of 55 2′-O-methyl and 2′-fluoro residues, as well as twelve riboses. Those twelve riboses are not phosphorothioated, but they are all buried in the interior of the protein and therefore largely protected from RNases when loaded into the SpyCas9 RNP. These chemical modifications in C20 and T2 do not impair the genome editing efficiencies by RNP in mammalian cells (Mir et al., 2018a).
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were conducted with at least three biological replicates. Number of biological replicates are reported in the individual Figure Legends. Error bars represent the standard deviations (Figures 2A, 2B, and 2D).
DATA AND CODE AVAILABILITY
This study did not generate unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Gibson Assembly Master mix | New England Biolabs | #E2611 |
| Phusion High-Fidelity DNA Polymerase | ThermoFisher Scientific | #F530 |
| DpnI-FD | ThermoFisher Scientific | #FD1703 |
| BsaI-HFv2 | New England Biolabs | #R3733 |
| Hifi-Assembly DNA Assembly | New England Biolabs | #E5520 |
| SYBR™ Gold | ThermoFisher Scientific | #S11494 |
| T7 Endonuclease 1 | New England Biolabs | #M0302L |
| DMEM (Medium for mammalian cell culture) | GIBCO | #11965092 |
| Fetal Bovine Serum (For mammalian cell culture) | Sigma Aldrich | #F4135 |
| Penicillin-Streptomycin (For mammalian cell culture) | Sigma Aldrich | #P4333 |
| PolyFect transfection reagent | QIAGEN | #3011 |
| High Fidelity 2X PCR Master Mix | New England Biolabs | #M0541S |
| DreamTaq Green PCR Master Mix 2X | ThermoFisher Scientific | #K1081 |
| DNA Gel Elution Buffer | New England Biolabs | #E7324A |
| SUPERase· In RNase Inhibitor | ThermoFisher Scientific | #AM2696 |
| Critical Commercial Assays | ||
| DNA Clean & Concentrator-5 | Zymo Research | #D4004 |
| NEBNext Small RNA Library Prep Set for Illumina | New England Biolabs | #E7330 |
| DNeasy Blood and Tissue Kit | QIAGEN | #69504 |
| Ni-NTA agarose resin | QIAGEN | #30210 |
| Superdex 200 10/300 | GE Healthcare | #28990944 |
| Experimental Models: Cell Lines | ||
| Neuro-2a | ATCC | CCL-131 |
| Human HEK293T | ATCC | ATCC CRL-3216 |
| HEK293T-TLR-MCV1 variant | This study | N/A |
| HEK293T-TLR-MCV1-AcrIIA5 | This study | N/A |
| Oligonucleotides | ||
| See Table S1 for sequences of sgRNAs used | This study | N/A |
| N66H70N73aA5F GAAATCCGTCTGTCCAATgcCAGTGCTGATgcTAAATACgcTGATCTTGAGAATGGGCG | Integrated DNA Technologies | N/A |
| N66H70N73aA5R CGCCCATTCTCAAGATCAgcGTATTTAgcATCAGCACTGTcATTGGACAGACGGATTTC | Integrated DNA Technologies | N/A |
| D50R62aA5F AAGTATGAAGGcTTCTGCCTATAAAGACTTTGGAAAATATGAAATCgcTCTGTCCAATCA | Integrated DNA Technologies | N/A |
| D50R62aA5R TGATTGGACAGAgcGATTTCATATTTTCCAAAGTCTTTATAGGCAGAAgCCTTCATACTT | Integrated DNA Technologies | N/A |
| D74K8588aA5F AAATACCATGcTCTTGAGAATGGGCGTCTTATCGTGAACATCgcAGCATCAgcGCTGAAC | Integrated DNA Technologies | N/A |
| D74K8588aA5R GTTCAGCgcTGATGCTgcGATGTTCACGATAAGACGCCCATTCTCAAGAgCATGGTATTT | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pGeoCas9-sgRNAMu | Harrington et al., 2017 | https://benchling.com/s/seq-IXSEQMlloSIZmGtpNsld/edit |
| pHpaCas9sgRNAMu | Lee et al., 2018 | N/A |
| pNme1Cas9sgRNAMu | Thavalingam et al., 2019 | N/A |
| pCjeCas9sgRNAMu | Thavalingam et al., 2019 | N/A |
| pBoeCas9sgRNAMu | This study | N/A |
| pKlaCas9sgRNAMu | This study | N/A |
| pCdiCas9sgRNAMu | This study | N/A |
| pSauCas9sgRNAMu | This study | N/A |
| pSpyCas9sgRNAMu | This study | N/A |
| pFnoCas9sgRNAMu | This study | N/A |
| pCDF-1b | Novagen | #71330-3 |
| pCDF-ACRE2 | Harrington et al., 2017 | N/A |
| pCDF-ACRIIC1Nme | Harrington et al., 2017 | N/A |
| pCDF-ACRIIC2Nme | Thavalingam et al., 2019 | N/A |
| pCDF-ACRIIC3Nme | This study | N/A |
| pCDF-ACRIIC4Hpa | Lee et al., 2018 | N/A |
| pCDF-ACRIIC5Smu | Lee et al., 2018 | N/A |
| pCDF-ACRIIA2Lmo | This study | N/A |
| pCDF-ACRIIA3Lmo | This study | N/A |
| pCDF-ACRIIA4Lmo | This study | N/A |
| pCDF-ACRIIA5SthD4276 | This study | N/A |
| pCDF-ACRIIA5SthD1126 | This study | N/A |
| pCDF-ACRIIA5Efa | This study | N/A |
| pCDF-ACRIIA5Dpi | This study | N/A |
| pCDF-ACRIIA5Lsa | This study | N/A |
| pCDF-ACRIIA5G572 | This study | N/A |
| pCRISPathBrick | Cress et al., 2015 | Addgene #65006 |
| pCM-str | J. Nodwell Lab | N/A |
| TOPO® vector | ThermoFisher Scientific | #K4500-01 |
| 6x-His-tagged Nme1Cas9-sgRNA in pMCSG7 | Pawluk et al., 2016 | N/A |
| 6x-His-tagged SpyCas9-sgRNA | This study | N/A |
| SpyCas9-sgRNA | Ran et al., 2013 | Addgene #62988 |
| SauCas9-sgRNA | Ran et al., 2015 | Addgene #61591 |
| pEJS24-pCSDest2-SpyCas9-NLS-3XHA-NLS | Bolukbasi et al., 2015 | Addgene #69220 |
| pEJS424-pCSDest2-NmeCas9-NLS-3XHA-NLS | Pawluk et al., 2016 | Addgene #87448 |
| pEJS485-pCSDest2-SauCas9 | S. Wolfe lab | N/A |
| pX404-CjeCas9 | F. Zhang lab | Addgene #68338 |
| pEJS433 pCSDest2-AcrIIC1 | Pawluk et al., 2016 | Addgene #85679 |
| p611-pCSDest2-AcrIIA4 | This study | N/A |
| pEJS1004-pCSDest2-AcrIIA5-NLS-FLAG | This study | N/A |
| pEJS1005-Lenti-mammalian c.o.AcrIIA5-FLAG-NLS-HygR | This study | N/A |
| Software and Algorithms | ||
| FigTree 1.4.3 | Institute of Evolutionary Biology, University of Edinburgh | http://tree.bio.ed.ac.uk/software/ |
| FlowJo® v10.4.1. | FlowJo LLC | N/A |
| Gen5 2.09 | BioTek Instruments Inc | https://www.biotek.com/products/software-robotics-software/gen5-microplate-reader-and-imager-software/software/ |
Highlights.
Anti-CRISPR AcrIIA5 potently inhibits all Cas9 homologs used in genome editing
AcrIIA5 functions well in a variety of mammalian cell genome-editing applications
The AcrIIA5 functional mechanism leads to sgRNA cleavage
ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Institutes for Health Research (CIHR) to A.R.D. (FDN-15427) and K.L.M. (PJT-152918) and by a grant from the U.S. NIH (GM125797) to A.R.D. and E.J.S. C.N.T. was funded through a CIHR fellowship. U.S. and J.M.C. were supported by NSERC (RGPIN-418683).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.017.
DECLARATION OF INTERESTS
E.J.S. is a co-founder of Intellia Therapeutics. The other authors declare no competing interests.
SUPPORTING CITATIONS
The following reference appears in the Supplemental Information: Edgar, 2004.
REFERENCES
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltimore D, Berg P, Botchan M, Carroll D, Charo RA, Church G, Corn JE, Daley GQ, Doudna JA, Fenner M, et al. (2015). Biotechnology. A prudent path forward for genomic engineering and germline gene modification. Science 348, 36–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolukbasi MF, Gupta A, Oikemus S, Derr AG, Garber M, Brodsky MH, Zhu LJ, and Wolfe SA (2015). DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat. Methods 12, 1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AL, Davidson AR, and Bondy-Denomy J (2017). The Discovery, Mechanisms, and Evolutionary Impact of Anti-CRISPRs. Annu. Rev. Virol 4, 37–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, and Scharenberg AM (2011). Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 8, 671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella P, Ronzitti G, and Mingozzi F (2017). Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev 8, 87–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DB, Platt RJ, and Zhang F (2015). Therapeutic genome editing: prospects and challenges. Nat. Med 21, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cress BF, Toparlak OD, Guleria S, Lebovich M, Stieglitz JT, Englaender JA, Jones JA, Linhardt RJ, and Koffas MA (2015). CRISPathBrick: Modular Combinatorial Assembly of Type II-A CRISPR Arrays for dCas9-Mediated Multiplex Transcriptional Repression in E. coli. ACS Synth. Biol 4, 987–1000. [DOI] [PubMed] [Google Scholar]
- Dong D, Guo M, Wang S, Zhu Y, Wang S, Xiong Z, Yang J, Xu Z, and Huang Z (2017). Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature 546, 436–39. [DOI] [PubMed] [Google Scholar]
- Dong L, Guan X, Li N, Zhang F, Zhu Y, Ren K, Yu L, Zhou F, Han Z, Gao N, and Huang Z (2019). An anti-CRISPR protein disables type V Cas12a by acetylation. Nat. Struct. Mol. Biol 26, 308–314. [DOI] [PubMed] [Google Scholar]
- Doudna JA, and Charpentier E (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz VM, and Bier E (2015). Genome editing. The mutagenic chain reaction: a method for converting heterozygous to homozygous mutations. Science 348, 442–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond A, Galizi R, Kyrou K, Simoni A, Siniscalchi C, Katsanos D, Gribble M, Baker D, Marois E, Russell S, et al. (2016). A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat. Biotechnol 34, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LB, Doxzen KW, Ma E, Liu JJ, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, et al. (2017). A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 170, 1224–1233.e1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess GT, Tycko J, Yao D, and Bassik MC (2017). Methods and Applications of CRISPR-Mediated Base Editing in Eukaryotic Genomes. Mol. Cell 68, 26–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann MD, Aschenbrenner S, Grosse S, Rapti K, Domenger C, Fakhiri J, Mastel M, Börner K, Eils R, Grimm D, and Niopek D (2019). Cell-specific CRISPR-Cas9 activation by microRNA-dependent expression of anti-CRISPR proteins. Nucleic Acids Res. 47, e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes AP, Rousseau GM, Lemay ML, Horvath P, Romero DA, Fremaux C, and Moineau S (2017). An anti-CRISPR from a virulent streptococcal phage inhibits Streptococcus pyogenes Cas9. Nat. Microbiol 2, 1374–1380. [DOI] [PubMed] [Google Scholar]
- Hynes AP, Rousseau GM, Agudelo D, Goulet A, Amigues B, Loehr J, Romero DA, Fremaux C, Horvath P, Doyon Y, et al. (2018). Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat. Commun 9, 2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraheim R, Song CQ, Mir A, Amrani N, Xue W, and Sontheimer EJ (2018). All-in-one adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome Biol. 19, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, and Miyata T (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ, Jung MH, Kim S, et al. (2017). In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun 8, 14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, Thornton BW, Lobba MJ, Liu JJ, Al-Shayeb B, Watters KE, and Doudna JA (2019). Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat. Struct. Mol. Biol 26, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Badran AH, and Liu DR (2017). CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Cradick TJ, and Bao G (2016). The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol. Ther 24, 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Mir A, Edraki A, Garcia B, Amrani N, Lou HE, Gainetdinov I, Pawluk A, Ibraheim R, Gao XD, et al. (2018). Potent Cas9 Inhibition in Bacterial and Human Cells by AcrIIC4 and AcrIIC5 Anti-CRISPR Proteins. MBio 9, e02321–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Mou H, Ibraheim R, Liang SQ, Liu P, Xue W, and Sontheimer EJ (2019). Tissue-restricted Genome Editing in vivo Specified by MicroRNA-repressible Anti-CRISPR Proteins. RNA 25, 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Psatha N, Gil S, Wang H, Papayannopoulou T, and Lieber A (2018). HDAd5/35++ Adenovirus Vector Expressing Anti-CRISPR Peptides Decreases CRISPR/Cas9 Toxicity in Human Hematopoietic Stem Cells. Mol. Ther. Methods Clin. Dev 9, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, and Pederson T (2016). CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol 214, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R, Maxwell CS, Collins SP, Jacobsen T, Luo ML, Begemann MB, Gray BN, January E, Singer A, He Y, et al. (2018). Rapid and Scalable Characterization of CRISPR Technologies Using an E. coli Cell-Free Transcription-Translation System. Mol. Cell 69, 146–157.e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda H, and Inouye M (2017). Toxins of Prokaryotic Toxin-Antitoxin Systems with Sequence-Specific Endoribonuclease Activity. Toxins (Basel) 9, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir A, Alterman JF, Hassler MR, Debacker AJ, Hudgens E, Echeverria D, Brodsky MH, Khvorova A, Watts JK, and Sontheimer EJ (2018a). Heavily and fully modified RNAs guide efficient SpyCas9-mediated genome editing. Nat. Commun. 9, 2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir A, Edraki A, Lee J, and Sontheimer EJ (2018b). Type II-C CRISPR-Cas9 Biology, Mechanism, and Application. ACS Chem. Biol 13, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Srinivasan P, Chavez M, Carter MA, Dominguez AA, La Russa M, Lau MB,Abbott TR, Xu X, Zhao D, et al. (2019). Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun 10, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, et al. (2016). Naturally Occurring Off-Switches for CRISPR-Cas9. Cell 167, 1829–1838.e1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Davidson AR, and Maxwell KL (2018). Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol 16, 12–17. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J,Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, and Bondy-Denomy J (2017). Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell 168, 150–158.e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau BA, Hou Z, Gramelspacher MJ, and Zhang Y (2018). Programmable RNA Cleavage and Recognition by a Natural CRISPR-Cas9 System from Neisseria meningitidis. Mol. Cell 69, 906–914.e904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Jiang F, Liu JJ, Bray NL, Rauch BJ, Baik SH, Nogales E, Bondy-Denomy J, Corn JE, and Doudna JA (2017). Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv 3, e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J, Biegert A, and Lupas AN (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thavalingam A, Cheng Z, Garcia B, Huang X, Shah M, Sun W, Wang M, Harrington L, Hwang S, Hidalgo-Reyes Y, et al. (2019). Inhibition of CRISPR-Cas9 ribonucleoprotein complex assembly by anti-CRISPR AcrIIC2. Nat. Commun 10, 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe RV, van der Helm E, Misiakou MA, Lee SW, Kol S, and Sommer MOA (2019). Discovery and Characterization of Cas9 Inhibitors Disseminated across Seven Bacterial Phyla. Cell Host Microbe. 25, 233–241.e235. [DOI] [PubMed] [Google Scholar]
- Winson MK, Swift S, Hill PJ, Sims CM, Griesmayr G, Bycroft BW, Williams P, and Stewart GS (1998). Engineering the luxCDABE genes from Photorhabdus luminescens to provide a bioluminescent reporter for constitutive and promoter probe plasmids and mini-Tn5 constructs. FEMS Microbiol. Lett 163, 193–202. [DOI] [PubMed] [Google Scholar]
- Yang H, and Patel DJ (2017). Inhibition Mechanism of an Anti-CRISPR Suppressor AcrIIA4 Targeting SpyCas9. Mol. Cell 67, 117–127.e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, and Zhang Y (2015). Protein Structure and Function Prediction Using I-TASSER. Curr. Protoc. Bioinformatics 52, 5.8.1–5.8.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Rajan R, Seifert HS, Mondragón A, and Sontheimer EJ (2015). DNase H Activity of Neisseria meningitidis Cas9. Mol. Cell 60, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li Z, Daczkowski CM, Gabel C, Mesecar AD, and Chang L (2019). Structural Basis for the Inhibition of CRISPR-Cas12a by Anti-CRISPR Proteins. Cell Host Microbe. 25, 815–826.e814. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate unique datasets or code.