

Abstract
The application of photoredox catalysis to atom-transfer radical polymerization (ATRP) has resulted in the development of strongly reducing organic photoredox catalysts (PCs) that are some of the most reducing catalysts known. The objectives of this review are to highlight these PCs with regard to their development and applications in polymer and organic synthesis, as well illuminate aspects of these PCs that remain to be studied further.
Keywords: phenothiazines, dihydrophenazines, phenoxazines, organic photoredox catalysis, O-ATRP, strong excited-state reductant, light-driven polymerization, precious-metal-free, oxidative quenching
1. Introduction
Photoredox catalysis has gained increasing attention because it provides chemists with the ability to perform challenging transformations by harnessing the reactivity of excited state photoredox catalysts (PCs) under mild reaction conditions. This ability is particularly important in synthetic organic chemistry, where photoinduced electron transfer (PET), between a PC and either a donor (reductive quenching) or acceptor (oxidative quenching), and energy transfer have facilitated previously challenging reactions and enabled a myriad of novel transformations.1-7 Among the best known PCs are those containing precious metals such as ruthenium and iridium.2,4 However, these PCs present an issue with regards to sustainability,8 as Ru and Ir are among the rarest metals on earth. To this end, several organic PC families have been reported as alternatives—including anthracenes,9-11 benzophenones,12-14 acridiniums,15-17 xanthene dyes,18-20 perylene diimides,21 and many more3—that are capable of mediating various synthetic transformations. However, most organic PCs operate through a reductive quenching pathway, and strongly reducing organic PCs are less common.
Given the wide range of transformations to which photoredox catalysis has been applied, it is not surprising that it has also found use in polymer synthesis.22 For example, in the free radical polymerization of methacrylates reported in 2011, Ru(bpy)32+ operated as a PC through a reductive quenching mechanism requiring the use of a sacrificial electron donor.23 Notably, the controlled radical polymerization of methyl methacrylate (MMA) in the presence of fac-[Ir(ppy)3] (1) under visible light irradiation was reported a year later.24 Operating by an atom-transfer radical polymerization (ATRP) mechanism, control in this class of reactions is achieved by reversible deactivation, most commonly with a bromide chain end group, which can be iteratively removed and reinstalled on the polymer chain via reduction and oxidation, respectively. As this process minimizes the number of reactive radicals in solution at any given time, it reduces bimolecular radical termination processes,25 thus enabling control over the polymerization as evidenced by (1) linear first-order kinetics, (2) linear growth of polymer molecular weight (MW) as a function of monomer conversion, (3) relatively low- to low-molecular-weight dispersity (Ð < 1.2 and Ð < 1.1, respectively), and (4) achievement of initiator efficiency (I* = Mn[theoretical]/Mn[experimentai]; Mn = number average molecular weight) near 100%. Notably, this system efficiently polymerized MMA employing low catalyst loadings (as little as 50 ppm of 1) while maintaining good control over the polymerization, something that has been challenging even in the traditional ATRP.26 Moreover, temporal control over the polymerization was demonstrated by cycling the light source on and off, showing that the polymerization could be started and stopped without loss of the bromide end-group functionality.24
Despite these achievements, concerns regarding the sustainability of this PC remained, motivating the development of an ATRP method employing organic PCs, or organocatalyzed ATRP (O-ATRP), instead. Furthermore, the purification of polymers presents challenges, and trace contamination by metal residues could impede application of these materials in biomedical devices, electronic applications, and multistep syntheses.27 Thus, shortly after the seminal paper by Fors and Hawker,24 O-ATRP was demonstrated in two concurrent reports, polymerizing MMA using perylene (2)28 or 10-phenylphenothiazine (3).29 Of these two catalysts, the superior capability of 3 to mediate a controlled polymerization was evidenced in its ability to synthesize polymers with lower dispersity (Ð) compared to 2. This difference was attributed to 3’s significantly stronger excited state reduction potential [E0(2PC•+/3PC*) = −0.6 V for 2,30 E0(2PC•+/1PC*) = −2.1 V for 3,29 both vs SCE]. However, 2 could operate using visible light irradiation, whereas 3 required the use of UV light, raising concerns for potential side reactions that might result from UV absorption by the organic molecules in solution.4 Thus, the development of strongly reducing and visible-light-absorbing organic PCs has been pursued, yielding a variety of PCs based on the phenothiazine (PhenS), dihydrophenazine (PhenN), and phenoxazine (PhenO) scaffolds (Figures 1 and 2).
Figure 1.

Examples Highlighting the Evolution of Strongly Reducing Organic PCs Based on PhenS, PhenN, and PhenO.
Figure 2.

Further Examples Highlighting the Evolution of Strongly Reducing Organic PCs Based on PhenS, PhenN, and PhenO.
To date, a number of reviews have been published detailing the applications of photoredox catalysis in organic and polymer synthesis.1-4,22,31,32 However, due to their relatively recent development, reviews of PCs based on PhenS, PhenN, and PhenO are relatively few.3,30,33 Therefore, this review will focus on these PCs, highlighting their development, reactions, and mechanisms in hope of demonstrating their broad utility in synthetic organic and polymer chemistry. Moreover, this review will discuss future research directions regarding these PCs in the hope of accelerating their development, improvement, and utilization in the coming years.
2. Development of Strongly Reducing Organic Photoredox Catalysts
While their application scope has since grown, the original motivation for using PhenS, PhenN, and PhenO as strongly reducing organic PCs came from O-ATRP. Inspired by the seminal report on photoredox catalyzed ATRP using 1,24 this method was developed as a metal-free variant of ATRP to eliminate metal contamination of polymer products for metal-sensitive applications. Originally, it made use of either 228 or 329 to polymerize methacrylates in a controlled fashion, but differences between these two organic PCs as well as the drawbacks of each soon became apparent, prompting the development of strongly reducing organic PCs that are capable of establishing a high degree of control over polymerizations by using a visible light source so as to avoid possible side reactions caused by UV light.34
In the proposed mechanism, the PC operates through oxidative quenching (Scheme 1),28,29 in which the photoexcited PC reduces an electron acceptor to generate the PC radical cation (2PC•+), followed by oxidation of an electron donor by 2PC•+ to regenerate the ground state catalyst. In the case of O-ATRP, the acceptor is typically an alkyl bromide initiator or a bromide-capped polymer chain-end, whereas the donor is the propagating radical formed by activation of the C─Br bond. With this mechanism in mind, several desirable characteristics were targeted in the search for new O-ATRP catalysts:34 (i) Strong visible-light absorption (high molar absorptivity); (ii) long-lived excited state; (iii) sufficiently negative excited state reduction potential [E0(2PC•+/PC*), singlet or triplet excited state] for the reduction of common alkyl bromide ATRP initiators [E0(C─Br/C─Br•−) = −0.6 to −0.8 V vs SCE35]; (iv) sufficiently oxidizing 2PC•+ [E0ox = E0(2PC•+/1PC)] for oxidation of the propagating radical [E0(C─Br/C─Br•−) = −0.6 to −0.8 V vs SCE]; (v) redox reversibility, i.e., a stable 2PC•+ that does not partake in degradative side reactions; (vi) low reorganization energy for the transition from PC* to 2PC•+ to JPC; and (vii) photoinduced charge-transfer excited states resulting from spatially separated singly occupied molecular orbitals (SOMOs).
Scheme 1.

A General, Oxidative Quenching Mechanism by Which Strongly Reducing PCs Operate. First, an Electron Is Donated by the Excited State PC (PC*) to an Acceptor (A), Followed by Extraction of an Electron from an Electron Donor (D) to Regenerate the Ground State PC. (Ref. 28,29)
Following these design principles and with guidance from computational methods, two new families of PCs with favorable properties for O-ATRP were discovered: N,N-diaryldihydrophenazines (PhenN’s) (e.g., 5 and 6)34 and N-arylphenoxazines (PhenO’s) (e.g., 7 and 8).36 PhenN’s improved upon previous generations of PCs by accessing more reducing triplet excited states [computationally predicted E0(2PC•+/3PC*) < −2.0 V vs SCE] while maintaining visible light absorption.34 In turn, polymerizations with PhenN’s produced polymers with dispersities (Ð) as low as 1.10 (for PC 5), although with consistently moderate initiator efficiencies, presumably due to side reactions of the PCs with propagating radicals. A major conclusion in this report was that PhenN PCs bearing electron-withdrawing groups (EWGs) or an extended π system on the N-aryl substituents appeared to consistently outperform other PCs, prompting an investigation into the cause of these observed differences.
As all of the PCs investigated in this study were sufficiently reducing (as PC*) and oxidizing (as 2PC•+) to mediate O-ATRP, density functional theory (DFT) calculations were used to elucidate the differences in their electronic structure that might influence PC performance.34 These calculations revealed that all of the PC triplet excited states studied featured low-lying SOMOs localized on the PhenN core, whereas the nature of the high-lying SOMOs was dependent on the functionality on the N-aryl substituents. Specifically, PCs bearing electronically neutral or donating groups exhibited population of high-lying SOMOs on the PhenN core. In contrast, PCs bearing EWGs or extended π systems on the N-aryl substituents showed high-lying SOMOs localized onto the N-aryl group, suggesting photoinduced intramolecular charge transfer (CT). With these properties in mind, PC 6 was computationally predicted to contain spatially separated SOMOs and, experimentally, 6 exhibited enhanced performance in O-ATRP, producing polymers with Ð as low as 1.03.
More recently, 10-phenylphenoxazine (7) was also predicted and demonstrated to have favorable properties for use as an O-ATRP catalyst.36 While 3 and 7 differ only by their chalcogenide, the difference in size between O and S was hypothesized to have significant impacts on the comparative performance of these PCs, as the ground state structure of 3 is noticeably bent while the ground state structure of 7 is more planar, and computations predict that the radical cation of both compounds is nearly planar. As such, owing to the smaller size of O and more planar core structure, DFT calculations predicted PhenO’s would have lower reorganization energies than PhenS when transitioning from the triplet excited state to the radical cation and back to the ground state. As these PCs have been proposed to operate via an outer-sphere electron-transfer mechanism, this lower penalty for structural reorganization was hypothesized to result in enhanced PC performance due to more favorable electron-transfer processes.
To investigate whether photoinduced intramolecular CT might also be accessible in PhenO’s, derivatives possessing different N-aryl substituents were synthesized and investigated in O-ATRP.36 Computations predicted that PhenO’s possessing either a 1- or 2-naphthyl substitution at the N-aryl position could access photoinduced CT excited states, and experimentally these PCs were observed to produce, under UV irradiation, polymers with Ð < 1.3. While these studies were useful in determining structural influences on PC properties, and these PCs were successful in O-ATRP, neither 7 nor the N-naphthylphenoxazines absorb light in the visible spectrum. Therefore, structural modifications of 1-naphthylphenoxazine were undertaken to red-shift its absorption while maintaining a strong excited state reduction potential as well as CT character. Thus, 8 was introduced, bearing biphenyl core-substituents that effectively red-shifted the absorption into the visible range as well as increased the molar extinction coefficient to 26,600 M−1cm−1 (Table 1).35,36 It should be noted that although the wavelength of maximum absorption of 8 is still in the UV range (λmax = 388 nm), its absorption tails significantly into the visible range, resulting nonetheless in strong visible light absorption. Gratifyingly, the use of 8 in the polymerization of MMA under white light irradiation resulted in polymers possessing relatively low Ð’s (Ð = 1.13) and achieving nearly quantitative I*. With regards to PhenS’s, similar efforts have been made to red-shift their absorption. For example, N-phenylbenzo[b]phenothiazine (9) has been reported, which featured a nearly 50 nm red-shifted absorption relative to 3, allowing it to absorb in the visible region.37 Additionally, methods to synthesize visible-light-absorbing PhenS derivatives by substitution of the PhenS core with 4-n-butylphenyl groups (PCs 10 and 11) have been reported.38,39
Table 1.
Tunable Properties of PhenO’s through Facile Synthetic Modifications of the Core Structure (Ref. 35,36)
| PC | 7 | 8 | 12 |
|---|---|---|---|
| E0(2PC•+/3PC*)a | −2.11 (−2.48)d | −1.70 (−1.80)d | −1.42 (−1.75)d |
| E0(2PC•+/1PC)a | 0.58 (0.68)e | 0.42 (0.65)e | 0.62 (0.69)e |
| λmax,absb | 324 nm | 388 nm | 411 nm |
| εmax,absc | 7,700 M−1cm−1 | 26,600 M−1cm−1 | 22,300 M−1cm−1 |
DFT-predicted redox potentials reported in V vs SCE.
Maximum absorption wavelengths.
Molar absorptivities at λmax.
Values in parentheses are experimental E0(2PC•+/1PC*) values (V vs SCE), where the lowest excited singlet energies were estimated from the maximum wavelength of emission.
Values in parentheses are experimental E1/2 values (V vs SCE) determined using cyclic voltammetry.
The tunability of PhenO-based PCs has also been demonstrated,35 as synthetic variations have been systematically made to tune PC absorption, CT in the excited state, and redox properties. While the former two have already been discussed in the context of various PCs, of more interest is the latter, which expanded on previous findings34 and yielded a library of PhenO PCs with DFT-predicted E0(2PC•+/3PC*) values spanning −1.42 V (for PC 12) to −2.11 V (for PC 7) and E0(2PC•+/1PC) spanning 0.30 V to 0.62 V (all vs SCE, Table 1).35,36 Additionally, some work has also been reported on synthetically tuning the PhenN34,40-42 and PhenS38,39,43-45 core structures. In particular, Matyjaszewski and co-workers investigated the influence of a number of N-aryl substituents on PC properties and reactivity in O-ATRP for a variety of PhenS-based PCs, such as 4.43
Finally, in an effort to improve PC recyclability, which remains one of the limitations of these PCs, a variant of 3 attached to a polymer support was developed that could be repeatedly added and removed from reaction mixtures simply with a set of tweezers.46 As a result, the PC could be used in multiple polymerizations (up to 6 times) without any observable loss in performance, thus providing a means of catalyst recycling.
3. Applications in Polymerization Reactions
3.1. O-ATRP
Broadly speaking, O-ATRP occurs by a mechanism similar to that of traditional ATRP, in that a catalyst mediates an equilibrium between “active” (Pn•) and “dormant” (PnBr) polymer chains, repeatedly activating and deactivating polymers by reversible removal and addition of a halide end group, often a bromide (Scheme 2). Key to this process is that deactivation is favored over activation and propagation, thus maintaining a low concentration of reactive radicals in solution to minimize bimolecular coupling and other termination reactions.25 In thermally driven ATRP, activation occurs via an inner sphere electron-transfer (ISET) mechanism, in which the halide is transferred from the polymer chain-end (or alkyl halide initiator) to the catalyst species at the same time as electron transfer. By contrast, activation in O-ATRP occurs through an outer sphere electron-transfer (OSET) mechanism, where an excited state PC (PC*) directly reduces an alkyl bromide (either an initiator or a polymer chain-end) to generate Br−, 2PC•+, and an active, propagating radical. 2PC•+ may subsequently associate with Br− to form the ion pair 2PC•+Br−.40 Upon deactivation, 2PC•+Br− oxidizes the radical chain-end and reinstalls the bromide to reform the dormant polymer species and regenerate the ground state PC. While specific mechanistic details are still under investigation and may vary between individual PCs, the current understanding of this mechanism will be discussed in greater detail in a later section (see Section 5).
Scheme 2.

Proposed O-ATRP Mechanism. (Ref. 40)
In initial reports on O-ATRP using either 228 or 329 in the presence of the initiator ethyl α-bromophenylacetate (EBP), the viability of the method was demonstrated through polymerization of MMA (eq 1).28,29 With the potential of this method established, the homo-polymerizations of several methacrylates were demonstrated, including benzyl methacrylate,29,34,36 tert-butyl methacrylate,29 isobutyl methacrylate,36 isodecyl methacrylate,36 trimethylsilylhydroxyethyl methacrylate,34 2,2,2-trifluoroethyl methacrylate,34 and di(ethylene glycol) methacrylate (Figure 3).34 Notably, the polymerization of 2-(dimethylamino)ethyl methacrylate to produce polymers with relatively low Ð’s was unsuccessful using 1, but was realized with PhenS catalysts.29 In subsequent reports, the monomer scope of these PCs was expanded to methacrylates bearing long alkyl chains,37,47 extended aromatic groups,45 and heterocyclic functionalities.45
Figure 3.

Monomers Successfully Polymerized via O-ATRP by Using PhenS, PhenN, and PhenO Photoredox Catalysts. (Ref. 29,34,36,45,47-49,51)
In addition to these more traditional monomers, the polymerizations of biomass-derived methacrylate monomers using PC 3 have also been reported, demonstrating that both homo- and co-polymers of these monomers could be achieved via O-ATRP.48 The polymerization of acrylonitrile by O-ATRP using 3 and other PhenS catalysts, albeit with relatively high Ð (Ð = 1.42) compared to traditional ATRP (Ð = 1.04), has also been reported.49,50 While methacrylic acid has been polymerized using 3,51 control over this polymerization was not evaluated, necessitating further future studies. Very interestingly, the polymerization of methacrylates with pendant furan-protected maleimides using 5 has also been reported.52 This report not only provides a strategy for post-polymerization modification, but also demonstrates that these PCs tolerate a wide array of functionalities. However, despite various efforts, the monomer scope of O-ATRP beyond methacrylates remains largely unestablished and, as such, an important future direction of O-ATRP is to define the monomer scope and capabilities of this polymerization platform.
In all ATRP methods, the choice of initiator can also play an influential role in controlling a given polymerization. Thus, various initiators have been investigated for use with all three PC families (see eq 1), especially traditional alkyl bromides such as EBP,29,34,36,49 diethyl 2-bromo-2-methyl malonate (DBMM),36 methyl α-bromoisobutyrate (MBiB),34,36,43 ethyl α-bromoisobutyrate (EBiB),29 methyl 2-bromopropionate (MBP),34,36 and 2-bromopropionitrile (BPN).34,49 In addition, several alkyl chloride initiators have been investigated with PhenS catalysts, though with less success.43 Although these PCs are capable of activating alkyl chlorides due to their strong excited state reduction potentials, they seem to be inefficient at deactivating the propagating radicals in conjunction with chloride initiators, resulting in less control during polymerizations. Perhaps the most interesting development in O-ATRP initiators thus far has been the introduction of aromatic sulfonyl halides by Chen and co-workers.39 In their report, nearly 20 sulfonyl bromides were investigated in polymerizations mediated by 11, achieving Ð’s as low as 1.21 in the polymerization of MMA, and in polymerizations of several other methacrylates and acrylates with varying levels of control. Of particular note is that this new initiating system allows for the post-polymerization modification of polystyrene (PS) via O-ATRP, as sulfonyl chlorides can be installed on the aromatic rings of PS to initiate an O-ATRP for grafting-from brush synthesis (see Section 3.3).
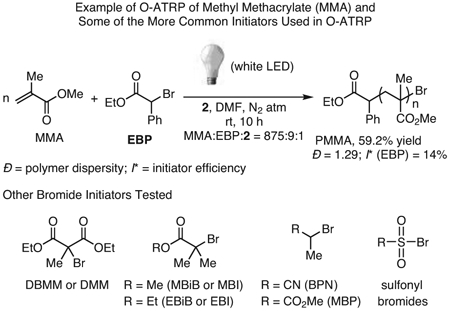 |
eq 1 |
One drawback of general photoredox catalysis is the difficulty of scaling photochemical reactions,53,54 as reactions in batch reactors must be performed on a small scale to ensure uniform irradiation throughout the reaction vessel. To overcome this obstacle, flow reactors have been implemented, in which a reaction mixture is pumped through a transparent, narrow tube wrapped around a light source to achieve both uniform irradiation as well as facile scalability.53 In an effort to extend these benefits to O-ATRP, the polymerizations of various methacrylates using PCs 5, 6, and 8 were undertaken in a flow setup, resulting in the ability to synthesize the polymers on a gram scale while maintaining relatively low Ð.48 Moreover, due to the improved irradiation conditions offered by continuous flow, enhanced PC performance was observed, allowing for a tenfold reduction in catalyst loading without significant loss of performance.
3.2. PET-RAFT
Although the primary application of PhenS, PhenN, and PhenO catalysts has been in O-ATRP, some applications of these PCs to PET-RAFT (photo-induced electron/energy transfer-reversible addition fragmentation chain-transfer) polymerizations have been reported.55-59 Much like traditional RAFT, PET-RAFT makes use of a chain-transfer agent (CTA), often a thiocarbonylthio compound, to mediate a controlled radical polymerization (Scheme 3).55-58 However, where traditional RAFT commonly utilizes thermal initiators, PET-RAFT makes use of a PC to mediate this process,55-58 minimizing the formation of dead chains from the reaction of initiator radicals with active polymers.59 Similar to what is seen in O-ATRP, PC* activates a dormant polymer-CTA bond, generating a radical that can engage in polymerization (Pn•), 2PC•+, and the respective thiocarbonylthiolate (in the case of a thiocarbonylthio CTA). The active Pn• radical can either propagate or undergo reversible deactivation by one of two pathways: in the first, the radical reacts with 2PC•+ to undergo oxidation and reinstallation of the CTA end group. In the second, the active radical can undergo chain-transfer with another CTA-capped polymer, resulting in the deactivation of one chain and the activation of another chain (Scheme 3).
Scheme 3.

Although originally reported using iridium PC 1,60 PET-RAFT was later expanded to PhenS, when 3 was employed to polymerize N-isopropylacrylamide, N,N-dimethylacrylamide, tert-butyl acrylate, and ethylene glycol methyl ether acrylate (Figure 4) with relatively low to low Ð’s and temporal control.59 This monomer scope was recently extended to various other acrylates and methacrylates while also introducing a method for catalyst recycling by using a polymer-supported PC that is based on 3.46 Finally, the ability of PhenS catalysts to polymerize partially fluorinated monomers has also been demonstrated, producing a variety of partially fluorinated polymers with generally low Ð’s.38
Figure 4.

Monomers Polymerized via PET-RAFT by Using PhenN and PhenS Catalysts. (Ref. 38,46,59)
To demonstrate the utility of PET-RAFT in materials manufacturing, existing polymer-based gels were homogeneously modified in an example of living additive manufacturing.61 Since the gels consisted of polymer networks with trithiocarbonate iniferters embedded in the polymer backbone, chain extensions could be achieved upon irradiation by infiltrating the gels with N-isopropylacrylamide and 3. Importantly, since this method involved modification of the existing polymer network, the resulting material was homogeneous in nature in contrast to the heterogeneous materials obtained by simply growing one material on another using previous methods.
Moreover, as these PCs have displayed the ability to mediate polymerizations both via O-ATRP and PET-RAFT, some work has combined these two reaction manifolds into a stepwise synthesis of copolymers of acrylates and methacrylates using 5.62 Capitalizing on the strengths (and weaknesses) of both methods, a multifunctional initiator bearing a trithiocarbonate moiety and an alkyl bromide moiety was synthesized, in which the former functional group would only react during PET-RAFT and the latter during O-ATRP. Thus, the polymerization of methyl acrylate was achieved via PET-RAFT, followed by the polymerization of methyl methacrylate via O-ATRP, resulting in a block copolymer that otherwise would have been challenging to prepare by either method alone.
3.3. Complex Polymer Architectures
One of the hallmarks of a controlled polymerization is the ability to synthesize complex polymer architectures.63-65 For the methods described above, this ability has been demonstrated at various levels, through the synthesis of linear block-copolymers,29,34,36,45,62,66 brushes,39,67 and even star68 polymers (Figure 5). For example, all three original reports on O-ATRP mediated by PhenS, PhenN, and PhenO catalysts showed that PMMA synthesized with these PCs could be isolated and used as a macroinitiator for the synthesis of various block copolymers.29,34,36 Moreover, a report by Xu et al. made use of 5 to synthesize block copolymers of N-isopropylacrylamide and tert-butyl methacrylate, albeit with poor control over Ð.69 Notably, in addition to chain-extending polymers synthesized via O-ATRP, polymers synthesized by other methods can be synthetically modified for use as O-ATRP macroinitiators. Thus, block copolymers containing poly(3-hexylthiophene)45 and poly(ethylene glycol)66 have also been realized, demonstrating the ability of these PCs to tolerate a wide range of functional groups.
Figure 5.

Polymer Architectures Synthesized Using PhenS, PhenN, and PhenO Photoredox Catalysts.
Expanding on this notion of modifying existing polymers, several methods have been reported for synthesizing brush polymers from existing linear homopolymers. One approach that has already been discussed to some extent is that in which sulfonyl halides were installed on the phenyl groups of polystyrene to enable grafting of poly(methyl acrylate) chains.39 Furthermore, fluorinated polymers with chloride-functionalized backbones have been modified using PC 3 to synthesize macromolecules with interesting dielectric properties for electronics applications (eq 2).67,70 Capitalizing on the presence of a chloride moiety in poly(vinylidene fluoride-co-chlorotrifluoroethylene) [P(VDF-co-CTFE)],71 grafting of various acrylates and methacrylates onto P(VDF-co-CTFE) was achieved while also avoiding metal contamination that occurs in traditional ATRP methods.70 The impact of this reduced metal contamination was also evaluated and will be discussed further in a later section (see Section 3.4).67 Finally, the ability of these PCs to synthesize star polymers in the presence of multifunctional initiators was investigated, yielding a range of complex star architectures with up to 8 arms that are composed of either homo- or block-copolymers.68
Thus, a variety of polymeric architectures have become accessible by using PhenS, PhenN, and PhenO photoredox catalysts. In particular, something that should be emphasized is the number of methods using these PCs to modify existing polymer structures and yield increasingly complex architectures. While a single method to polymerize any monomer is ideal, the reality is that most methods have associated strengths and weaknesses. However, the ability of PhenS, PhenN, and PhenO photoredox catalysts to tolerate various functional groups allows methods with complementary strengths to be combined, whether it be in a one-pot synthesis62 or in a multiple-step synthetic sequence, giving rise to polymer architectures that might not be possible by any of these methods alone.
3.4. Avoiding Metal Contamination for Sensitive Applications
Often cited as one of the major advantages of O-ATRP,28,29,34,36 is the use of PhenS’s, PhenN’s, and PhenO’s for the synthesis of polymers without the metal contamination associated with traditional ATRP. Although significant advances have been made toward reducing catalyst loadings72-74 and purifying polymers synthesized by traditional ATRP,75-78 even trace metal contamination remains problematic in polymers for electronics applications.67,70,71 In particular, grafting insulators, such as PMMA, to poly(vinylidene fluoride)-based polymers (PVDF) has shown promise to yield materials suitable for high-pulse capacitors, whereas residual metal ions from traditional ATRP can result in significant dielectric loss.79 As this loss has been attributed to ion migration under an applied electric field, using an organic PC to mediate the grafting process can eliminate this issue, since any catalyst remaining in the polymer should be in the ground state and would thus not be influenced by an applied field.
In this regard, the method developed for the modification of P(VDF-co-CTFE) using 3 (see eq 2) was shown to be capable of activating the C─Cl bond toward hydrogenation71 as well as O-ATRP.70 In a later report, the impact of employing an organic PC versus a traditional copper catalyst was evaluated, demonstrating that polymers prepared via O-ATRP exhibited a far reduced ion mobility compared to polymers prepared using traditional ATRP.67 Moreover, when comparing the materials properties of these two samples, the former exhibited both enhanced discharge energy density and discharge efficiency over a range of applied electric fields, suggesting that 3 can yield these desirable materials with reduced impact on their performance.
In addition to electronics applications, biological applications of polymers have also been cited as potentially metal-sensitive to warrant the investigation of PhenS’s,66 PhenN’s,34 and PhenO’s.36 To this end, the ability of 3 to synthesize amphiphilic diblock copolymers was investigated,66 as these materials have attracted attention for drug and gene delivery.80,81 By modifying poly(ethylene glycol) for use as an ATRP macroinitiator, copolymers of ethylene glycol and glycidyl methacrylate could be obtained, albeit with Ð values well above 1.5. It should be noted, however, that organic PCs for biologically relevant polymers may be unnecessary, as copper is vital to human life and copper dietary supplements have even been used to mediate traditional ATRP.82 Moreover, while these PCs have been shown to be biologically active molecules,83-90 their toxicity in humans has not been investigated and should warrant further study.
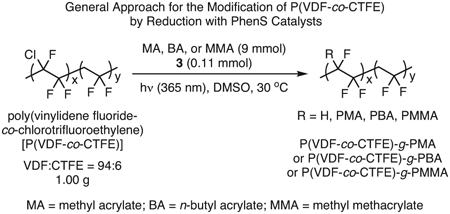 |
eq 2 |
3.5. Surface Modifications
Surface-initiated polymerizations represent a versatile approach for the production of hybrid organic–inorganic materials with interesting surface properties.91 In particular, surface-initiated ATRP (SI-ATRP) has emerged as an important technique capable of yielding such materials with precisely controlled architectures.92 However, until recently, the production of patterned surfaces by SI-ATRP remained a challenge, requiring the use of advanced lithographic and printing methods. To overcome this challenge, a method was developed that capitalizes on the spatiotemporal control achieved in O-ATRP and allows the use of binary photomasks to produce patterned polymer coatings on functionalized silicon surfaces in a single step (eq 3).92 Notably, features at even the micron scale could be produced reliably, demonstrating the high level of precision obtainable by this method.
In addition to the modification of flat surfaces, SI-ATRP photocatalyzed by 3 has also been reported using functionalized silica nanoparticles, which were simultaneously used to investigate parameters influencing PC control in the grafting process.93,94 For example, the effects of various initiating moieties were investigated for both small (16 nm) and large (120 nm) silica nanoparticles, revealing that 2-bromo-2-phenylacetate based tetherable initiators exhibited superior performance to those with 2-bromoisobutyrate moieties.93 Moreover, this work was extended to determine the impact of the tetherable initiator spacer length on the grafted polymer properties. Thus, it was determined that for O-ATRP, increasing the initiator spacer length results in both lower Ð polymer chains and higher grafting density (number of chains per unit of area).93
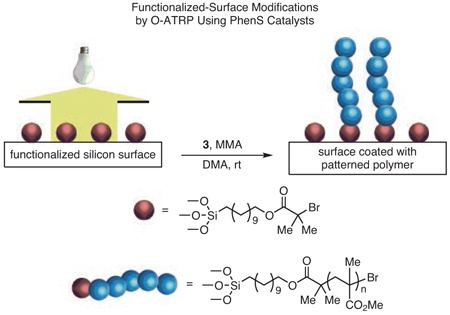 |
eq 3 |
(Ref. 92)
Other work in this area has focused on surfaces of other materials, including iron(III) oxide nanoparticles95 and europiumdoped hydroxyapatite,96 capitalizing on the ability of these PCs to yield controlled polymer grafts without introduction of unwanted metal ions. Moreover, surface grafting of methacrylic acid onto a silicon wafer has also been achieved,51 demonstrating compatibility between this monomer and 3. As controlling the polymerization of methacrylic acid has historically been challenging for ATRP,97 future work should undoubtedly include investigation of the ability of PhenS’s, PhenN’s, and PhenO’s to yield well-defined poly(methacrylic acid).
3.6. Post-Polymerization Modifications
In addition to the ability to synthesize well-defined polymers for various applications, the ability to alter polymers post-polymerization is also desirable. One such example that has already been discussed is the one in which polystyrene was modified to enable grafting of methyl acrylate chains from the aromatic pendants.39 In another example, researchers reported on the ability of 3 to remove chloride, bromide, and trithiocarbonate end-groups from various polymers, enhancing their long-term stability by removing these reactive functionalities.98 Moreover, this method was extended to dehalogenate tethered initiators on functionalized silicon surfaces, allowing for patterns to be prepared on functionalized surfaces prior to use in SI-ATRP. Thus, the utility of these PCs has been demonstrated in a diverse range of polymer-based applications, including polymer synthesis and modification.
4. Applications in Small-Molecule Transformations
Although the primary application thus far of these strongly reducing organic PCs has been in polymer synthesis, several reports have emerged on their application in small-molecule transformations, demonstrating their broader utility as catalysts for diverse chemical reactions.
4.1. Carbon-Carbon Bond Formations
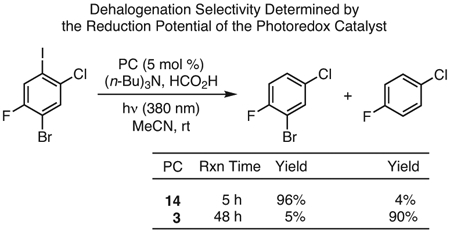
PhenS catalysts have been reported as PCs in the dehalogenation of various organic molecules (eq 4).44 Much like the activation of alkyl halides in O-ATRP, the dehalogenation of organic halides—including aromatic iodides, aromatic and alkyl bromides, and aromatic chlorides—was demonstrated using PC 3.99 While these reactions were originally limited to dehalogenations followed by hydrogenations, the ability of 3 to mediate a radical cyclization provided evidence for a radical mechanism. Thus, this reactivity was later exploited to form C─C bonds with several substrates using PCs 3 and 14.44 In addition, by tuning PC reduction potentials [E0(2PC•+/1PC*) = −2.1 V and −1.5 V vs SCE for 3 and 14, respectively], selectivity for certain halides over others was achieved. For example, using PC 14, iodo functionalities in multi-halide substrates could be targeted. In contrast, and as PC 3 is more reducing, both iodo and bromo functionalities could be targeted while leaving chloro and fluoro groups intact.
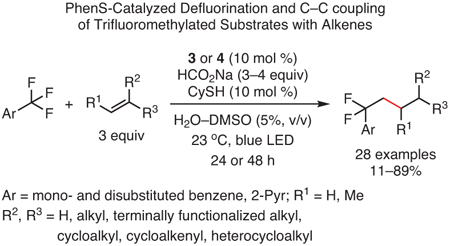
Interestingly, the ability of PhenS catalysts to activate carbon-halogen bonds was also extended to fluorides. Using PC 3 and cyclohexanethiol (CySH) as co-catalyst, C─F bonds in various trifluoromethylarenes were activated for reaction with unactivated alkenes, allowing for the alkylation of several substrates under mild conditions (eq 5).100 Although this activation approaches the thermodynamic limit of PC 3’s reducing ability [−2.07 V vs SCE for 1,3-bis(trifluoromethyl)-benzene; E0(2PC•+/1PC*) = −2.1 V vs SCE for 3], quenching of PC* by 1,3-bis(trifluoromethyl)benzene was demonstrated using Stern–Volmer analysis. Thus, it was proposed that such substrates could be activated to form a radical species capable of mesolytic cleavage of a C─F bond, which would then lead to reaction with alkenes to effect the desired transformations.
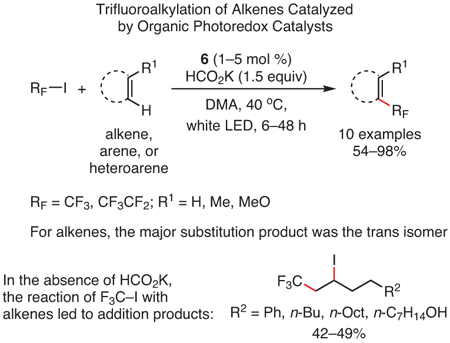
Finally, the trifluoromethylation of several aromatic and olefinic compounds has been reported using PC 6 under visible light irradiation of F3C─I (eq 6),101 as PC 6’s excited state is sufficiently reducing to directly reduce CF3I and generate CF3• for the trifluoromethylation reaction. While such transformations were previously accessible by photoredox catalysis, they required the use of polypyridyl Ru and Ir PCs such as fac-[Ir(ppy)3] (1),102-106 as few PCs possess the excited state reduction potentials necessary to mediate these reactions. However, due to the strongly reducing excited states accessible by PhenS’s, PhenN’s, and PhenO’s, transformations such as these have become accessible without the need for these precious metal PCs,101 demonstrating the potential of these organic PCs as sustainable alternatives to precious metal catalysts.
4.2. Other Coupling Reactions
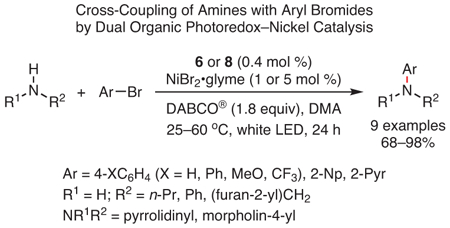
In addition to the C─C bond formations described above, methodologies for C─N and C─S cross-couplings have also been reported using these strongly reducing organic PCs. For example, through the use of a dual photoredox/nickel catalytic system, the coupling of various primary and secondary amines with aryl bromides was achieved in the presence of PCs 6 or 8 (eq 7).101 Furthermore, using PC 8 at 10 times less catalyst loading than the Ir PC used in the seminal report by Oderinde, Johannes, and co-workers,107 a similar approach was employed to couple thiols to aryl bromides, yielding a range of products in moderate-to-high yields (eq 8).101 It should be noted that, while similar C─S coupling reactions were reported using aryl iodides with an Ir PC,107 aryl bromide coupling partners were ineffective in this system. Thus, this reaction (eq 8) represents an example in which these organic PCs have enabled transformations previously inaccessible using precious metals.
 |
eq 4 |
(Ref. 44)
 |
eq 5 |
(Ref. 100)
 |
eq 6 |
(Ref. 101)
 |
eq 7 |
(Ref. 101)
4.3. Selective Decarboxylative Olefinations
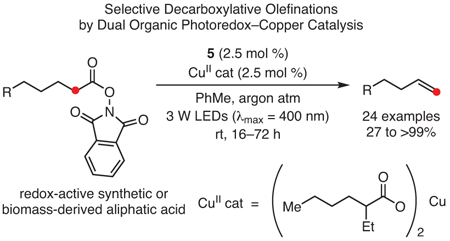
In another example of reactivity enabled by these strongly reducing PCs, PhenN’s were employed in visible-light-mediated decarboxylative olefinations to yield terminal alkenes (eq 9).108 The use of 5 in conjunction with a copper catalyst enabled these transformations to be performed with high selectivity (as preventing isomerization to an internal alkene had previously been challenging), under mild conditions and without the use of precious-metal catalysts. Furthermore, this method was demonstrated for a range of activated aliphatic acids, including some derived from biomass feedstocks—showing that the reaction tolerates a variety of functional groups within the substrates.
4.4. Photocatalytic Phosgene Generation for Organic Synthesis
PC 8 can generate fluorophosgene photocatalytically in situ for the synthesis of carbonates, carbamates, and urea derivatives (Scheme 4).109 While the ability of phosgene derivatives to perform such transformations was previously understood, such syntheses required special equipment for handling phosgene due to its severe toxicity.110,111 Alternatives to this class of reagents do exist, but they tend to be far less effective,111 requiring one to choose between an effective synthesis and the safety of the associated reagents. As such, the ability of this method to generate a phosgene derivative in situ using photocatalysis is highly attractive, since the phosgene reacts quickly once generated, minimizing the risk of exposure and thus the safety concerns surrounding this reagent.
 |
eq 8 |
(Ref. 101)
 |
eq 9 |
(Ref. 108)
Scheme 4.

Photocatalytic Generation of Fluorophosgene in Situ for the Synthesis of Carbonates, Carbamates, and Urea Derivatives. (Ref. 109)
5. Mechanistic Insights Guiding Catalyst Development
Since PCs based on PhenS, PhenN, and PhenO were originally developed for use in O-ATRP, mechanistic work surrounding these PCs has primarily focused on their function in O-ATRP. Thus, mechanistic discussions in this section will be made in the context of this method, although the implications of these discoveries likely extend beyond O-ATRP.
5.1. Photoexcitation, Activation, and Deactivation in O-ATRP
As with any photoredox-catalyzed reaction, the absorption of light is the first important step to the operation of the PC. In the context of O-ATRP, visible-light absorption is preferred over absorption of UV light, as the latter has the potential to initiate undesirable side reactions. To achieve this property, synthetic modifications have been reported for various catalyst families, allowing for the design of strongly reducing but also visible-light absorbing PCs (see Section 2). Since the intensity of a light source can often be tuned with ease, this external stimulus can also be manipulated to influence light absorption by the PC (and thereby the reaction it mediates). This principle was demonstrated by polymerizing MMA in the presence of 8 under various irradiation conditions, where the emission intensity of the light source was modulated.111,112 As a result of decreasing light intensity, molecular-weight growth during polymerization became less controlled and Ð increased, indicating a loss of control over the polymerization. This result is consistent with a decrease in deactivation efficiency, as decreased light intensity yields less PC* and thereby less PC•+Br− to deactivate reactive radicals in solution. Significantly, the performance of 8 appeared to be influenced to a lesser extent than that of 2, suggesting that tolerance to varying reaction conditions can be designed into these PCs.
Once a PC is photoexcited, the lifetime of the desired excited state must be long enough to allow energy or electron transfer to occur with the substrate.5-7 In the case of PhenO’s and PhenN’s, activation has been proposed to occur from 3PC*. As such, the lifetime of 3PC* has been measured for some of the PCs in these families, including 6 (4.3 ± 0.5 μs) and 8 (480 ± 50 μs).101 Interestingly, these lifetimes are competitive with, or even exceed, those of traditional precious-metal-containing PCs (e.g., 1.9 μs for 1),113,114 although only 8 has a comparable quantum yield for the triplet excited state (Φt = 2% for 6 and Φt = 90% for 8).101
However, whether these PCs operate predominately via the 1PC* or 3PC* excited state remains to be determined. Recently, an investigation of electron transfer between photoexcited PhenN’s and methyl 2-bromopropionate (MBP) was presented, suggesting 1PC* may be the most important excited state in regards to catalysis for this family of PCs.115 Similarly, mechanistic investigations related to the dehalogenation of aryl halides have suggested that 3 can operate efficiently from 1pc*.99 on the other hand, others have argued that these PCs likely operate predominately from 3PC* in O-ATRP, as these states tend to be much longer-lived than 1PC*.116 Thus, further studies are required regarding which excited state species of the PC is most pertinent to catalysis, something that may prove to be case-specific.
Regardless of the nature of the excited state, the importance of photoexcitation has been demonstrated with 3.43 For example, upon irradiation, 3 activates methyl 2-bromoisobutyrate (MBiB) with a rate constant kact = 5.8 × 108 M−1 S−1, whereas in the absence of irradiation kact = 1.0 × 10−14 M−1 S−1, demonstrating that ground state 3 is essentially incapable of performing the necessary reduction for activation. Moreover, a comparison of the activation of various initiators suggests similar trends are observable as in traditional ATRP, such as ethyl α-bromophenylacetate (EBP) (kact = 2.0 × 1010 M−1 S−1) being a faster acting initiator than MBiB, while MCiB (the chloride analogue of MBiB) is a slower acting initiator (kact = 1.5 × 106 M−1 S−1) than MBiB.
With regard to deactivation, initial reports proposed this step may occur via bimolecular reaction between 2PC•+Br− and the propagating radical, requiring 2PC•+ to pre-associate with Br prior to deactivation.38,39,34,36,43 Alternatively, other work has suggested that this process may in fact proceed via a termolecular mechanism.43 Using derived activation parameters, the rates of various deactivation pathways were calculated according to Marcus theory and compared to the rate of termination for evaluation of their viability. Based on these calculations, a termolecular deactivation was predicted to be more viable than other pathways involving ISET, OSET, and dissociative ET. However, these calculations did not explicitly account for the entropic penalty associated with a three-body collision, which makes termolecular reactions unfavorable,117 especially considering the species involved are at very low concentrations in O-ATRP. Alternatively, the influence of ion pairing on deactivation in O-ATRP has been reported, supporting a bimolecular mechanism in which PC•+ and Br− form an ion pair prior to deactivation.40
5.2. Intramolecular Charge Transfer in the Excited State
During early investigations of PhenN’s, it was observed that PCs bearing N-aryl substituents with EWGs or extended π systems exhibited noticeably better performance in O-ATRP (especially in regards to producing polymers possessing lower Ð’s) than those bearing electron-donating or electron-neutral N-aryl substituents.34 Through the aid of computational chemistry, it was discovered that the electronic properties of these substituents could influence electron density distribution in the 3PC* excited state, giving rise to intramolecular charge transfer (CT) from the PhenN core to the N-aryl substituent containing EWGs or extended conjugation. Computationally, this property can be observed by the presence of spatially separated SOMOs in 3PC* (Figure 6), as well as by visualizing the shift in electron density upon photoexcitation using electrostatic-potential-mapped electron density diagrams (Scheme 5, Part (a)).40 Experimentally, the effects of CT can be observed (i) visually through the solvatochromism of these PCs, (Scheme 5, Part (b)) and (ii) by using fluorescence spectroscopy for quantitative analysis.35,40 Notably, this intramolecular CT is analogous to the metal-to-ligand CT,30 which is observed in many successful metal-based PCs.118
Figure 6.
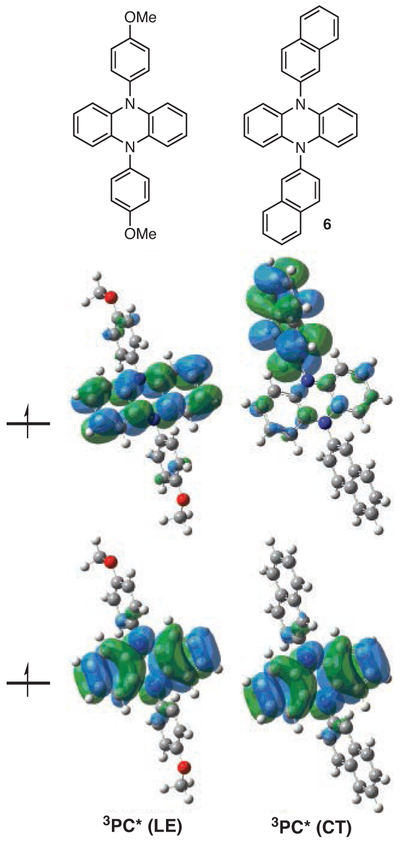
PhenN’s Computed to Have Spatially Separated SOMOs (Right) Were Observed to Perform Better as O-ATRP Catalysts than Those That Possessed Localized SOMOs. (Ref. 40)
Scheme 5.

Comparison of PhenN’s without (Top) and with (Bottom) CT Excited States. (A) Computed Electrostatic-Potential-Mapped Electron- Density Diagrams Portraying the Distribution of Electron Density within PCs upon Photoexcitation to an Excited State, with Red Signifying Larger Populations of Electron Density. (B) Charge Transfer PCs Exhibit Large Solvatochromic Shifts in Their Emissions in Solvents of Different Polarity, While Non-CT PCs Do Not. Solvents of Increasing Polarity from Left to Right: 1-Hexene, Benzene, Dioxane, THF, Pyridine, and DMF. (Ref. 35,40)
After the correlation of these CT properties and their influence on the performance of the PC, several studies have been reported on ways to manipulate CT in favor of improving polymerization control in O-ATRP. For example, following the discovery that PCs with CT character could operate in a range of solvents (whereas non-CT PCs could not), solvent optimization was performed under O-ATRP conditions for a wide range of PCs, including 5 and 6.41 As result, it was discovered that switching the solvent from N,N-dimethylacetamide (DMA) to ethyl acetate (EtOAc) can yield improved control over the polymerization of MMA, as observed through more linear growth of polymer molecular weight and lower Ð (1.08 in EtOAc vs 1.17 in DMA for PC 5). In addition, a recent investigation into the photophysical properties of PCs with and without CT character has suggested that a CT excited state with perpendicular geometry and appropriate energy (e.g., PC 8) can aid intersystem crossing (ISC) to the 3PC*,119 allowing for PCs with favorable photophysical properties to be targeted synthetically. Notably, these findings can be used to explain the observed differences in performance between CT and non-CT PCs, as improved ISC would yield a larger concentration of the active catalytic species in O-ATRP (assuming the PC operates via the 3PC* excited state and not the 1PC*).
5.3. Excimer Formation and Reactivity
The possibility of these PCs forming excimers has also been investigated as a means of understanding their reactivity in thermodynamically challenging reductions.109 In an attempt to prepare carbonates, carbamates, and urea derivatives, it was observed that PC 8 was capable of reducing 4-(trifluoromethoxy)-benzonitrile (15), which was surprising given that this PC should not be thermodynamically capable of reducing this substrate [E*red(2PC•+/3PC*) = −1.7 V whereas E0red(15/15−) = −2.1 V, both vs SCE]. To explain this observation, it was proposed that PC 8 might form excimers under reaction relevant concentrations, leading to the formation of a PC radical anion and radical cation upon photoexcitation. In the absence of an electron donor, these species likely undergo a comproportionation reaction to generate two ground state PC molecules. However, upon addition of an electron donor such as an amine, it was proposed that the PC radical cation could be quenched, resulting in a longer-lived radical anion capable of reducing a substrate. Supporting these hypotheses, quenching of the radical cation upon addition of an amine was observed using transient absorption spectroscopy, and the E0red(1PC/2PC•−) of the radical anion of 8 was measured to be about −2.5 V vs SCE, which is sufficient to reduce 15.109 Thus, excimers of these PCs may offer a means of enhancing their reducing power to access more challenging transformations in the future.
6. Conclusion and Outlook
Until recently, few PCs with strongly reducing excited states existed, especially organic PCs. Thus, PhenS’s, PhenN’s, and PhenO’s represent a unique subset of molecules that are capable of performing challenging reductions catalytically without the use of precious metals. Capitalizing on their strong excited state reduction potentials, these PC families have been widely applied to the synthesis of polymers with controlled molecular weights, low dispersities, and complex architectures. Furthermore, their ability to operate via several mechanisms (e.g., O-ATRP and PET-RAFT) has also been demonstrated. Moreover, the ability of these PC families to mediate a variety of small-molecule transformations has been reported, the scope of which will undoubtedly expand in coming years. To promote this expansion, future investigations focusing on the mechanisms of these PCs in a range of applications will be crucial, allowing for their design principles to be refined to target desired, selective transformations. Moreover, these PCs have a unique potential to increase the long-term sustainability of transformations currently mediated by precious metal catalysts. However, the sustainability of these PCs is currently hindered by the fact that all of the PCs discussed herein to date are synthesized via palladium-catalyzed transformations. Thus, future efforts should also include the development of more sustainable PC syntheses that are not dependent on precious metals.
7. Acknowledgments
The authors are thankful for financial support from Colorado State University, the American Chemical Society Petroleum Research Fund, and the National Institute of General Medical Sciences of the National Institutes of Health (Award Number R35GM119702). D. A. Corbin is grateful for financial support from a CSU Chemistry Graduate Fellowship and Professor Louis S. Hegedus Fellowship. C.-H. Lim is grateful for an NIH F32 Postdoctoral Fellowship (F32GM122392). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Biography

Daniel A. Corbin received his B.S. degree in chemistry in May of 2017 from James Madison University, where he performed research with Professor Brycelyn Boardman synthesizing and investigating metallopolymers with potential as hybrid photovoltaic materials. In the fall of 2017, he moved to Colorado State University in Fort Collins to pursue a doctoral degree under the direction of Professor Garret Miyake. Currently, his research is focused on developing a better understanding of photocatalysts that are used in the organocatalyzed atom-transfer radical polymerization (O-ATRP) in order to improve their performance and expand the boundaries of O-ATRP.

Chern-Hooi Lim is a postdoctoral researcher in the group of Professor Garret Miyake in the Chemistry Department at Colorado State University. He earned his Ph.D. degree in December of 2015 with Professor Charles Musgrave in the Chemical and Biological Engineering Department at the University of Colorado, Boulder. His research interests include computation-driven design of organic photoredox catalysts, photoredox- and nickel-catalyzed cross-coupling reactions, and mechanistic studies on photoredox-catalyzed reactions.

Garret M. Miyake performed his undergraduate studies at Pacific University and earned his Ph.D. degree from Colorado State University under the direction of Professor Eugene Y.- X. Chen. He was a Camille and Henry Dreyfus Environmental Chemistry Postdoctoral Fellow at the California Institute of Technology with Professor Robert H. Grubbs. The Miyake group’s current research interests focus on photoredox catalysis, organocatalyzed atom-transfer radical polymerizations, the synthesis of block copolymers and their self-assembly into photonic crystals, and polymeric composite materials. He has received a Cottrell Scholar Award, a Sloan Research Fellowship, and the 2017 ACS Division of Polymer Chemistry Mark Young Scholar Award.
Footnotes
Trademarks. DABCO® (Evonik Degussa GmbH).
8. References
- (1).Narayanam JMR; Stephenson CRJ Chem. Soc. Rev 2011, 40, 102. [DOI] [PubMed] [Google Scholar]
- (2).Prier CK; Rankic DA; MacMillan DWC Chem. Rev 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Romero NA; Nicewicz DA Chem. Rev 2016, 116, 10075. [DOI] [PubMed] [Google Scholar]
- (4).Shaw MH; Twilton J; MacMillan DWC J. Org. Chem 2016, 81, 6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Blum TR; Miller ZD; Bates DM; Guzei IA; Yoon TP Science 2016, 354, 1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lu Z; Yoon TP Angew. Chem., Int. Ed 2012, 51, 10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Welin ER; Le C; Arias-Rotondo DM; McCusker JK; MacMillan DWC Science 2017, 355, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ludwig JR; Schindler CS Chem 2017, 2, 313. [Google Scholar]
- (9).Nakamura M; Dohno R; Majima T J. Org. Chem 1998, 63, 6258. [DOI] [PubMed] [Google Scholar]
- (10).Hintz S; Mattay J; van Eldik R; Fu W-F Eur. J. Org. Chem 1998, 1998, 1583. [Google Scholar]
- (11).Pandey G; Laha R Angew. Chem., Int. Ed 2015, 54, 14875. [DOI] [PubMed] [Google Scholar]
- (12).Manfrotto C; Mella M; Freccero M; Fagnoni M; Albini A J. Org. Chem 1999, 64, 5024. [DOI] [PubMed] [Google Scholar]
- (13).Dondi D; Protti S; Albini A; Carpio SM; Fagnoni M Green Chem 2009, 11, 1653. [Google Scholar]
- (14).Xia J-B; Zhu C; Chen C J. Am. Chem. Soc 2013, 135, 17494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kotani H; Ohkubo K; Fukuzumi S J. Am. Chem. Soc 2004, 126, 15999. [DOI] [PubMed] [Google Scholar]
- (16).Ohkubo K; Mizushima K; Iwata R; Souma K; Suzuki N; Fukuzumi S Chem. Commun 2010, 46, 601. [DOI] [PubMed] [Google Scholar]
- (17).Xiang M; Meng Q-Y; Li J-X; Zheng Y-W; Ye C; Li Z-J; Chen B; Tung C-H; Wu L-Z Chem.—Eur. J 2015, 21, 18080. [DOI] [PubMed] [Google Scholar]
- (18).Keshari T; Yadav VK; Srivastava VP; Yadav LDS Green Chem. 2014, 16, 3986. [Google Scholar]
- (19).Yang W; Yang S; Li P; Wang L Chem. Commun 2015, 51, 7520. [DOI] [PubMed] [Google Scholar]
- (20).Meyer AU; Jäger S; Hari DP; König B Adv. Synth. Catal 2015, 357, 2050. [Google Scholar]
- (21).Ghosh I; Ghosh T; Bardagi JI; König B Science 2014, 346, 725. [DOI] [PubMed] [Google Scholar]
- (22).Corrigan N; Shanmugam S; Xu J; Boyer C Chem. Soc. Rev 2016, 45, 6165. [DOI] [PubMed] [Google Scholar]
- (23).Zhang G; Song IY; Ahn KH; Park T; Choi W Macromolecules 2011, 44, 7594. [Google Scholar]
- (24).Fors BP; Hawker CJ Angew. Chem., Int. Ed 2012, 51, 8850. [DOI] [PubMed] [Google Scholar]
- (25).Matyjaszewski K Macromolecules 2012, 45, 4015. [Google Scholar]
- (26).Krys P; Matyjaszewski K Eur. Polym. J 2017, 89, 482. [Google Scholar]
- (27).Shanmugam S; Boyer C Science 2016, 352, 1053. [DOI] [PubMed] [Google Scholar]
- (28).Miyake GM; Theriot JC Macromolecules 2014, 47, 8255. [Google Scholar]
- (29).Treat NJ; Sprafke H; Kramer JW; Clark PG; Barton BE; de Alaniz JR; Fors BP; Hawker CJ J. Am. Chem. Soc 2014, 136, 16096. [DOI] [PubMed] [Google Scholar]
- (30).Theriot JC; McCarthy BG; Lim CH; Miyake GM Macromol. Rapid Commun 2017, 38, 1700040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pitre SP; McTiernan CD; Scaiano JC Acc. Chem. Res 2016, 49, 1320. [DOI] [PubMed] [Google Scholar]
- (32).Majek M; von Wangelin AJ Acc. Chem. Res 2016, 49, 2316. [DOI] [PubMed] [Google Scholar]
- (33).Discekici EH; Anastasaki A; de Alaniz JR; Hawker CJ Macromolecules 2018, 51, 7421. [Google Scholar]
- (34).Theriot JC; Lim C-H; Yang H; Ryan MD; Musgrave CB; Miyake GM Science 2016, 352, 1082. [DOI] [PubMed] [Google Scholar]
- (35).McCarthy BG; Pearson RM; Lim C-H; Sartor SM; Damrauer NH; Miyake GM J. Am. Chem. Soc 2018, 140, 5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Pearson RM; Lim C-H; McCarthy BG; Musgrave CB; Miyake GM J. Am. Chem. Soc 2016, 138, 11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Dadashi-Silab S; Pan X; Matyjaszewski K Chem.—Eur. J 2017, 23, 5972. [DOI] [PubMed] [Google Scholar]
- (38).Gong H; Zhao Y; Shen X; Lin J; Chen M Angew. Chem., Int. Ed 2018, 57, 333. [DOI] [PubMed] [Google Scholar]
- (39).Zhao Y; Gong H; Jiang K; Yan S; Lin J; Chen M Macromolecules 2018, 51, 938. [Google Scholar]
- (40).Lim C-H; Ryan MD; McCarthy BG; Theriot JC; Sartor SM; Damrauer NH; Musgrave CB; Miyake GM J. Am. Chem. Soc 2017, 139, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ryan MD; Theriot JC; Lim C-H; Yang H; Lockwood AG; Garrison NG; Lincoln SR; Musgrave CB; Miyake GM J. Polym. Sci., Part A: Polym. Chem 2017, 55, 3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Singh VK; Yu C; Badgujar S; Kim Y; Kwon Y; Kim D; Lee J; Akhter T; Thangavel G; Park LS; Lee J; Nandajan PC; Wannemacher R; Milian-Medina B; Luer L; Kim KS; Gierschner J; Kwon MS Nat. Catal 2018, 1, 794. [Google Scholar]
- (43).Pan X; Fang C; Fantin M; Malhotra N; So WY; Pateanu LA; Isse AA; Gennaro A; Liu P; Matyjaszewski K J. Am. Chem. Soc 2016, 138, 2411. [DOI] [PubMed] [Google Scholar]
- (44).Poelma SO; Burnett GL; Discekici EH; Mattson KM; Treat NJ; Luo Y; Hudson ZM; Shankel SL; Clark PG; Kramer JW; Hawker CJ; de Alaniz JR J. Org. Chem 2016, 81, 7155. [DOI] [PubMed] [Google Scholar]
- (45).Nguyen TH; Nguyen L-TT; Nguyen VQ; Phan LNT; Zhang G; Yokozawa T; Phung DTT; Nguyen HT Polym. Chem 2018, 9, 2484. [Google Scholar]
- (46).Chen M; Deng S; Gu Y; Lin J; MacLeod MJ; Johnson JA J. Am. Chem. Soc 2017, 139, 2257. [DOI] [PubMed] [Google Scholar]
- (47).Wang J; Yuan L; Wang Z; Rahman MA; Huang Y; Zhu T; Wang R; Cheng J; Wang C; Chu F; Tang C Macromolecules 2016, 49, 7709. [Google Scholar]
- (48).Ramsey BL; Pearson RM; Beck LR; Miyake GM Macromolecules 2017, 50, 2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pan X; Lamson M; Yan J; Matyjaszewski K ACS Macro Lett. 2015, 4, 192. [DOI] [PubMed] [Google Scholar]
- (50).Matyjaszewski K; Jo SM; Paik H; Gaynor SG Macromolecules 1997, 30, 6398. [Google Scholar]
- (51).Ramakers G; Krivcov A; Trouillet V; Welle A; Mobius H; Junkers T Macromol. Rapid Commun 2017, 38, 1700423. [DOI] [PubMed] [Google Scholar]
- (52).Discekici EH; Amant AH; Nguyen SN; Lee I-H; Hawker CJ; de Alaniz JR J. Am. Chem. Soc 2018, 140, 5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hook BDA; Dohle W; Hirst PR; Pickworth M; Berry MBB ; Booker-Milburn KI J. Org. Chem 2005, 70, 7558. [DOI] [PubMed] [Google Scholar]
- (54).Lu H; Schmidt MA; Jensen KF Lab Chip 2001, 1, 22. [DOI] [PubMed] [Google Scholar]
- (55).Shanmugam S; Xu J; Boyer C J. Am. Chem. Soc 2015, 137, 9174. [DOI] [PubMed] [Google Scholar]
- (56).Xu J; Jung K; Boyer C Macromolecules 2014, 47, 4217. [Google Scholar]
- (57).Shanmugam S; Xu J; Boyer C Macromolecules 2014, 47, 4930. [Google Scholar]
- (58).Xu J; Shanmugam S; Duong HT; Boyer C Polym. Chem 2015, 6, 5615. [Google Scholar]
- (59).Chen M; MacLeod MJ; Johnson JA ACS Macro Lett. 2015, 4, 566. [DOI] [PubMed] [Google Scholar]
- (60).Xu J; Jung K; Atme A; Shanmugam S; Boyer C J. Am. Chem. Soc 2014, 136, 5508. [DOI] [PubMed] [Google Scholar]
- (61).Chen M; Gu Y; Singh A; Zhong M; Jordan AM; Biswas S, Korley LTJ; Balazs AC; Johnson JA ACS Cent. Sci 2017, 3, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Theriot JC; Miyake GM; Boyer CA ACS Macro. Lett 2018, 7, 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Quirk RP; Lee B Polym. Int 1992, 27, 359. [Google Scholar]
- (64).Grubbs RB; Grubbs RH Macromolecules 2017, 50, 6979. [Google Scholar]
- (65).Hawker CJ; Wooley KL Science 2005, 309, 1200. [DOI] [PubMed] [Google Scholar]
- (66).Kim YA; Park GS; Son K Polym. Int 2018, 67, 127. [Google Scholar]
- (67).Tan S; Xiong J; Zhao Y; Liu J; Zhang Z J. Mater. Chem. C 2018, 6, 4131. [Google Scholar]
- (68).Buss BL; Beck LR; Miyake GM Polym. Chem 2018, 9, 1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Xu Y; Li G; Hu Y; Wang Y Macromol. Chem. Phys 2018, 219, 1800192. [Google Scholar]
- (70).Hu X; Zhang Y; Cui G; Zhu N; Guo K Macromol. Rapid Commun 2017, 38, 1700399. [DOI] [PubMed] [Google Scholar]
- (71).Tan SB; Zhao YF; Zhang WW; Gao P; Zhu WW; Xhang ZC Polym. Chem 2018, 9, 221. [Google Scholar]
- (72).Matyjaszewski K; Jakubowski W; Min K; Tang W; Huang J; Braunecker WA; Tsarevsky NV Proc. Natl. Acad. Sci. U. S. A 2006, 103, 15309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Jakubowski W; Min K; Matyjaszewski K Macromolecules 2006, 39, 39. [Google Scholar]
- (74).Konkolewicz D; Wang Y; Zhong M; Krys P; Isse AA; Gennaro A; Matyjaszewski K Macromolecules 2013, 46, 8749. [Google Scholar]
- (75).Matyjaszewski K; Pintauer T; Gaynor S Macromolecules 2000, 33, 1476. [Google Scholar]
- (76).Zhang H; Abeln CH; Fijten MWM; Schubert US e-Polym. 2006, 6, 90. [Google Scholar]
- (77).Faucher S; Okrutny P; Zhu S Macromolecules 2006, 39, 3. [Google Scholar]
- (78).Canturk F; Karagoz B; Bicak NJ Polym. Sci., Part A: Polym. Chem 2011, 49, 3536. [Google Scholar]
- (79).Yang L; Allahyarov E; Guan F; Zhu L Macromolecules 2013, 46, 9698. [Google Scholar]
- (80).Ranger M; Jones M-C; Yessine M-A; Leroux J-C J. Polym. Sci., Part A: Polym. Chem 2001, 39, 3861. [Google Scholar]
- (81).Siegwart DJ; Oh JK; Matyjaszewski K Prog. Polym. Sci 2012, 37, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Nikolaou V; Anastasaki A; Alsubaie F; Simula A; Fox DJ; Haddleton DM Polym. Chem 2015, 6, 3581. [Google Scholar]
- (83).Gilman H; Shirley DA J. Am. Chem. Soc 1944, 66, 888. [Google Scholar]
- (84).Massie SP Chem. Rev 1954, 54, 797. [Google Scholar]
- (85).Shirley DA; Lehto EA J. Am. Chem. Soc 1957, 79, 3481. [Google Scholar]
- (86).Dietrich LEP; Teal TK; Price-Whelan A; Newman DK Science 2008, 321, 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Price-Whelan A; Dietrich LEP; Newman DK Nat. Chem. Biol 2006, 2, 71. [DOI] [PubMed] [Google Scholar]
- (88).Palmer BD; Rewcastle GW; Atwell GJ; Baguley BC; Denny WA J. Med. Chem 1988, 31, 707. [DOI] [PubMed] [Google Scholar]
- (89).Laursen JB; Nielsen J Chem. Rev 2004, 104, 1663. [DOI] [PubMed] [Google Scholar]
- (90).Mavrodi DV; Blankenfeldt W; Thomashow LS Annu. Rev. Phytopathol 2006, 44, 417. [DOI] [PubMed] [Google Scholar]
- (91).Barbey R; Lavanant L; Paripovic D; SchÜwer N; Sugnaux C; Tugulu S; Klok H-A Chem. Rev 2009, 109, 5437. [DOI] [PubMed] [Google Scholar]
- (92).Discekici EH; Pester CW; Treat NJ; Lawrence J; Mattson KM; Narupai B; Toumayan EP; Luo Y; McGrath AJ; Clark PG; de Alaniz JR; Hawker CJ ACS Macro Lett. 2016, 5, 258. [DOI] [PubMed] [Google Scholar]
- (93).Yan J; Pan X; Wang Z; Zhang J; Matyjaszewski K Macromolecules 2016, 49, 9283. [Google Scholar]
- (94).Yan J; Pan X; Schmitt M; Wang Z; Bockstaller MR; Matyjaszewski K ACS Macro Lett. 2016, 5, 661. [DOI] [PubMed] [Google Scholar]
- (95).Wang X; You N; Lan F; Fu P; Cui Z; Pang X; Liu M; Zhao Q RSC Adv. 2017, 7, 7789. [Google Scholar]
- (96).Zeng G; Liu M; Heng C; Huang Q; Mao L; Huang H; Hui J ; Deng F; Zhang X; Wei Y Appl. Surf. Sci 2017, 399, 499. [Google Scholar]
- (97).Fantin M; Isse AA; Venzo A; Gennaro A; Matyjaszewski K J. Am. Chem. Soc 2016, 138, 7216. [DOI] [PubMed] [Google Scholar]
- (98).Mattson KM; Pester CW; Gutekunst WR; Hsueh AT; Discekici EH; Luo Y; Schmidt BVKJ; McGrath AJ; Clark PG; Hawker CJ Macromolecules 2016, 49, 8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Discekici EH; Treat NJ; Poelma SO; Mattson KM; Hudson ZM; Luo Y; Hawker CJ; de Alaniz JR Chem. Commun 2015, 51, 11705. [DOI] [PubMed] [Google Scholar]
- (100).Wang H; Jui NT J. Am. Chem. Soc 2018, 140, 163. [DOI] [PubMed] [Google Scholar]
- (101).Du Y; Pearson RM; Lim C-H; Sartor SM; Ryan MD; Yang H; Damrauer NH; Miyake GM Chem.—Eur. J 2017, 23, 10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Pham PV; Nagib DA; MacMillan DWC Angew. Chem., Int. Ed 2011, 50, 6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Nguyen JD; Tucker JW; Konieczynska MD; Stephenson ARJ J. Am. Chem. Soc 2011, 133, 4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Iqbal N; Choi S; Ko E; Cho EJ Tetrahedron Lett. 2012, 53, 2005. [Google Scholar]
- (105).Wallentin C-J; Nguyen JD; Finkbeiner P; Stephenson CRJ J. Am. Chem. Soc 2012, 134, 8875. [DOI] [PubMed] [Google Scholar]
- (106).Koike T; Akita M Top. Catal 2014, 57, 967. [Google Scholar]
- (107).Oderinde MS; Frenette M; Robbins DW; Aquila B; Johannes JW J. Am. Chem. Soc 2016, 138, 1760. [DOI] [PubMed] [Google Scholar]
- (108).Tlahuext-Aca A; Candish L; Garza-Sanchez RA; Glorius F ACS Catal. 2018, 8, 1715. [Google Scholar]
- (109).Petzold D; Nitschke P; Brandl F; Scheidler V; Dick B; Gschwind RM; König B Chem.—Eur. J 2019, 25, 361. [DOI] [PubMed] [Google Scholar]
- (110).Schneider W; Diller W Phosgene In Ullmann’s Encyclopedia of Industrial Chemistry, 5th ed.; Elvers B, Hawkins S, Schulz G, Eds.; VCH: Weinheim, 1991; Vol. A 19, pp 411–420. [Google Scholar]
- (111).Ryan MD; Pearson RM; French TA; Miyake GM Macromolecules 2017, 50, 4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Cotarca L; Geller T; Répási J Org. Process Res. Dev 2017, 21, 1439. [Google Scholar]
- (113).Flamigni L; Barbieri A; Sabatini C; Ventura B; Barigelletti F Photochemistry and Photophysics of Coordination Compounds: Iridium In Photochemistry and Photophysics of Coordination Compounds II; Balzani V, Campagna S, Eds.; Topics in Current Chemistry Series, Vol. 281; Springer: Berlin, Heidelberg, 2007; pp 143–203. [Google Scholar]
- (114).King KA; Spellane PJ; Watts RJ J. Am. Chem. Soc 1985, 107, 1431. [Google Scholar]
- (115).Koyama D; Dale HJA; Orr-Ewing AJ J. Am. Chem. Soc 2018, 140, 1285. [DOI] [PubMed] [Google Scholar]
- (116).Jockusch S; Yagci Y Polym. Chem 2016, 7, 6039. [Google Scholar]
- (117).Espenson JH Chemical Kinetics and Reaction Mechanisms, 2nd ed.; McGraw-Hill Series in Advanced Chemistry; McGraw-Hill: New York, NY, 2002. [Google Scholar]
- (118).Arias-Rotondo DM; McCusker JK Chem. Soc. Rev 2016, 45, 5803. [DOI] [PubMed] [Google Scholar]
- (119).Sartor SM; McCarthy BG; Pearson RM; Miyake GM; Damrauer NH J. Am. Chem. Soc 2018, 140, 4778. [DOI] [PMC free article] [PubMed] [Google Scholar]