

Plasmon-like interactions stabilize folded protein states and offer a long-range interaction mechanism for solvated biomolecules.
Abstract
Quantum-mechanical van der Waals dispersion interactions play an essential role in intraprotein and protein-water interactions—the two main factors affecting the structure and dynamics of proteins in water. Typically, these interactions are only treated phenomenologically, via pairwise potential terms in classical force fields. Here, we use an explicit quantum-mechanical approach of density-functional tight-binding combined with the many-body dispersion formalism and demonstrate the relevance of many-body van der Waals forces both to protein energetics and to protein-water interactions. In contrast to commonly used pairwise approaches, many-body effects substantially decrease the relative stability of native states in the absence of water. Upon solvation, the protein-water dispersion interaction counteracts this effect and stabilizes native conformations and transition states. These observations arise from the highly delocalized and collective character of the interactions, suggesting a remarkable persistence of electron correlation through aqueous environments and providing the basis for long-range interaction mechanisms in biomolecular systems.
INTRODUCTION
Water is an essential basis of life. It provides the environment in which the biomolecular machinery can exist and function. By screening and stabilizing static electronic multipoles, water crucially alters the structure, stability, and dynamics of biomolecules (1–3). The favorable exposure of moieties with static electronic multipoles to water and the corresponding burying of nonpolar residues into a hydrophobic core are also an important twofold driving force for protein folding: First, the (short-range) interaction with the aqueous environment is optimized, and second, the disruption of the dynamic hydrogen bond network of the surrounding water by hydrophobic residues is minimized (3–5). While the importance of this “hydrophobic effect” and the pivotal role of water for biomolecular systems are under no dispute (4, 5), the underlying fundamental physics of solvated (bio)molecular systems is still not fully explored and understood. In particular, here, we focus on the quantum-mechanical nature of solute-solvent interactions. It has already been shown that polarization effects and the many-body character of bonded interactions and hydrogen bond networks play an important role for solvated systems (3, 6–8); but long-range van der Waals (vdW) dispersion interactions also form an essential component for water and for both intraprotein and protein-water interactions. This vdW component, however, has not been investigated in full detail or on a fundamental level up to now. In the present study, we address exactly this intricate issue and find that these quantum-mechanical interactions can account for up to 30% of the total solvation energy. Together with their essential role for intraprotein and interprotein interactions, this calls for a more complete microscopic understanding of vdW dispersion forces under physiological conditions, which is imperative to shed light on the physics of proteins in aqueous solvation.
Noncovalent vdW dispersion interactions arise from instantaneous, correlated fluctuations of the electron density and, hence, are inherently quantum-mechanical and many-body in nature. However, current solvation models or molecular mechanics force fields include them only in a phenomenological manner via pairwise additive potentials. Recent studies demonstrate that, for a variety of systems including polypeptides (8, 9), such approximations can fundamentally fail and that one has to explicitly account for the intrinsic many-body character of vdW interactions (9–18)—even for the properties of pristine water (19) and its surfaces (20). Moreover, pairwise or phenomenological models only provide a very approximate description of the energetic aspects of vdW forces and no description of the underlying quantum mechanics. Such insights, however, can be vital to comprehend and conceptually understand vdW interactions as recently illustrated for hybrid and nanostructured systems (21, 22) or π-π stacked molecules (15). Note that the dielectric permittivity of water has an expected value around 2.3 (23) at the frequencies of the electronic fluctuations, which are responsible for dispersion interactions, i.e., at a few petahertz. Therefore, in contrast to static electronic multipoles, vdW interactions are not strongly screened by aqueous environments and, thus, can give rise to long-range interactions also in solvated systems. As such, long-range correlation forces may play an important role for the long-range ordering often observed in biological systems or form the quantum-mechanical basis for the emergence of coherent molecular vibrations (13, 24). Such collective nuclear behavior has been proposed to play an important role in long-distance recognition among biological macromolecules (25–28). Within the conventional view of solvated proteins, however, the basis for long-range recognition under physiological conditions is still controversially discussed. Recent studies also suggest connections between collective electronic fluctuations—the basis of vdW dispersion interactions—and enzymatic action on DNA (29) or pharmaceutical activity (30).
In this work, we aim at a more complete microscopic description of solvated proteins and report an explicitly quantum-mechanical study, which accounts for many-body dispersion interactions as well as for electrostatics, polarization, and hydrogen bonding. Molecular mechanics approaches are often found to reliably capture the latter three, (semi)classical interactions. This, however, usually holds true only for certain conditions (typically designed for the liquid phase at room temperature and ambient pressure), which shows that the physical description is incomplete. In addition, conventional classical potentials show limited transferability with respect to the conformational space, and results can vary widely among different force fields, when trying to cover both folded and unfolded states or intrinsically disordered proteins (31–34). ‘As especially the quantum-mechanical, nonlocal dispersion interactions, in particular, are often described insufficiently and inconclusively by conventional approaches (9, 35), we focus on a comprehensive description of vdW interactions and collective electronic behavior to highlight the role of water for the vdW energetics of prototypical fast-folding proteins. In this context, previous studies have pointed out that traditional molecular mechanics potentials and water models likely provide an unbalanced description of vdW interactions for proteins in water (36), which typically results in an overcompaction of unfolded states (31, 32, 34). Typically, this unbalanced description is approached by adapting the pairwise vdW interaction coefficients for the intraprotein, water-water, or protein-water interaction (32, 34, 36). However, such adaptations can still be highly system dependent especially when also considering disordered proteins (33). In this work, we seek to understand the fundamental basis for the failure of the traditional models in a bottom-up approach. Such understanding is essential to pave the way toward a more accurate and balanced description of the relevant conformational space of biomolecules, which, in the end, plays a key role for predicting folding mechanisms, barriers, and potential misfolds. Our calculations have been carried out using a combined approach (37) of the self-consistent charge density-functional tight-binding (DFTB) method (38) and accurate ab initio dispersion models, which allows for a robust, albeit approximate, quantum-mechanical treatment on an atomistic level. An ab initio description of vdW interactions is the only way to study the role of the solvent, as force field methods are typically strongly limited in their transferability between gas and liquid phase due to their high degree of parameterization. In our study, we focus on a comparison of a pairwise additive description of vdW interactions and an accurate, quantum-mechanical many-body treatment. The pairwise vdW models are represented by the vdW(TS) (39) as well as Grimme’s D2 (40) and D3 (41) approaches. Such an approximate, pairwise formalism represents the basis for the standard phenomenological description of long-range correlation in biomolecular simulations via Lennard-Jones potentials. For comparison, we study the vdW interaction within the Many-Body Dispersion formalism (MBD) (10, 42, 43), which accounts for the many-body character of vdW dispersion interactions to infinite order in perturbation theory within an interatomic framework and has been proven to provide quantitative improvements and a better qualitative understanding compared to the pairwise additive approximation in numerous studies (9–16). In the case of polarizable supramolecular complexes and complex molecular crystals, which show similar bonding features as biomolecular systems, MBD allows reaching a quantitative agreement to within 1 and 0.5 kcal/mol, respectively, when compared to (near-)exact quantum-mechanical methods and experiments (12, 15, 44). Yet, its computational efficiency, together with modern implementations and the ever-growing availability of computational resources, allows for treatment of systems consisting of several thousands of atoms. The MBD formalism therefore represents the physically most-complete microscopic vdW method for investigating the fundamental physics of vdW dispersion interactions in solvated proteins and biomolecules. Moreover, it also represents a model for collective electronic fluctuations (15, 21, 22)—a molecular analog to the plasmon pseudoparticle in metallic systems—which we will use to further characterize the protein-water interaction.
RESULTS
We exemplify our findings in detail for the Fip35 Hpin1 WW domain (Fip35-WW) and further showcase their general validity for the de novo chignolin variant “cln025” and the fast-folding Nle/Nle double mutant of the villin headpiece (HP35-NleNle). The folding trajectories of the three proteins have been obtained in atomistic detail and explicit solvent in previous molecular dynamics simulations by Shaw et al. (45), Lindorff-Larsen et al. (46), and Ensign et al. (47), respectively.
Intraprotein interactions and many-body dispersion effects
We start out by investigating the Fip35-WW trajectory by artificially removing the surrounding solvent from a molecular dynamics trajectory in explicit water; i.e., we first focus on intraprotein interactions, where dispersion forces represent one of the main sources of interaction within the protein core. Accordingly, we observe an increased magnitude of the vdW energy while this core is being formed and particularly during the hydrophobic collapse in all applied dispersion models (see Fig. 1 and the Supplementary Materials). Notably, in comparison to the results obtained within the pairwise approaches, many-body dispersion effects consistently decrease the relative stability of the native state for the isolated protein by 6 kcal/mol on average, cf. Fig. 1 (bottom). The outliers of this general behavior observed around 15 and 26 μs correspond to transient, partly folded intermediates. The relative destabilization by beyond-pairwise contributions can be explained by an overestimation of the intracore vdW interactions (“overcorrelation”) in the pairwise approximation. By reducing the interaction to pairwise additive potentials, a two-body formulation assumes ideal correlation between all pairs of atoms and, with that, neglects the complex geometrical arrangement within the protein core. Such geometrical constraints limit the emergence of correlated fluctuating dipole patterns and thus lower the interaction energy as already observed for a wide variety of systems (11, 12, 14, 15, 48). For small peptides, such effects have been found to be mostly negligible (35, 49). Our findings show that for larger biomolecules, however, a many-body treatment of vdW interactions is essential, which is in line with the findings of Schubert et al. (9) for 20-residue peptides.
Fig. 1. Intraprotein vdW interaction energy along the folding trajectory of Fip35-WW in solvated geometry.
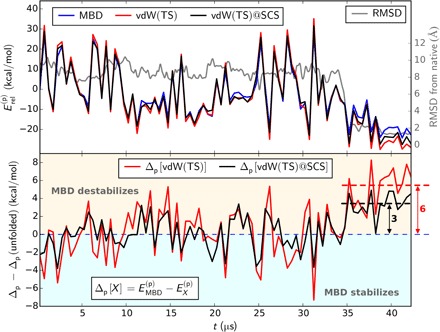
Top: vdW energetics as obtained with MBD and the pairwise approaches vdW(TS) and vdW(TS)@SCS, i.e., vdW(TS) with self-consistent screening. RMSD from native state shown in grey. Bottom: Beyond-pairwise contributions, as given by the difference between many-body and pairwise treatment. For a more comprehensive version, see Supplementary Materials.
In the MBD formalism, we make use of a two-step procedure: We obtain effective, screened atomic polarizabilities from self-consistent electrodynamic screening to account for the presence of multiple fluctuating dipoles in the system and then solve a many-body Hamiltonian, which is defined in terms of these polarizabilities, to capture many-body vdW interactions. To study the effect of each step, we combined vdW(TS) with the self-consistent screening procedure. In this variant, which we refer to as vdW(TS)@SCS, screened interaction coefficients enter the pairwise additive potentials instead of the hybridized chemical analog used in vdW(TS). In this way, we account for the effects on atomic polarizabilities due to the local field of the surrounding dipoles but do not include long-range many-body interactions. Within vdW(TS)@SCS, we already capture some part of the destabilization of native states amounting to 3 kcal/mol (cf. Fig. 1). Thus, half of the overstabilization in vdW(TS) is from neglecting the presence of multiple dipoles in the system and half is from the many-body character of dispersion interactions. This also implies that for a proper description of gas phase proteins, one has to account for the screening of polarizabilities and the many-body nature of vdW interactions.
vdW solvation energy
Figure 2 (top) shows the vdW contribution to the solvation energy obtained with MBD and the vdW(TS) model, as defined by
| (1) |
with ps referring to Fip35-WW in solvation, p referring to Fip35-WW in gas phase, and s referring to the pristine solvent. As an artifact of the abovementioned overcorrelation within the pairwise approach, we find a consistent overestimation of the dispersion contribution in vdW(TS). In terms of the relative solvation energy, however, pairwise and many-body treatment show the same general trend, which qualitatively follows the inverse root mean square deviation (RMSD) from the native state with a step coinciding with the collapse of the protein into the native, more globular shape. This finding can be explained by the removal of hydrophobic residues from the protein-water interface and thus decreasing their interaction with the solvent. The average dispersion contribution to the solvation energy drops by 29 kcal/mol (15%) at the hydrophobic collapse. The step-like behavior of the vdW solvation energy along the trajectory is even more distinct than for the intraprotein vdW interaction in the gas phase and almost resembles a two-state model of folded and unfolded states. As such, the vdW solvation energy strongly correlates with the protein’s deviation from the folded conformation. This feature has been found for all dispersion models considered here: the MBD formalism and the pairwise approaches vdW(TS), D2, and D3.
Fig. 2. Relative vdW solvation energy, , during the folding process of the Fip35-WW.
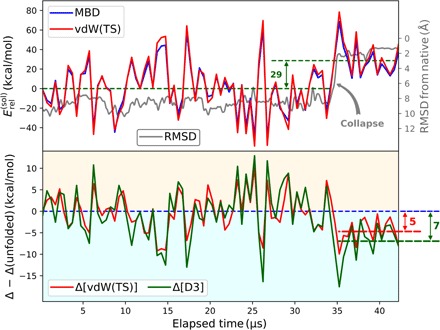
Top: Backbone RMSD from final conformation illustrating the hydrophobic collapse around 35 μs (gray). The vdW contribution to the relative solvation energy is shown for the pairwise vdW(TS) model (red) and MBD (blue). Bottom: Difference in the relative stabilization by the solvent between MBD and the pairwise vdW(TS) and D3 referenced to the unfolded state.
Comparing many-body and pairwise treatment of dispersion interactions, Fip35-WW no longer features a consistent change in the relative stability of native versus non-native conformations once embedded in an aqueous environment. Thus, beyond-pairwise effects in the protein-water vdW interaction stabilize folded conformations. Correspondingly, we see a clear increase in the relative vdW solvation energy for native and native-like states, when comparing the pairwise models to MBD [5 kcal/mol for vdW(TS) and 7 kcal/mol for D3]. This shift is due to the lack of a systematic many-body (de)stabilization in the total vdW energy of solvated Fip35-WW and the water box during the whole folding trajectory, combined with an inversion of the behavior observed for the isolated protein shown in Fig. 1 (see Eq. 1 and the Supplementary Materials). This implies that the protein-water interaction compensates for the destabilization of native states via many-body dispersion effects, observed in vacuo. In summary, besides screening permanent electronic multipoles, water also provides the necessary environment to stabilize native conformations via beyond-pairwise vdW interactions, which counteracts the destabilizing effect that such many-body terms have on the intraprotein interaction.
The plasmon-like character of protein-water vdW interactions
As has been shown previously, MBD also provides a model for the intrinsic electronic fluctuations (15, 21, 22): In analogy to nuclear quantum effects, the electronic fluctuations, which ultimately give rise to vdW interactions, can be understood as the zero-point oscillations around the average electron density. The MBD formalism gives access to an orthonormal decomposition of this zero-point fluctuation, which can be interpreted as “eigenmodes” of the electron density. A detailed analysis of these electronic eigenmodes reveals that the number of very localized high-frequency fluctuations, formerly mainly located on the solute, substantially decreases upon coupling to the surrounding water. This implies a delocalization of electronic fluctuations and an increase in the collectivity of electronic behavior. This plasmon-like character and the delocalization over protein and solvent form the fundamental reason for the stabilization of native states via many-body dispersion effects in the protein-water interaction. The role of the surrounding solvent can be seen as providing weakly structured polarizable matter, which attenuates the destabilizing many-body effects observed in vacuo for native and partially folded states. To gain further insight into the characteristics of protein-water vdW interactions, we additionally obtain the contribution of individual electronic fluctuations to the solvation energy: The main contributions to the vdW solvation energy are due to highly collective, plasmon-like electronic fluctuations around 2.5 PHz (120 nm), such as the one depicted in Fig. 3A. Their contribution to the total protein-water interaction is 14 to 16% (≈41 kcal/mol) and exceeds 20% (≈6 kcal/mol) for relative solvation energies.
Fig. 3. Characteristics of correlated electronic fluctuations.
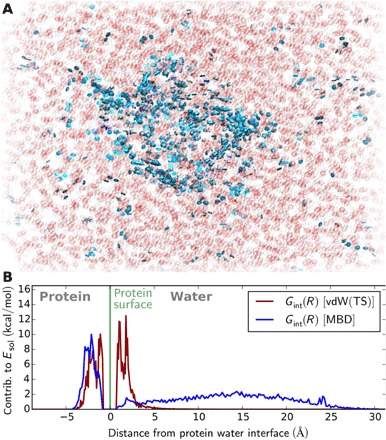
(A) Illustration of low-frequency plasmon-like fluctuations in solvated Fip35-WW, which show the largest contribution to the protein-water interaction (solvent shown in atomistic detail: oxygen, red; hydrogen, white). The arrows (blue) depict the direction of simultaneous electron density deformations (eigenmode of the electron density). If no arrow is shown, the given atom does not contribute notably to the eigenmode. (B) Contributions to the vdW solvation energy within the pairwise vdW(TS) approach and the MBD formalism as radial distribution functions.
As demonstrated in Fig. 3A, such fluctuations commonly feature large charge displacements along the polarizable protein backbone coupled to electronic fluctuations throughout the solvent. While a number of these plasmon-like fluctuations remain largely on the protein, many reach from the protein backbone inside the hydrophobic core far into the aqueous environment. Comparing the radial distribution of the contributions to the vdW solvation energy between the pairwise vdW(TS) and MBD models, as shown in Fig. 3B, reveals a notable difference in the interaction range within the two treatments: In the pairwise model, the contribution of solvent atoms to the vdW solvation energy subsides beyond 6 Å, i.e., roughly twice the sum of the vdW radii of carbon and oxygen. Accounting for many-body dispersion effects, on the other hand, shows that electronic correlation between the protein and solvent atoms up to 25 Å from the protein-water interface is still relevant for the protein-water interaction. This reflects the weakness of the screening of dispersion forces by the solvent and is in evident contrast to the often assumed locality of vdW interactions in solvated systems. While such a range is unprecedented in the context of solvated systems, similar and larger interaction ranges have already been found for molecular crystals (16) or nanostructures (11). From a different point of view, Fig. 3B represents a radial analysis of the change in the distribution and frequency of electronic fluctuations introduced by embedding the protein in water. It thus demonstrates that, while the atomistic structure and the local dynamics of water typically remain largely unperturbed beyond a few solvation layers (3, 50), the instantaneous electronic structure can indicate the presence of a protein over large distances. Such long-range collective electronic behavior and the ensuing nonlocal nuclear dynamics could be experimentally measured thanks to recent advances in terahertz spectroscopy (51–53), as we will further discuss below.
Effect of secondary structure
Fip35-WW is a showcase example for the formation of β sheets. To test the general validity of our hypotheses, we carried out the same analysis for the modified villin headpiece, HP35-NleNle (formation of α-helical entities), and the cln025 variant of the de novo protein chignolin (plain β-hairpin formation). Our analysis confirmed our early findings for Fip35-WW. The vdW contribution to the solvation energy reflects the trend of the inverse RMSD from the native structure with a drop of 15% at the hydrophobic collapse. Again, the protein-water interaction counteracts the destabilizing many-body dispersion effects observed in gas phase and increases the relative stabilization of native states with respect to unfolded structures. In addition, collective plasmon-like electronic fluctuations have been found to show a major contribution to the total and relative solvation energy for both the hairpin-forming cln025 and the helix-forming HP35-NleNle. We would like to point out that the above conclusions remain unaltered, when studying structures obtained from simulations using more recent and accurate molecular mechanics approaches, which avoid the spurious overcompaction of unfolded protein states [namely, a99SB-disp with the TIP4P-D water model (32, 34); see the Supplementary Materials].
Figure 4 summarizes the abovementioned features and highlights the general validity of the present findings in the biomolecular context. Independent of the secondary structure to be formed, the vdW solvation energy captures the hydrophobic collapse in the form of a sizable jump in stabilization (“ΔEsol at collapse”) for all considered proteins. In addition, the consistent increase in the relative stability of native states due to the many-body character of protein-water vdW interactions and the significance of low-frequency, plasmon-like electronic fluctuations [as characterized by their contribution to relative solvation energies, ]turned out to be independent of the final secondary structure. We note that the magnitudes of the different quantities are not necessarily representative for secondary structure elements as also the system size varies from 6000 to 14,000 atoms. The systematic investigation of the relation between secondary structure elements and the magnitude of the present observations and characteristic features of fluctuation patterns, as shown in Fig. 3A, is beyond the scope of the present publication and subject to ongoing studies.
Fig. 4. Characteristics of protein-water dispersion interactions.
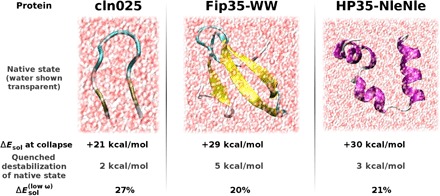
Independent of the secondary structure, the vdW solvation energy captures the hydrophobic collapse in the form of a 20–30 kcal/mol jump (“ΔEsol at collapse”), and many-body protein-water vdW interactions consistently stabilize native states in solvation. Low-frequency, collective electronic fluctuations contribute substantially to the relative solvation energy [] in all cases.
DISCUSSION AND OUTLOOK
In conclusion, we have shown that many-body dispersion effects lead to a considerable relative destabilization (≈4.5 kcal/mol for the proteins studied here) of the native state of solvated proteins in terms of the bare intraprotein interaction. Here, we find that the screening of the instantaneous dipoles due to the surrounding dipole field and many-body interactions contribute in similar parts to the destabilization. Notably, this effect is of a comparable order of magnitude as estimates for the zero-point vibrational and entropic contribution found for the folding of isolated polypeptides (54, 55). A detailed analysis and comparison of these effects is the subject of future work. The destabilization via many-body dispersion effects can play an important role in explaining why proteins often do not adopt the same folded conformation in the gas phase and in solvation. It also indicates how the neglect of the inherent many-body character of dispersion interactions in traditional vdW approaches (and molecular mechanics force fields) can lead to a spurious description of intraprotein interactions in general.
In aqueous solvation, the vdW contribution to the solute-solvent interaction of (small) proteins closely tracks the conformational changes of the folding process. The hydrophobic collapse of the protein is accompanied by a jump of about 15% (20 to 30 kcal/mol) in the vdW solvation energy. The total electronic energy of solvation, for comparison, does not provide such clear insight (see the Supplementary Materials)—only the free energy of solvation does. The beyond-pairwise contributions to the protein-water vdW interaction favor folded states, and thus, the many-body aspect of solvation leads to a considerable stabilization of native conformations. So, a pairwise vdW model overestimates the internal interaction as a driving force of folding (pairwise additive potentials in general favor minimal atom-atom distances and thus globular shapes), while the protein-water vdW interaction should have a larger stabilizing effect on the native state than the pairwise formalism suggests. Careful measurements of the solvation enthalpy of folded and disordered proteins would allow one to further study the correctly balanced description of intraprotein and protein-water interactions and to estimate the effect of beyond-pairwise vdW interactions via complementary simulations. The lack of stabilization in the pairwise approach is the result of a distinct many-body character of the protein-water dispersion interaction in the form of delocalization and a high degree of collectivity of electronic fluctuations across protein and solvent. This plasmon-like character of vdW interactions in solvated systems can also be pivotal for other quantities, as demonstrated for the protein-water interaction range in Fig. 3B.
Collective electronic petahertz fluctuations should also directly manifest in the system’s (response) properties. As such, the plasmon-like fluctuations are, in principle, directly accessible via (vacuum) ultraviolet spectroscopy. However, this phenomenon would need to be carefully disentangled from ionization and other excitation processes in this spectral range, e.g., the Rydberg series of water. Our study shows that the findings summarized above can be generalized for helix-, β sheet–, or hairpin-forming proteins and are, thus, independent of secondary structure motifs. So, an accurate description of solvated proteins generally requires capturing the subtle balance between beyond-pairwise effects on the intraprotein vdW interaction (destabilizing native states) and the highly collective, plasmon-like character of protein-water interactions (stabilizing native states). With increasing system size and complexity, finding this balance without explicit account for the quantum-mechanical many-body nature of vdW interactions is an intricate task, and failure to do so can contribute to the fundamental origin of the previously reported (31–34, 36) unbalanced description of vdW forces by traditional pairwise molecular mechanics potentials and water models.
The previously proposed remedies for this shortcoming effectively involve a general adaptation of the relative magnitudes of protein-protein and protein-water dispersion interactions (31, 32, 34). In contrast to these “static fixes,” our findings suggest that conventional pairwise potentials actually lack a conformation-dependent adaptation of intraprotein and protein-water vdW interactions due to the neglect of many-body effects (cf. Figs. 1 and 2). In the spirit of a static fix, a global rescaling of pairwise additive vdW energetics considerably improves the obtained protein-water interaction (see the Supplementary Materials and fig. S5). Nevertheless, this still shows considerable deviations and is highly system, conformation, and size dependent (no rescaling provides optimal performance for small molecular systems). This renders the simple rescaling without conformation- and configuration-dependent adaptations insufficient for treating (bio)molecules on different length scales, as required for describing assembly or docking processes, for example. Further analysis of the relation between structure and the many-body (de)stabilization represents a promising avenue toward more transferable and first principles–motivated “fixes” to conventional potentials and is subject to ongoing research. Last, we have also performed our analysis based on a new sampling of the folded and unfolded states of the chignolin variant cln025, which has been obtained using the recently developed a99SB-disp force field in conjunction with the TIP4P-D water model (32, 34). This modified approach has been designed and shown to provide a more balanced description of intraprotein and protein-water interactions and thus avoid the spurious overcompaction of unfolded states. The additional analysis confirmed the results reported above and shows that the present conclusions are not an artifact of an unphysical sampling of unfolded states but truly represent the effect of neglecting many-body vdW physics in pairwise additive approaches (for further details, see the Supplementary Materials).
In a broader perspective, our findings imply that an effective pairwise additive treatment of vdW interactions and derivative molecular mechanics potentials can provide accurate energetics for a particular application, but only explicitly quantum-mechanical models, such as the one used here, allow one to attain an unbiased microscopic understanding of biomolecular interactions in a more general context. On the basis of Fig. 3B, for instance, we expect that for several solutes or a larger solute, the approximation of pairwise additivity can fail on a fundamental level. The persistence and collectivity of electronic fluctuations through the solvent can mediate long-range correlation forces between individual solutes or moieties of solvated macromolecules, and an effective pairwise description might not be able to reproduce the subtle balance between long-range correlation on all these scales. As such, plasmon-like interactions can also substantially affect molecular assembly and the formation of tertiary structures. In addition, complex long-range fluctuations of the electron density are less sensitive to the instantaneous solvent structure and thus to thermal fluctuations, which makes them an ideal contender for biomolecular recognition. In this form of recognition, the solvent provides electron density that serves as a mediator for long-range interaction, while the actual atomistic structure and the nuclear dynamics of the solvent do not necessarily have to be altered in the process, which has been concluded from a number of experiments (3, 50). It is worthwhile to mention, however, that most of these experiments probed rather local interactions and dynamics. Recent terahertz spectroscopy experiments, for instance, show that the presence of a solute can have a considerable effect on the long–time scale dynamics and long-range polarization of water (51–53, 56). Obviously, such correlations require a long-range interaction mechanism, which is not present in the traditional, classical view of biomolecular systems, but represented by the plasmon-like, many-body vdW forces shown in this work. From previous studies, we expect the observed long-range persistence of electron correlation through aqueous environments to mainly manifest in the longer–time scale nuclear dynamics of the system. Such behavior has been observed in crystalline molecular systems, where many-body dispersion effects particularly affect low-frequency (“slow”) phonon modes (13, 24). Long-range electronic correlation between (solvated) biomolecules can also form the quantum-mechanical basis for correlated collective nuclear motion within the respective partners. Such concerted motion is essential for the emergence of coherent molecular vibrations, a promising explanation for long-range recognition through electrodynamic interaction of the resulting oscillating molecular dipoles (27, 28), or coordinated enzymatic action (29). Likewise, the presented long-range interaction mechanism can also provide a qualitatively new explanation of allosteric regulation as it is observed in various biomolecular systems, e.g., hemeproteins or the lac operon. Together, our findings apply in a broader context of biomolecular interactions—not just in the case of protein folding as exemplified here.
Obviously, the stability and functionality of biomolecules are ultimately determined by their free energy. Hence, this work represents a first step toward a more fundamental understanding of the physics of proteins in water, but to accurately address the implications of plasmon-like features within biological systems, we need to extend our study to free energy at finite temperature. It is already known that the many-body character of vdW dispersion interactions can give rise to low-frequency, high entropy vibrational modes in organic matter (13, 24). Experimentally, a wealth of such vibrations has been observed also for proteins. While classical simulations often do predict a notable vibrational eigenmode density in the low-terahertz domain, they still lack several qualitative features, and a final confirmation by correctly reproducing actual experimental spectra is rarely provided (57). Given that the quantum-mechanical interaction mechanism presented in this work is strongly delocalized over many atoms including solvent, we suggest that it represents an essential component for the emergence of collective vibrations and the feature-rich terahertz signal of (solvated) biomolecules. In the case of crystalline aspirin, for instance, the lowest-frequency phonon band arises only because of many-body dispersion interactions. These low-frequency vibrations then lead to a selective, relative entropic stabilization of one of the polymorphs by 0.6 kcal/mol per molecule at room temperature (13). This can be seen as an estimate for the effect on a protein’s relative entropy per residue. As the dynamics and functionality of a biomolecule can be strongly related to its eigenmodes (58), the impact on collective vibrational modes also hints at an unrevealed role of plasmon-like interaction mechanisms for the functionality and coordination in the biochemical machinery. In summary, our work presents the quantum-mechanical basis for a long-range interaction mechanism for (solvated) biomolecular systems, which is proposed to play important roles in long-distance recognition, enzymatic action (29), and pharmaceutical activity (30). Our DFTB+MBD framework provides a robust formalism for the investigation of such further-reaching implications as it allows for a fully quantum-mechanical many-body treatment of large-scale systems in atomistic detail.
METHODS
For each snapshot along the folding trajectories, we obtained effective atomic polarizabilities, αA, as used within MBD and vdW(TS), in a seamless and nonempirical formalism via net atomic populations as described in (37). Electronic structure calculations were carried out in the DFTB framework with self-consistent charge (38) using a locally modified version of DFTB+ (59, 60). In vdW(TS), the effective isotropic polarizabilities define effective C6 interaction coefficients for pairwise additive interaction potentials (37, 39). In MBD then, αA is subject to short-range electrodynamic screening and then enters the coupled fluctuating dipole model (10). So, electron density fluctuations in a system consisting of N atoms are modeled by a set of N three-dimensional, isotropic quantum harmonic oscillators (QHOs) in dipole coupling defined by the Hamiltonian
| (2) |
| (3) |
In Eq. 2, is the kinetic energy, is the potential energy matrix, and ζ is the direct sum of mass-weighted displacements of the individual QHOs. is defined by the characteristic excitation frequencies, ω, the screened atomic polarizabilities, , and the long-range dipole coupling tensor (10, 43). In summary, the main approximations within MBD are the coarse graining of the system’s response to an atom-centered framework and the dipole approximation for the coupling between electronic fluctuations. Unitary transformation of the Hamiltonian 2 to a new set of collective variables, ξ = C ζ, such that
| (4) |
transforms Eq. 2 into an uncoupled set of 3N one-dimensional QHOs with frequencies and displacements ξi. It therefore provides a model for intrinsic collective charge density fluctuations—a molecular analog to the plasmon pseudoparticle in metallic systems. The dispersion energy can then be shown to equal the zero-point interaction energy of this set of QHOs (42), given by
| (5) |
The contribution of an individual collective electronic fluctuation, ξi, to the total vdW solvation energy, as defined in Eq. 1, is obtained as the ith element of the vector
| (6) |
The transformation matrix, , is given by the element-wise absolute square of (Cp ⊕ Cs)† Y. C corresponds to the transformation matrix used in Eq. 4, and corresponds to the vector of characteristic frequencies. The subscript “sub” always refers to the direct sum of the corresponding quantity for the isolated protein and the pristine solvent, e.g., ωsub = ωp ⊕ ωs. Note that the above transformation preserves energy and, thus, the sum of all elements in εint equals the total solvation energy. For further information on the procedure, see (15). Using the above definitions, we may also obtain the spatial distribution of plasmon-like interactions relevant for the vdW solvation energy. The radial MBD interaction range, Gint(R)[MBD], shown in Fig. 3B, is calculated via
| (7) |
Here, is the contribution of mode ξi to Esol, i.e., the ith element of εint as defined by Eq. 6, and δ corresponds to a generalized Kronecker delta on . denotes the elements of the transformation matrix Cps, which define the x, y, and z components of the subvector of ξi that resides on atom A. Note that integrating Gint(R)[MBD] or Gint(R)[vdW(TS)] yields the corresponding interaction (solvation) energy. Further details and in-depth analysis of the physics of plasmon-like electronic fluctuations in solvated (bio)molecular systems will be provided in a future publication. For additional theoretical background and computational details, see the Supplementary Materials.
Supplementary Material
Acknowledgments
We thank D. E. Shaw Research for providing the trajectories of cln025 and Fip35-WW plus the force field definition of a99SB-disp as used in this work and J. Hermann for helpful discussions. Funding: M.S. acknowledges financial support from the Fonds National de la Recherche, Luxembourg (AFR PhD Grant CNDTEC). A.T. was supported by the European Research Council (ERC-CoG Grant BeStMo). The results presented in this publication have been obtained using the HPC facilities of the University of Luxembourg. Author contributions: Both authors designed the research and contributed to preparing the manuscript. M.S. performed the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaax0024/DC1
Section S1. Computational details and vdW models
Section S2. Summary of gas-phase energetics for Fip35-WW
Section S3. vdW energetics in detail
Section S4. Correlation and rescaling of vdW solvation energies
Section S5. Total electronic energy of solvation
Section S6. Effect of overcompaction of unfolded states
Fig. S1. vdW interaction energy of Fip35-WW in gas phase.
Fig. S2. vdW energetics of Fip35-WW in solvation.
Fig. S3. Relative vdW solvation energy of cln025.
Fig. S4. Relative vdW energetics of cln025 in absence of solvent.
Fig. S5. Correlation of rescaled relative vdW solvation energies as obtained from pairwise models in comparison to the results obtained from many-body treatment.
Fig. S6. Total electronic energy of solvation of the Fip35-WW as obtained with DFTB in conjunction with the MBD model.
Fig. S7. Total electronic energy of solvation of the chignolin variant cln025 as obtained with DFTB in conjunction with the MBD model.
Fig. S8. Intraprotein vdW interaction energy of cln025 based on improved structural sampling.
Fig. S9. Relative vdW solvation energy of cln025 based on improved structural sampling.
REFERENCES AND NOTES
- 1.Xu F., Cross T. A., Water: Foldase activity in catalyzing polypeptide conformational rearrangements. Proc. Natl. Acad. Sci. U.S.A. 96, 9057–9061 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patriksson A., Adams C. M., Kjeldsen F., Zubarev R. A., van der Spoel D., A direct comparison of protein structure in the gas and solution phase: The Trp-cage. J. Phys. Chem. B 111, 13147–13150 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Bellissent-Funel M.-C., Hassanali A., Havenith M., Henchman R., Pohl P., Sterpone F., van der Spoel D., Xu Y., Garcia A.-E., Water determines the structure and dynamics of proteins. Chem. Rev. 116, 7673–7697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kauzmann W., Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 14, 1–63 (1959). [DOI] [PubMed] [Google Scholar]
- 5.Chandler D., Interfaces and the driving force of hydrophobic assembly. Nature 437, 640–647 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Shvab I., Sadus R. J., Intermolecular potentials and the accurate prediction of the thermodynamic properties of water. J. Chem. Phys. 139, 194505 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Dannenberg J. J., The importance of cooperative interactions and a solid-state paradigm to proteins: What peptide chemists can learn from molecular crystals. Adv. Protein Chem. 72, 227–273 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Rossi M., Fang W., Michaelides A., Stability of complex biomolecular structures: van der Waals, hydrogen bond cooperativity, and nuclear quantum effects. J. Phys. Chem. Lett. 6, 4233–4238 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Schubert F., Rossi M., Baldauf C., Pagel K., Warnke S., von Helden G., Filsinger F., Kupser P., Meijer G., Salwiczek M., Koksch B., Scheffler M., Blum V., Exploring the conformational preferences of 20-residue peptides in isolation: Ac-Ala19-Lys + H+ vs. Ac-Lys-Ala19 + H+ and the current reach of DFT. Phys. Chem. Chem. Phys. 17, 7373–7385 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Tkatchenko A., DiStasio R. A. Jr., Car R., Scheffler M., Accurate and efficient method for many-body van der Waals interactions. Phys. Rev. Lett. 108, 236402 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Gobre V. V., Tkatchenko A., Scaling laws for van der Waals interactions in nanostructured materials. Nat. Commun. 4, 2341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambrosetti A., Alfè D., DiStasio R. A. Jr., Tkatchenko A., Hard numbers for large molecules: Toward exact energetics for supramolecular systems. J. Phys. Chem. Lett. 5, 849–855 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Reilly A. M., Tkatchenko A., Role of dispersion interactions in the polymorphism and entropic stabilization of the aspirin crystal. Phys. Rev. Lett. 113, 055701 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Reilly A. M., Tkatchenko A., van der Waals dispersion interactions in molecular materials: Beyond pairwise additivity. Chem. Sci. 6, 3289–3301 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hermann J., Alfè D., Tkatchenko A., Nanoscale π–π stacked molecules are bound by collective charge fluctuations. Nat. Commun. 8, 14052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoja J., Reilly A. M., Tkatchenko A., First-principles modeling of molecular crystals: Structures and stabilities, temperature and pressure. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 7, e1294 (2017). [Google Scholar]
- 17.Dobson J. F., Gould T., Vignale G., How many-body effects modify the van der Waals interaction between graphene sheets. Phys. Rev. X 4, 021040 (2014). [Google Scholar]
- 18.Kronik L., Tkatchenko A., Understanding molecular crystals with dispersion-inclusive density functional theory: Pairwise corrections and beyond. Acc. Chem. Res. 47, 3208–3216 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Jones A. P., Cipcigan F. S., Sokhan V. P., Crain J., Martyna G. J., Electronically coarse-grained model for water. Phys. Rev. Lett. 110, 227801 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Sokhan V. P., Jones A. P., Cipcigan F. S., Crain J., Martyna G. J., Signature properties of water: Their molecular electronic origins. Proc. Natl. Acad. Sci. U.S.A. 112, 6341–6346 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao W., Chen Y., Jiang Q., Discriminating between metallic and semiconducting single-walled carbon nanotubes using physisorbed adsorbates: Role of wavelike charge-density fluctuations. Phys. Rev. Lett. 117, 246101 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Ambrosetti A., Ferri N., DiStasio R. A. Jr., Tkatchenko A., Wavelike charge density fluctuations and van der Waals interactions at the nanoscale. Science 351, 1171–1176 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Yada H., Nagai M., Tanaka K., The intermolecular stretching vibration mode in water isotopes investigated with broadband terahertz time-domain spectroscopy. Chem. Phys. Lett. 473, 279–283 (2009). [Google Scholar]
- 24.Folpini G., Reimann K., Woerner M., Elsaesser T., Hoja J., Tkatchenko A., Strong local-field enhancement of the nonlinear soft-mode response in a molecular crystal. Phys. Rev. Lett. 119, 097404 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Fröhlich H., Long-range coherence and energy storage in biological systems. Int. J. Quantum Chem. 2, 641–649 (1968). [Google Scholar]
- 26.Acbas G., Niessen K. A., Snell E. H., Markelz A. G., Optical measurements of long-range protein vibrations. Nat. Commun. 5, 3076 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Preto J., Pettini M., Tuszynski J. A., Possible role of electrodynamic interactions in long-distance biomolecular recognition. Phys. Rev. E 91, 052710 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Nardecchia I., Torres J., Lechelon M., Giliberti V., Ortolani M., Nouvel P., Gori M., Meriguet Y., Donato I., Preto J., Varani L., Sturgis J., Pettini M., Out-of-equilibrium collective oscillation as phonon condensation in a model protein. Phys. Rev. X 8, 031061 (2018). [Google Scholar]
- 29.Kurian P., Dunston G., Lindesay J., How quantum entanglement in DNA synchronizes double-strand breakage by type II restriction endonucleases. J. Theor. Biol. 391, 102–112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Craddock T. J. A., Kurian P., Preto J., Sahu K., Hameroff S. R., Klobukowski M., Tuszynski J. A., Anesthetic alterations of collective terahertz oscillations in tubulin correlate with clinical potency: Implications for anesthetic action and post-operative cognitive dysfunction. Sci. Rep. 7, 9877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Best R. B., Zheng W., Mittal J., Balanced protein–water interactions improve properties of disordered proteins and non-specific protein association. J. Chem. Theory Comput. 10, 5113–5124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piana S., Donchev A. G., Robustelli P., Shaw D. E., Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 119, 5113–5123 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Rauscher S., Gapsys V., Gajda M. J., Zweckstetter M., de Groot B. L., Grubmüller H., Structural ensembles of intrinsically disordered proteins depend strongly on force field: A comparison to experiment. J. Chem. Theory Comput. 11, 5513–5524 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Robustelli P., Piana S., Shaw D. E., Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U.S.A. 115, E4758–E4766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baldauf C., Rossi M., Going clean: Structure and dynamics of peptides in the gas phase and paths to solvation. J. Phys. Condens. Matter 27, 493002 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Nerenberg P. S., Jo B., So C., Tripathy A., Head-Gordon T., Optimizing solute-water van der Waals interactions to reproduce solvation free energies. J. Phys. Chem. B 116, 4524–4534 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Stöhr M., Michelitsch G. S., Tully J. C., Reuter K., Maurer R. J., Communication: Charge-population based dispersion interactions for molecules and materials. J. Chem. Phys. 144, 151101 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Elstner M., Porezag D., Jungnickel G., Elsner J., Haugk M., Frauenheim T., Suhai S., Seifert G., Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 58, 7260–7268 (1998). [Google Scholar]
- 39.Tkatchenko A., Scheffler M., Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 073005 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Grimme S., Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Grimme S., Antony J., Ehrlich S., Krieg H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Tkatchenko A., Ambrosetti A., DiStasio R. A. Jr., Interatomic methods for the dispersion energy derived from the adiabatic connection fluctuation-dissipation theorem. J. Chem. Phys. 138, 074106 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Ambrosetti A., Reilly A. M., DiStasio R. A. Jr., Tkatchenko A., Long-range correlation energy calculated from coupled atomic response functions. J. Chem. Phys. 140, 18A508 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Hoja J., Ko H.-Y., Neumann M. A., Car R., DiStasio R. A. Jr., Tkatchenko A., Reliable and practical computational description of molecular crystal polymorphs. Sci. Adv. 5, eaau3338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw D. E., Maragakis P., Lindorff-Larsen K., Piana S., Dror R. O., Eastwood M. P., Bank J. A., Jumper J. M., Salmon J. K., Shan Y., Wriggers W., Atomic-level characterization of the structural dynamics of proteins. Science 330, 341–346 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Lindorff-Larsen K., Piana S., Dror R. O., Shaw D. E., How fast-folding proteins fold. Science 334, 517–520 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Ensign D. L., Kasson P. M., Pande V. S., Heterogeneity even at the speed limit of folding: Large-scale molecular dynamics study of a fast-folding variant of the villin headpiece. J. Mol. Biol. 374, 806–816 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang S., Jiang Y., Li S., Liu W., Many-body dispersion effects on the binding of TCNQ and F4-TCNQ with graphene. Carbon 111, 513–518 (2017). [Google Scholar]
- 49.Rossi M., Chutia S., Scheffler M., Blum V., Validation challenge of density-functional theory for peptides—Example of Ac-Phe-Ala5-LysH+. J. Phys. Chem. A 118, 7349–7359 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Persson F., Söderhjelm P., Halle B., The spatial range of protein hydration. J. Chem. Phys. 148, 215104 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Ebbinghaus S., Kim S. J., Heyden M., Yu X., Heugen U., Gruebele M., Leitner D. M., Havenith M., An extended dynamical hydration shell around proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 20749–20752 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meister K., Ebbinghaus S., Xu Y., Duman J. G., DeVries A., Gruebele M., Leitner D. M., Havenith M., Long-range protein-water dynamics in hyperactive insect antifreeze proteins. Proc. Natl. Acad. Sci. U.S.A. 110, 1617–1622 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu Y., Havenith M., Perspective: Watching low-frequency vibrations of water in biomolecular recognition by THz spectroscopy. J. Chem. Phys. 143, 170901 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Tkatchenko A., Rossi M., Blum V., Ireta J., Scheffler M., Unraveling the stability of polypeptide helices: Critical role of van der Waals interactions. Phys. Rev. Lett. 106, 118102 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Rossi M., Scheffler M., Blum V., Impact of vibrational entropy on the stability of unsolvated peptide helices with increasing length. J. Phys. Chem. B 117, 5574–5584 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Duboisset J., Brevet P.-F., Salt-induced long-to-short range orientational transition in water. Phys. Rev. Lett. 120, 263001 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Plusquellic D. F., Siegrist K., Heilweil E. J., Esenturk O., Applications of terahertz spectroscopy in biosystems. ChemPhysChem 8, 2412–2431 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Levy R. M., Perahia D., Karplus M., Molecular dynamics of an α-helical polypeptide: Temperature dependence and deviation from harmonic behavior. Proc. Natl. Acad. Sci. U.S.A. 79, 1346–1350 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aradi B., Hourahine B., Frauenheim T., DFTB+, a sparse matrix-based implementation of the DFTB method. J. Phys. Chem. A 111, 5678–5684 (2007). [DOI] [PubMed] [Google Scholar]
- 60.The development version of the DFTB+ code, which has been used in the current work (mbd branch); https://github.com/aradi/dftbplus.git.
- 61.Gaus M., Cui Q., Elstner M., DFTB3: Extension of the self-consistent-charge density-functional tight-binding method (SCC-DFTB). J. Chem. Theory Comput. 7, 931–948 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaus M., Goez A., Elstner M., Parametrization and benchmark of DFTB3 for organic molecules. J. Chem. Theory Comput. 9, 338–354 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Gaus M., Lu X., Elstner M., Cui Q., Parameterization of DFTB3/3OB for sulfur and phosphorus for chemical and biological applications. J. Chem. Theory Comput. 10, 1518–1537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kubillus M., Kubař T., Gaus M., Řezáč J., Elstner M., Parameterization of the DFTB3 method for Br, Ca, Cl, F, I, K, and Na in organic and biological systems. J. Chem. Theory Comput. 11, 332–342 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Grimme S., Ehrlich S., Goerigk L., Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011). [DOI] [PubMed] [Google Scholar]
- 66.Clark S. J., Segall M. D., Pickard C. J., Hasnip P. J., Probert M. I. J., Refson K., Payne M. C., First-principles methods using CASTEP. Z. Kristallogr. Cryst. Mater. 220, 567–570 (2005). [Google Scholar]
- 67.Bahn S. R., Jacobsen K. W., An object-oriented scripting interface to a legacy electronic structure code. Comput. Sci. Eng. 4, 56–66 (2002). [Google Scholar]
- 68.B. Aradi, GitHub—aradi/dftd3-lib: Library Version of S. Grimme’s DFTD3 code (2016).
- 69.Berendsen H. J. C., van der Spoel D., van Drunen R., GROMACS: A message-passing parallel molecular dynamics implementation. Comp. Phys. Commun. 91, 43–56 (1995). [Google Scholar]
- 70.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaax0024/DC1
Section S1. Computational details and vdW models
Section S2. Summary of gas-phase energetics for Fip35-WW
Section S3. vdW energetics in detail
Section S4. Correlation and rescaling of vdW solvation energies
Section S5. Total electronic energy of solvation
Section S6. Effect of overcompaction of unfolded states
Fig. S1. vdW interaction energy of Fip35-WW in gas phase.
Fig. S2. vdW energetics of Fip35-WW in solvation.
Fig. S3. Relative vdW solvation energy of cln025.
Fig. S4. Relative vdW energetics of cln025 in absence of solvent.
Fig. S5. Correlation of rescaled relative vdW solvation energies as obtained from pairwise models in comparison to the results obtained from many-body treatment.
Fig. S6. Total electronic energy of solvation of the Fip35-WW as obtained with DFTB in conjunction with the MBD model.
Fig. S7. Total electronic energy of solvation of the chignolin variant cln025 as obtained with DFTB in conjunction with the MBD model.
Fig. S8. Intraprotein vdW interaction energy of cln025 based on improved structural sampling.
Fig. S9. Relative vdW solvation energy of cln025 based on improved structural sampling.