

Abstract
Bacterial meningitis is a medical emergency requiring highly bactericidal antibiotics to achieve cure. Many challenges exist to achieving optimal patient outcome. First, antibiotics must pass the blood brain barrier. Once in the subarachnoid space, achieving bactericidal therapy involves circumventing antibiotic resistance and more commonly, antibiotic tolerance arising from the slow growth of bacteria in the nutrient poor cerebrospinal fluid. Finally, bactericidal therapy is most often bacteriolytic and debris from lysis is highly inflammatory. Controlling damage from lytic products may require adjunctive therapy to prevent neuronal death. These challenges are an extreme example of the different requirements for treating infections in different body sites.
Keywords: tolerance, pneumococcus, adjunctive therapy, autolysis
Graphical Abstract
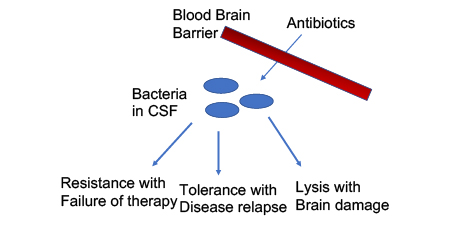
Bacterial meningitis, one of the oldest infections recorded in human history, remains a significant cause of mortality and morbidity. High rates of fatality occur even in treated patients and life altering sequelae occur in up to half of survivors. Studies conducted 40 years ago identified five primary bacterial pathogens responsible for over 80% of community acquired acute bacterial meningitis: Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae, Neisseria meningitidis (meningococcus), Lancefield Group B streptococcus, and Listeria monocytogenes.1,2 Vaccination campaigns against Haemophilus type b (Hib), pneumococci, and meningococci, have been massively successful at the prevention of community acquired meningitis, such that only cases of community acquired pneumococcal meningitis outnumber cases of nosocomial meningitis.3
The problem: Victories in prevention belie the continued poor prognosis of patients presenting with community acquired acute bacterial meningitis (ABM), which remains as devastating today as two decades ago.4 Today the challenge remains to improve both survivability and recovery from ABM. This perspective will highlight some of the advancements in our understanding of central nervous system (CNS) responses to damaging components of bacteria with an eye on opportunities for future adjunctive therapeutic interventions. We will then discuss the microbiology of ABM, challenges to understanding how antibiotics function in eliminating (or failing to eliminate) infection in model systems and in clinical practice and perspectives on new antibiotics and drug delivery systems to the CNS.
Acute Bacterial Meningitis: pneumococcus stands out as a treatment failure
The modern era of treatment of ABM dates to the mid-20th century when penicillin was first used. In a landmark study, meningococcal meningitis treated with penicillin resulted in 1 death amongst 71 cases.5 At the same time, 70% of patients treated with penicillin for pneumococcal meningitis died. By the 1950s, the combined use of sulfonamides (meningococcus, Haemophilus influezae), chloramphenicol (H. influenzae), and high dose penicillin (pneumococcus) as standard therapies decreased the case fatality rate to single digits for H. influenzae and meningococcus.6 Yet the fatality rate after pneumococcal meningitis remained close to 40.7
Third generation cephalosporins, developed in the late 1970s and 1980s, showed a remarkably favorable pharmacodynamic profile for treatment of pneumococcal ABM: high levels of CSF accumulation, bactericidal activity against the pneumococcus, and a largely susceptible population of isolates.8,9 These drugs are the current mainstay of treatment of ABM which usually entails intravenous administration for 7 to 10 days. Yet despite this improved profile, the fatality rate has dropped only modestly to around 30%. Thus, the current challenge to ABM therapy is the continued poor prognosis, specifically for pneumococcal meningitis. This presents a new paradigm: killing bacteria is not enough to improve outcome.
The failure of antibiotics with excellent pharmacodynamic properties to improve outcome of pneumococcal ABM has been partially explained by studies in model systems. As rapid bacterial killing progresses, cell wall fragments are produced by lysis and are liberated in high amounts. These components retain their inflammatory capacity and exacerbate the symptoms of meningitis even as the bacterial load decreases. When administered intracisternally in animal models, they elicit pronounced inflammatory cytokine production followed by neuronal death.10–12 Thus, the first few hours of bactericidal treatment appear to unleash a wave of inflammation before clearance of bacterial debris. Efforts to attenuate this inflammation with adjunct therapies has become a major focus of modern experimental therapy. Choosing the right agents depends on understanding the complex pathways by which the CNS responds to eliminate bacteria and how neurons die in the process.
The current overall picture of ABM indicates that bacterial pathogens gain access to the CNS space through penetration of the blood brain barrier (BBB) or blood cerebrospinal fluid barrier (BCFB). The preponderance of the pneumococcus, meningococcus, and Haemophilus pathogens in cases of community acquired ABM is likely due to their high colonization rates of humans and shared molecular mechanisms of BBB invasion (reviewed extensively elsewhere13). Once in the CNS, pathogens are relatively shielded from the clearance mechanisms of innate and adaptive immunity present in the blood and most tissues. The immune privilege of the CNS is maintained through the barrier function of the BBB and BCFB.14 Thus, successful treatment of meningitis requires the choice of an actively bactericidal antibiotic with CNS specific dosing or administration route. If all of these parameters are met, then 1 in 3 patients still have a poor outcome. Currently, the effort to decrease inflammation and bystander killing of neurons has been limited to adjunct therapy with dexamethasone. The use of dexamethasone before or together with the first antibiotic dose improved outcome in animals and was associated with modest improvements in sequelae in patients.15–18 Adjunct anti-inflammatory therapy is an option in current clinical guidelines.19
The rise of adjunctive therapy
Microglia: the good and bad of activating the innate immune defense of the CNS
Traditionally, the CNS has been termed an “immune-privileged” compartment.20 The lack of circulating leukocytes and low protein levels (specifically immunoglobulins and acute phase reactants) in healthy cerebrospinal fluid suggested to many that the CNS compartment lacked immune surveillance and robust responses. Infiltration of leukocytes and increased proteins levels in CSF is a pathological indicator of disease, caused by the breakdown of the barrier functions of the BBB and BCFB, and not generally considered a productive response towards disease resolution as leukocytes phagocytose poorly in fluids. This model was revised by several observations. In animal models, bacterial molecules injected directly into the CSF lead to recruitment of leukocytes and increases in protein concentration, suggesting a programmed response arising in the CNS can occur in the absence of direct damage to the BBB.10–12 It was also found that these same bacterial products lead to direct damage of neurons; infiltration of neutrophils is not necessary for neuronal death in ABM.21 In response to these findings, a revised definition of the immune capabilities of the CNS was formulated. Healthy CNS tissue could sense, respond, and be damaged by bacterial products directly without the involvement of an immune infiltrate originating in blood and passing through the BBB.
Pathogens are sensed by host cells through specific receptors, termed pattern recognition receptors (PRRs), which recognize either pathogen derived molecules (Pathogen Associated Molecular Patterns, PAMPs), or signals of cellular damage induced by infectious pathologies (Danger Associated Molecular Patterns, DAMPs). The Toll-like receptor family, a PAMP-sensing PRR, is comprised of 10 members in humans. Surprisingly all 10 TLR family members are expressed in CNS tissues and upon binding to an appropriate PAMP, signal through the adapter protein MyD88 (reviewed in 14,22,23). In innate immune cells, the MyD88 signaling feeds through to NF-kB activation, leading to the production of pro-inflammatory cytokines and defenses (reviewed in 24–26). In the case of ABM, the main TLRs involved are TLRs 2 and 4, which recognize bacterial cell wall fragments and LPS respectively.27–31 These are the predominant PAMPs involved in neuronal damage. In models of both bacterial sepsis and ABM, TLR2 deficient mice showed notably less neuronal damage.20,28,31
In addition to TLRs, other PRRs have been described to contribute to neuronal damage in response to ABM. The nucleotide-binding oligomerization domain (NOD) and NOD-like receptor (NLR) proteins serve as intracellular sensors of peptidoglycan fragments and DAMPs respectively (reviewed in 32,33). Like TLR2, NOD signaling in innate immune cells leads to NF-kB activation and an increase in inflammatory cytokine production. NOD expression in the brain has been described for astrocytes and microglia (NOD2) and pericytes (NOD1). Of these, NOD2 expression has been linked to damage in response to meningococcal and pneumococcal meningitis.34,35 NLRs are a large family of proteins in humans, however only a few NLRs and corresponding inflammasomes have been linked to damage during meningitis. In pneumococcal meningitis release of cellular DAMPs through the activity of the pore forming toxin pneumolysin leads to activation of the NLRP3 inflammasome, and inhibition of this inflammasome can ameliorate some tissue damage.36
Microglia, the resident innate immune cell of the CNS, express TLRs 1–9, NOD1/2, and NLRs 1–3/6/12. 33,37 The activation of microglial PRRs in response to ABM leads to inflammatory signaling and production of damaging molecules leading to neuronal damage (reviewed in 38). In response to common bacterial products, microglia produce TNFα and IL-1ß, both classical inflammatory cytokines that can recruit inflammatory cells to migrate into the CNS and can directly bind to the receptors expressed on neurons, inducing neuronal death by programmed cell death pathways. 23,39,40 Microglia respond by producing reactive oxygen species through iNOS (nitric oxide production) and PHOX (superoxide production). Both these molecules are implicated in microglial damage of neurons.41,42 This damage can directly lead to neuronal death or creation of neuronal DAMPs which lead to microglial phagocytosis of neurons.
Downstream of microglial activation through TLR2 or 4, matrix metalloproteinases are activated that further damage neurons.31,43 Inhibition of MMPs during experimental ABM has been shown to reduce the neuronal damage and improve long term neurological function.44,45 Combination of MMP inhibition with treatment with a non-lytic antibiotic (preventing the release of TLR2 ligands from dying bacteria, discussed below) was additively protective in this animal model suggesting that inhibiting MMP pharmacologically as well as preventing MMP activation by reducing TLR agonists is a viable avenue for protecting against the worst neuronal damage.45
Despite contributing to direct damage, microglia also sit at an axis of immune surveillance and repair that can be leveraged in both treatment and recovery of ABM. Thus, pathways exist that can prime the innate immune mechanisms in the CNS while bypassing inflammatory signaling. Further studies are needed to assess the ability of these types of treatments to act not just as prophylaxis, but also as adjunctive therapy during ABM to clear acute phase pathogens and PAMPs liberated during treatment and resolution without inflammation. One example is to boost the little protection that microglia provide in the absence of neutrophils.46 Such strategies include pretreatment of microglia with TLR agonists to increase phagocytosis and intracellular killing of ABM pathogens. In a recent study, stimulation of microglia with CpG combined with Activin A, a TGFß family member cytokine, led to a 750% increase in phagocytosis of E. coli but with reduced nitrite production compared to stimulation with LPS.47 Thus, it is possible to prime microglia to phagocytose pathogens in conjunction with treatments that bias downstream signaling away from inflammation. A second example is the use of palmitoylethanolamide, a natural anti-inflammatory lipid produced endogenously in the CNS. It has also been shown to stimulate the phagocytic and intracellular killing activity of rat and mouse microglia without subsequent upregulation of TNFα and IL-1ß.48
Boosting protective effects of microglia without inducing inflammation remains experimental. Given the large number of PRRs that are expressed in the CNS, blocking all inflammatory signaling will be a challenge in clinical practice. Beyond conserved PAMPs, such as peptidoglycan and LPS, bacterial pathogens also produce species specific PAMPs and toxins that trigger further damage. Thus, as our knowledge of the immune capabilities of the CNS has grown, our ability to rationally combat all the various routes of ABM damage has diminished. Microglia represent both the first line of defense and the initiators of damage repair during ABM. Adjunctive and post-meningitis therapies focusing on harnessing both the protective and reparative functions of microglia while minimizing inflammatory damage are promising future avenues for ABM therapies.
Neuronal death: how to target such a complex process?
While microglia are central in the response to meningeal pathogens, it is damage to the neurons in the cortex and hippocampus that results in most acute and post-infectious sequelae. Neurons are generally damaged by three mechanisms during ABM: 1) Neurons are damaged as bystanders during the inflammatory immune response mediated by microglia and recruited leukocytes; 2) Acute traumatic injury to neurons due to high intracranial pressure; and 3) Direct activation of programmed death pathways in neurons through sensing of DAMPs and PAMPs.12, 28 A fourth mechanism, intoxication by bacterially produced toxins, is largely pathogen dependent, and development of toxin neutralization strategies is not a general mechanism of treatment for ABM. In addition to the strategies discussed above to control the inflammatory process, there are parallel studies ongoing to directly intercede in neuronal death pathways as well as to promote neuro-regeneration after ABM.
Damaged neurons often undergo apoptosis due to the triggering of inflammasomes. Human neurons express functional NLRP1 and 3 and it is clear in many models of neurodegeneration that inflammasomes within neurons can be triggered to cause programmed cell death. The pharmacological NLRP3 inhibitor MCC950 has been shown to be protective against neuronal loss after traumatic brain injury.49,50 NLRP3 plays a central role in damage during ABM, particularly in pneumococcal meningitis. In NLRP3 knockout mice, neuronal damage assessed after pneumococcal meningitis was found to be greatly reduced. 36 In an LPS neuronal damage model, the protective role of pretreatment with cinnamaldehyde was found to be at least partially mediated through the inhibition of the NLRP3 inflammasome.51
Once damaged, neurons have a limited ability to repair and regenerate themselves. After activation, microglia can secrete neurotrophic molecules like brain-derived neurotrophic factor (BDNF) which encourages repair and limited regrowth of neurons. The application of BDNF in a pneumococcal meningitis model leads to reduced caspase induction and significantly decreased neuronal death in the cortex and hippocampus.52–54 After pneumococcal ABM, it had been noted that levels of BDNF naturally rise as the brain attempts repair mechanisms. However in a recent study it was found that neural precursor cells do not fully differentiate after pneumococcal ABM, and that addition of exogenous BDNF helped with the differentiation to more mature and therefore likely functional neurons.55
Death versus proliferation and differentiation of neural precursor cells in response to PAMPs is an active area of research for both developmental and infectious disease biology. In animal models, activation of TLR2 by pneumococcal cell wall pieces initiates apoptosis.56 In contrast, embryonic exposure to bacterial cell wall fragments causes proliferation of neuronal progenitor cells giving rise to significantly more cortical neurons.57 This response is dependent on TLR2, suggesting that in specific developmental stages TLR2 signaling in neuronal precursors drives a proliferative response. The plasticity of the outcome of TLR signaling is likely to give rise to future interventions preventing damage and protecting neuronal integrity during ABM.
The future of antibiotics for ABM
While investigation continues in the area adjunctive therapy for meningitis, current antibiotics such as third generation cephalosporins are the standard of care. The future success of antibiotics in treating ABM relies on optimizing four major areas (Inset): Penetration of the BBB, bactericidal effect, minimizing lytic release of pro-inflammatory bacterial molecules, avoidance of known resistance mechanisms, and overcoming tolerance (phenotypic and genotypic). Penetration from blood through the BBB is optimized through chemical modifications of antibiotic structure. Alternatively, drug delivery systems under development, including nanoparticles, liposomes, and methods to disrupt the BBB, may improve pharmacokinetics for many antibiotics. Minimizing the lead time to achieving the minimum inhibitory concentration (MIC) of antibiotics in CSF is a concern of both future drug development and delivery systems. Depending on the mechanism of action, long build up times for antibiotics can lead to worse treatment outcomes.58 Thus, while the first two hurdles are being aggressively addressed by innovative technologies. Points 3, 4 and 5 still remain major problems.
Death without lysis
Understanding bactericidal mechanisms of antibiotics and how they relate to release of pro-inflammatory bacterial molecules, despite decades or research, is still poorly understood. Most antibiotics are clinically characterized as either bacteriostatic or bactericidal. Bactericidal drugs cause a reduction in the number of viable bacteria under tested conditions; the MIC for bactericidal drugs is close to the minimum bactericidal concentration (MBC), the concentration of antibiotic at or above which bacteria die.59 Treating infections like meningitis, where innate immune clearance mechanisms are poor, requires reaching the MBC of bactericidal antibiotics. Antibiotics that are bactericidal trigger programmed cell death pathways, particularly for the major meningeal pathogens that are known to respond by autolysis.60,61 The molecular events downstream of β-lactam antibiotics that lead to lysis are largely unknown and represent an active field of research. The autolytic response is conserved amongst many diverse bacteria and results in the rapid release of pro-inflammatory molecules from cell envelope degradation.62 Importantly, as discussed above, this release of bacterial PAMPs is one of the major pathologic processes in ABM. Preclinical models have shown convincingly that use of non-lytic but still bactericidal antibiotics reduces the neuronal damage as the release of cell envelope derived PAMPs is reduced.63,64
While cell wall active antibiotics lead to autolysis, most RNA/protein synthesis and DNA replication inhibitors do not. Chloramphenicol, rifampicin, and clindamycin have been shown to be bactericidal against the pneumococcus in models of meningitis without inducing autolysis. 58, 64–70 Chloramphenicol serves as an excellent example of an ideal meningitis drug: highly bactericidal to major meningeal pathogens, nonlytic, rapid penetration into the CNS, oral bioavailability and low prevalence of resistance. It was used in the clinic extensively in the past, however it is currently discouraged due to the side effects of bone marrow suppression.69–72 In pneumococcal infection models, the use of clindamycin to treat both sepsis and meningitis markedly improves CNS pathology64,65,68 in part because clindamycin appears to induce programmed cell death pathways leading to filamentation rather than lysis.73 However, all protein synthesis inhibitors are not necessarily good meningitis drugs. Both rifampicin and clindamycin have relatively high spontaneous resistance rates. Another less well appreciated aspect, is that inhibitors of RNA and protein synthesis have been shown to result in persister bacterial cells in several models (reviewed in 74). Persistence is a form of antibiotic escape, where a small proportion of a bacterial population does not die.75 When the antibiotic is removed, the persisting cells begin to grow and the infection slowly relapses. Persister cells are generally thought to form through the de-energization of the bacterial cell, a depletion of ATP and cessation of metabolic function. These cells would appear dead by many assays, however, upon removal of antibiotic, a portion of persister cells will re-emerge. In bacterial infections, such as meningitis, where sterilization of the CNS is almost entirely dependent on the bactericidal efficacy of the antibiotic, the induction of persister cells is not a trivial concern.
Considering these factors, new antibiotic regimens designed to improve long term effects of meningitis will likely use combination therapies, some of which have been studied in preclinical models. Treatment with either rifampicin or daptomycin together with a third-generation cephalosporin has been shown to be effective at clearing pneumococci as well as attenuating the release of pro-inflammatory molecules.69,77 Besides combination therapy, the future development of antibiotics effective against ABM pathogens is relatively limited. Tigecycline, which has been used clinically against highly resistant Acinetobacter infections, is a poor empiric choice due to high spontaneous resistance rates while its good CNS pharmacokinetics may make it a useful adjunctive antibiotic.78–80 The discovery of a new peptide derived antibiotic, the founding member of a new class of drugs, Teixobactin, is exciting as it provides both a new structure for use in combinatorial chemistry approaches, but also targets lipid II.81 Other lipid II targeting antibiotics, nisin and vancomycin, have relatively low levels of resistance, and resistance requires horizontal gene transfer events which tend to be rare (reviewed in 82–84). However oral bioavailability and penetration of the CNS are notoriously poor for peptide derivative antibiotics, and therefore much work will have to be done to develop teixobactin as a drug for meningitis.
Phenotypic tolerance: the slower they grow, the slower they die
Generally, the MIC and the MBC are quite close in value for most bacteria/antibiotic combinations. In contrast, antibiotic tolerance is a state where an entire population of bacteria stop growing at MIC, as expected, but then do not die (MBC > MIC) (Fig 2). In the microbiology lab, tolerant strains stop growing at the same MIC as sensitive strains and thus are missed as a source of antibiotic failure. Only when bacterial viability is assessed, is it clear that the cessation of growth did not lead to actual killing and that the MBC is much higher for a tolerant strain than a sensitive one. Tolerance is generally exhibited by an entire population of bacteria rather than the concept of persisters which refers to a small subpopulation. While resistance is universally accounted for in both drug design and clinical usage, tolerance is rarely considered as it is never reported from the lab. In the case of meningitis, tolerance is as important as resistance in treatment outcome (Fig 3). Treatment failure as a result of tolerance is clinically documented, yet generally underappreciated.
Fig 2. Death kinetics of tolerant and sensitive bacteria.
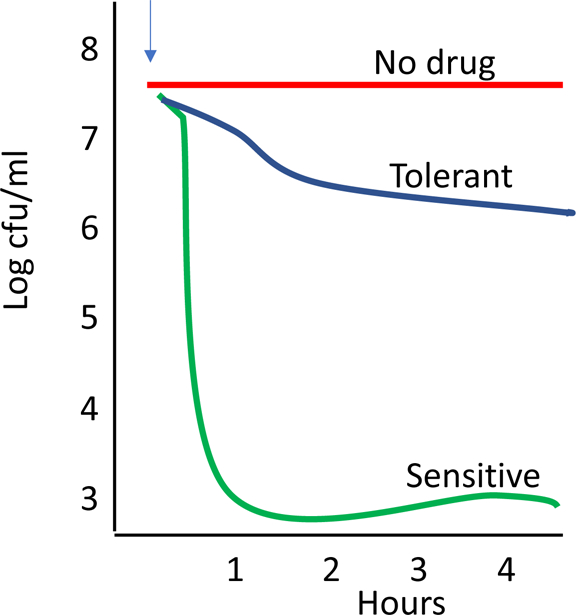
A schematic representation of bacterial cell death kinetics in vitro. Sensitive bacterial strains (green) are rapidly killed by antibiotics (added at arrow). In contrast, tolerant bacteria (blue) stop growing but do not die.
Fig 3. Effect of tolerance on survival from treated infection.
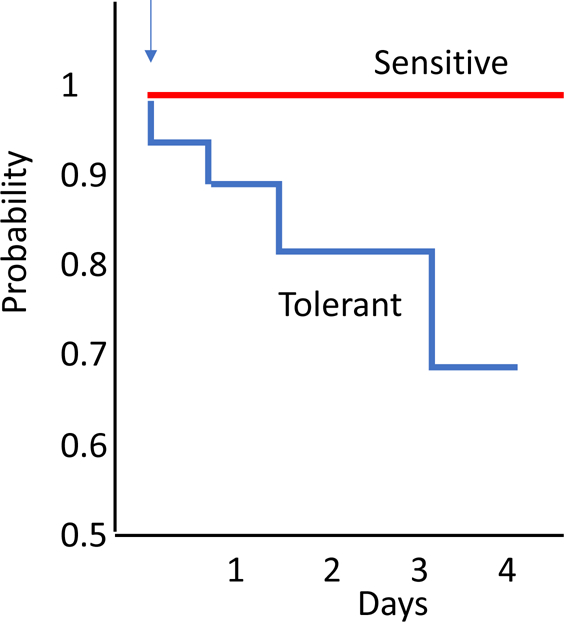
The probability of surviving meningitis based on the efficacy of bacterial killing has been examined in animal models. In vitro testing of bacterial strains from human cases of meningitis where treatment failed has revealed tolerance to antibiotic-induced killing as in Figure 2. When tested in animal models, these tolerant strains stop multiplying during antibiotic therapy but when therapy is stopped, infection relapses leading to treatment failure (blue).
Phenotypic tolerance is a property of survival shared by all bacteria under specific environmental conditions.76 Bacteria elude death when they are not growing at the time of antibiotic exposure.85 This response leads to the need for prolonged therapy for infections in nutrient poor body sites, such as endocarditis, osteomyelitis, and abscesses. Once in the CNS, all antibiotics face the obstacle of phenotypic tolerance. The CSF is an exceptionally poor growth medium and thus bacterial growth rate is drastically reduced as bacteria move from blood to CSF. This response is universal and mediated by the RelA stringent response pathway whereby bacteria slow metabolism in response to limited nutrients.85–87 Triggering the stringent response slows the rate of cell death: the slower they grow, the slower they die.86 This universal feature complicates virtually all therapies for meningitis by all pathogens. Few antibiotics can break the stringent response rule. One exception is chloramphenicol and another is the penem family of antibiotics. 87–90 Although the mechanism is unknown, the ability of these drugs to retain some ability to kill non- and slow-growing bacteria suggests it may be possible to improve clinical outcome by more rapidly eliminating the CSF bacterial load. However, efforts to overcome the stringent response and kill slowly growing pathogens, preferably without lysis, is yet to be successful.
Genotypic tolerance: the ultimate stealth countermeasure to antibiotics
While all bacteria invoke phenotypic tolerance transiently as they slow down growth while entering the CSF, it has become evident that bacteria can also acquire mutations that result in a permanent tolerant phenotype regardless of environment. These strains are genotypically tolerant and have been linked to patients with relapsing meningitis where antibiotic killing, that is critical to cure, fails. The first description of genotypic tolerance was demonstrated in 1970 for the pneumococcus. For this bacterium, antibiotic induced death is due to lysis by the main autolytic enzyme, LytA. In the lab, LytA null mutants treated with cell wall active antibiotics arrest growth at a normal MIC but then don’t die.91 Rather, they enter a relatively quiescent state with extended viability, i.e. they are genotypically tolerant. In this case, the MIC is achieved but the MBC is not.11 In model infections with LytA mutants, antibiotics stop bacterial replication with an apparent good response to therapy but infection reappears as the antibiotic therapy is discontinued. Given constant antibiotic pressure in the clinic, it would be expected that bacteria would quickly delete this suicidal enzyme and become genotypically tolerant. Yet, LytA mutations are not seen in clinical isolates. This conundrum was recently solved as it was found that LytA has a previously unrecognized role in capsule shedding which protects bacteria from antimicrobial peptides. 92 Thus, the suicidal enzyme is under selective pressure to remain in the genome under strong regulation. If not due to loss of the lytic enzyme itself, then genotypic tolerance must arise by alterations in the poorly understood control mechanisms for autolysis. One mechanism of genotypic tolerance is acquisition of mutations that extend the lag time before bacteria grow in the presence of drug.93 This plays into the concept of nongrowing bacteria escaping death.
In the clinic, strains demonstrating genotypic tolerance have been clearly described although the genetic basis is mostly unknown. First, during the early detailed analysis of pneumococci emerging as resistant to penicillin, it was noted that virtually all resistant isolates are also tolerant.94 Not only is the MIC higher (i.e. resistant) but also these strains don’t die readily and have an even higher MBC (i.e. tolerant); death requires much higher amounts of antibiotic (Fig 4). To explain this association of tolerance and resistance, it is reasoned that bacteria under antibiotic pressure would first acquire the capacity to survive treatment (tolerance) and then go on to acquire the genes for the ability to actually grow in the presence of antibiotic (resistance). Thus, the high rate of tolerance in resistant strains appears to make sense. Another instance of genotypic tolerance is exemplified by isolates from patients that failed therapy.75,95,96 Several strains showing abnormal autolysis to ß lactams have been isolated from cases of relapsing meningitis. Furthermore, tolerance to vancomycin was shown to be an emerging phenotype: the meningitis strain Tupelo survives both vancomycin and cephalosporins despite a normal LytA autolysin.97 The prevalence of tolerance to vancomycin became more widely recognized and its impact on clinical practice was noted as vancomycin became the antibiotic of last resort for penicillin resistant pneumococcal meningitis.98,99
Fig 4. Schematic of different possible responses to antibiotics.
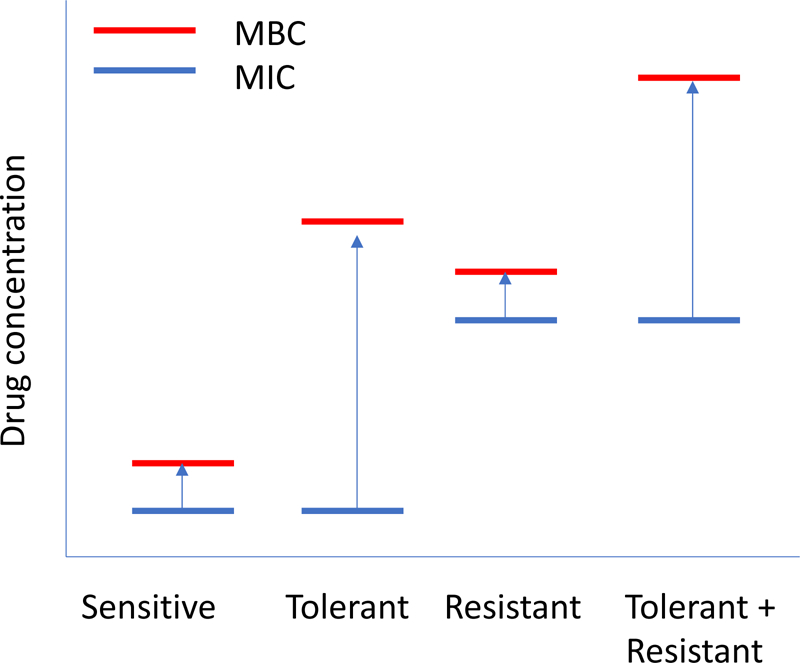
Sensitive bacterial strains have a minimal inhibitory concentration that is near the minimum bactericidal concentration (MIC=MBC). Tolerant strains stop growing at the normal MIC but require a much higher drug concentration to induce killing (MBC>MIC). Resistant strains have a high MIC but the MBC is still close to the MIC (high MIC=highMBC). Strains that are both resistant and tolerant have a high MIC but a very much higher MBC (high MBC>>high MIC).
Summary
In the near future the best hope for improved outcomes in antibacterial ABM treatment is development of new antibiotic classes and delivery methods. Currently, highly bactericidal antibiotics are the mainstay and trials may eventually couple them to adjunctive therapies that dampen the complex host response and preserve neurons. Combination treatment with rifampicin or quinolones during therapy with third generation cephalosporins is attractive as it may reveal less bacterial lysis and improved outcomes but this remains to be tested. The development of new antibiotics based on the teixobactin backbone is exciting but still requires improvement to achieve high CNS concentrations. Prevention of meningitis through vaccination remains the best strategy for gains in overall public health.
Fig 1. Bacterial lytic products induce inflammation and neuronal death.
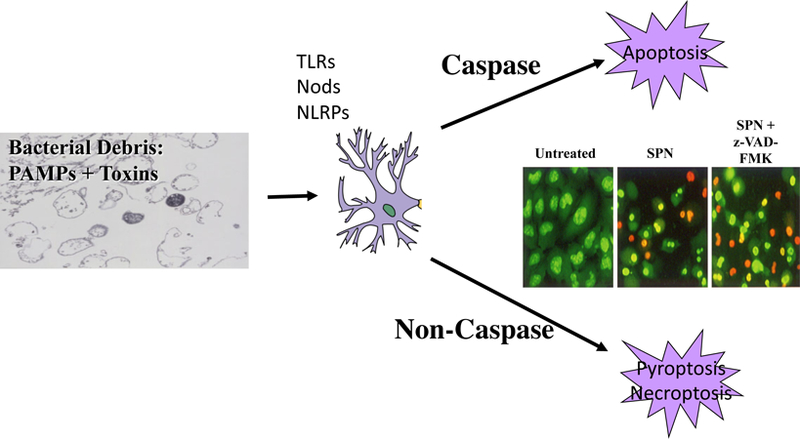
Based on meningitis models in rabbits and mice, the inflammatory and cell death pathways operative in meningitis are extensive. Antibiotics induce lysis of bacteria releasing cell wall and intracellular debris which contain a library of inflammatory components: Pathogen Associated Molecular Patterns (PAMPS). These are recognized by innate immune receptors on many brain cells including neurons: surface Toll-like receptors (TLR), intracellular Nod receptors and inflammasomes (NLRP). The resulting inflammatory cascade can induce cell death in many ways: apoptosis, pyroptosis, necroptosis etc. The insert shows in vitro neurons (green) ± exposure to pneumococcal lytic products (SPN). Orange indicates dead neurons. Note that the z-VAD-FMK, an inhibitor of caspase dependent apoptosis, blocks death of only half the neurons indicating non-caspase mediated death is not blocked.
Neuron image via Wikimedia: DDhp1080, svg adaptation by Actam
[CC BY-SA 3.0https://creativecommons.org/licenses/by-sa/3.0)]
Requirements for successful meningitis therapy.
Bactericidal antibiotic activity
Antibiotic penetration across the blood brain barrier
Limiting inflammation from bacterial debris
Requirements for new antibiotics for meningitis.
Improved penetration of the BBB
Avoid resistance
Bactericidal without lysis
Overcome phenotypic tolerance
Overcome genotypic tolerance
REFERENCES
- 1.Wenger JD; Hightower AW; Facklam RR; Gaventa S; Broome CV (1990) Bacterial Meningitis in the United States, 1986: Report of a Multistate Surveillance Study. J. Infect. Dis 162 (6), 1316–1323. DOI: 10.1093/infdis/162.6.1316 [DOI] [PubMed] [Google Scholar]
- 2.Schlech WF, Bacterial Meningitis in the United States, 1978 Through 1981. (1985) JAMA 253 (12). DOI: 10.1001/jama.1985.03350360075022 [DOI] [PubMed] [Google Scholar]
- 3.Castelblanco RL; Lee M; Hasbun R (2014) Epidemiology of bacterial meningitis in the USA from 1997 to 2010: a population-based observational study. Lancet. Infect. Dis 14 (9), 813–819. DOI: 10.1016/s1473-3099(14)70805-9 [DOI] [PubMed] [Google Scholar]
- 4.Thigpen MC; Whitney CG; Messonnier NE; Zell ER; Lynfield R; Hadler JL; Harrison LH; Farley MM; Reingold A; Bennett NM; Craig AS; Schaffner W; Thomas A; Lewis MM; Scallan E; Schuchat A; (2011) Emerging Infections Programs: Bacterial meningitis in the United States, 1998–2007. N. Engl. J. Med 364 (21), 2016–25. DOI: 10.1056/NEJMoa1005384 [DOI] [PubMed] [Google Scholar]
- 5.Keefer CS (1943) Penicillin in the Treatment of Infections. JAMA 122 (18), 1217 DOI: 10.1001/jama.1943.02840350001001 [DOI] [Google Scholar]
- 6.Swartz MN (2004) Bacterial meningitis--a view of the past 90 years. N. Engl. J. Med 351 (18), 1826–8. DOI: 10.1056/NEJMp048246 [DOI] [PubMed] [Google Scholar]
- 7.Dowling HF; Sweet LK; (1949) The treatment of pneumococcic meningitis with massive doses of systemic penicillin. Am. J. Med. Sci 217 (2), 149–56. URL https://www.ncbi.nlm.nih.gov/pubmed/18109275 [PubMed] [Google Scholar]
- 8.Tauber MG; Hackbarth CJ; Scott KG; Rusnak MG; Sande MA (1985) New cephalosporins cefotaxime, cefpimizole, BMY 28142, and HR 810 in experimental pneumococcal meningitis in rabbits. Antimicrob Ag Chemother, 27 (3), 340–342. DOI: 10.1128/aac.27.3.340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosley GS; Elliott JA; Oxtoby MJ; Facklam RR (1987) Susceptibility of relatively penicillin-resistant Streptococcus pneumoniae to newer cephalosporin antibiotics. Diag. Microb. Infect. Dis 7 (1), 21–27. DOI: 10.1016/0732-8893(87)90065-4 [DOI] [PubMed] [Google Scholar]
- 10.Tuomanen E; Liu H; Hengstler B; Zak O; Tomasz A (1985) The induction of meningeal inflammation by components of the pneumococcal cell wall. J. Infect. Dis 151 (5), 859–868. DOI: 10.1093/infdis/151.5.859 [DOI] [PubMed] [Google Scholar]
- 11.Tuomanen E; Tomasz A; Hengstler B; Zak O (1985) The relative role of bacterial cell wall and capsule in the induction of inflammation in pneumococcal meningitis. J. Infect. Dis 151 (3), 535–40. DOI: 10.1093/infdis/151.3.53 [DOI] [PubMed] [Google Scholar]
- 12.Braun JS; Novak R; Herzog KH; Bodner SM; Cleveland JL; Tuomanen EI (1999) Neuroprotection by a caspase inhibitor in acute bacterial meningitis. Nat. Med 5 (3), 298–302. DOI: 10.1038/6514 [DOI] [PubMed] [Google Scholar]
- 13.Doran KS; Fulde M; Gratz N; Kim BJ; Nau R; Prasadarao N; Schubert-Unkmeir A; Tuomanen EI; Valentin-Weigand P (2016) Host-pathogen interactions in bacterial meningitis. Acta Neuropathol 131 (2), 185–209. DOI: 10.1007/s00401-015-1531-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lampron A; Elali A; Rivest S (2013) Innate immunity in the CNS: redefining the relationship between the CNS and its environment. Neuron 78 (2), 214–32. DOI: 10.1016/j.neuron.2013.04.005 [DOI] [PubMed] [Google Scholar]
- 15.Wald ER; Kaplan SL; Mason EO; Sabo D; Md LR; Arditi M; Wiedermann BL; Barson W; Kim KS; Yogev R; Hofkosh D(1995) Dexamethasone therapy for children with bacterial meningitis. Pediatrics 95 (1), 21–28. URL http://pediatrics.aappublications.org/content/pediatrics/95/1/21.full.pdf [PubMed] [Google Scholar]
- 16.de Gans J; van de Beek D; European Dexamethasone in Adulthood Bacterial Meningitis Study, I. (2002) Dexamethasone in adults with bacterial meningitis. N. Engl. J. Med 347 (20), 1549–56. DOI: 10.1056/NEJMoa021334 [DOI] [PubMed] [Google Scholar]
- 17.Tuomanen E (1990) Adjuncts to the therapy of bacterial meningitis. Pediatr. Infect. Dis. J 9 (10), 782–3. URL https://www.ncbi.nlm.nih.gov/pubmed/2235165 [PubMed] [Google Scholar]
- 18.Odio CM; Faingezicht I; Paris M; Nassar M; Baltodano A; Rogers J; Saez-Llorens X; Olsen KD; McCracken GH Jr. (1991) The beneficial effects of early dexamethasone administration in infants and children with bacterial meningitis. N. Engl. J. Med 324 (22), 1525–31. DOI: 10.1056/NEJM199105303242201 [DOI] [PubMed] [Google Scholar]
- 19.Kaplan SL (2019) https://www.uptodate.com/contents/bacterial-meningitis-in-children-dexamethasone-and-other-measures-to-prevent-neurologic-complications?search=bacterial%20meningitis%20children%20dexamethasone&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1.
- 20.Aarli J (1983) The immune system and the nervous system. J. Neurol 229 (3), 137–54. [DOI] [PubMed] [Google Scholar]
- 21.Orihuela CJ; Fillon S; Smith-Sielicki SH; El Kasmi KC; Gao G; Soulis K; Patil A; Murray PJ; Tuomanen EI (2006) Cell wall-mediated neuronal damage in early sepsis. Infect. Immun 74 (7), 3783–9. DOI: 10.1128/IAI.00022-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bsibsi M; Ravid R; Gveric D; van Noort JM (2002) Broad expression of Toll-Like receptors in the human central nervous system. (2002) J. Neuropath. Exp. Neurol 61 (11), 1013–1021. DOI: 10.1093/jnen/61.11.1013 [DOI] [PubMed] [Google Scholar]
- 23.Hanke ML; Kielian T (2011) Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci 121 (9), 367–87. DOI: 10.1042/CS20110164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deguine J; Barton GM (2014) MyD88: a central player in innate immune signaling. F1000 Prime Rep 6, 97 DOI: 10.12703/P6-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda K; Akira S (2004) TLR signaling pathways. Seminars Immunol 16 (1), 3–9. DOI: 10.1016/j.smim.2003.10.003 [DOI] [PubMed] [Google Scholar]
- 26.Janssens S; Beyaert R (2002) A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci 27 (9), 474–482. DOI: 10.1016/s0968-0004(02)02145-x [DOI] [PubMed] [Google Scholar]
- 27.Koedel U (2009) Toll-like receptors in bacterial meningitis. Curr. Top. Microbiol. Immunol 336, 15–40. DOI: 10.1007/978-3-642-00549-7_2 [DOI] [PubMed] [Google Scholar]
- 28.Hoffmann O; Braun JS; Becker D; Halle A; Freyer D; Dagand E; Lehnardt S; Weber JR (2007) TLR2 mediates neuroinflammation and neuronal damage. J. Immunol 178 (10), 6476–6481. DOI: 10.4049/jimmunol.178.10.6476 [DOI] [PubMed] [Google Scholar]
- 29.Lehnardt S; Wennekamp J; Freyer D; Liedtke C; Krueger C; Nitsch R; Bechmann I; Weber JR; Henneke P (2007) TLR2 and caspase-8 are essential for Group B Streptococcus-induced apoptosis in microglia. J. Immunol 179 (9), 6134–6143. DOI: 10.4049/jimmunol.179.9.6134 [DOI] [PubMed] [Google Scholar]
- 30.Yoshimura A; Lien E; Ingalls RR; Tuomanen E; Dziarski R; Golenbock D (1999) Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol 163 (1), 1–5. URL https://www.ncbi.nlm.nih.gov/pubmed/10384090 [PubMed] [Google Scholar]
- 31.Sellner J; Grandgirard D; Gianinazzi C; Landmann RM; Leib SL (2009) Effects of Toll-like receptor 2 agonist Pam(3)CysSK(4) on inflammation and brain damage in experimental pneumococcal meningitis. J. Neuroimmunol 206 (1–2), 28–31. DOI: 10.1016/j.jneuroim.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 32.Motta V; Soares F; Sun T; Philpott DJ (2015) NOD-like receptors: versatile cytosolic sentinels. Physiol. Rev 95 (1), 149–78. DOI: 10.1152/physrev.00009.2014 [DOI] [PubMed] [Google Scholar]
- 33.Rosenzweig HL; Planck SR; Rosenbaum JT (2011) NLRs in immune privileged sites. Curr. Opin. Pharmacol 11 (4), 423–8. DOI: 10.1016/j.coph.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chauhan VS; Sterka DG Jr.; Furr SR; Young AB; Marriott I (2009) NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia 57 (4), 414–23. DOI: 10.1002/glia.20770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X; Chauhan VS; Young AB; Marriott I (2010) NOD2 mediates inflammatory responses of primary murine glia to Streptococcus pneumoniae. Glia 58 (7), 839–47. DOI: 10.1002/glia.20968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoegen T; Tremel N; Klein M; Angele B; Wagner H; Kirschning C; Pfister HW; Fontana A; Hammerschmidt S; Koedel U (2011) The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J. Immunol 187 (10), 5440–51. DOI: 10.4049/jimmunol.1100790 [DOI] [PubMed] [Google Scholar]
- 37.Olson JK; Miller SD (2004) Microglia Initiate Central Nervous System Innate and Adaptive Immune Responses through Multiple TLRs. J. Immunol 173 (6), 3916–3924. DOI: 10.4049/jimmunol.173.6.3916 [DOI] [PubMed] [Google Scholar]
- 38.Brown GC; Vilalta A (2015) How microglia kill neurons. Brain Res 1628 (Pt B), 288–297. DOI: 10.1016/j.brainres.2015.08.031 [DOI] [PubMed] [Google Scholar]
- 39.Salter MW; Stevens B (2017) Microglia emerge as central players in brain disease. Nat. Med 23 (9), 1018–1027. DOI: 10.1038/nm.4397 [DOI] [PubMed] [Google Scholar]
- 40.Glass CK; Saijo K; Winner B; Marchetto MC; Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140 (6), 918–34. DOI: 10.1016/j.cell.2010.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheret C; Gervais A; Lelli A; Colin C; Amar L; Ravassard P; Mallet J; Cumano A; Krause KH; Mallat M (2008) Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J. Neurosci 28 (46), 12039–51. DOI: 10.1523/JNEUROSCI.3568-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy S (2000) Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia 29 (1), 1–13. DOI: [DOI] [PubMed] [Google Scholar]
- 43.Bai Y; Zhu Z; Gao Z; Kong Y (2014) TLR2 signaling directs NO-dependent MMP-9 induction in mouse microglia. Neurosci. Lett 571, 5–10. DOI: 10.1016/j.neulet.2014.04.025 [DOI] [PubMed] [Google Scholar]
- 44.Liechti FD; Grandgirard D; Leppert D; Leib SL (2014) Matrix metalloproteinase inhibition lowers mortality and brain injury in experimental pneumococcal meningitis. Infect. Immun 82 (4), 1710–8. DOI: 10.1128/IAI.00073-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muri L; Grandgirard D; Buri M; Perny M; Leib SL (2018) Combined effect of non-bacteriolytic antibiotic and inhibition of matrix metalloproteinases prevents brain injury and preserves learning, memory and hearing function in experimental paediatric pneumococcal meningitis. J. Neuroinflam 15 (1), 233 DOI: 10.1186/s12974-018-1272-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiavolini D; Tripodi S; Parigi R; Oggioni MR; Blasi E; Cintorino M; Pozzi G; Ricci S (2004) Method for inducing experimental pneumococcal meningitis in outbred mice. BMC Microbiol 4, 36 DOI: 10.1186/1471-2180-4-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diesselberg C; Ribes S; Seele J; Kaufmann A; Redlich S; Bunkowski S; Hanisch UK; Michel U; Nau R; Schutze S(2018) Activin A increases phagocytosis of Escherichia coli K1 by primary murine microglial cells activated by toll-like receptor agonists. J. Neuroinflam 2018, 15 (1), 175 DOI: 10.1186/s12974-018-1209-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Redlich S; Ribes S; Schutze S; Czesnik D; Nau R (2012) Palmitoylethanolamide stimulates phagocytosis of Escherichia coli K1 and Streptococcus pneumoniae R6 by microglial cells. J. Neuroimmunol 244 (1–2), 32–4. DOI: 10.1016/j.jneuroim.2011.12.013 [DOI] [PubMed] [Google Scholar]
- 49.Primiano MJ; Lefker BA; Bowman MR; Bree AG; Hubeau C; Bonin PD; Mangan M; Dower K; Monks BG; Cushing L; Wang S; Guzova J; Jiao A; Lin LL; Latz E; Hepworth D; Hall JP (2016) Efficacy and Pharmacology of the NLRP3 Inflammasome Inhibitor CP-456,773 (CRID3) in Murine Models of Dermal and Pulmonary Inflammation. J. Immunol 197 (6), 2421–33. DOI: 10.4049/jimmunol.1600035 [DOI] [PubMed] [Google Scholar]
- 50.Ismael S; Nasoohi S; Ishrat T (2018) MCC950, the selective inhibitor ofnucleotide oligomerization domain-like receptor protein-3 inflammasome, protects mice against traumatic brain injury. J. Neurotrauma 35 (11), 1294–1303. DOI: 10.1089/neu.2017.5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu F; Wang F; Wen T; Sang W; Wang D; Zeng N (2017) Inhibition of NLRP3 inflammasome: a new protective mechanism of cinnamaldehyde in endotoxin poisoning of mice. Immunopharmacol. Immunotoxicol 39 (5), 296–304. DOI: 10.1080/08923973.2017.1355377 [DOI] [PubMed] [Google Scholar]
- 52.Bifrare YD; Kummer J; Joss P; Tauber MG; Leib SL (2005) Brain-derived neurotrophic factor protects against multiple forms of brain injury in bacterial meningitis. J. Infect. Dis 191 (1), 40–5. DOI: 10.1086/426399 [DOI] [PubMed] [Google Scholar]
- 53.Li L; Shui QX; Liang K; Ren H (2007) Brain-derived neurotrophic factor rescues neurons from bacterial meningitis. Pediatr. Neurol 36 (5), 324–9. DOI: 10.1016/j.pediatrneurol.2007.01.007 [DOI] [PubMed] [Google Scholar]
- 54.Xu D; Lian D; Wu J; Liu Y; Zhu M; Sun J; He D; Li L (2017) Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J. Neuroinflamm 14 (1), 156 DOI: 10.1186/s12974-017-0930-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lian D; He D; Wu J; Liu Y; Zhu M; Sun J; Chen F; Li L (2016) Exogenous BDNF increases neurogenesis in the hippocampus in experimental Streptococcus pneumoniae meningitis. J. Neuroimmunol 294, 46–55. DOI: 10.1016/j.jneuroim.2016.03.014 [DOI] [PubMed] [Google Scholar]
- 56.Mitchell L; Smith SH; Braun JS; Herzog KH; Weber JR; Tuomanen EI (2004) Dual phases of apoptosis in pneumococcal meningitis. J. Infect. Dis 190 (11), 2039–46. DOI: 10.1086/425520 [DOI] [PubMed] [Google Scholar]
- 57.Humann J; Mann B; Gao G; Moresco P; Ramahi J; Loh LN; Farr A; Hu Y; Durick-Eder K; Fillon SA; Smeyne RJ; Tuomanen EI (2016) Bacterial peptidoglycan traverses the placenta to induce fetal neuroproliferation and aberrant postnatal behavior. Cell Host Microbe 19 (3), 388–99. DOI: 10.1016/j.chom.2016.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mellado MC; Rodriguez-Contreras R; Mariscal A; Luna JD; Delgado Rodriguez M; Galvez-Vargas R (1991) Effect of penicillin and chloramphenicol on the growth and endotoxin release by N. meningitidis. Epidemiol. Infect 106 (2), 283–8. URL https://www.ncbi.nlm.nih.gov/pubmed/1902182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pankey GA; Sabath LD (2004) Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin. Infect. Dis 38 (6), 864–70. DOI: 10.1086/381972 [DOI] [PubMed] [Google Scholar]
- 60.Lewis K (2000) Programmed death in bacteria. Microbiol. Molecbiol. Rev 64 (3), 503–514. DOI: 10.1128/mmbr.64.3.503-514.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tomasz A(1974) The role of autolysins in cell death. Ann. N.Y. Acad. Sci 235, 439–447. DOI: 10.1111/j.1749-6632.1974.tb43282.x [DOI] [PubMed] [Google Scholar]
- 62.Henriques-Normark B; Tuomanen EI (2013) The pneumococcus: Epidemiology, microbiology, and pathogenesis. Cold Spring Harbor Persp. Med 3, 7–14. DOI: 10.1101/cshperspect.a010215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spreer A; Kerstan H; Bottcher T; Gerber J; Siemer A; Zysk G; Mitchell TJ; Eiffert H; Nau R (2003) Reduced release of pneumolysin by Streptococcus pneumoniae in vitro and in vivo after treatment with nonbacteriolytic antibiotics in comparison to ceftriaxone. Antimicrob. Agents Chemother 47 (8), 2649–54. DOI: 10.1128/AAC.47.8.2649-2654.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bottcher T; Ren H; Goiny M; Gerber J; Lykkesfeldt J; Kuhnt U; Lotz M; Bunkowski S; Werner C; Schau I; Spreer A; Christen S; Nau R (2004) Clindamycin is neuroprotective in experimental Streptococcus pneumoniae meningitis compared with ceftriaxone. J. Neurochem 91 (6), 1450–60. DOI: 10.1111/j.1471-4159.2004.02837.x [DOI] [PubMed] [Google Scholar]
- 65.Klepser ME; Nicolau DP; Quintiliani R; Nightingale CH (1997) Bactericidal activity of low-dose clindamycin administered at 8- and 12-hour intervals against Staphylococcus aureus, Streptococcus pneumoniae, and Bacteroides fragilis. Antimicrob. Ag. Chemother 41 (3), 630–635. DOI: 10.1128/aac.41.3.630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ribes S; Taberner F; Domenech A; Cabellos C; Tubau F; Linares J; Fernandez Viladrich P; Gudiol F (2005) Evaluation of ceftriaxone, vancomycin and rifampicin alone and combined in an experimental model of meningitis caused by highly cephalosporin-resistant Streptococcus pneumoniae ATCC 51916. J. Antimicrob. Chemother 56 (5), 979–82. DOI: 10.1093/jac/dki323 [DOI] [PubMed] [Google Scholar]
- 67.Sauermann R; Schwameis R; Fille M; Ligios ML; Zeitlinger M (2008) Antimicrobial activity of cefepime and rifampicin in cerebrospinal fluid in vitro. J. Antimicrob. Chemother 62 (5), 1057–60. DOI: 10.1093/jac/dkn312 [DOI] [PubMed] [Google Scholar]
- 68.Spreer A; Lugert R; Stoltefaut V; Hoecht A; Eiffert H; Nau R (2009) Short-term rifampicin pretreatment reduces inflammation and neuronal cell death in a rabbit model of bacterial meningitis. Crit. Care Med 37 (7), 2253–8. DOI: 10.1097/CCM.0b013e3181a036c0 [DOI] [PubMed] [Google Scholar]
- 69.Rahal JJ; Simberkoff MS (1979) Bactericidal and bacteriostatic action of chloramphenicol against meningeal pathogens. Antimicrob. Ag. Chemother 16 (1), 13–18. 10.1128/Aac.16.1.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scheld WM; Sande MA (1983) Bactericidal versus bacteriostatic antibiotic therapy of experimental pneumococcal meningitis in rabbits. J. Clin. Invest 71 (3), 411–9. DOI:https://dx.doi.org/10.1172%2FJCI110785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Erslev AJ; Iossifides IA(1962) In vitro action of chloramphenicol and chloramphenicol-analogues on the metabolism of human immature red blood cells. Acta Haematol 28, 1–19. DOI: 10.1159/000207232 [DOI] [PubMed] [Google Scholar]
- 72.Dinos GP; Athanassopoulos CM; Missiri DA; Giannopoulou PC; Vlachogiannis IA; Papadopoulos GE; Papaioannou D; Kalpaxis DL (2016) Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions. Antibiotics 5 (2). DOI: 10.3390/antibiotics5020020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Silvestro EM; Nakano V; Arana-Chavez VE; Marques MV; Avila-Campos MJ (2006) Effects of subinhibitory concentrations of clindamycin on the morphological, biochemical and genetic characteristics of Bacteroides fragilis. FEMS Microbiol. Lett 257 (2), 189–94. DOI: 10.1111/j.1574-6968.2006.00162.x [DOI] [PubMed] [Google Scholar]
- 74.Lewis K (2010) Persister cells. Annu. Rev. Microbiol 64, 357–72. DOI: 10.1146/annurev.micro.112408.134306 [DOI] [PubMed] [Google Scholar]
- 75.Tuomanen E; Durack DT; Tomasz A (1986) Antibiotic tolerance among clinical isolates of bacteria. Antimicrob. Ag. Chemother 30 (4), 521–527. URL http://www.ncbi.nlm.nih.gov/pmc/articles/PMC176473/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tuomanen E (1986) Phenotypic tolerance - the search for Beta-lactam antibiotics that kill nongrowing bacteria. Rev. Infect. Dis 8, S279–S291. DOI, <Go to ISI>://WOS:A1986D587700004 [DOI] [PubMed] [Google Scholar]
- 77.Grandgirard D; Oberson K; Buhlmann A; Gaumann R; Leib SL (2010) Attenuation of cerebrospinal fluid inflammation by the nonbacteriolytic antibiotic daptomycin versus that by ceftriaxone in experimental pneumococcal meningitis. Antimicrob. Ag. Chemother 54 (3), 1323–6. DOI: 10.1128/AAC.00812-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang Y; Zhao L; Jia B; Wu L; Li Y; Curthoys N; Zheng JC(2011) Glutaminase dysregulation in HIV-1-infected human microglia mediates neurotoxicity: relevant to HIV-1-associated neurocognitive disorders. J. Neurosci 31 (42), 15195–204. DOI: 10.1523/JNEUROSCI.2051-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li X; Mu X; Yang Y; Hua X; Yang Q; Wang N; Du X; Ruan Z; Shen X; Yu Y (2016) Rapid emergence of high-level tigecycline resistance in Escherichia coli strains harbouring blaNDM-5 in vivo. Int. J. Antimicrob. Agents 47 (4), 324–7. DOI: 10.1016/j.ijantimicag.2016.01.005 [DOI] [PubMed] [Google Scholar]
- 80.Hammerstrom TG; Beabout K; Clements TP; Saxer G; Shamoo Y (2015) Acinetobacter baumannii repeatedly evolves a hypermutator phenotype in response to Tigecycline that effectively surveys evolutionary trajectories to resistance. PLoS One 10 (10), e0140489 DOI: 10.1371/journal.pone.0140489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Homma T; Nuxoll A; Gandt AB; Ebner P; Engels I; Schneider T; Gotz F; Lewis K; Conlon BP (2016) Dual targeting of cell wall precursors by Teixobactin leads to cell lysis. Antimicrob. Ag. Chemother 60 (11), 6510–6517. DOI: 10.1128/AAC.01050-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Breukink E; de Kruijff B (2004) Lipid II as a target for antibiotics. Nat. Rev. Drug Discov 5 (4), 321–32. DOI: 10.1038/nrd2004 [DOI] [PubMed] [Google Scholar]
- 83.Oppedijk SF; Martin NI; Breukink E (2016) Hit ‘em where it hurts: The growing and structurally diverse family of peptides that target lipid-II. Biochim. Biophys. Acta 1858 (5), 947–57. DOI: 10.1016/j.bbamem.2015.10.024 [DOI] [PubMed] [Google Scholar]
- 84.Ulm H; Schneider T (2016) Targeting bactoprenol-coupled cell envelope precursors. Appl. Microbiol. Biotechnol 100 (18), 7815–25. DOI: 10.1007/s00253-016-7732-0 [DOI] [PubMed] [Google Scholar]
- 85.Cozens RM; Tuomanen E; Tosch W; Zak O; Suter J; Tomasz A (1986) Evaluation of the bactericidal activity of beta-lactam antibiotics on slowly growing bacteria cultured in the chemostat. Antimicrob. Ag. Chemother 29 (5), 797–802.URL https://www.ncbi.nlm.nih.gov/pubmed/3089141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tuomanen E; Cozens R; Tosch W; Zak O; Tomasz A (1986) The rate of killing of Escherichia coli by beta-lactam antibiotics is strictly proportional to the rate of bacterial growth. J. Gen. Microbiol 132 (5), 1297–304. DOI: 10.1099/00221287-132-5-1297 [DOI] [PubMed] [Google Scholar]
- 87.Kusser W; Ishiguro EE (1985) Involvement of the relA gene in the autolysis of Escherichia coli induced by inhibitors of peptidoglycan biosynthesis. J. Bacteriol 164 (2), 861–5. URL https://www.ncbi.nlm.nih.gov/pubmed/3902801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kusser W; Ishiguro EE (1986) Lysis of nongrowing Escherichia coli by combinations of beta-lactam antibiotics and inhibitors of ribosome function. Antimicrob. Ag. Chemother 29 (3), 451–5. URL https://www.ncbi.nlm.nih.gov/pubmed/2424368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cozens RM; Markiewicz Z; Tuomanen E (1989) Role of autolysins in the activities of imipenem and CGP 31608, a novel penem, against slowly growing bacteria. Antimicrob. Ag. Chemother 33 (10), 1819–21. URL https://www.ncbi.nlm.nih.gov/pubmed/2574024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tuomanen E; Schwartz J (1987) Penicillin-binding protein 7 and its relationship to lysis of nongrowing Escherichia coli. J. Bacteriol 1987, 169 (11), 4912–5. URL https://www.ncbi.nlm.nih.gov/pubmed/3312163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tomasz A; Albino A; Zanati E (1970) Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature 227 (5254), 138–40. URL http://www.ncbi.nlm.nih.gov/pubmed/4393335 [DOI] [PubMed] [Google Scholar]
- 92.Kietzman CC; Gao G; Mann B; Myers L; Tuomanen EI (2016) Dynamic capsule restructuring by the main pneumococcal autolysin LytA in response to the epithelium. Nat. Comm 7, 10859 DOI: 10.1038/ncomms10859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fridman O; Goldberg A; Ronin I; Shoresh N; Balaban NQ (2014) Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 513 (7518), 418–21. DOI: 10.1038/nature13469 [DOI] [PubMed] [Google Scholar]
- 94.Liu HH; Tomasz A (1985) Penicillin Tolerance in Multiply Drug-Resistant Natural Isolates of Streptococcus pneumoniae. J. Infect. Dis 152 (2), 365–372. DOI: 10.1093/infdis/152.2.365 [DOI] [PubMed] [Google Scholar]
- 95.Tuomanen E; Pollack H; Parkinson A; Davidson M; Facklam R; Rich R; Zak O (1988) Microbiological and clinical significance of a new property of defective lysis in clinical strains of pneumococci. J. Infect. Dis 158 (1), 36–43. DOI: 10.1093/infdis/158.1.36 [DOI] [PubMed] [Google Scholar]
- 96.Handwerger S; Tomasz A(1985) Antibiotic tolerance among clinical isolates of bacteria. Rev. Infect. Dis 1985, 25, 349–80. DOI: 10.1146/annurev.pa.25.040185.002025 [DOI] [PubMed] [Google Scholar]
- 97.McCullers JA; English BK; Novak R (2000) Isolation and characterization of vancomycin-tolerant Streptococcus pneumoniae from the cerebrospinal fluid of a patient who developed recrudescent meningitis. J. Infect. Dis 2000, 181 (1), 369–73. DOI: 10.1086/315216 [DOI] [PubMed] [Google Scholar]
- 98.Novak R; Henriques B; Charpentier E; Normark S; Tuomanen E (1999) Emergence of vancomycin tolerance in Streptococcus pneumoniae. Nature 399 (6736), 590–3. DOI: 10.1038/21202 [DOI] [PubMed] [Google Scholar]
- 99.Henriques Normark B; Novak R; Ortqvist A; Kallenius G; Tuomanen E; Normark S (2001) Clinical isolates of Streptococcus pneumoniae that exhibit tolerance of vancomycin. Clin. Infect. Dis 32 (4), 552–8. DOI: 10.1086/318697 [DOI] [PubMed] [Google Scholar]