

Abstract
The inflammasomes are multi-molecular platforms that are activated in host cell cytoplasm when the innate immune cells are infected with pathogens or exposed to damage signals. Many independent groups reported that Leishmania infection trigger activation of the NLRP3 inflammasome in macrophages for restriction of intracellular parasite replication. Accordingly, Leishmania can dampen NLRP3 activation as an evasion strategy. In vivo, the NLRP3 inflammasome can promote parasite clearance, but the failure to eliminate parasites in the tissues together with sustained inflammasome activation can promote IL-1β-mediated disease pathology. In this review, we discuss the recent data regarding activation of the NLRP3 inflammasome in response to Leishmania and the beneficial and detrimental effects of the inflammasome during development of Leishmaniasis.
Introduction
The inflammasomes are multiprotein complexes that are assembled in the cytoplasm of innate immune cells when intracellular pattern recognition receptors detect pathogenic microorganisms or damage signals. The most studied inflammasome is the nucleotide-binding domain leucine-rich repeat protein (NLR) family member, NLRP3, which is activated in response to multiple pathogens and molecules that induce damage in the host cell membranes (Reviewed in [1]). Upon activation, NLRP3 oligomerizes and promotes the polymerization of ASC via Pyrin-Pyrin interaction, exposing multiple CARD domains of ASC in the filaments, which can recruit Caspase-1 via CARD/CARD interaction to promote caspase-1 activation (Figure 1). Importantly, two independent signals are required for activation of NLRP3. The first signal (also called priming) occurs when microbial components (or TNF-α) stimulate TLRs (or TNFR) leading to transcription of many inflammatory genes, including Nlrp3, Casp11 and II1b. Although the first signal is essential, it is not sufficient, and a required second signal occurs with formation of pores or rupture in the macrophage membranes, which allows for the decrease in intracellular K+. The induction of membrane pores by microbial toxins, the activation of K+ channels or membrane lysis by crystals or noxious molecules allow K+ efflux and the so called canonical activation of the NLRP3 inflammasome [1,2]. Besides the efflux of potassium, the production of ROS and lysosomal cathepsins are also important for canonical NLRP3 activation (reviewed in [1]). In contrast, activation of caspase-11 promotes the non-canonical activation of the NLRP3 inflammasome. Mechanistically, caspase-11 is activated when bacterial LPS is present in the cytoplasm and promotes proteolytic cleavage of Gasdermin-D. The active N-terminal domain of Gasdermin-D is further inserted in the macrophage membrane and forms a pore that allows K+ efflux and the non-canonical activation of the NLRP3 inflammasome (reviewed in [1]). The requirement for the two signals provides a regulated control of the inflammasome activation that prevents unnecessary inflammation in the absence of infection or noxious molecules. Not surprisingly, intracellular pathogens have developed mechanisms to inhibit these signals, as discussed below in the context of Leishmania.
Figure 1. Activation of NLRP3 inflammasome in response to pathogens, crystals and pore-forming molecules.
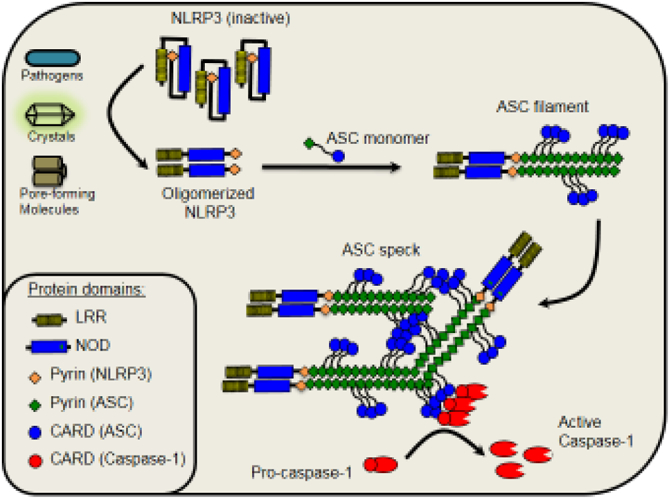
NLRP3 is activated when pathogens or specific molecules (crystals, toxins, purinergic receptors, etc.) promote perturbation of the host cell membranes. Once activated NLRP3 oligomerizes and promotes the polymerization of ASC via Pyrin-Pyrin interaction. Polymerized ASC forms a long filament exposing multiple CARD domains. This filament can bind to other ASC filaments to form the ASC speck. This platform recruits pro-caspase-1 via CARD/CARD interaction to promote caspase-1 activation and cleavage.
Leishmanial diseases represent a spectrum of neglected tropical diseases that are endemic in 88 countries worldwide, with over 1 million new cases each year and over 300 million people at risk. There are different forms of clinical disease in humans, ranging from self-limiting cutaneous lesions, to tissue destructive mucosal involvement, to visceral dissemination that is fatal in the absence of treatment. These diverse clinical outcomes are caused by different species of protozoan parasites in the genus Leishmania that are in each case transmitted to their mammalian hosts by the bite of an infected sand fly vector. Leishmania have a digenetic life cycle consisting of extracellular, flagellated promastigotes replicating in the sand fly, and intracellular aflagellate amastigotes replicating in phagocytic cells, primarily macrophages, in the mammalian host. There are no effective vaccines for use in humans, despite the fact that primary infection, especially those involving cutaneous strains, often result in strong, long lasting acquired immunity. As would be expected for immunity against pathogens that reside in phagosomes, T helper 1 (Th1) responses are a crucial component of the protective response, with strong T cell responses associated with healing, and weak T cell responses associated with high parasite burdens and disseminating infections. This dichotomy is not always so clear, however, as mucosal patients present with an exaggerated T cell response that controls the infection but drives the chronic tissue destruction. The severity of even localized cutaneous lesions reflects a balance between immunoprotective and immunopathologic processes that can be difficult to distinguish. This review focuses on central components of the innate response, NLRP3 inflammasome activation and secretion of IL-1β, that have been found to contribute to both protective and pathologic outcomes in experimental Leishmaniasis.
Activation of NLRP3 Inflammasome and restriction of Leishmania replication in macrophages
The initial demonstration that Leishmania infection triggers the activation of inflammasomes occurred in 2013 when it was shown that bone marrow-derived macrophages infected with L. amazonensis, L. major and L. braziliensis trigger Caspase-1 activation and IL-1β production. This process was abolished in the absence of NLRP3 or ASC, indicating that the NLRP3 inflammasome is responsible for caspase-1 activation in response to infection [3]. This study also demonstrated that the inflammasome activation in macrophages favored the restriction of parasite replication in macrophages and in mouse by mechanisms dependent on IL-1R signaling. Another study published in the same year reported that L.infantum induced Caspase-1 and IL-1β production through Dectin-1 and mannose receptor, which stimulated p47phox and arachidonic acid-NADPH oxidase signaling for production of ROS and restriction of parasite replication via Caspase-1 and IL-1β [4]. In agreement to these findings, it was demonstrated that L.amazonensis induced Dectin-1 activation, leading to activation of Syk kinase and ROS production by NADPH oxidase, a process that is critical for activation of NLRP3 in macrophages [5]. Importantly, inhibition of ROS and NADPH oxidase during the early stages of infection was sufficient to abrogate inflammasome activation, indicating that the initial signals for inflammasome activation occurs during the phagocytosis of the parasites [5]. These studies emphasize the participation of ROS in the activation of NLRP3 during Leishmania infection. More recently, lipophosphoglycan (LPG) from many Leishmania species was shown to trigger caspase-11 activation in macrophages [6], demonstrating non-canonical activation of the NLRP3 inflammasome, although a caspase-11-independent pathway for inflammasome activation was also detected [6]. The demonstration that caspase-11 is involved in activation of NLRP3 in response to Leishmania provided clues to explain the requirement of priming for inflammasome activation in BMDMs infected with Leishmania in vitro. Similarly to Pro-IL-1β, Caspase-11 is expressed in lower levels in steady state cells and transcriptional responses are required for protein expression [7]. According to these observations, a recent report demonstrated that because of the low levels of Caspase-11 and Pro-IL-1β in steady state BMDMs, priming is required for detection of Caspase-1 cleavage (assessed by western blot) and IL-1β activation (assessed by ELISA) in response to Leishmania amazonensis infection [8]. However, priming was dispensable for assembly of the NLRP3 inflammasome (assessed by ASC specks), Caspase-1 activation (assessed by FAM-YVAD) and restriction of parasite replication via the NLRP3 inflammasome [8]. During in vivo infection, priming may occur via TNF-α, INF-γ or when bacteria contaminate the cutaneous lesions developed in patients skins. It was also reported that gut bacteria from the sand fly may be egested into host skin with Leishmania parasites [9], and may thus be the source of priming allowing robust inflammasome activation when the parasites infect the macrophages.
It was also reported that the mitochondrial phosphatase phosphoglycerate mutase family member 5, a protein that plays a role in the restriction of Leishmania replication in macrophages, is important for IL-1β production in response to Leishmania, implying the participation of this protein in the activation of the NLRP3 inflammasome [10]. Compounds that trigger inflammasome activation and promote IL-1β production have also been shown to restrict parasite replication in macrophages, including nanoparticles of polyester poly (lactide-co-glycolide acid) loaded with an 11kDa Leishmania antigen, [11], the antiprotozoal drug diterpene kaurenoic acid, and the leishmanicidal drug Amphotericin B [12]. Administration of anti-IL-1β reduced the parasite clearance by AmpB in a model of visceral Leishmaniasis [13]. Collectively, these studies unequivocally demonstrate that the inflammasome can be activated in response to Leishmania infection and plays an important role in restricting Leishmania replication in macrophages.
Inhibition of NLRP3 Inflammasome by Leishmania as an evasion strategy
Given its role as an innate immune defense against Leishmania infection, it is not surprising that Leishmania have evolved mechanisms to inhibit or limit the activation of the NLRP3 inflammasome in macrophages. Shio et al. demonstrated that L.mexicana and L.major use the virulence factor GP63 to inhibit the production of IL-1β production in human THP-1 cells. The mechanisms of inhibition are still unclear but may involve inhibition of ROS production and also a direct cleavage of inflammasome components [14]. L.donovani inhibits the NLRP3 inflammasome by manipulating A20 (a negative regulator of NF-kB) and UCP2 (mitochondrial uncoupling protein 2). A20 was thought to inhibit the first signal (priming) whereas UCP2 inhibits of the second signal, involving the ROS production [13]. Upregulation of A20 as a mechanism to inhibition inflammasome activation was also evident in experiments using L. guyanensis [15]. Two recent studies involving L. amazonensis and L. donovani showed transcriptional inhibition of inflammasome components in infected macrophages [16,17]. Collectively, these findings do not necessarily conflict with the articles cited above reporting inflammasome activation in response to Leishmania infection. The inhibitory effect in certain conditions may reduce/limit the magnitude of inflammasome activation, but may not fully block the activation. The consensus is that the magnitude of inflammasome activation in response to Leishmania is reduced as compared to infection with bacteria or other protozoan parasites such as Toxoplasma gondii and Trypanosoma cruzi (reviewed in [18]). This may occur because Leishmania inhibits inflammasome activation and may explain why some groups fail to detect inflammasome activation in response to Leishmania infection.
Inflammasome activation and IL-1β can promote pathology and parasite growth in vivo
In vivo studies performed with L.amazonensis in mice indicated that the inflammasome were important for protective host response and restriction of parasite replication in the tissues. This was demonstrated using Nlrp3−/−, Asc−/−, Casp1/11−/−, Casp11−/−and II1r−/− mice in C57BL/6 genetic background and using Casp1/11−/−mice in A/J background [3,5,6]. However, studies performed with other Leishmania species reported a detrimental effect of the NLRP3 inflammasome. Infection of C57BL/6 mice with many L.major strains results in a healing lesion with minimal pathology at the site of inoculation in the skin. By contrast, using a low dose infection with a strain of L. major (Lm Sd) isolated from a patient with chronic cutaneous lesions, C57BL/6 mice developed severe, non-healing dermal lesions and failed to effectively control tissue parasite loads despite a strong TH1 response [19]. Infection was associated with elevated early and sustained levels of IL-1β mRNA and IL-1β+ cells in the inoculation site, and a strong neutrophil infiltrate that persisted throughout the course of lesion development [20]. Critically, mice deficient in IL-1R, IL-1β, ASC, caspase-1/11, and NLRP3, each showed minimal pathology and healed their Lm Sd infection. While a contribution of the inflammasome/IL-1β axis to the severe pathology might be expected, the pathology appeared to be secondary to the inability to control the infection, resulting in the persistence of organisms able to drive the inflammatory response. Similar observations have been made in mice lacking the NLRP3 inflammasome on a Leishmania-susceptible background (BALB/c), which exhibited better control over L.major growth in the footpad [21], and in C57BL/6 and BALB/c mice treated with exogenous IL-1β, that in both cases led to increased L.major parasite burdens and lesion progression [22]. The severity of experimental visceral leishmaniasis following transmission of L.donovani by bites of infected sand flies has also been linked to the activation of the NLRP3 inflammasome and IL-1β production, thought to be exacerbated by gut microbiota from the fly that were egested along with the parasites into the skin [9].
How might the NLRP3 inflammasome and IL-1β function to compromise host defense against Leishmania? Sustained recruitment of neutrophils is characteristic of each of these skin inoculations sites, and their ability to sequester the infection [23], or to be captured by macrophages and dendritic cells to suppress the microbicidal and antigen presentation function of these cells, has been described [24,25]. IL-1β has also been shown to promote the expansion of Th2 and Th17 cells, at least in the mouse [26]. The inflammasome deficit in the L. major infected C57BL/6 and BALB/c mice resulted in diminished levels of Th2 cytokines, though in the later case IL-18 was thought to regulate the Th1/Th2 balance observed [21]. IL-1β can also promote the proliferation and maturation of group 2 innate lymphoid cells (ILC2) [27], which could act by promoting Th2 development. The type 2 cytokines produced by ILC2 are also crucial to sustain eosinophils and alternatively activated macrophages in local tissue environments, such as adipose tissue [28]. This may explain the loss of the population of embryonic derived, alternatively activated, dermis resident macrophages in the inflammasome and IL-1β deficient mice, whose preferential infection by the Lm Sd strain was found to be essential to the development of the non-healing phenotype [29]. Importantly, in contrast to the infected, monocyte-derived cells recruited to the site, the infected, dermis resident macrophages did not produce IL-1β.
Clinically, IL-1β expression has been correlated with disease severity in patients with cutaneous leishmaniasis due to L. mexicana [30], and monocytes from patients with CL due to L. braziliensis expressed NLRP3, and secreted IL-1β in levels that correlated with areas of necrosis and lesion progression [31]. More directly, blockade of the NLRP3 inflammasome using the small molecule inhibitor glyburide prevented IL-1β release in lesion biopsies of L. braziliensis patients [32]. Transcriptionally, it was found that the levels of IL-1β expression in biopsied skin lesions positively correlated with genes involved in both inflammasome activation and cytotoxicity, including GZMB, GZMA and PRF1 [32,33]. A clear link between cytotoxic CD8+ T cells and NLRP3 inflammasome activation and IL-1β production was established in a mouse model of L. braziliensis infection in which RAG deficient mice that were reconstituted with CD8+ T cells developed immunopathology that was ameliorated using CD8+ T cells from perforin knockout mice, or by pharmacological inhibition of NLRP3 or IL-1β [32]. Thus, inflammasome activation by dead cells, or danger associated molecular patterns (DAMPS), in conjunction with the tissue parasites themselves, could augment the pathogenesis. Severe pathology in this model was again associated with the persistent recruitment of neutrophils, which were themselves a source of IL-1β, but importantly was not associated with increased tissue parasite loads.
Conclusions and perspectives
Work performed in the last 5 years by independent groups reported activation of the NLRP3 inflammasome in response to different Leishmania species. Inflammasome activation can be triggered as a protective host response and accordingly, Leishmania was reported to actively limit the inflammasome activation in macrophages. Nonetheless, when innate immune cells fail to clear parasites in the tissues, sustained inflammasome activation promotes exacerbated IL-1β production and inflammation, which can lead to tissue damage and aggravation of the disease. In experimental infections involving L. major, inflammasome activation can indirectly contribute to parasite growth and persistence, likely due to the ability of the inflammasome / IL-1β axis to promote Th2, ILC2, and/or neutrophil responses. The inability to appreciate an inflammasome driven host protective response in L.major infected mice might be explained by the relative strength of the T cell response that supersedes any direct effect of inflammasome activation on the innate response. Collectively, these observations highlight the importance of the inflammasome in the pathophysiology of Leishmaniasis and raises promising therapeutic approaches to reduce the clinical manifestations of the disease in certain cases.
Many other important aspects of NLRP3 inflammasome activation by Leishmania remain unclear, including at the most basic level how an intraphagosomal parasite triggers this cytosolic sensor. The production of ROS via NADPH oxidase and Syk kinase, as well as the efflux of K+ appear to be important for the NLRP3 response to infection [4,5,13,14]. The involvement of the parasite glycoconjugate LPG in triggering Caspase-11 [6] suggests that Leishmania-induced inflammasome activation occurs, at least partially, via the non-canonical pathway (Figure 2), although how LPG reaches the macrophage cytoplasm to trigger caspase-5/11 is still unclear. It is also possible that the release of ATP by dying macrophages favors the canonical activation the NLRP3 via purinergic receptors such as P2X7 and P2Y [34,35]. Finally, understanding the pathogenic effects of IL-1β/IL-1R in the tissues requires further investigation. The comprehensive understanding of the roles of NLRP3 inflammasome during Leishmaniasis is key for our understanding of the molecular basis of this important disease.
Figure 2. Mechanisms of inflammasome activation in macrophages infected with Leishmania.
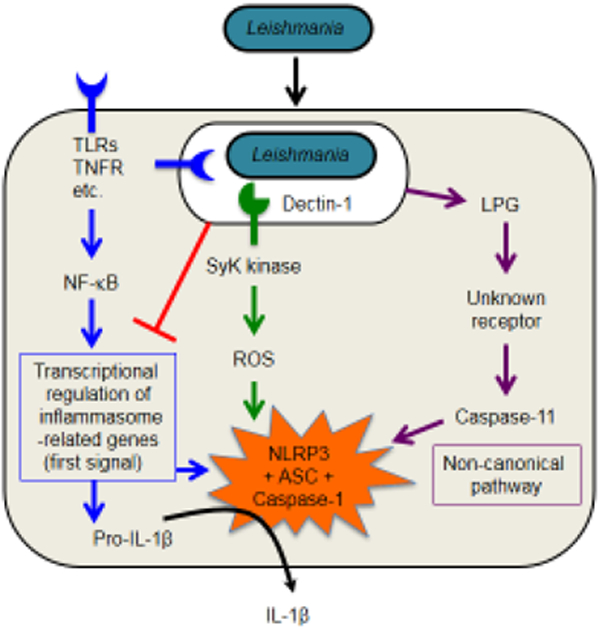
NLRP3 activation requires the first signal (or priming) that can be achieved when Toll-like receptors (TLRs) ligands, TNF-α (or other cytokines) induce transcriptional upregulation of inflammasome-related genes (indicated in blue). This process usually occurs via nuclear factor-κB (NF-κB)-mediated gene expression. Leishmania is able to inhibit the inflammasome activation at the priming stage by multiple mechanisms (indicated in red). Leishmania trigger Dectin-1, a C-type lectin receptor that signals via spleen tyrosine kinase (Syk) to induce ROS, which is critical for activation of the NLRP3 inflammasome (pathway shown in green). Leishmania Lipophosphoglycan (LPG) present in the macrophage cytoplasm triggers Caspase-11 activation indirectly. This pathway feeds in the non-canonical pathway for activation of the NLRP3 inflammasome (indicated in purple).
Highlights.
Leishmania infection trigger activation of NLRP3 inflammasome in macrophages
Inflammasome activation in macrophages restricts intracellular parasite replication
Leishmania can partially inhibit inflammasome activation as an evasion strategy
Inflammasome activation can promote pathology/parasite growth in certain conditions.
Acknowledgments
We are grateful to many colleagues in the field for the helpful discussions about Inflammasomes and Leishmania. Research in the Zamboni Lab is supported by grants from PEW, Training in Tropical Diseases/World Health Organization (TDR/WHO), Conselho Nacional de Desenvolvimento Científico e Tecnológico (Grants 401577/2014-7 and 445881/2014-3) and Fundação de Amparo à Pesquisa do Estado de São Paulo (Grants CRID/FAPESP 2013/08216-2, and FAPESP 2014/04684-4). Work in the Sacks lab is supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors declare no conflict of interest.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as
* of special interest
** of outstanding interest
- 1.Broz P, Dixit VM: Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016, 16:407–420. [DOI] [PubMed] [Google Scholar]
- 2.Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, Mittal M, Hong Z, Kanneganti TD, Rehman J, et al. : The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-lnduced Inflammation. Immunity 2018, 49:56–65 e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva AL, Mineo TW, Gutierrez FR, Bellio M, Bortoluci KR, Flavell RA, et al. : Inflammasome-derived IL-1beta production induces nitric oxide-mediated resistance to Leishmania. Nat M 2013, 19:909–915.** This manuscript was the first to demonstrate activation of the NLRP3 inflammasome in response to Leishmania. It was reported that inflammasome is activated in response to L. amazonensis, L. braziliensis and L. infantum and promoted a protective effect in macrophages and in vivo.
- 4.Lefevre L, Lugo-Villarino G, Meunier E, Valentin A, Olagnier D, Authier H, Duval C, Dardenne C, Bernad J, Lemesre JL, et al. : The C-type lectin receptors dectin-1, MR, and SIGNR3 contribute both positively and negatively to the macrophage response to Leishmania infantum. Immunity 2013, 38:1038–1049** This manuscript reported the effect of Dectin-1 and Manose receptor in microbicidal response to Leishmania infantum, linking these receptors with ROS production and Syk kinase signalling for caspase-1-induced IL-1β secretion.
- 5.Lima-Junior DS, Mineo TWP, Calich VLG, Zamboni DS: Dectin-1 Activation during Leishmania amazonensis Phagocytosis Prompts Syk-Dependent Reactive Oxygen Species Production To Trigger Inflammasome Assembly and Restriction of Parasite Replication. J Immunol 2017, 199:2055–2068.* This manuscript reported that during the phagocytosis of L. amazonensis, the activation of Dection-1, Syk kinase and NADPH oxidase promotes ROS that is required for activation of NLRP3 inflammasome.
- 6.de Carvalho RVH, Andrade WA, Lima-Junior DS, Dilucca M, de Oliveira CV, Wang K, Nogueira PM, Rugani JN, Soares RP, Beverley SM, et al. : Leishmania Lipophosphoglycan Triggers Caspase-11 and the Non-canonical Activation of the NLRP3 Inflammasome. Cell Rep 2019, 26:429–437 e425.** This manuscript revealed that LPG from many Leishmania species trigger Caspase-11 activation, leading to the non-canonical pathway for NLRP3 activation. It advances on our undestanding of the molecular mechanisms by which the NLRP3 is activated in response to Leishmania infection in macrophages.
- 7.Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R: Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem 2002, 277:41624–41630. [DOI] [PubMed] [Google Scholar]
- 8.de Carvalho RVH, Silva AlN, Santos LL, Andrade WA, de Sa KSG, Zamboni DS: Macrophage priming is dispensable for NLRP3 inflammasome activation and restriction of Leishmania amazonensis replication. J Leukoc Biol 2019, May 7. doi: 10.1002/JLB.MA1118-471R. [DOI] [PubMed] [Google Scholar]
- 9.Dey R, Joshi AB, Oliveira F, Pereira L, Guimaraes-Costa AB, Serafim TD, de Castro W, Coutinho-Abreu IV, Bhattacharya P, Townsend S, et al. : Gut Microbes Egested during Bites of Infected Sand Flies Augment Severity of Leishmaniasis via Inflammasome-Derived IL-1beta. Cell Host Microbe 2018, 23:134–143 e136.* This manuscript demonstrated that microbiota delivered by sand flie bite during mammalian infection promotes the first signal (priming) for further activation of the inflammasome by the parasites.
- 10.Farias Luz N, Balaji S, Okuda K, Barreto AS, Bertin J, Gough PJ, Gazzinelli R, Almeida RP, Bozza MT, Borges VM, et al. : RIPK1 and PGAM5 Control Leishmania Replication through Distinct Mechanisms. J Immunol 2016, 196:5056–5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santos DM, Carneiro MW, de Moura TR, Soto M, Luz NF, Prates DB, Irache JM, Brodskyn C, Barral A, Barral-Netto M, et al. : PLGA nanoparticles loaded with KMP-11 stimulate innate immunity and induce the killing of Leishmania. Nanomedicine 2013, 9:985–995. [DOI] [PubMed] [Google Scholar]
- 12.Miranda MM, Panis C, da Silva SS, Macri JA, Kawakami NY, Hayashida TH, Madeira TB, Acquaro VR Jr., Nixdorf SL, Pizzatti L, et al. : Kaurenoic Acid Possesses Leishmanicidal Activity by Triggering a NLRP12/IL-1beta/cNOS/NO Pathway. Mediators Inflamm 2015, 2015:392918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta AK, Ghosh K, Palit S, Barua J, Das PK, Ukil A: Leishmania donovani inhibits inflammasome-dependent macrophage activation by exploiting the negative regulatory proteins A20 and UCP2. FASEB J 2017, 31:5087–5101.* This manuscript reported an inhibitory effect of Leishmania donovani in the activation of the inflammasome. It was demonstrated that the inhibition of inflammasome activation occur at the priming stage (first signal).
- 14.Shio MT, Christian JG, Jung JY, Chang KP, Olivier M: PKC/ROS-Mediated NLRP3 Inflammasome Activation Is Attenuated by Leishmania Zinc-Metalloprotease during Infection. PLoS Negl Trop Dis 2015, 9:e0003868.** This manuscript was the first to report that Leishmnaia inhibits the activation of the inflammasome in macrophages. Inhibition occurs by multiple mechanisms, including inhibition of ROS production and GP63-mediated degradation of inflammasome components.
- 15.Hartley MA, Eren RO, Rossi M, Prevel F, Castiglioni P, Isorce N, Desponds C, Lye LF, Beverley SM, Drexler SK, et al. : Leishmania guyanensis parasites block the activation of the inflammasome by inhibiting maturation of IL-1beta. Microb Cell 2018, 5:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giraud E, Rouault E, Fiette L, Colle JH, Smirlis D, Melanitou E: Osteopontin in the host response to Leishmania amazonensis. BMC Microbiol 2019, 19:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saha G, Khamar BM, Singh OP, Sundar S, Dubey VK: Leishmania donovani evades Caspase 1 dependent host defense mechanism during infection. Int J Biol Macromol 2019, 126:392–401. [DOI] [PubMed] [Google Scholar]
- 18.Zamboni DS, Lima-Junior DS: Inflammasomes in host response to protozoan parasites. Immunol Rev 2015, 265:156–171. [DOI] [PubMed] [Google Scholar]
- 19.Anderson CF, Mendez S, Sacks DL: Nonhealing infection despite Th1 polarization produced by a strain of Leishmania major in C57BL/6 mice. J Immunol 2005, 174:2934–2941. [DOI] [PubMed] [Google Scholar]
- 20.Charmoy M, Hurrell BP, Romano A, Lee SH, Ribeiro-Gomes F, Riteau N, Mayer-Barber K, Tacchini-Cottier F, Sacks DL: The Nlrp3 inflammasome, IL-1beta, and neutrophil recruitment are required for susceptibility to a nonhealing strain of Leishmania major in C57BL/6 mice. Eur J Immunol 2016, 46:897–911.** This manuscript reported the detrimental effect of the inflammasome during infection with L. major Seidman strain in mice. The authors demonstrate that NLRP3-mediated IL-1β production favor neutrophil recruitment and promote patology and parasite replication in the tissues.
- 21.Gurung P, Karki R, Vogel P, Watanabe M, Bix M, Lamkanfi M, Kanneganti TD: An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. J Clin Invest 2015, 125:1329–1338.** This manuscript reported the detrimental effect of the inflammasome during infection with L. major in BALB/c mice. It was reported that inflammasome-mediated IL-18 production favour the generation of a Th2 response that promotes the disease.
- 22.Patil T, More V, Rane D, Mukherjee A, Suresh R, Patidar A, Bodhale N, Mosser D, Dandapat J, Sarkar A: Pro-inflammatory cytokine lnterleukin-1beta (IL-1beta) controls Leishmania infection. Cytokine 2018, 112:27–31. [DOI] [PubMed] [Google Scholar]
- 23.Hurrell BP, Schuster S, Grun E, Coutaz M, Williams RA, Held W, Malissen B, Malissen M, Yousefi S, Simon HU, et al. : Rapid Sequestration of Leishmania mexicana by Neutrophils Contributes to the Development of Chronic Lesion. PLoS Pathog 2015, 11:e1004929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laskay T, van Zandbergen G, Solbach W: Neutrophil granulocytes as host cells and transport vehicles for intracellular pathogens: apoptosis as infection-promoting factor. Immunobiology 2008, 213:183–191.* This manuscript was the first to propose how neutrophils can promote the infectious process in Leishmaniasis.
- 25.Ribeiro-Gomes FL, Peters NC, Debrabant A, Sacks DL: Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-leishmania response. PLoS Pathog 2012, 8:e1002536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE: IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A 2009, 106:7119–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, Copenhaver AM, Humbles AA, Liu YJ: IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol 2016, 17:646–655. [DOI] [PubMed] [Google Scholar]
- 28.Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, Chawla A, Locksley RM: Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 2013, 210:535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SH, Charmoy M, Romano A, Paun A, Chaves MM, Cope FO, Ralph DA, Sacks DL: Mannose receptor high, M2 dermal macrophages mediate nonhealing Leishmania major infection in a Th1 immune environment. J Exp Med 2018, 215:357–375.* This manuscript demonstrated that inflammasome activation and IL-1 receptor signaling are required to maintain the population of alternatively activated, dermis resident macrophages in mice infected with L. major.
- 30.Fernandez-Figueroa EA, Rangel-Escareno C, Espinosa-Mateos V, Carrillo-Sanchez K, Salaiza-Suazo N, Carrada-Figueroa G, March-Mifsut S, Becker I: Disease severity in patients infected with Leishmania mexicana relates to IL-1beta. PLoS Negl Trop Dis 2012, 6:e1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santos D, Campos TM, Saldanha M, Oliveira SC, Nascimento M, Zamboni DS, Machado PR, Arruda S, Scott P, Carvalho EM, et al. : IL-1beta Production by Intermediate Monocytes Is Associated with Immunopathology in Cutaneous Leishmaniasis. J Invest Dermatol 2018, 138:1107–1115.* This manuscript associate disease severity with IL-1β production in humans, providing evidence that IL-1β promotes immunopatology in patients.
- 32.Novais FO, Carvalho AM, Clark ML, Carvalho LP, Beiting DP, Brodsky IE, Carvalho EM, Scott P: CD8+ T cell cytotoxicity mediates pathology in the skin by inflammasome activation and IL-1beta production. PLoS Pathog 2017, 13:e1006196.* This manuscript reported that NLRP3 activation and IL-1β release are a detrimental consequence of a cytotoxic CD8+ T cell response to L. brazlllensls infection.
- 33.Novais FO, Carvalho LP, Passos S, Roos DS, Carvalho EM, Scott P, Beiting DP: Genomic profiling of human Leishmania braziliensis lesions identifies transcriptional modules associated with cutaneous immunopathology. J Invest Dermatol 2015, 135:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Figliuolo VR, Chaves SP, Savio LEB, Thorstenberg MLP, Machado Salles E, Takiya CM, D'Imperio-Lima MR, de Matos Guedes HL, Rossi-Bergmann B, Coutinho-Silva R: The role of the P2X7 receptor in murine cutaneous leishmaniasis: aspects of inflammation and parasite control. Purinergic Signal 2017, 13:143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thorstenberg ML, Rangel Ferreira MV, Amorim N, Canetti C, Morrone FB, Alves Filho JC, Coutinho-Silva R: Purinergic Cooperation Between P2Y2 and P2X7 Receptors Promote Cutaneous Leishmaniasis Control: Involvement of Pannexin-1 and Leukotrienes. Front Immunol 2018, 9:1531. [DOI] [PMC free article] [PubMed] [Google Scholar]