

Abstract
The genomes of diverse mycobacterial species encode multiple proteins with the canonical L,D-transpeptidase (Ldt) sequence motif. The reason for this apparent redundancy is not well understood, but evidence suggests paralogous Ldts may serve niche roles in maintaining and/or remodeling mycobacterial peptidoglycan. We examined 323 mycobacterial Ldts and determined these enzymes cluster into six clades. We identified a variably represented yet distinct Ldt class (class 6) containing Mycobacterium smegmatis (Msm) LdtF and built a homology model of Msm LdtF toward elucidating class 6 structural and functional differences. We report class 6 Ldts have structurally divergent catalytic domains containing a 10-residue insertion near the active site, and additionally determined that meropenem preferentially acylates LdtF. Our data demonstrate an evolutionary basis for mycobacterial Ldt multiplicity that lends support to the idea that paralogous Ldts serve non-redundant roles in vivo and suggests class 6 Ldts can be selectively targeted by specific carbapenem antibiotics.
Keywords: l,d-transpeptidase; Ldt; carbapenem; Mycobacterium smegmatis; Mycobacterium tuberculosis; peptidoglycan
Graphical Abstract
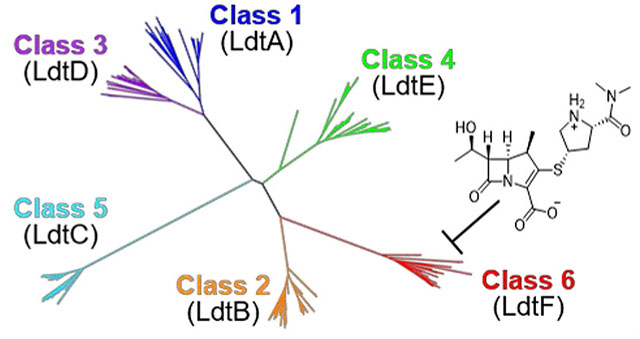
The Mycobacterium genus is diverse; its members range from harmless soil dwellers to causative agents of debilitating diseases including tuberculosis and leprosy. Mycobacteria can be classified on the basis of their colony morphology, growth rates, pigmentation, clinical, and/or epidemiological traits. Despite their unique characteristics, as members of the Corynebacterineae family, all mycobacteria possess complex cell walls comprised of an outer membrane-like layer of mycolic acids anchored to a middle arabinogalactan layer that is tethered to the ubiquitous peptidoglycan (PG) layer. The PG layer contains alternating N-acetylglucosamine and modified muramic acid residues, and stemming from the muramic acid residues are L-alanyl-D-iso-glutaminyl-meso-diaminopimelyl-D-alanyl-D-alanine peptides of varying lengths. These peptides are crosslinked to one another by a peptide bond, thereby creating a meshwork that encapsulates the cell and provides strength and rigidity. The PG layer likewise acts as a protective barrier and helps the cell withstand intracellular and extracellular osmotic pressure differences.1 Peptide crosslinks occur between the third amino acid of one stem (meso-diaminopimelic acid, mDAP) and the fourth of another (D-alanine) or between the third amino acid and the third amino acid of a nearby peptide stem (mDAP-mDAP), resulting in 4→3 or 3→3 crosslinks, respectively. The 4→3 crosslink is the predominant form among most species and this transpeptidation reaction is catalyzed by D,D-transpeptidases (Ddts) that are the canonical targets of the β-lactam class of antibiotics. Conversely, 3→3 crosslink formation is catalyzed by the L,D-transpeptidases (Ldts). Members of the Ldt enzyme family are not efficiently inhibited by most β-lactam classes, although they are acylated by carbapenems, penems, [herein collectively referred to as (carba)penems], and, to a lesser extent, cephalosporins.2–5 In both cases, enzyme inactivation by β-lactams occurs through acylation of the catalytic serine (Ddts) or cysteine (Ldts).
PG is present in virtually all bacteria and has a generally conserved architecture. However, subtle chemical features of the PG vary between taxonomic groups. In mycobacteria, the muramic acid residues from which the peptide stem extends can be N-glycolylated6 or N-acetylated,7 the first amino acid of the stem peptide is a glycine in Mycobacterium leprae,8 and both mDAP and D-glutamate residues are extensively amidated to mDAPNH2 and D-glutamine,9 respectively. Further distinguishing mycobacterial PG from other bacterial species is the high degree of Ldt-catalyzed 3→3 crosslinks,10,11 and Ldt enzymes appear to preferentially require differentially modified PG component substrates.12 Significantly, a Mycobacterium tuberculosis (Mtb) mutant lacking a functional copy of its predominant Ldt (LdtMt2) displays aberrant growth morphology, is attenuated in a mouse model and susceptible to amoxocillin-clavulanate,13 highlighting the importance of the Ldt enzyme class in mycobacterial physiology, virulence, and β-lactam resistance.
In addition to catalyzing 3→3 crosslinks, Ldts serve other roles across diverse bacterial species, and the genomes of these species encode for multiple gene products with the signature Ldt motif HXX14–17[S/T]HGChN, where h is a hydrophobic residue. In Escherichia coli, Ldts additionally catalyze the attachment of Braun lipoprotein to PG,14 and in Gram-positive (including mycobacterial species) and Gram-negative species, Ldts can exchange non-canonical D-amino acids in PG.4,15 Mycobacterial species have multiple putative Ldts, and studies suggest paralogous mycobacterial enzymes likely serve unique roles in the proper maintenance of bacterial PG. For example, Mtb possesses five Ldts (LdtMt1 − LdtMt5) which, excepting LdtMt3, catalyze 3→3 crosslink formation and can exchange D-alanine for D-methionine in PG fragments in vitro.4 Unlike LdtMt1 − LdtMt4, LdtMt5 is not readily acylated by carbapenem antibiotics.4,16 Recent studies demonstrate paralogous mycobacterial Ldts remodel older PG at the cell sidewall,17,18 indicating Ldts likely play significant and distinct roles in remodeling as the cell grows and divides. However, there does appear to be functional redundancy and the specific roles of paralogous Ldts are poorly understood.
Previously, a classification of Mtb and Mycobacterium smegmatis (Msm) Ldts was proposed based on sequence homology of Ldts in 18 translated mycobacterial genomes.19 Ldt sequences were aligned and enzyme families were identified in ClustalX. However, the explicit method and parameters within ClustalX used to assign gene families and the 18 organisms from which the Ldt sequences in their analysis were obtained were not described, nor was it clear how the classification was validated or benchmarked. Using this approach, researchers concluded Msm has two class 2 Ldts, LdtB and LdtF, while Mtb has one (LdtMt2). Despite a high degree of sequence similarity (68%), Mtb LdtMt3 and Msm LdtG were placed into separate classes. While clustering within the ClustalX webserver is a convenient method for rapidly visualizing evolutionary relationships, we sought to provide a detailed scheme for taxonomic classification of mycobacterial Ldts, complete with benchmarking and a dataset large enough to capture monophyletic enzyme classes. We believe this to be a critical step in organizing and designing functional experiments to probe either the niche roles or the functional redundancy of these enzymes. Here, we report a sequence-phylogeny based classification scheme of 323 Ldts from 58 translated mycobacterial genomes to map the evolutionary divergence within the mycobacterial Ldt family, followed by structural and biochemical analyses of Msm Ldts. We find that Msm LdtF is represented in a monophyletic clade and contains divergent features in its catalytic domain that likely promote its preferential acylation by the carbapenem meropenem.
Our phylogenetic analysis revealed six distinct clades (Figure 1A), five of which validate Ldt classes previously discovered using ClustalX (classes 1 – 5)19 (Figure 1B). A comprehensive list of the species examined and a detailed phylogenetic tree is available in Supporting Information (Figure S1).
Figure 1.

Phylogenetic analysis and reclassification of mycobacterial Ldts based on structure, sequence, and phylogeny. A) Mycobacterial Ldts cluster into six clades that confirm five previously identified Ldt classes: class 1 (blue), class 2 (orange), class 3 (purple), class 4 (green) and class 5 (cyan). Msm LdtF clusters into a distinctive sixth clade (red). Mav = Mycobacterium avium, Mab = Mycobacterium abscessus. B) Reclassification and cartoon depiction of mycobacterial Ldts. CD = catalytic domain, BIg = bacterial Ig-like domain, R2 = Region 2 sequence (Figures S2 and S3), PRR = proline-rich region, CTSD = C-terminal subdomain, ex-CTSD = extension of the CTSD. Yellow CDs indicate the presence of Region 1 sequence (Figure S3). Class 6 Ldts contain a 10-residue insertion (red cylinder in CD). Classes 2, 5, and 6 contain an N-terminal lipobox.19 Asterisk refers to reference 19. Classes are arranged to indicate increasing apparent structural complexity.
Msm LdtG was previously annotated as a class 6 Ldt and the presence of a proline/glutamate-rich 73 amino acid insertion within the catalytic domain was discussed as a distinguishing feature.19 However, our analysis of a larger dataset of mycobacterial Ldts shows this insertion, present in Mycobacterium goodie and Msm homologs, is a longer variation of Region 2 (Figure S2) found in all class 3 Ldts. Msm mc2155, Msm NCTC 8159, and M. goodie X7B LdtD cluster neatly with Mtb LdtMt3 and other LdtD enzymes in the clade corresponding to class 3 Ldts (Figures 1A and S1). Thus, we are proposing Msm LdtG be renamed LdtD and be described as a class 3 Ldt (Figure 1B). To our knowledge, no biochemical studies have been conducted on Msm LdtD (formerly LdtG), and we do not anticipate this re-designation will promote confusion in the Ldt community.
Our phylogenetic analysis indicates class 1 and 3 enzymes are closely related yet distinctively monophyletic (Figure 1A), supporting the initial classification proposed by Sanders, et al. Msm LdtE has one BIg domain and an N-terminal proline-rich region (PRR) of approximately 100 amino acid that appears to be unique to class 4 Ldts (Figure 1B). There are no structural data on class 4 Ldts, but the N-termini of class 4 Ldts are predicted to be intrinsically disordered and mediate a protein-protein interaction (Figure S4). Class 5 Ldts appear to be the most evolutionarily divergent among mycobacterial Ldts.
We note a unique clade of Ldt enzymes containing Msm LdtF. Msm LdtF was previously annotated as a class 2 Ldt and has a modest 43% and 42% sequence identity to Msm LdtB and Mtb LdtMt2, respectively (Figures 1B, 2A). Based on our comprehensive phylogenetic analysis, we have re-designated members of the LdtF-containing clade as class 6 Ldts. The genomes of approximately half of the species we examined encode for class 6 Ldt enzymes, and class 6 Ldts are equally represented among slow-growing and rapidly-growing mycobacteria (Figure S5). This finding implies that, barring horizontal gene transfer, a class 6 Ldt was present in a common ancestor of the slow-growing and rapidly-growing nontuberculous mycobacteria (NTM) phylogenies. The genomes of all Mycobacterium avium complex (MAC) species analyzed in this study encode for a class 6 Ldt. MAC species are opportunistic pathogens known to infect immunocompromised patients, including those infected with HIV.20 A class 6 Ldt also appears to be conserved among members of the Mycobacterium fortuitum-vaccae clade (Figure S1), some of which are known to cause soft tissue infections, particularly post-surgery.21
Figure 2.

Comparison of mycobacterial class 2 and class 6 Ldt catalytic domains. A) A sequence alignment of Msm LdtB and LdtF and Mtb LdtMt2. The LdtF 10-residue insertion is indicated by a maroon bar and the rest of the loop between β-strands 20 and 21 is indicated by a blue bar. The Ldt motif is boxed in green, and catalytic cysteine and histidine residues are noted with green stars. The conserved tryptophan residue (an aspartate in LdtF) is noted with an orange star, and additional residues discussed are highlighted with purple stars. The CTSD is boxed in purple. The alignment was performed using Molsoft ICM-Chemist-Pro26 and visualized with ESPript 3.0.27 B) Homology models of LdtB-faropenem (left) and LdtF-meropenem (right) indicate the location of the LdtF insertion (red). The LdtF insertion is near the active site, as indicated by the modeled (carba)penems. Faropenem and meropenem are shown as yellow balls and sticks.
Members of the class 6 clade share their most recent common ancestor with those of the class 2 clade (Figure 1A). Msm LdtF has a 10-residue insertion within the catalytic domain (Figures 1B, 2A, LdtF residues 289–298). To characterize the distinguishing features of class 2 and class 6 Ldts, we built a homology model of Msm LdtF. The 10-residue insertion of Msm LdtF is between β-strands 20 and 21, near the active site (Figure 2B). The N-terminal end of the loop contains Glu289, which is close to the active site and could contact an LdtF substrate, and the C-terminal portion of the loop contains aromatic residues Phe298 and Trp299 adjacent to the aromatic rich, C-terminal structural domain (CTSD) (Figure 2A). The CTSD is approximately 20 amino acids in length and contains three conserved tryptophan residues that pack with residues on the second BIg domain (BIgB, Figures 1B, 2A), as evidenced in the LdtMt222 and LdtMt516 crystal structures. The role of the CTSD has been postulated to fix the orientations of the catalytic domain and the structural BIg domain.23 However, LdtMt1 and LdtMt3 lack CTSDs and their crystal structures indicate nearly identical domain orientations to LdtMt2 and LdtMt5,24–25 suggesting the CTSD could instead be affecting the behavior and dynamics of the two domains relative to one other in solution. LdtF insertion residues Phe298 and Trp299 could interact with and modulate the structure and/or dynamic behavior of the largely aromatic CTSD, and structural biology studies may shed light on the role of the insertion and/or its potential interaction with the CTSD.
We performed ultraperformance liquid chromatography (UPLC) high resolution mass spectrometry (MS) analyses to determine whether the Msm Ldts are acylated by (carba)penems. Our data show that Msm LdtA, LdtB, LdtD (formerly LdtG), and LdtE (classes 1−4, respectively) are fully acylated by the (carba)penems faropenem, biapenem, doripenem, and meropenem (Figure 3). Only low levels of acylated LdtC, a class 5 Ldt, were detected after 5 hours, as observed in LdtMt5,4,16 although Msm LdtC is fully acylated by faropenem after 24 hours (Figure S6). Faropenem (molar mass = 284.3 g/mol) is degraded on the enzyme after acylation to yield a Δm = +86 Da.28 To determine the relative ability of these drugs to acylate each Ldt, a competition experiment was performed where each Ldt enzyme was incubated with a cocktail of equimolar concentrations of the four (carba)penems, and the relative amounts of each drug-Ldt species were determined by UPLC-MS. LdtA, LdtB, LdtD and LdtE are all preferentially acylated by faropenem, a penem with a neutral side chain (Figure 3). Minor amounts of the biapenem, less of the doripenem, and none of the meropenem adduct masses were observed in these competition experiments. These data are in agreement with a previous report indicating faropenem rapidly acylates Mtb LdtMt2.22
Figure 3.

Class 1− 4 Ldts are preferentially acylated by faropenem. Faropenem, biapenem, doripenem, and meropenem acylate Msm LdtA, LdtB, LdtD, and LdtE. When incubated with an equimolar concentration of a cocktail of the same drugs (orange traces), these Ldts are preferentially acylated by faropenem, as indicated by the +86 Da shift in mass. A Δm = −44 Da is also observed with biapenem, doripenem, and meropenem, and this is the result of the retro-aldol scission of the hydroxyethyl side chain on the carbapenems at C6.29
In contrast to class 1−4 Ldts in which complete acylation of these enzymes were observed with all drugs (Figure 3), small amounts of apo-LdtF remained following incubation with biapenem, doripenem, and faropenem, and only meropenem completely acylates LdtF under the same conditions (Figure 4). When LdtF was incubated with (carba)penems in competition, the meropenem-LdtF species was the most abundant. This finding is atypical relative to the profile of class 1−4 Ldts examined here, where the faropenem-Ldt adduct is the predominant species (Figure 3).
Figure 4.

Meropenem fully and preferentially acylates LdtF.
While much of the LdtF active site is conserved, three substitutions occur near the outer pocket of the catalytic domain in our model, Ala330 in LdtB is a leucine (Leu351) in LdtF, Pro331 in LdtB is an asparagine in LdtF (Asn352), and the highly conserved Trp332 of LdtB is an aspartate (Asp353) (Figure 2A). The carba(penem) class of drugs inhibits Ldts by forming covalent adducts with the catalytic cysteine residue, and their side chains make contacts with outer pocket residues of LdtMt2 in X-ray crystal structures.22,30,31 We hypothesized that Asp353 could electrostatically interact with a localized cationic side chain like that of meropenem (Figure 3). Faropenem has a neutral side chain (Figure 3), which conversely may interact more favorably with hydrophobic amino acids like Trp332 of LdtB. To determine if these favorable interactions between (carba)penems and LdtB or LdtF are possible, we covalently docked faropenem and meropenem to homology models of LdtB and LdtF, respectively (Figure S7). In the case of LdtB, we indeed observe strong contacts between the hydrophobic face of the tetrahydrofuran ring of faropenem and Trp332 in our model (Figure S7). In the LdtF-meropenem model, both hydrogens of the protonated secondary pyrrolidine nitrogen form a bidentate hydrogen bond with Asp353 (Figure S7).
While the class 1−4 Msm Ldts all contain relatively conserved active sites, we hypothesized that the substitution of a conserved outer pocket aromatic tryptophan residue with an anionic aspartate in LdtF could change the relative ability of each drug to acylate class 6 Ldts. Toward understanding the role of Asp353 in LdtF (carba)penem binding and acylation, we performed site-directed mutagenesis to generate the LdtF D353W variant. We anticipated substitution of Asp353 with tryptophan would shift the propensity of the enzyme to be acylated by faropenem as seen with LdtB. However, meropenem preferentially acylated the LdtF D353W variant in a competition experiment (Figure S8). We additionally generated the D353A and D353N LdtF variants and determined that substitution of Asp353 with alanine or asparagine likewise resulted in acylation by meropenem in a competition experiment with faropenem (Figure S9). Interestingly, substitution of Asp353 with tryptophan nearly abolishes acylation by biapenem but does not affect acylation by faropenem, doripenem, or meropenem (Figure S8). These unexpected results from our LdtF variant experiments exemplify the need for LdtF structural studies toward understanding how the unique sequence elements of class 6 Ldts affect structure and function.
We considered two hypotheses as to why meropenem preferentially acylates LdtF: 1) the rates of meropenem binding and covalent adduct formation are faster than that of faropenem binding and adduct formation, or 2) faropenem is hydrolyzed more rapidly from LdtF and the LdtF-meropenem adduct is stable. Toward addressing these hypotheses, we performed competition experiments and measured LdtF acylation by faropenem and meropenem over the course of five hours. While LdtB is rapidly and fully acylated by faropenem in a competition experiment after 20 minutes (Figure S10) and remains acylated by faropenem throughout the duration of the competition experiment (Figure 3), LdtF is initially acylated by both faropenem and meropenem (Figure 5). However, the LdtF-faropenem species accounts for only 30% of the total species present after 90 minutes, and the abundance of this species decreases throughout the remainder of the experiment while the LdtF-meropenem mass increases. Thus, we conclude that while both faropenem and meropenem initially acylate LdtF, faropenem is hydrolyzed off the enzyme at a faster rate than meropenem is. This conclusion is reinforced by the observation that after 20 minutes, the ratio of LdtF-meropenem to LdtF-faropenem is approximately 1.5-fold. However, by 5 hours, the ratio of LdtF-meropenem to LdtF-faropenem has gradually increased to approximately 3-fold. Due to mass action, as the concentration of drug-bound LdtF increases, the relative susceptibility of the two adducts to hydrolysis becomes a more significant determinant of species abundance. Thus, the concentration of LdtF-faropenem begins to decrease relative to LdtF-meropenem concentrations. Significantly, this is in sharp contrast to what is observed with LdtB; LdtB is rapidly acylated by faropenem, and the LdtB-faropenem adduct mass remains most abundant relative to the carbapenems over the course of our experiment.(Figure 3).
Figure 5.

Meropenem forms a stable adduct with LdtF while the faropenem adduct is hydrolyzed from the enzyme. This experiment was performed in triplicate and all replicates are plotted.
Previous kinetic studies of LdtMt1 show an LdtMt1-faropenem hydrolysis rate constant approximately four-fold higher than that of LdtMt1-meropenem, yet the rate of LdtMt1 acylation by faropenem is significantly higher than the corresponding acylation rate by meropenem.32 It appears from our data that both the slower inactivation rate of LdtF by faropenem and the faster hydrolysis of LdtF-faropenem lead to the relative selectivity of meropenem in the inactivation of LdtF. It should be noted that both faropenem and meropenem take significantly longer to fully acylate LdtF compared to the observed full acylation of LdtB by faropenem in 20 minutes (Figure S10) leaving much room for optimization of LdtF inhibitors. Likewise, additional studies fully characterizing the acylation and hydrolysis kinetics of inhibitor binding are warranted.
While the class 2 LdtMt2 is the dominant Ldt in Mtb, data suggest the class 5 Msm LdtC is the dominant Ldt in Msm; a ΔldtC strain is the only single ldt-deletion mutant strain to display enhanced susceptibility to the carbapenem imipenem.19 Interestingly, LdtF complementation alone can restore the phenotype of an Msm ΔldtB,ΔldtF,ΔldtC triple knockout strain to that of the ΔldtC mutant, while LdtB complementation could not fully compensate for the loss of both LdtF and LdtB in the triple knockout strain,19 indicating the importance of class 6 Ldts in Msm physiology. Therefore, we hypothesized that Msm would display enhanced susceptibility to meropenem relative to the other (carba)penems evaluated here due to the presence of LdtF. We determined the minimum inhibitory concentrations (MICs) of (carba)penems against two Msm strains that express LdtF. As indicated in Table 1, Msm is more susceptible to meropenem than faropenem, and is as susceptible to biapenem and doripenem. Ampicillin was included as a control, because it does not acylate Msm Ldts (data not shown). We find it interesting that Msm, a rapidly-growing organism with a class 6 Ldt, is more susceptible to meropenem than faropenem, whereas CDC1551 and H37Rv Mtb strains that lack a class 6 Ldt are reliably more susceptible to faropenem.4,32–34 Notably, Mtb is slow-growing, and while many factors play into drug susceptibility, these data suggest further understanding the role of LdtF in cell wall biosynthesis may provide opportunities for selectively targeting class 6 Ldts.
Table 1.
MIC data of β-lactam antibiotics against Msm mc2155 and Msm ATCC 19420 (NCTC 1859). Data are reported in μg/mL. aKaushik. et al. Antimicrob. Agents Chemother. 2015, 59, 6561. NR = not reported.
| ampicillin | biapenem | doripenem | faropenem | meropenem | |
|---|---|---|---|---|---|
| Msm mc2155 | >32 | 4–8 | 4–8 | >32 | 4–8 |
| Msm ATCC 19420 | >32 | 4–8 | 4–8 | >32 | 4–8 |
| Mtb H37Rva | NR | 2.5–5 | 2.5–5 | 2.5–5 | 5–10 |
In this study, we used phylogenetics to validate and build upon the mycobacterial classification proposed by Sanders, et al., and dissected the biochemical redundancy of the Msm Ldt family with respect to antibiotic susceptibility. We demonstrate that faropenem, one of the most potent Ldt inhibitors, and meropenem acylate the class 6 Ldt, LdtF. However, while the faropenem-Ldt adduct persists in class 1−4 Ldts, faropenem is hydrolyzed from LdtF. Given the unique antibiotic inhibition profile, the distinct LdtF catalytic domain features, and the variability in PG structure among mycobacterial species, we speculate that class 6 Ldts may utilize unique PG substrates. Our analyses highlight the uniqueness of class 6 Ldts, and we anticipate that differences in the catalytic domains of class 6 Ldts can be exploited in the development of novel drugs targeting pathogenic or opportunistic mycobacterial species such as M. avium and M. fortuitum.
Methods.
General -
DNA primers were purchased from Integrated DNA Technologies. (Carba)penems were purchased from Sigma-Aldrich with an HPLC-determined purity of 98%. UPLC-high resolution MS samples were analyzed on a Waters Acquity H-Class ultraperformance LC system equipped with a multiwavelength UV-violet diode array detector in conjunction with a Waters Acquity BEH-300 ultraperformance LC column packed with a C4 stationary phase (2.1 × 50 mm; 1.7 μm) in tandem with high resolution MS analysis by a Waters Xevo-G2 quadrupole-TOF electrospray ionization mass spectrometer.
Phylogenetic Analysis of Mycobacterial Ldts -
Only complete mycobacterial genomes that were accepted by Refseq were used in our analysis as a quality control. In total, 58 mycobacterial genomes following the above criteria were downloaded from the NCBI. BLAST+ tools were used to build custom blast databases of translated protein sequences for each genome and these sequences were mined for Ldt enzymes using Mtb LdtMt2 as a template, with an E-value of 0.0001. Protein sequences were formatted into a human-readable FASTA file with an in-house Python script and aligned with Clustal Omega.26 The resulting sequence alignment was trimmed of regions with less than 95% coverage and 201 sites in the multiple sequence alignment were used to build a phylogenetic tree in MEGAX 10.0.535 with a maximum likelihood algorithm. The resultant phylogenetic tree was validated with 500 bootstrap replicates. The phylogenetic tree graphics were generated with the Interactive Tree of Life 4.4.2.36
Homology Models -
The homology models of Msm LdtB and LdtF were built using carbapenem-bound LdtMt2 (PDB ID: 5K69) as a template with SWISS-MODEL. The placement of the loop insertion between β20 and β21 in LdtF was determined by adjusting the alignment to preserve secondary structure and minimize clashes in the resulting homology model. These models were subsequently used to predict the structure of the inhibited enzyme by covalently docking faropenem to LdtB and meropenem to LdtF in ICM-Chemist-Pro with the 3D ligand editor’s covalent docking tool,37,38 and poses were selected from the lowest three energy structures.
Cloning, Overexpression, and Purification of M. smegmatis Ldts -
Truncated versions of each ldt gene (Table S1) were amplified by PCR (1 × New England Biolabs GC reaction buffer, 200 μM dNTPs, 2 ng/μL mc2155 Msm genomic DNA, 500 nM primers, 1 unit of Phusion DNA polymerase, and 3% DMSO), digested with NdeI and either XhoI (ldtA, ldtB, ldtD, ldtE, and ldtF) or HindIII (ldtC), and cloned into a modified pET28b vector that encodes for a TEV-cleavable N-terminal His6 tag.23 Escherichia coli BL21(DE3) cells harboring one of the ldt-pET28b plasmids were grown in LB broth to an OD600 of ~0.5 at 20 °C with shaking. Protein overexpression was induced with 100 μM isopropyl 1-thio-β-d-galactopyranoside (IPTG), and following induction cells were incubated with shaking at 20 °C for an additional 14–20 h. Cells were harvested at 4 °C, resuspended in protein purification buffer (25 mM Tris, pH 8.0, 400 mM NaCl, and 10% glycerol), and lysed by ultrasonication. Cell debris was removed by centrifugation (15,000 rpm, 30 min) at 4 °C. The supernatants were incubated with washed nickel-nitrilotriacetic acid (Ni-NTA) resin at 4 °C with gentle rocking for 1–2 hrs, and His-tagged Ldts were eluted with imidazole. Fractions containing Ldt (as determined by SDS-PAGE) were combined, and protein concentration was determined using the Bio-Rad Protein Assay with bovine serum albumin (BSA) as a standard. Samples were dialyzed overnight at 4 °C against 50 mM Tris, pH 8.0, 100 mM NaCl, and 10% glycerol in the presence of TEV protease (1:100 w/w TEV:Ldt). Following dialysis, the TEV-treated samples were incubated with fresh Ni-NTA resin at 4 °C with gentle rocking for 1–2 hrs. The cleaved Ldts were collected as flow-through and were subjected to a second dialysis against 50 mM Tris, pH 8.0, 100 mM NaCl, 10% glycerol, and 1 mM tris(2-carboxyethyl)phosphine (TCEP). Ldts were concentrated to at least 1 mg/mL prior to being flash frozen in liquid N2 and were stored long-term at −80 °C.
Mass Spectrometry Analyses -
Ldts (2 μM) were incubated in the presence or absence of 50 μM drug in 25 mM Tris, pH 8.0 for 5 hrs at 20 °C. In the case of LdtC, 200 μM drug was used and enzyme was incubated additionally for 24 hrs. Samples were quenched with trifluoroacetic acid (final concentration 0.1% v/v) and analyzed by UPLC-high resolution MS. A cocktail stock solution containing faropenem, biapenem, doripenem and meropenem (LdtA, LdtB, LdtD, LdtE, LdtF, and LdtF D353W), or faropenem and meropenem (LdtF, LdtF D353N, and LdtF D353A) was prepared and diluted into a solution of 2 μM enzyme such that the final concentration of each drug was 50 μM. LdtF time-course competition experiments with meropenem and faropenem were performed in triplicate by the addition of a cocktail of both drugs (final concentration 50 μM) to a solution of LdtF (final concentration 2 μM). Reactions were quenched with TFA (final concentration 0.1% v/v) at each time-point and were immediately analyzed by UPLC-high resolution MS. Enzyme samples were separated at 60 °C to enhance peak resolution with a flow-rate of 0.3 mL/min and the following mobile phase: 0–1 min 90% water, 10% ACN, 0.1% formic acid (FA); 1–7.5 min gradient up to 20% water, 80% ACN, 0.1% FA; 7.5–8.4 min 20% water, 80% ACN, 0.1% FA; 8.4–8.5 min gradient up to 90% water, 10% ACN, 0.1% FA; 8.5–10 min 90% water + 10% ACN 0.1% FA. MaxEnt1 from MassLynx was used to deconvolute each m/z spectrum, and the resulting average mass spectrum was normalized to the sum of the intensities. Biopharmalynx was additionally used to combine the intensity of both meropenem-LdtB adducts followed by percentage determination of apo-LdtF, meropenem-bound LdtF, and faropenem-bound LdtF.
Determination of Minimum Inhibitory Concentration (MIC) -
(Carba)penems MICs were determined using the standard broth dilution method39 in a 96-well plate (Fisher, catalog number 08-772-54). Drug stocks were prepared fresh in 7H9 broth supplemented with 0.2% glycerol and 10% (v/v) albumin/dextrose/saline (herein referred to as 7H9 complete media), filter sterilized, and serially diluted in 7H9 complete media in a 96-well plate. Then, ~105 Msm bacilli were inoculated into 7H9 complete media (final volume of 150 uL). Plates were sealed with sterile film to minimize evaporation and prevent contamination. The cultures were incubated at 37 °C without shaking and were evaluated for growth by visual inspection after 36 hours. MIC values are reported of duplicate experiments.
Supplementary Material
Acknowledgement
We gratefully acknowledge Dr. Craig A. Townsend for helpful discussions and for constructive feedback on this work. We kindly thank Dr. Phil Mortimer (JHU) for his assistance with UPLC-MS experiments, Dr. Gyanu Lamichhane for supplying (carba)penems used in UPLC-MS experiments, Midshipmen Kelsey Melinoksy and Rachel Parker for their assistance with MIC experiments, Dr. Jason Kwan for discussion of phylogenetics data, and Midshipmen Grace Lane and Katherine Marapese for their assistance in generating Msm LdtF variants. We additionally thank Drs. Virginia Smith and Danny Morse for careful critiques of this manuscript and Dr. Alicia A. DeColli for her help transporting samples. This work was supported in part by the National Institutes of Health grant R01AI137329 (TAZ). This work was also supported by the Department of Defense Threat Reduction Agency (DTRA) Service Academy Research Initiative grant number CB3260 (LABB, RLM, and PKS) and the Naval Academy Research Council grant numbers N0001416WX00796 and N0001418WX00709 (LABB).
Abbreviations:
- PG
peptidoglycan
- Mtb
Mycobacterium tuberculosis
- Msm
Mycobacterium smegmatis
- Ldt
L,D-transpeptidase
- Ddt
D,D-transpeptidase
- NTM
nontuberculous mycobacteria
- R1
Region 1 sequence
- R2
Region 2 sequence
- CD
catalytic domain
- CTSD
C-terminal sub-domain
- ex-CTSD
extension of the CTSD
- PRR
proline-rich region
Footnotes
Supporting Information
Msm Ldts used in this study; complete phylogenetic tree and mycobacterial classification information; sequence alignments; Msm LdtC acylation profiles; IUPred2a/ANCHOR2 analyses; experimental methods for generating Msm LdtF variants and UPLC-MS data.
Notes: The authors declare no competing financial interest.
References
- (1).Vollmer W; Blanot D; De Pedro MA Peptidoglycan Structure and Architecture. FEMS Microbiol. Rev 2008, 32 (2), 149–167. 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- (2).Bastien Triboulet S; Dubé V; Lecoq L; Bougault C; Mainardi J-L; Rice LB; Ve-Quelquejeu E; Gutmann L; Marie A; Dubost L; Hugonnet J-E; Simorre J-P; Arthur M Kinetic Features of L,D-Transpeptidase Inactivation Critical for b-Lactam Antibacterial Activity. 2013. 10.1371/journal.pone.0067831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dubée V; Triboulet S; Mainardi JL; Ethève-Quelquejeu M; Gutmann L; Marie A; Dubost L; Hugonnet JE; Arthur M Inactivation of Mycobacterium Tuberculosis L,D-Transpeptidase Ldt Mt1 by Carbapenems and Cephalosporins. Antimicrob. Agents Chemother 2012. 10.1128/AAC.00665-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cordillot M; Dubée V; Triboulet S; Dubost L; Marie A; Hugonnet JE; Arthur M; Mainardia JL In Vitro Cross-Linking of Mycobacterium Tuberculosis Peptidoglycan by L,D-Transpeptidases and Inactivation of These Enzymes by Carbapenems. Antimicrob. Agents Chemother 2013, 57 (12), 5940–5945. 10.1128/AAC.01663-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Arthur M; Mainardi J-L; Veckerlé C; Dubée V; le NGuekam-Moumi A; Gutmann L; Rice LB; Hugonnet J-E Inactivation Kinetics of a New Target Of-Lactam Antibiotics * □ S Sé Bastien Triboulet Downloaded From. NUMBER 26 J. Biol. Chem 2011, 286 (26), 22777–22784. 10.1074/jbc.M111.239988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Azuma I; Thomas DW; Adam A; Ghuysen J-M; Bonaly R; Petit J-F; Lederer E Occurrence of N-Glycolylmuramic Acid in Bacterial Cell Walls: A Preliminary Survey. Biochim. Biophys. Acta - Gen. Subj 1970, 208 (3), 444–451. 10.1016/0304-4165(70)90217-5. [DOI] [PubMed] [Google Scholar]
- (7).Mahapatra S; Crick DC; Mcneil MR; Brennan PJ Unique Structural Features of the Peptidoglycan of Mycobacterium Leprae. J. Bacteriol 2008, 190 (2), 655–661. 10.1128/JB.00982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Draper P; Kandler O; Darbre A Peptidoglycan and Arabinogalactan of Mycobacterium Leprae. J. Gen. Microbiol 1987, 133 (5), 1187–1194. [DOI] [PubMed] [Google Scholar]
- (9).Pavelka MS Jr.; Mahapatra S; Crick DC Genetics of Peptidoglycan Biosynthesis. Microbiol. Spectr 2014, 2 (4). 10.1128/microbiolspec.mgm2-0034-2013. [DOI] [PubMed] [Google Scholar]
- (10).Wietzerbin J; Wietzerbin J; Lederer E; Das BC; Lederer E; Leyh-Bouille M Occurrence of D-Alanyl-(d)-Meso-Diaminopimelic Acid and Meso-Diaminopimelyl-Meso-Diaminopimelic Acid Interpeptide Linkages in the Peptidoglycan of Mycobacteria. Biochemistry 1974, 13 (17), 3471–3476. 10.1021/bi00714a008. [DOI] [PubMed] [Google Scholar]
- (11).Lavollay M; Arthur M; Fourgeaud M; Dubost L; Marie A; Veziris N; Blanot D; Gutmann L; Mainardi J-L The Peptidoglycan of Stationary-Phase Mycobacterium Tuberculosis Predominantly Contains Cross-Links Generated by L,D-Transpeptidation. J. Bacteriol 2008, 190 (12), 4360–4366. 10.1128/JB.00239-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ngadjeua F; Braud E; Saidjalolov S; Iannazzo L; Schnappinger D; Ehrt S; Hugonnet JE; Mengin-Lecreulx D; Patin D; Ethève-Quelquejeu M; Fonvielle M; Arthur M Critical Impact of Peptidoglycan Precursor Amidation on the Activity of l,d-Transpeptidases from Enterococcus Faecium and Mycobacterium Tuberculosis. Chem. - A Eur. J 2018. 10.1002/chem.201706082. [DOI] [PubMed] [Google Scholar]
- (13).Gupta R; Lavollay M; Mainardi JL; Arthur M; Bishai WR; Lamichhane G The Mycobacterium Tuberculosis Protein Ldt Mt2 Is a Nonclassical Transpeptidase Required for Virulence and Resistance to Amoxicillin. Nat. Med 2010, 16 (4), 466–469. 10.1038/nm.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Magnet S; Bellais S; Dubost L; Fourgeaud M; Mainardi J-L; Petit-Frè S; Marie A; Mengin-Lecreulx D; Arthur M; Gutmann L Identification of the L,D-Transpeptidases Responsible for Attachment of the Braun Lipoprotein to Escherichia Coli Peptidoglycan Downloaded From. J. Bacteriol 2007, 189 (10), 3927–3931. 10.1128/JB.00084-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cava F; De Pedro MA; Lam H; Davis BM; Waldor MK Distinct Pathways for Modification of the Bacterial Cell Wall by Non-Canonical D-Amino Acids. EMBO J. 2011, 30, 3442–3453. 10.1038/emboj.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Brammer Basta LA; Ghosh A; Pan Y; Jakoncic J; Lloyd EP; Townsend CA; Lamichhane G; Bianchet MA Loss of a Functionally and Structurally Distinct Ld-Transpeptidase, LdtMt5, Compromises Cell Wall Integrity in Mycobacterium Tuberculosis. J. Biol. Chem 2015, 290 (42), 25670–25685. 10.1074/jbc.M115.660753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).García-Heredia A; Pohane AA; Melzer ES; Carr CR; Fiolek TJ; Rundell SR; Chuin Lim H; Wagner JC; Morita YS; Swarts BM; Siegrist MS Peptidoglycan Precursor Synthesis along the Sidewall of Pole-Growing Mycobacteria. Elife 2018, 7 10.7554/elife.37243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Baranowski C; Welsh MA; Sham L-T; Eskandarian HA; Lim HC; Kieser KJ; Wagner JC; McKinney JD; Fantner GE; Ioerger TR; Walker S; Bernhardt TG; Rubin EJ; Rego EH Maturing Mycobacterium Smegmatis Peptidoglycan Requires Non-Canonical Crosslinks to Maintain Shape. Elife 2018. 10.7554/elife.37516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sanders AN; Wright LF; Pavelka MS Genetic Characterization of Mycobacterial L,D-Transpeptidases. Microbiol. (United Kingdom) 2014, 160, 1795–1806. 10.1099/mic.0.078980-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Horsburgh CR Mycobacterium Avium Complex Infection in the Acquired Immunodeficiency Syndrome. N. Engl. J. Med 1991, 324, 1332–1338. 10.1056/nejm199105093241906. [DOI] [PubMed] [Google Scholar]
- (21).Henkle E; Winthrop KL Nontuberculous Mycobacteria Infections in Immunosuppressed Hosts. Clin. Chest Med 2015, 36 (1), 91–99. 10.1016/j.ccm.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kumar P; Kaushik A; Lloyd EP; Li S-G; Mattoo R; Ammerman NC; Bell DT; Perryman AL; Zandi TA; Ekins S; Ginell SL; Townsend CA; Freundlich JS; Lamichhane G Non-Classical Transpeptidases Yield Insight into New Antibacterials. Nat. Chem. Biol 2017, 13 (1), 54–61. 10.1038/nchembio.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Erdemli SB; Gupta R; Bishai WR; Lamichhane G; Amzel LM; Bianchet MA Targeting the Cell Wall of Mycobacterium Tuberculosis: Structure and Mechanism of L,D-Transpeptidase 2. Structure 2012, 20 (12), 2103–2115. 10.1016/j.str.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Correale S; Ruggiero A; Pedone E; Berisio R Expression, Purification, Crystallization and Preliminary X-Ray Crystallographic Analysis of the l,d-Transpeptidase LdtMt1 from Mycobacterium Tuberculosis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun 2013, F69 10.1107/S1744309112052141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Libreros-Zúniga GA; Dos Santos Silva C; Salgado Ferreira R; Dias MVB Structural Basis for the Interaction and Processing of β-Lactam Antibiotics by L,D-Transpeptidase 3 (LdtMt3) from Mycobacterium Tuberculosis. ACS Infect. Dis 2019, 5 (2), 260–271. 10.1021/acsinfecdis.8b00244. [DOI] [PubMed] [Google Scholar]
- (26).Abagyan RA and Batalov S Do aligned sequences share the same fold? J. Mol. Bio 1997, 1, 355–368. 10.1006/jmbi.1997.1287. [DOI] [PubMed] [Google Scholar]
- (27).Robert X; Gouet P Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42 (W1), W320–W324. 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lohans CT; Chan HTH; Malla TR; Kumar K; Kamps JJAG; McArdle DJB; van Groesen E; de Munnik M; Tooke CL; Spencer J; Paton RS; Brem J; Schofield CJ Non-Hydrolytic β-Lactam Antibiotic Fragmentation by l,d-Transpeptidases and Serine β-Lactamase Cysteine Variants. Angew. Chemie - Int. Ed 2019, 58 (7), 1990–1994. 10.1002/anie.201809424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hugonnet JE; Tremblay LW; Boshoff HI; Barry CE; Blanchard JS Meropenem-Clavulanate Is Effective against Extensively Drug-Resistant Mycobacterium Tuberculosis. Science (80-.). 2009, 323 (5918), 1215–1218. 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kumar P; Chauhan V; Silva JRA; Lameira J; d’Andrea FB; Li S-G; Ginell SL; Freundlich JS; Alves CN; Bailey S; Cohen KA; Lamichhane G Mycobacterium Abscessus l,d-Transpeptidases Are Susceptible to Inactivation by Carbapenems and Cephalosporins but Not Penicillins. Antimicrob. Agents Chemother 2017, 61 (10), e00866–17. 10.1128/AAC.00866-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bianchet MA; Pan YH; Basta LAB; Saavedra H; Lloyd EP; Kumar P; Mattoo R; Townsend CA; Lamichhane G Structural Insight into the Inactivation of Mycobacterium Tuberculosis Non-Classical Transpeptidase LdtMt2 by Biapenem and Tebipenem. BMC Biochem. 2017, 18 (1), 8 10.1186/s12858-017-0082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dhar N; Dubeé V; Ballell L; Cuinet G; Hugonnet JE; Signorino-Gelo F; Barros D; Arthur M; McKinney JD Rapid Cytolysis of Mycobacterium Tuberculosis by Faropenem, an Orally Bioavailable β-Lactam Antibiotic. Antimicrob. Agents Chemother 2015. 10.1128/AAC.03461-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Rullas J; Dhar N; McKinney JD; García-Pérez A; Lelievre J; Diacon AH; Hugonnet J-E; Arthur M; Angulo-Barturen I; Barros-Aguirre D; Ballell L Combinations of β-Lactam Antibiotics Currently in Clinical Trials Are Efficacious in a DHP-I-Deficient Mouse Model of Tuberculosis Infection. Antimicrob. Agents Chemother 2015. 10.1128/aac.01063-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kaushik A; Gupta C; Fisher S; Story-Roller E; Galanis C; Parrish N; Lamichhane G Combinations of Avibactam and Carbapenems Exhibit Enhanced Potencies against Drug-Resistant Mycobacterium Abscessus. Future Microbiol. 2017, 12 (6), 473–480. 10.2217/fmb-2016-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kumar S; Stecher G; Li M; Knyaz C; Tamura K MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 2018, 35 (6), 1547–1549. 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Letunic I; Bork P Interactive Tree Of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res 2019. 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Abagyan R; Totrov M Biased Probability Monte Carlo Conformational Searches and Electrostatic Calculations for Peptides and Proteins. J. Mol. Biol 1994, 235 (3), 983–1002. 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- (38).Katritch V; Byrd CM; Tseitin V; Dai D; Raush E; Totrov M; Abagyan R; Jordan R; Hruby DE Discovery of Small Molecule Inhibitors of Ubiquitin-like Poxvirus Proteinase I7L Using Homology Modeling and Covalent Docking Approaches. J. Comput. Aided. Mol. Des 2007. 10.1007/s10822-007-9138-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cynamon MH; Speirs RJ; Welch JT In Vitro Antimycobacterial Activity of 5-Chloropyrazinamide. Antimicrob. Agents Chemother 1998, 42 (2), 462–463. 10.1128/AAC.42.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.