

Abstract
Here we report that the approach of metal-templated ligand synthesis can be applied to construct a dimeric protein assembly (BMOERIDC12), which is stabilized by non-covalent interactions and flexible covalent crosslinkers around the Zn templates. Despite its flexibility, BMOERIDC12 selectively binds ZnII over other divalent metals and undergoes dimerization upon metal binding. Such simultaneous fulfillment of plasticity and selectivity is a hallmark of cellular signaling events that involve ligand/metalinduced protein dimerization.
Graphical abstract
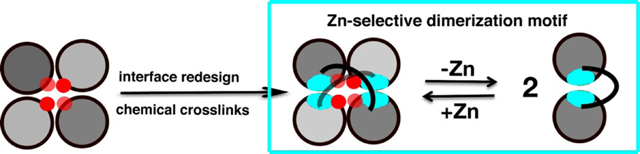
Described here is the metal-templated construction of a dimeric protein assembly (BMOERIDC12), which is stabilized by non-covalent interactions and flexible covalent crosslinkers around the Zn templates. BMOERIDC12 selectively binds ZnII over other divalent metals and undergoes dimerization upon metal binding.
INTRODUCTION
We describe the metal-templated construction of a protein complex that undergoes dimerization in selective response to Zn binding. In previous examples of designed molecular recognition, chemists have devised small organic building blocks that self-assemble into discrete supramolecular structures upon selective binding of organic or inorganic substrates.1 Such systems include Rebek2 and Gibb’s3 cavitands that dimerize into nanocapsules via hydrocarbon sequestration, and Lehn4 and Raymond’s5 helicates that selectively self-organize into higher-order assemblies upon metal complexation. Our work represents an extension of such synthetic work into the realm of protein building blocks, while simultaneously demonstrating that a traditional approach in coordination chemistry (metal-templated ligand synthesis) can readily lead to the emergence of a selective, chemically-induced protein dimerization event, which is a cornerstone of cellular signaling pathways.6
Proteins are nature’s premier ligand scaffolds for constructing functional metal centers, as they can afford an exquisite tuning of metal selectivity and reactivity through a combination of covalent and noncovalent interactions. An absolute requirement for the generation of any metallic (or non-metallic) active site with a specialized function is that it is internalized within a network of such interactions.7 We have proposed that a straightforward route for engineering functional metal sites may proceed through a metalmediated nucleation of multiprotein complexes and a subsequent stabilization of the resulting protein interfaces around the now-internalized metal centers.8,9 This approach was termed Metal-Templated Interface Redesign (MeTIR). The possibility of an evolutionary pathway similar to MeTIR has previously been suggested by Bolin10 and Armstrong11 for the vicinal oxygen chelate (VOC) superfamily of metalloenzymes and by Hol12 for hemocyanin and other dioxygen binding proteins. In further support, the inspection of many sophisticated metal active sites or binding pockets for non-metallic substrates reveal that they are formed in the interfaces between protein domains or subunits,13,14 which likely were independently folded units.15
A synthetic analogy for MeTIR is provided by the time-proven method of metal-templated ligand synthesis, first reported by Busch16 and adopted by others,17-19 in which a macrocyclic chelate is first organized and then covalently assembled around a metal ion. Inspired by this approach, we recently reported that a ZnII-mediated, D2-symmetrical, tetrameric superstructure of a monomeric protein, cytochrome cb562 (Zn4:MBPC14, Fig. 1a and b), can be converted into a stable, unimolecular assembly (C96RIDC14, Fig. 1c) through the installment of stable covalent and non-covalent bonds into the interfaces surrounding the four Zn centers.20 C96RIDC14 is not only one of the most stable multiprotein complexes ever engineered, but also displays impressive ZnII-selectivity over other divalent ions including CuII and couples ZnII binding to a large conformational change akin to a metal-dependent signaling protein.20 The seemingly contradictory traits of stability–both in terms of oligomerization and metal binding–and flexibility are afforded by the combination of fluid hydrophobic interactions incorporated into one set of interfaces (interface 1, i1) and short disulfide linkages (Cys96-Cys96’) built into a second set (i2) (Fig. 1c). In this study, we envisioned that the lengthening of the interfacial covalent linkages may further increase the flexibility of the assembly and result in the formation of alternative oligomeric forms while still retaining metal selectivity, which is the most basic but essential metal-based function. We report that such long covalent linkages across i3 result in the formation of a stable, folded structure consisting of two protomers (BMOERIDC12) that dimerizes into a tetrameric form (Zn4:BMOERIDC14) upon selective Zn binding (Fig. 1d).
Figure 1.

Templated construction of Zn-selective protein complexes. The metal-chelating variant of cyt cb562, MPBC-1 (a) undergoes Zn-mediated tetramerization into the D2-symmetrical Zn4:MBPC14 (b). (c) The combination of fluid, hydrophobic interactions installed into the i1 interface with a disulfide bond across i2 produces a tetramer 96CMBPC14 that undergoes a Zn-selective conformational change,20 whereas the combination of i1 interactions with a flexible linker across i3 (d) yields a Zn-selective dimerization motif (this study).
RESULTS AND DISCUSSION
The three C2-symmetric interfaces of Zn4:MBPC14 are highlighted in Fig. 1b and shown in detail in Fig. S1. Of these, i1 is the most extensive and closely packed, and readily lent itself to stabilization through the insertion of computationally prescribed and mostly hydrophilic-to-hydrophobic mutations (R34A/L38A/Q41W/K42S/D66W/V69I, blue patches in Fig. 1) to generate RIDC1.8 As the dihedral symmetry of the Zn-driven tetramer dictates, the combination of the stabilizing, non-covalent interactions in i1 with a short disulfide bond across the i2 interface (via the T96C mutation) should result in the formation of a stable tetramer (C96RIDC14), as was borne out through hydrodynamic and crystallographic investigations.20 i3, in contrast, is a minimal interface (490 Å2 buried surface) formed entirely by the inner Zn coordination sphere (Fig. S1) and is not amenable to redesign or crosslinking through a disulfide bond. We surmised that a long, flexible chemical linker incorporated across i3 into RIDC1 could result in the formation of either a folded-over dimer (stabilized by the hydrophobic mutations in i1) (Fig. 1d) or a D2symmetrical tetramer in the absence of Zn, in effect mimicking what nature accomplishes through genetically encoded peptide linkers for fusing proteins in order to realize the “chelate effect”.11,21
Toward this end, we replaced the symmetrically related pairs of Gly82 residues (Fig. S1) near the vertex of i3 with cysteines for chemical coupling through bis-maleimide functionalized crosslinkers.The crosslinkers used, bis-maleimidoethane (BMOE), bis-maleimidobutane (BMB) and bismaleimidohexane (BMH) (Fig. 2), have spacer arm lengths of 8.0 Å, 10.9 Å and 13.0 Å, respectively, and are all expected to readily cover the anticipated C82S-C82’S distance of 10–11 Å. The coupling reactions proceed cleanly, whereby the crosslinking agents are added slowly to the protein solution to promote the formation of cross-coupled dimers over singly-modified (saturated) monomers. The dimers, BMOERIDC12, BMBRIDC12 and BMHRIDC12, were separated from monomers through size-exclusion chromatography, and their compositions were confirmed by mass spectroscopy (12,349 Da for the parent 82CRIDC1; 24,918 Da, 24,946 Da, and 24,974 Da for the BMOE, BMB, and BMH crosslinked dimers, respectively).
Figure 2.

Cys-specific, bis-maleimide functionalized crosslinkers.
We first probed whether the crosslinked dimers assemble into the same Zn-mediated D2-symmetric tetrameric architecture that formed the basis of templated redesign. Sedimentation velocity (SV) experiments show that even at a concentration of 2.5 μM (lowest dimeric concentration measurable) BMOERIDC12, BMBRIDC12 and BMHRIDC12 all are entirely tetrameric in the presence of ZnII, with a peak sedimentation coefficient of approx. 4.5 S (Fig. 3), which matches that of the parent complex, Zn4:RIDC14.8 In comparison, the non-crosslinked RIDC-1 shows a distribution of 60% dimer and 40% tetramer under comparable solution conditions (5 μM RIDC1 and ZnII). Although SV is a non-equilbrium technique, this comparison nevertheless indicates the stabilization of Zn-induced tetramers due to crosslinking, likely due to entropic reasons. The crystal structures of the Zn-adducts of BMOERIDC12 (PDB ID: 3QVY), BMBRIDC12 (PDB ID: 3QW0) and BMHRIDC12 (PDB ID: 3QW1) were determined at 2.3 Å, 1.8 Å and 2.6 Å resolution, respectively. As shown in a structural superposition in Fig. 4a, the supramolecular architectures of the resulting complexes are identical to one another, and to Zn4:MBPC1422 and Zn4:RIDC14.8 Importantly, the Zn-coordination environments, which lie in close proximity to the crosslinking sites, are unchanged as intended by the use of flexible linkers (Fig. 4b). The electron density for the entire length of the BMOE linker is evident, while for the longer BMB and BMH linkers only the Cys82-thioether-succinamide moiety can be discerned (Fig. 4c).
Figure 3.

Hydrodynamic analysis of maleimide crosslinked constructs in the presence of ZnII. Sedimentation velocity (SV) profiles for various crosslinked (2.5 μM dimer) and a non-crosslinked construct (5 μM monomer) in the presence of 5 μM ZnII demonstrate almost full conversion to a ZnII induced tetramer of the three crosslinked constructs at relatively low protein and metal concentrations.
Figure 4.

Comparison of the supramolecular structures of the ZnII mediated tetramers of maleimide crosslinked constructs. a) Structural superposition of Zn4:RIDC14 (orange), Zn4:BMOERIDC14 (magenta), Zn4:BMBRIDC14 (cyan) and Zn4:BMHRIDC14 (marine). b) Superposition of the Zn coordination environments (same coloring scheme). c) Fo-Fc omit electron density maps (2.5 s) of the BMOE, BMB and BMH crosslinkers. For BMH, the hexane linker region was not modeled due to the lack of electron density.
Having verified their Zn-driven assembly into the desired “macrocyclic” architecture, we next investigated the structures of BMOERIDC12, BMBRIDC12 and BMHRIDC12 in the absence of Zn. SV measurements indicate that dimers are the predominant oligomeric form under the conditions tested for all crosslinkers. At low proteins concentrations (2.5 μM), a small population of possibly tetrameric species (4.5–4.8 S) is observed (Fig. 5a), while at higher concentrations (>25 μM), the SV distributions for such high-order aggregates become considerably broader and tail into larger sedimentation coefficients that are suggestive of linear polymeric chains (Fig, 5b). These results suggest that the crosslinked dimers prefer to adopt the folded-over conformation rather than a pre-organized tetramer reminiscent of the Zn-induced structure.
Figure 5.

Hydrodynamic analysis of metal free maleimide crosslinked constructs. a) Sedimentation velocity (SV) profiles for various crosslinked (2.5 μM dimer) and a non-crosslinked construct (5 μM monomer) in the absence of metals. b) Same as in (a), but at 25 μM and 50 μM concentrations, respectively, highlighting the presence of higher ordered species formed by the crosslinked constructs even in the absence of any metal ions. c) Hypothetical conformations for BMOERIDC12 and their calculated sedimentation coefficients. The rightmost structure is that of crystallographically characterized RIDC12 (non-crosslinked) with its calculated and experimentally determined sedimentation coefficient.
Despite extensive efforts, we could not crystallize the crosslinked dimers in the absence of metal ions, which likely is a result of their flexibility as well as their tendency to form large aggregates at high concentrations. In order to gain some insight into the structures of the crosslinked dimers, we compared their experimentally determined sedimentation coefficients–which are not only dependent on molecular weight but also shape–to those of various theoretical dimeric conformations of BMOERIDC12 (Fig. 5c) using Hydropro.23 These simulations show that as the BMOE-dimer transitions from a fully extended configuration to a compact form, its sedimentation coefficient increases from 2.2 to 2.6 S, which is the lower end of the observed range (2.6–2.8 S) for the crosslinked dimers. Validating this analysis, the maximal sedimentation coefficient of the structurally characterized, antiparallel dimer RIDC12 was determined to be ~2.75 S,8 which is the most compact structure shown in the series (Fig. 5c). Based on these findings, we conclude that the chemically-crosslinked dimers likely assume a folded structure in solution, driven by the interactions between hydrophobic residues installed for i1 redesign. Given that the crosslinked dimers in their folded form should have many metal-coordinating residues (His, Glu and Asp) in close proximity and be relatively flexible, one would expect them to promiscuously coordinate different transition metal ions. Indeed, CoII, NiII or CuII binding to BMOERIDC12 all induce the formation of a tetrameric species, with a maximal sedimentation coefficient of 4.5 identical to that of Zn4:BMOERIDC14 (Fig. 6a). However, since the redesign of i1 and the placement of crosslinkers across i3 were based on the four tetrahedral Zn templates, we anticipated that there should be increased affinity for ZnII binding relative to the other divalent metal ions. To test this possibility, metal binding competition assays were performed with the metal chelating fluorophore, MagFura-2,24 whereby increasing concentrations of CoII, NiII, CuII or ZnII were titrated into a mixture of BMOERIDC12 and MagFura-2, and the changes in MagFura2 fluorescence were monitored (Fig. 6b–d, Fig. S2). In parallel, titrations were performed to determine the affinity of Mag-Fura-2 for these metal ions (Fig. S3), which yielded similar values to those previously reported.25,26 Because BMOERIDC12 does not self-associate into a pre-formed tetramer in the absence of metal binding, and the titration solutions contain a mixture of BMOERIDC12 and BMOERIDC14 in their various metal-bound forms, it is not possible to extract accurate metal binding equilibrium constants. Nevertheless, the titrations show that BMOERIDC14 can accommodate four equivalents each of CoII, NiII, or ZnII with affinities that rival that of MagFura-2 for those ions. The competition isotherms can be reasonably well described by a 4-equivalent-binding-site model, yielding half-saturation concentrations of 9 μM for CoII, 0.5 μM for NiII and 0.04 μM for ZnII (Table 1). It should be noted that these are not true dissociation constants, but rather approximate metal concentrations where half of the starting BMOERIDC12 population is converted into the fully metal-bound, M4:BMOERIDC14 species. Given that i, i+4 bis-histidine motifs on a-helices–which constitute the metal binding motifs on the parent MBPC1 molecule–bind CoII, NiII, or ZnII with micromolar affinities that are within an order of magnitude of each other (Kd-Zn ≈ Kd-Ni < Kd-Co),27,28 our results clearly show an increased thermodynamic selectivity for ZnII coordination over the other two ions.
Figure 6.

BMOERIDC12 tetramerization mediated by various divalent metal ions with a preference for ZnII binding. a) SV profiles for 2.5 μM BMOERIDC12 dimer in the presence of 5 μM CoII, NiII. CuII, and ZnII, reflecting the formation of a tetrameric species on binding all four metal ions. (b-d) Binding isotherms for Mag-Fura-2 / BMOERIDC12 competition experiments with (b) Co2+, (c) Ni2+, and (d) Zn2+, with tick marks shown on the top x-axis that correspond to theoretical titration end points for the indicated number of binding sites per tetramer. Solid lines represent fits obtained using DynaFit to a four-equivalent-bindingsites model, with Kd values listed in Table 1. Cu2+ competition experiments (e) demonstrate 12 metal binding events.
Table 1.
Apparent Dissociation Constants for Divalent Metal Ion Adducts of Mag-Fura2 and BMOERIDC12.
| Kd (μM) | ||
|---|---|---|
| metal | Mag-Fura-2 | BMOERIDCl2 |
| Zn | 0.047 (0.005) | 0.041 (0.010) |
| Ni | 0.175(0.014) | 0.54 (0.055) |
| Co | 1.41 (0.22) | 9.29(1.02) |
The ultimate test for ZnII selectivity is provided by CuII, which lies atop the Irving-Williams series292 for the relative stabilities of divalent metal complexes (MnII < FeII < CoII < NiII < CuII > ZnII), and readily outcompetes ZnII for most natural and synthetic Zn-specific binding sites.30–32 This is particularly the case for flexible peptides or proteins that cannot impose steric constraints to offer selectivity between metalions.33 In accord with the high affinity of CuII for protein-based donor atoms, our titrations show that BMOERIDC14 has at least twelve binding sites for CuII that can compete with MagFura-2 (i.e., their Kd’s are within an order of magnitude of 8.5 nM)34 (Fig. 6e), some likely on the surface and some in the core. The high plurality of CuII binding to BMOERIDC14 and the availability of surface binding sites make it challenging to assess the ZnII vs. CuII selectivity of the internal coordination sites through titrations. We therefore cocrystallized BMOERIDC12 in the presence of a mixture of ZnII and CuII. The structure of the resulting complex (PDB ID: 3QVZ) was determined at 2.6 Å resolution, which revealed that it possesses the same supramolecular architecture as Zn4:BMOERIDC14, with four metals associated with the core and seven with the surface of the tetramer (Fig. 7). To identify these metal ions, anomalous difference maps were calculated using X-ray diffraction data collected at the Zn and Cu K-edges (1.265 and 1.377 Å). These maps indicate the core ions to be Zn, which display anomalous scattering only at the Zn K-edge and not at the Cu K-edge, and the majority (6 out of 7) of surface ions to be Cu. X-ray fluorescence scans further confirm the presence of both Cu and Zn in the thoroughly washed crystals (Fig. S4). Subsequently, we investigated through size exclusion chromatography (SEC) whether Zn4:BMOERIDC14 is a true thermodynamic product. The SEC elution profile of Zn4:BMOERIDC14 ran in the presence of Zn indicates the presence of both the tetrameric and the dimeric (as well as higher-order) species (Fig. S5).35 Although the observed dimeric species is a minor component, as would be predicted from the SV experiments (Fig. 3a), the SEC findings suggest that Zn4:BMOERIDC14 is in exchange with its dimeric form in solution, and that Zn vs. Cu selectivity observed under crystallization conditions likely is thermodynamically driven.36 We attribute the thermodynamic basis for Zn selectivity not only to the formation of a covalently and non-covalently stabilized macrocycle built around the tetrahedral Zn coordination sites, but also to the fact that this macrocyle has been programmed to house four Zn ions in its interior, all of which contribute to the overall stability of Zn4:BMOERIDC14 formation.
Figure 7.

Structure of Zn4:BMOERIDC14 grown in the presence of CuII. A ribbon representation of Zn4:BMOERIDC14 crystallized in the presence of CuSO4 is shown along with the anomalous difference maps generated from data collected (a) at the Cu K-edge (green, 5 s) and (b) at the Zn K-edge (blue, 5 s). The surface-associated Cu ions are shown with arrows in (a). The heme Fe centers, as well as well-ordered surface Cu ions have anomalous contributions at both energies, whereas the core Zn ions only contribute at the higher energy Zn K-edge.
In summary, we have shown here that a traditional approach in synthetic coordination chemistry (metaltemplated ligand synthesis) can be applied to the design of a large and dynamic protein assembly, which displays an extent of Zn selectivity that is challenging to achieve in small molecule systems. The advantage of proteins as ligand scaffolds lies in their chemically rich surfaces that enable the formation of an extensive network of stable, yet reversible non-covalent interactions around the metal centers. The ensuing, simultaneous fulfillment of plasticity and selectivity, such as that displayed in our system, is a hallmark of all cellular signaling systems, and particularly those that couple selective metal binding to conformational changes or oligomerization to activate downstream processes, such as the prototypical EF-hand signaling proteins (calmodulin,37 S10038) or any metalloregulatory gene transcription factor.39
EXPERIMENTAL SECTION
Protein Mutagenesis. Purification, Modification with Crosslinkers.
The G82C-RIDC1 cyt cb562 construct (C82RIDC1) was generated, expressed, and purified according to published procedures.22,40,41 Purified C82RIDC1 was buffer exchanged into 20 mM TRIS (pH 7) and 50 mM dithiothreitol (DTT) and concentrated using Amicon stirred cells with MW=10 kDa cutoff-filters. Concentrated protein was eluted through a 10 DG desalting column (BioRad) to eliminate excess DTT and diluted with an appropriate volume of 20 mM TRIS (pH 7) to yield a protein concentration of 100 μM. Concentrated stock solutions of BMOE, BMB, or BMH (Pierce) were added to the sample in ten equal aliquots every 30 sec over 5 min to give a final crosslinker concentration of 100 μM. The reaction was then allowed to proceed at room temperature under constant stirring for 30 min. Crosslinking reactions were quenched by the addition of 50 mM DTT and allowed to incubate at room temperature for 15 min. Crosslinking yields were approx. 50%. Crosslinked proteins were subsequently purified through multiple size exclusion chromatography runs using a Pall ACA54 resin (fractionation range = 5–70 kDa) using an elution buffer of 20 mM TRIS (pH 7) and 150 mM NaCl. Dimer purity was assessed by SDS-PAGE electrophoresis, and the compositions of the dimers were verified by MALDI mass spectrometry.
Analytical Ultracentrifugation and SV simulations.
Sedimentation velocity (SV) measurements were made on a Beckman XL-I Analytical Ultracentrifuge (Beckman-Coulter Instruments) using an An60 Ti rotor at 41,000 rpm for a total of 250 scans per sample. Data were collected at 415 nm for low concentration (2.5 μM crosslinked dimer, 5 μM RIDC1 monomer) samples, and 560 nm for higher concentrations (25 μM crosslinked dimer). All data were processed in SEDFIT42 with the following fixed parameters: buffer density (r) = 0.99764 g/ml; buffer viscosity = 0.0089485 poise; Vbar, which was calculated to be 0.7310 ml/g for BMOERIDC12 and BMBRIDC12; 0.7306 ml/g for BMHRIDC12. The buffer density (r) and viscosity for 20 mM TRIS (pH 7) were calculated through SEDNTERP (http://www.jphilo.mailway.com/default.htm). Theoretical sedimentation coefficients were calculated using Hydropro Version 7.C,23 using the same parameters employed in SV data processing.
Crystallography.
Crystals of all crosslinked dimers were obtained by sitting drop vapor diffusion at room temperature. The crystallization conditions for the four different crystal forms described in this study are listed in Table S1 and Table S2, along with corresponding data collection and refinement statistics. All protein stocks solutions were in 20 mM TRIS (pH 7) buffer. Appropriate crystals were transferred to a solution of mother liquor containing 20% glycerol as cryoprotectant and frozen in liquid nitrogen prior to data collection at 100 K.
Data were integrated using MOSFLM and scaled in SCALA,43 except for Zn4:BMBRIDC14, which was processed using SAINT and Bruker SADABS. All structures were determined through molecular replacement with MOLREP,44 using the RIDC1 monomer structure (PDB ID:3HNI) as the search model, followed by rigid-body, positional, thermal and TLS refinement with REFMAC45 using appropriate noncrystallographic symmetry restraints. The crystals for both forms of Zn4: BMOERIDC14 (crystallized in the presence and the absence of CuSO4) were found to have 48% and 44% twin fractions, respectively. Both data sets were refined using intensity-based twin refinement as implemented in REFMAC using the twin operator: −1.000h-1.000k, 1.000k, -l. All structures were manually built in COOT46 to produce the final models. All figures were produced using PYMOL.47
Metal Binding Titrations.
For Mag-Fura-2 competition titrations, 1 mg Mag-Fura-2 (Invitrogen) was resuspended in 1 ml Millipore purified water. Mag-Fura-2 concentration was determined using an extinction coefficient of 22,000 M−1cm−1 at 369 nm. The titrations for determining Mag-Fura-2 affinities for NiII and CoII were performed in a 1-cm cuvette with a 2 ml sample volume containing 0.5 μM MagFura-2 and 1 mM CaCl2 in Chelex-treated 20 mM 3-(N-morpholino)propanesulfonic acid (MOPS, pH 7) buffer with 150 mM NaCl. The sample was allowed to equilibrate with stirring for 3 min. after each addition of the competing metal stock before recording fluorescence excitation scans monitoring emission at 505 nm. ZnII titrations were performed with 1 mM NiII (sulfate salt) as the competing ion in place of CaCl2. Titration data (fluorescence excitation monitored at 330 nm) were fit to a 1-site binding model using Dynafit48 assuming a Mag-Fura-2:Ca2+ Kd of 25 μM, which was found to be constant over a wide variety of pH and ionic strength conditions.26 The resulting Kd values are listed in Table 1. The Mag-Fura2:ZnII Kd was back-calculated using the Mag-Fura-2:NiII Kd determined here.
BMOERIDC12/Mag-Fura-2 competition assays were performed in a 1-cm cuvette with a 1.5-ml sample volume containing 25 μM BMOERIDC12 and 10 μM Mag-Fura-2 in Chelex-treated MOPS (pH 7) buffer with 150 mM NaCl. As with the titrations described above, samples were allowed to equilibrate under stirring for 3 min. after the addition of each aliquot of the appropriate divalent metal stock before recording fluorescence excitation scans monitoring emission at 505 nm. NiII and CoII binding to MagFura-2 results in quenching of the florescence signal, which was subsequently followed using the emission readout using 372 nm excitation. ZnII binding results in an increased fluorescence excitation at 323 nm, which was used as the excitation wavelength. All titration curves could be adequately described with a minimal (4 ´ 1) binding model consisting of four binding sites with equivalent half-saturation values listed in Table 1. CuII titrations were performed similarly to NiII, CoII and ZnII, except Mag-Fura-2:CuII binding was monitored by the increase in absorbance at 300 nm, rather than by fluorescence.
Analytical Size Exclusion Chromatography (SEC).
Size exclusion chromatography was performed on a Biologic DuoFlow FPLC system (Bio-Rad) equipped with a UV detector set to monitor absorbance at 280 nm. For each run, 100 μl of 25 μM BMOERIDC12 in 20 mM TRIS (pH 7) buffer with 50mM NaCl and either 50 μM ZnCl2 or 5 mM EDTA were injected on an analytical SEC column packed with Pall ACA54 resin (fractionation range = 5–70 kDa). The samples were eluted from the column at 0.5 ml/min in 20 mM TRIS (pH 7) with 50 mM NaCl, and additionally, 5 mM EDTA for the metal free sample. The dead volume in Fig. S5 is based on a ferritin (MW ~ 500 kDa) standard run under conditions identical to the metal free sample.
Supplementary Material
Acknowledgments.
This work was supported by the National Science Foundation (CHE-0908115), the Hellman Foundation, the Sloan Foundation and the Arnold and Mabel Beckman Foundation. F.A.T. was in part supported by DOE Basic Energy Sciences, Biomolecular Materials Program (DE-FG0210ER46677, crystallographic analyses). Portions of this research were carried out at SSRL, operated by Stanford University on behalf of DOE.
Supporting Information Available http://pubs.acs.org: Detailed views of RIDC1 interfaces, spectra for metal-binding assays, X-ray fluorescence spectra of the ZnII-CuII cocrystal of BMOERIDC14, sizeexclusion chromatograms of BMOERIDC12, and tables for X-ray data collection and refinement statistics.
REFERENCES
- (1).Schmuck C, Angew. Chem. Int. Ed. 2007, 46, 5830–5833. [DOI] [PubMed] [Google Scholar]
- (2).Ajami D; Rebek J Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 16000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gibb CLD; Gibb BC J. Am. Chem. Soc. 2006, 128, 16498. [Google Scholar]
- (4).Kramer R; Lehn JM; Marquisrigault A Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Caulder DL; Raymond KN Acc. Chem. Res. 1999, 32, 975. [Google Scholar]
- (6).Klemm JD; Schreiber SL; Crabtree GR Ann. Rev. Immun. 1998, 16, 569. [DOI] [PubMed] [Google Scholar]
- (7).Mann S, Angew. Chem. Int. Ed. 2008, 47, 5306–5320. [DOI] [PubMed] [Google Scholar]
- (8).Salgado EN; Ambroggio XI; Brodin JD; Lewis RA; Kuhlman B; Tezcan FA Proc. Natl. Acad. Sci. USA 2010, 107, 1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Salgado EN; Radford RJ; Tezcan FA Acc. Chem. Res. 2010, 43, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bergdoll M; Eltis LD; Cameron AD; Dumas P; Bolin JT Prot. Sci. 1998, 7, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Armstrong RN Biochemistry 2000, 39, 13625. [DOI] [PubMed] [Google Scholar]
- (12).Volbeda A; Hol WG J. J. Mol. Biol. 1989, 206, 531. [DOI] [PubMed] [Google Scholar]
- (13).Rausell A; Juan D; Pazos F; Valencia A Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rees DC; Howard JB Curr. Op. Chem. Biol. 2000, 4, 559. [DOI] [PubMed] [Google Scholar]
- (15).Ali MH; Imperiali B Bioorg. Med. Chem. 2005, 13, 5013. [DOI] [PubMed] [Google Scholar]
- (16).Thompson MC; Busch DH J. Am. Chem. Soc. 1964, 86, 3651. [Google Scholar]
- (17).Creaser II; Geue RJ; Harrowfield JM; Herlt AJ; Sargeson AM; Snow MR; Springborg JJ Am. Chem. Soc. 1982, 104, 6016. [Google Scholar]
- (18).McMurry TJ; Raymond KN; Smith PH Science 1989, 244, 938. [DOI] [PubMed] [Google Scholar]
- (19).Dietrichbuchecker CO; Sauvage JP; Kern JM J. Am. Chem. Soc. 1984, 106, 3043. [Google Scholar]
- (20).Brodin JD; Medina-Morales A; Ni T; Salgado EN; Ambroggio XI; Tezcan FA J. Am. Chem. Soc. 2010, 132, 8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Goodsell DS; Olson AJ Annu. Rev. Biophys. Struct. 2000, 29, 105. [DOI] [PubMed] [Google Scholar]
- (22).Salgado EN; Faraone-Mennella J; Tezcan FA J. Am. Chem. Soc. 2007, 129, 13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).de la Torre JG; Huertas ML; Carrasco B Biophys. J. 2000, 78, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Raju B; Murphy E; Levy LA; Hall RD; London RE Am. J. Physiol. 1989, 256, C540. [DOI] [PubMed] [Google Scholar]
- (25).Simons TJ B. J. Biochem. Biophys. Meth. 1993, 27, 25. [DOI] [PubMed] [Google Scholar]
- (26).Golynskiy MV; Gunderson WA; Hendrich MP; Cohen SM Biochemistry 2006, 45, 15359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Krantz BA; Sosnick TR Nat. Struct. Biol. 2001, 8, 1042. [DOI] [PubMed] [Google Scholar]
- (28).Ghadiri MR; Choi CJ Am. Chem. Soc. 1990, 112, 1630. [Google Scholar]
- (29).Frausto da Silva JJR; Williams RJP The biological chemistry of the elements; Oxford University Press: Oxford, 2001. [Google Scholar]
- (30).Nolan EM; Ryu JW; Jaworski J; Feazell RP; Sheng M; Lippard SJ J. Am. Chem. Soc. 2006, 128, 15517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Walkup GK; Imperiali BJ Am. Chem. Soc. 1997, 119, 3443. [Google Scholar]
- (32).Hunt JA; Ahmed M; Fierke CA Biochemistry 1999, 38, 9054. [DOI] [PubMed] [Google Scholar]
- (33).Waldron KJ; Robinson NJ Nature Rev. Microbiol. 2009, 7, 25. [DOI] [PubMed] [Google Scholar]
- (34).Liu JB; Dutta SJ; Stemmler AJ; Mitra B Biochemistry 2006, 45, 763. [DOI] [PubMed] [Google Scholar]
- (35).The initial concentration of the Zn4:BMOERIDC14 sample applied to the SEC column is 25 μM (based on BMOERIDC12), at which no dimer would be expected to be present.
- (36).Based on the available results, we cannot completely discount the possibility that the apparent Zn over Cu selectivity is due to the kinetic stability of Zn4:BMOERIDC14 and that this species preferentially crystallizes out of a mixture of Zn-and Cu-adducts. Even if this were the case, our results on BMOERIDC12 represent a significant improvement of ZnII-selectivity over other divalent metal ions compared to the parent (pre-templated) species, MBPC1, which shows considerably higher affinities (as least 10-fold) for CuII and NiII over ZnII (E.N. Salgado, et al., J. Am. Chem. Soc., 129, 13374–13375; E.N. Salgado, et al., Inorg. Chem. 2009, 48, 2726–2728).
- (37).Nelson MR; Chazin WJ Prot. Sci. 1998, 7, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Fritz G; Botelho HM; Morozova-Roche LA; Gomes CM FEBS J. 2010, 277, 4578. [DOI] [PubMed] [Google Scholar]
- (39).Reyes-Caballero H; Campanello GC; Giedroc DP Biophys. J. 2011, 156, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Salgado EN; Lewis RA; Faraone-Mennella J; Tezcan FA J. Am. Chem. Soc. 2008, 130, 6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Salgado EN; Lewis RA; Mossin S; Rheingold AL; Tezcan FA Inorg. Chem. 2009, 48, 2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Schuck P Biophys. Chem. 2004, 108, 187. [DOI] [PubMed] [Google Scholar]
- (43).Collaborative Computational Project, Number 4. 1994. “The CCP4 Suite: Programs for Protein Crystallography”. Acta Cryst. D50, 760–763. [DOI] [PubMed] [Google Scholar]
- (44).Vagin A; Teplyakov AJ Appl. Cryst. 1998, 30, 1022. [Google Scholar]
- (45).Murshudov G; Vagin A; Dodson E Acta Cryst. 1996, D53, 240. [DOI] [PubMed] [Google Scholar]
- (46).Emsley P; Cowtan K Acta Cryst. 2004, D60, 2126. [DOI] [PubMed] [Google Scholar]
- (47).DeLano WL The PYMOL Molecular Graphics System (http://www.pymol.org), 2003. [Google Scholar]
- (48).Kuzmic P Anal. Biochem. 1996, 237, 260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.