

Abstract
C-Nucleosides are characterized by a C-C rather than a C-N linkage between the heterocyclic base and the ribofuranose ring. While the biosynthesis of pseudouridine-C-nucleosides has been studied, less is known about the pyrazole-C-nucleosides such as the formycins and pyrazofurin. Herein, genome screening of Streptomyces candidus NRRL 3601 led to the discovery of the pyrazofurin biosynthetic gene cluster pyf. In vitro characterization of gene product PyfQ demonstrated that it is able to catalyze formation of the C-glycoside carboxyhydroxypyrazole ribonucleotide (CHPR) from 4-hydroxy-1H-pyrazole-3,5-dicarboxylic acid and phosphoribosyl pyrophosphate (PRPP). Similarly, ForT, the PyfQ homologue in the formycin pathway, can catalyze the coupling of 4-amino-1H-pyrazole-3,5-dicarboxylic acid and PRPP to form carboxyaminopyrazole ribonucleotide. Finally, PyfP and PyfT are shown to catalyze amidation of CHPR to pyrazofurin 5′-phosphate thereby establishing the latter stages of both pyrazofurin and formycin biosynthesis.
Keywords: C-nucleosides, biosynthesis, biosynthetic gene cluster, C-glycosidation reaction, enzyme mechanism
Graphical Abstract
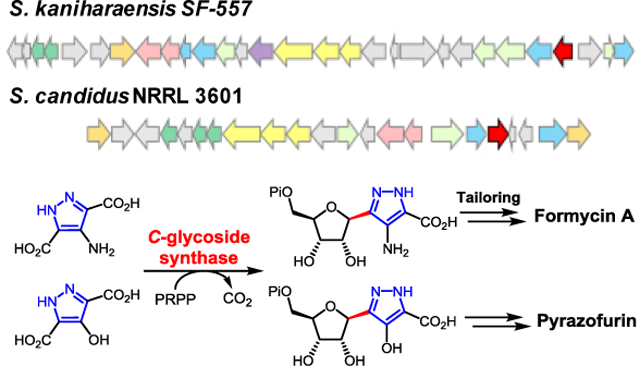
Enzymatic C-glycoside bond formation: The C-glycoside synthases encoded in the formycin and pyrazofurin biosynthetic gene clusters are identified and compared by genomic analysis of the producing strains. Both enzymes catalyze decarboxylative C-glycoside bond formation between phosphoribosyl pyrophosphate and a pyrazole nucleobase in reactions likely to involve electrophilic aromatic substitution.
The C-nucleosides are a noncanonical class of nucleosides in which the heterocyclic base and the ribofuranosyl core are connected through a C-C bond rather than the more common C-N linkage that is found in the canonical nucleosides.[1–3] A wide variety of nucleobases and functional groups are found in the naturally occurring C-nucleosides including uracil in pseudouridine (1), [4] malayamycin (2) [5,6] and pseudouridimycin (3), [7,8] the oxazine dione group in minimycin (4), [9,10] the maleimide unit in showdomycin (5), [11–14] and the pyrazole moiety in pyrazofurins (6) [15] and formycins (7 and 8) [16,17] (see Figure 1). Many C-nucleosides found in nature are known to have antibiotic and antiviral activities mainly due to their structural resemblance to the N-nucleosides that results in their interference with primary metabolic processes. [5,7,9,18–27] Their biological activities have thus inspired the synthesis of many nonnatural C-nucleosides, some of which have been demonstrated to be effective therapeutic agents. [2,3,28] However, how the C-nucleosides are biosynthesized in nature remains largely unexplored.
Figure 1.

Representatives of C-nucleoside antibiotics (1-8) and coformycin (9).
Early studies have shown that pseudouridine (1) is derived from uridine (10) in a reaction catalyzed by pseudouridine synthase. [29] The catalytic cycle of this enzyme is initiated by C-N bond cleavage to release uracil (11) and an oxocarbenium ion (12) (or a 1,2-glycal species after C2 deprotonation). Reorientation of 11 in the active site followed by nucleophilic addition back to 12 can then lead to C-glycosidic bond formation in 1 as shown in Scheme 1A. [30–32] PumJ, a pseudouridine synthase equivalent, has been demonstrated to catalyze the same reaction during the biosynthesis of pseudouridimycin (3). [8] The discovery of sdmA, which encodes a pseudouridine synthase homologue in the showdomycin (5) gene cluster, likewise suggested a similar mechanism of C-C bond formation in showdomycin assembly (see Scheme 1B). [14]
Scheme 1.

Proposed mechanism for pseudouridine synthase. The cosubstrate 13 in SdmA reaction is likely derived from L-glutamine. [11,12]
In contrast, little is known about the mode of C-glycosidic bond formation in the biosynthesis of pyrazofurin (6) and the formycins (7/8). As both carry a pyrazole-derived nucleobase that is different from those in 1-5, enzymes catalyzing the C-glycosidation in 6-8 likely involve distinct mechanisms from that of pseudouridine synthase. The formycin biosynthetic gene cluster (i.e., the for cluster) from Streptomyces kaniharaensis SF-557 has already been identified. [33] In vitro experiments showed that carboxyaminopyrazole ribonucleotide (CAPR, 14) is a late intermediate in the formycin A biosynthetic pathway and its maturation to formycin A 5’-phosphate (18) is sequentially catalyzed according to Scheme 2A by ForC, ForB, ForH, ForA, and ForB together with PurH, which originates in primary metabolism. [33,34] Importantly, the forJ, forK and forL genes in the for cluster were noted to be homologous to spb38, spb39 and spb40 in the gene cluster of s56-p1 (21). [35] Enzymes encoded by the latter set of genes have been shown to catalyze the transformation of lysine and glycine to give hydrazino acetic acid (20) enroute to s56-p1 (Scheme 2B). [35] Hence, hydrazine formation may also be involved in the construction of the pyrazole group of formycins[33] and perhaps pyrazofurin as well (Scheme 2C). If correct, this hypothesis suggested a criterion by which the pyrazofurin gene cluster might be identified in a genome-wide search of the producing strain. Towards this goal, a comparative genomics approach was employed to find the responsible enzymes and investigate the mechanisms of C-C coupling in 6-8. Reported herein is the discovery of novel enzyme-catalyzed C-glycosidation reactions that underlie biosynthesis of the pyrazole group in biological systems.
Scheme 2.

(A) Conversion of CAPR (14) to formycin A 5’-phospahte (18) in the formycin biosynthetic pathway and the intermediacy of CHPR (19) in pyrazofurin pathway. (B) Formation of hydrazino acetic acid (20) in the biosynthesis of s56-p1 (21).[35] (C) Possible involvement of ForJ/K/L and PyfG/H/I in hydrazine assembly in formycin and pyrazofurin biosynthesis, respectively.
The genome of Streptomyces candidus NRRL 3601 was sequenced (9.2 Mbp with 301 contigs) (N50: 152,140 bp), and the genes pyfG, pyfH, and pyfI, which are homologous to forJ, forK and forL, were found clustered on the k141_289 contig. This cluster, designated as the pyf cluster, spans a ~25 kbp region between two ABC transporters which are the likely boundary of the pyf cluster (see Figure 2). BLAST (basic local alignment search tool) comparison of the pyf and for clusters not only revealed the conservation of the putative hydrazine-assembly genes (forJ/forK/forL versus pyfG/pyfH/pyfI) in each cluster, but also the presence of forC and forB homologues, denoted here as pyfP and pyfT, respectively. These findings are consistent with the pyf cluster being responsible for pyrazofurin biosynthesis and suggest that formycins and pyrazofurin may share the same pyrazole forming machinery. In addition, carboxyhydroxypyrazole ribonucleotide (CHPR, 19) was considered to be a likely intermediate of pyrazofurin biosynthesis and specifically a substrate for sequential reactions catalyzed by the pyfP and pyfT gene products as shown in Scheme 2A.
Figure 2.

The pyrazofurin (pyf) biosynthetic gene cluster from S. candidus in comparison with the formycin (for) gene cluster from S. kaniharaensis. The coformycin biosynthetic genes (cof genes) are found adjacent to the main clusters in both the pyf and for clusters.
Also noted was the absence of forHAFI homologs in the pyf cluster (see dashed box in Figure 2). This is consistent with the observation that ForH and ForA are required only for the construction of the second heterocycle in formycins, which is absent in pyrazofurin (see Scheme 2A). [33,34] Furthermore, the coformycin (9) biosynthetic genes (i.e., the cof cluster) were found to be adjacent to both the for and pyf main clusters. [36] This flanking arrangement is not surprising, because formycins (7/8) are produced synergistically with coformycin (9), which is an inhibitor of adenosine deaminase (ADA) and can therefore prevent formycin A (7) deactivation by ADA. [16,37] Similarly, amide bond hydrolysis of pyrazofurin (e.g., 23 → 19) was also observed when pyrazofurin (6) was incubated with ADA (type X) (see Figure S2 in the Supporting Information). This bioinformatic analysis collectively suggested that the pyf cluster encodes the enzymes of pyrazofurin biosynthesis.
To test the hypothesis that the pyf cluster is indeed responsible for pyrazofurin biosynthesis, selected pyf gene products were expressed and characterized in vitro. In particular, PyfQ as well as ForT from the for cluster are annotated as encoding a β-ribofuranosylaminobenzene 5’-phosphate (β-RFA-P) synthase. β-RFA-P synthase has been demonstrated to catalyze formation of the C-glycoside 27 from phosphoribosyl pyrophosphate (PRPP, 24) and p-aminobenzoic acid (pABA, 25) during biosynthesis of methanopterin (Scheme 3A). [38–41] Furthermore, this reaction is believed to involve electrophilic aromatic substitution (EAS) of pABA (25) with the oxocarbenium ion (12) from PRPP (24, Scheme 3A). [38,39] Therefore, it was proposed that an analogous substitution reaction also occurs between PRPP and an aromatic nucleobase in the catalytic cycles of ForT and PyfQ such that these two enzymes are responsible for C-glycosidic bond formation in formycin[33] and pyrazofurin, respectively.
Scheme 3.

(A) Reaction catalyzed by β-ribofuranosylaminobenzene 5’-phosphate synthase. (B) and (C) Reactions catalyzed by ForT and PyfQ using PRPP (24) and different nitrogen-containing compounds as substrates.
In order to investigate these hypotheses, the putative six-membered pyridazinone[33] intermediate 22 was incubated with ForT and PyfQ, but no C-glycosidation product was observed (Scheme 3B and Figure S3). However, when 4-amino-1H-pyrazole-3-carboxylic acid (APA, 29) was prepared (see Supporting Information) and incubated with PRPP (24) and ForT for 36 h, two new products were detected (see Figure 3B, trace 4). These products were identified as aminopyrazole ribonucleotide (APR, 30) and CAPR (14) based on MS analysis (see Supporting Information) and HPLC coelution with the synthetic standards. Although the yield was poor (<5%), these results demonstrated that ForT catalyzes a C-glycosidation reaction. The production of both APR and CAPR suggested competing electrophilic addition of the putative oxocarbenium intermediate 12 at C3 versus C5 of 29 to yield APR (30) versus CAPR (14), respectively (see Figure 3A, routes a vs. b). While ForT appeared to promote addition at both C3 and C5, the same experiment with PyfQ demonstrate primarily decarboxylative EAS (route a) at C3 to yield APR (30) with essentially no addition at C5 (see Figure 3B, trace 5). Furthermore, APA (29) appeared to be a much better substrate for PyfQ compared to ForT showing significantly greater turnover during the incubation time period.
Figure 3.

(A) ForT/PyfQ reactions using APA (29) as substrate. (B) HPLC analysis of ForT/PyfQ reactions with APA (29).
The poor activity of ForT on APA indicated that APA may not be the correct substrate for ForT. The observed decarboxylation of APA (29) to APR (30) also implied that a C3 carboxylation reaction is required to convert APR to CAPR (14) or a derivative thereof in the biosynthesis of both formycins and pyrazofurin (see Figure 3A). However, no genes in either cluster were annotated to encode a carboxylase. Furthermore, neither PurE nor PurK, which catalyze an analogous carboxylation reaction in de novo purine biosynthesis, [42,43] led to CAPR (14) formation when incubated with APR (30) (see Figure S4, S5 in the Supporting Information). Moreover, prior labeling experiments have indicated that both the C6 of formycin A and the amide carbon of pyrazofurin originate from C1 of glutamate rather than bicarbonate. [44] Taken together, APA (29) is unlikely to be a biosynthetic intermediate in the formycin and pyrazofurin pathways.
If ForT and PyfQ are indeed the C-glycosidases of formycin and pyrazofurin biosynthesis, then the carboxylate group at C3 carbon of their pyrazole substrates should be retained. Therefore, 4-amino-1H-pyrazole-3,5-dicarboxylic acid (APDA, 31) and 4-hydroxy-1H-pyrazole-3,5-dicarboxylic acid (HPDA, 32) were prepared (see Supporting Information) and tested for activity with ForT and PyfQ (see Figure 4A). Indeed, incubation of APDA (31) with PRPP (24) in the presence of either ForT or PyfQ led to quantitative conversion of APDA to CAPR (14) within 1 h (see Figure 4D). Similarly, PyfQ catalyzes complete conversion of HPDA (32) and PRPP (24) to CHPR (19) within 1 h of incubation (see Figure 4E); however, 32 was not found to be a substrate for ForT. These results strongly support correct assignment of the clusters and indicate that ForT exhibits greater substrate specificity compared to PyfQ. Moreover, no reaction was observed with either ForT or PyfQ when 1H-pyrazole-3,5-dicarboxylic acid (PDA, 33) was supplied as the substrate (Figure 4C and Figure S6). This implies that an electron donating group is required to increase the nucleophilicity of the heterocycle and is consistent with a C-glycosidation mechanism that involves EAS.
Figure 4.

ForT/PyfQ reactions with (A) APDA (31); (B) with HPDA (32); (C) with PDA (33). (D) HPLC analysis of ForT/PyfQ reactions with APDA (31). (E) HPLC analysis of ForT/PyfQ reactions with HPDA (32).
To further investigate the pyf gene cluster, the CHPR (19) product of the PyfQ-catalyzed reaction with HPDA (32) was investigated as a substrate for the pyfP-encoded gene product. PyfP is annotated as a phosphoribosylaminoimidazole-succino-carboxamide (SAICAR) synthetase and thus predicted to catalyze amidation of 19 with aspartate (see Figure 5A). When purified PyfP was incubated with 19, L-Asp, and ATP, a single product was produced (see Figure 5B, trace 3) and displayed a mass consistent with succinyl-hydroxypyrazole carboxamide ribonucleotide (SHPCAR, 34) (calcd m/z for C13H17N3O13P [M - H]− 454.0499; found 454.0529). Furthermore, addition of purified PyfT, a putative adenylsuccinate lyase, to PyfP previously incubated with 19, l-Asp and ATP, led to consumption of 34 and formation a new compound (Figure 5B, trace 4) having the expected mass 338.0384 of pyrazofurin 5’-phosphate (23, calcd m/z for C9H13N3O9P [M - H]− 338.0389). Confirmation of this assignment was obtained when the PyfQ/PyfP/PyfT product from 32 was isolated, treated with calf intestinal phosphatase and found to coelute with a standard of pyrazofurin (6) on HPLC (see Figure S7 in the Supporting Information). It was also found that ForC and PurC, which are homologues of PyfP in the formycin and de novo purine pathways, also catalyzed the conversion of 19 to 34 (see Figure S8 in the Supporting Information).
Figure 5.

(A) Reactions of CHPR (19) with PyfP and PyfT. (B) HPLC analysis of the PyfP and PyfT reactions.
In summary, PyfQPT encoded in the pyf cluster [45] are able to catalyze the conversion of HPDA (32), PRPP (24), and l-aspartate to pyrazofurin 5′-phosphate (23). Similarly, ForT catalyzes the coupling of APDA (31) and PRPP (24) to produce CAPR (14), which is subsequently converted to formycin A 5′-phosphate (18) in reactions catalyzed by ForABCH and PurH. [33,34] In particular, PyfQ as well as ForT are C-glycoside synthases [46] and are likely characterized by catalytic cycles that involve EAS reactions. These observations indicate that the for and pyf clusters are indeed responsible for formycin and pyrazofurin biosynthesis and thus establish the latter stages of both biosynthetic pathways. Nevertheless, the early stages of these pathways involving the assembly of the pyrazole rings in APDA (31) and HPDA (32) remain to be determined and may involve a hydrazine-generating process (see Scheme S3 in Supporting Information) analogous to that found in the s56-p1 biosynthetic pathway. [35]
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health (GM035906, GM040541) and the Welch Foundation (F-1511).
Footnotes
Experimental Section
Experimental procedures, genome sequencing and analysis, chemical syntheses of enzymatic substrates, and supporting figures and tables are available in Supporting Information. The genome sequence reported in this paper has been deposited into the GenBank database under the accession number MN305320.
References
- [1].Suhadolnik RJ “Nucleoside Antibiotics,” Wiley-Interscience, New York, N.Y., 1970. [Google Scholar]
- [2].Štambasky J, Hocek M, Kočovský P, Chem. Rev 2009, 109, 6729–6764. [DOI] [PubMed] [Google Scholar]
- [3].De Clercq E, J. Med. Chem 2016, 59, 2301–2311. [DOI] [PubMed] [Google Scholar]
- [4].Charette M, Gray MW, IUBMB Life, 2000, 49, 341–351. [DOI] [PubMed] [Google Scholar]
- [5].Li W, Csukai M, Corran A, Crowley P, Solomon PS, Oliver RP, Pest. Manag. Sci 2008, 64, 1294–1302. [DOI] [PubMed] [Google Scholar]
- [6].Hong H, Samborskyy M, Zhou Y, Leadley PF, Cell Chem. Biol 2019, 26, 493–501. [DOI] [PubMed] [Google Scholar]
- [7].Maffioli SI, Zhang Y, Degen D, Carzaniga T, Del Gatto G, Serina S, Monciardini P, Mazzetti C, Guglierame P, Candiani G, Chiriac AI, Facchetti G, Kaltofen P, Sahl HG, Dehò G, Donadio S, Ebright RH, Cell, 2017, 169, 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sosio M, Gaspari E, Iorio M, Pessina S, Medema MH, Bernasconi A, Simone M, Maffioli SI, Ebright RH, Donadio S, Cell Chem. Biol 2018, 25, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kusakabe Y, Nagatsu J, Shibuya M, Kawaguchi O, Hirose C, J. Antibiot 1972, 25, 44–47. [DOI] [PubMed] [Google Scholar]
- [10].Isono K, Suhadolnik RJ, Ann. N. Y. Acad. Sci 1975, 255, 390–401. [DOI] [PubMed] [Google Scholar]
- [11].Elstner EF, Suhadolnik RJ, Biochemistry 1971, 10, 3608–3614. [DOI] [PubMed] [Google Scholar]
- [12].Elstner EF, Suhadolnik RJ,. Biochemistry 1972, 11, 2578–2584. [DOI] [PubMed] [Google Scholar]
- [13].Buchanan JG, Hamblin MR, Kumar A, Wightman RH, J. C. S. Chem. Commun 1984, 1515–1517. [Google Scholar]
- [14].Palmu K, Rosenqvist P, Thapa K, Ilina Y; Siitonen V, Baral B, Mäkine J, Belogurov G, Virta P, Niemi J;,Metsä-Ketelä M, ACS Chem. Biol 2017, 12, 1472–1477. [DOI] [PubMed] [Google Scholar]
- [15].Gutowski GE, Sweeney MJ, DeLong DC, Hamill RL, Gerzon K, Dyke RW, Ann. N. Y. Acad. Sci 1975, 255, 544–551. [DOI] [PubMed] [Google Scholar]
- [16].Hori M, Ito E, Takita T, Koyama G, Takeuchi T, Umezawa H A New Antibiotic, Formycin. J. Antibiotics. Ser. A 1964, 17, 96–99. [PubMed] [Google Scholar]
- [17].Ishizuka M, Sawa T, Hori S, Takayama H, Takeuchi T, Umezawa H, J. Antibiot 1968, 21, 5–12. [DOI] [PubMed] [Google Scholar]
- [18].Buchanan JG in Fortschritte der Chemie organischer Naturstoffe / Progress in the Chemistry of Organic Natural Products, Vol. 44 Spring0er, Vienna, 1983, pp. 243–299 [DOI] [PubMed] [Google Scholar]
- [19].Bzowska A, Kulikowska E, Shugar D, Biochim. Biophys. Act 1992, 1120, 239–247. [DOI] [PubMed] [Google Scholar]
- [20].Kierdaszuk B, Modrak-Wójcik A, Wierzchowski J, Shugar D, Biochim. Biophys. Act 2000, 1476, 109–128. [DOI] [PubMed] [Google Scholar]
- [21].Long MC, Parker WB, Biochem. Pharmacol 2006, 71, 1671–1682. [DOI] [PubMed] [Google Scholar]
- [22].Takeuchi T, Iwanaga J, Aoyagi T, Umezawa H, Antibiot J., Ser. A 1966, 19, 286–287. [PubMed] [Google Scholar]
- [23].Dapp MJ, Bonnac L, Patterson SE, Mansky LM, J. Virol 2014, 88, 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sweeney MJ, Davis FA, Gutowski GE, Hamill RL, Hoffman DH, Poore GA, Cancer Res 1973, 33, 2619–2623. [Google Scholar]
- [25].Plagemann PG, Behrens M, Cancer Res 1976, 36, 3807–3812. [PubMed] [Google Scholar]
- [26].Dix DE, Lehman CP, Jakubowski A, Moyer JD, Handschumacher RE, Cancer Res, 1979, 39, 4485–4490. [PubMed] [Google Scholar]
- [27].Komatsu Y, Tanaka K, Agri. Biol. Chem 1968, 32, 1021–1027. [Google Scholar]
- [28].Yang Y, Yu B, Chem. Rev 2017, 117, 12281–12356. [DOI] [PubMed] [Google Scholar]
- [29].Hamma T; Ferré-D’Amaré AR, Chem. Biol 2006, 13, 1125–1135. [DOI] [PubMed] [Google Scholar]
- [30].Huang L; Pookanjanatavip M; Gu X; Santi DV, Biochemistry, 1998, 37, 334–351. [DOI] [PubMed] [Google Scholar]
- [31].Gu X; Liu Y; Santi DV, Proc. Natl. Acad. Sci. U. S. A 1999, 96, 14270–14275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Veerareddygari GR, Singh SK, Mueller EG, J. Am. Chem. Soc 2016, 138, 7852–7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang S-A; Ko Y; Zeng J; Geng Y; Ren D; Ogasawara Y; Irani S; Zhang Y; Liu H.-w., J. Am. Chem. Soc 2019, 141, 6127–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ko Y; Wang S-A; Ogasawara Y; Ruszczycky MW, Liu H.-w., Org. Lett 2017, 19, 1426–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Matsuda K, Tomita T, Shin-ya K, Wakimoto T, Kuzuyama T, Nishiyama M, J. Am. Chem. Soc 2018, 140, 9083–9086. [DOI] [PubMed] [Google Scholar]
- [36].Wu P, Wan D, Xu G, Wang G, Ma H, Wang T, Gao Y, Qi J, Chen X, Zhu J, Li Y-Q, Deng Z, Chen W, Cell Chem. Biol 2017, 24, 171–181. [DOI] [PubMed] [Google Scholar]
- [37].Sawa T, Fukagawa Y, Homma I, Takeuchi T, Umezawa H, J. Antibiot. Ser. A 1967, 20, 227–231. [PubMed] [Google Scholar]
- [38].Rasche ME, White RH, Biochemistry 1998, 37, 11343–11351 [DOI] [PubMed] [Google Scholar]
- [39].Dumitru RV, Ragsdale SW, J. Biol. Chem 2004, 279, 39389–39395. [DOI] [PubMed] [Google Scholar]
- [40].White RH, Biochemistry 2011, 50, 6041–6052. [DOI] [PubMed] [Google Scholar]
- [41].Bechard ME, Farahani P, Greene D, Pham A, Orry A, Rasche ME, AIMS Microbiol. 2019, 5, 186–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mueller EJ, Meyer E, Rudolph J, Davisson VJ, Stubbe J, Biochemistry 1994, 33, 2269–2278. [DOI] [PubMed] [Google Scholar]
- [43].Meyer E, Kappock TJ, Osuji C, Stubbe J, Biochemistry 1999, 38, 3012–3018. [DOI] [PubMed] [Google Scholar]
- [44].Buchanan JG, Hamblin MR, Sood GR, Wightman RH, J. Chem. Soc. Chem. Commun 1980, 917–918. [Google Scholar]
- [45].During peer-review of this manuscript, identification of the pyrazofurin biosynthetic gene cluster was also reported by Du and coworkers(; Zhao G, Yao S, Rothchild KW, Liu T, Liu Y, Lian J, He H-Y, Ryan KS, Du Y-L, The biosynthetic gene cluster of the C-nucleoside antibiotic pyrazomycin with a rare pyrazole moiety, ChemBioChem 2019, Just accepted). [DOI] [PubMed] [Google Scholar]
- [46].The C-glycosidase function of PyfQ and ForT in the biosynthesis of pyrazofurin and formycin, respectively, were recently described in a bioRxiv preprint:; Zhang M, Zhang P, Xu G, Zhou W, Gao Y, Gong R, Cai Y-S, Cong H, Deng Z, Price NPJ, Mao X, Chen W, 2019, bioRxiv 10.1101/728154 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.