

Abstract
Background
Non-small cell lung cancer (NSCLC) is the main histologic form of lung cancer that affects human health, but biomarkers for therapeutic diagnosis and prognosis of the disease are currently lacking.
Material/Methods
The gene expression profile GSE18842 was downloaded from the Gene Expression Omnibus database in this prospective study, which consisted of 46 tumors and 45 controls. After screening differentially expressed genes (DEGs), we conducted functional enrichment analysis and KEGG analysis with upregulated differentially expressed genes (uDEGs) and downregulated differentially expressed genes (dDEGs), respectively. Protein–protein interaction (PPI) networks among DEGs and corresponding coding protein complexes, constructed using the STRING database, were analyzed using Cytoscape. Kaplan-Meier method was used to verify survival associated with hub genes. The GEPIA webserver was used to plot the gene expression level heat map of hub genes between NSCLC and adjacent lung tissues in the TCGA database.
Results
We identified 368 DEGs (168 uDEGs and 200 dDEGs) in NSCLC samples relative to control samples after gene integration. We established a PPI network for the DEGs, which had 249 nodes and 1472 edges protein pairs. Ten undefined hub genes with the highest connectivity degree (CDK1, UBE2C, AURKA, CCNA2, CDC20, CCNB1, TOP2A, ASPM, MAD2L1, and KIF11) were verified by survival analysis, and 9 of them were associated with poorer overall survival in NSCLC. The expression reliability of hub genes was verified by use of the GEPIA web tool.
Conclusions
The results suggested that UBE2C, AURKA, CCNA2, CDC20, CCNB1, TOP2A, ASPM, MAD2L1, and KIF11 are inherent key biomarkers for diagnosis and prognosis, while KEGG analysis results showed the mitotic cell cycle pathway is a probable signaling pathway contributing to NSCLC progression. These genes could be promising biomarkers for diagnosis and provide a new approach for developing targeted therapeutic NSCLC drugs.
MeSH Keywords: Biological Markers; Carcinoma, Non-Small-Cell Lung; Gene Expression Profiling; Transcriptome
Background
Lung cancer is the deadliest malignant tumor in both developing and developed countries, with less than 20% 5-year survival rate; most patients are diagnosed at a point at which surgery is not feasible [1]. Lung cancer is generally divided into non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) according to WHO criteria for lung tumors classification and diagnosis, and NSCLC is the main histological form of lung cancer [2]. At present, many patients are diagnosed at an advanced stage, when they are no longer suitable for surgical treatment and can only receive radiotherapy, chemotherapy, or targeted therapy; therefore, the survival of most patients with advanced lung cancer is short and quality of life is poor. Surgery is the ideal choice for early NSCLC patients, but there is high risk of metastasis or recurrence [3]. Nonetheless, it has been reported that survival benefits have been achieved in patients with NSCLC by using small-molecule tyrosine kinase inhibitors and immunotherapy [4]. With accumulating research on NSCLC treatment, we have deepened our understanding of the biology of the disease and the mechanism of tumor progression and promoted early detection and multimodal therapy [5]. The overall cure rate and survival rate of NSCLC remain low, especially in underperforming and elderly patients, as these groups require special treatment and there are still no established standards for new targeted treatment [6]. It is therefore important to explore possible targets and targeted therapeutic drugs to expand the range of clinical benefits and improve the prognosis of patients with NSCLC.
With the continuous advancement of high-throughput sequencing technology and calculation methods, especially RNA sequencing, hundreds of thousands of RNA-seq have been identified. Growing evidence shows that the expression profile in tumor tissues is different from that in adjacent non-tumor tissues in many types of malignant tumors [7]. We rationally presume that differentially expressed genes (DEGs) can affect the promotion of a variety of diseases, including malignant tumors. Some RNA-seq are insensitive to ribonuclease due to its unique structure, but different genes can exist in tissues and serum, making it a biomarker of cancer. Lung cancer is a molecularly heterogeneous disease, and understanding its biological characteristics is important in improving clinical treatment outcomes.
In the present study, we downloaded the gene expression profile GSE18842 from the Gene Expression Omnibus database and conducted a bioinformatics analysis to study the differentially expressed genes (DEGs) between non-small cell carcinoma tumor tissues and normal lung tissues. We performed function and pathways analyses, as well as protein–protein interaction (PPI) network analysis, and overall survival associated with hub genes was also assessed with the Kaplan-Meier method. The expression reliability of hub genes was verified after visualization in the TCGA database using the GEPIA web tool. We attempted to identify the biomarkers as diagnostic and prognostic indicators, or as potential targets for precision biotherapy and find pathways involved in the progression of NSCLC and to reveal the underlying molecular mechanisms.
Material and Methods
Microarray data
The gene expression profile GSE18842 based on GPL570 (HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0 Array was downloaded from the NCBI GEO database (http://www.ncbi.nlm.nih.gov/geo/, National Center for Biotechnology Information, Gene Expression Omnibus), a public depository database of gene expression data [8]. GSE18842 consisted of 91 NSCLC samples to establish the gene markers of primary adenocarcinoma and squamous cell carcinoma, to determine the differentially expressed genes at different stages of the disease, and to determine the sequences that are of biological significance to the progression of the tumor. Sanchez-Palencia et al. deposited GSE18842 [9]; their study was performed according to the protocol approved by the Ethics Committee of the University of Granada School of Medicine.
Data preprocessing and identification of differentially expressed genes by GEO2R
GEO2R (www.ncbi.nlm.nih.gov/geo/geo2r/), a web tool performing comparisons based on limma and GEOquery R packages of the Bioconductor project, was used to identify the DEGs in postoperative NSCLC samples and normal lung tissues samples. The cutoff criteria were set as P value <0.001 and |log FC| (|log2Fold Change|) >2.8.
Gene ontology (GO) terms and Kyoto encyclopedia of genes and genomes (KEGG) pathway analyses of DEGs
Gene ontology (GO), which provides comprehensive information about the gene function of individual genome products through ontology, is a frequently used bioinformatics tool. The functional enrichment analysis of upregulated differentially expressed genes (uDEGs) and downregulated differentially expressed genes (dDEGs) identified in GSE18842 was performed using the Database for Annotation, Visualization, and Integrated Discovery (https://david.ncifcrf.gov/, DAVID) bioinformatics resources online tool, including molecular functional (MF), biological process (BP), and cellular component (CC) [10]. The cutoff criterion was set as p<0.05.
PPI network construction and analysis of modules
The STRING (http://string-db.org/, the Search Tool for the Retrieval of Interacting Genes) database provides crucial information on the correlation of protein–protein interactions [11]. We used Cytoscape to visualize the PPI network [12]. The PPI network formed by DEGs was analyzed through the STRING database, and then Cytoscape was applied for visualization of the network. CytoHubba, a Cytoscape plug-in, was used to discover the key targets, subnetworks of complex networks, and the central elements in the network. According to the standard of combined score >0.9, the top 10 genes were selected using the 12 topological analysis methods. Subsequently, the molecular complex was obtained by using Molecular Complex Detection (MCODE), a Cytoscape plug-in, to detect the global PPI network module with Cutoff degree=2, Cutoff Node Score=0.2, Haircut=true, Fluff=false, K-Core=2, Max, and Depth from Seed=100. The functional annotation of DEGs in the identified module was investigated with the DAVID bioinformatics resources. P <0.05 was set as the cutoff criterion.
Survival analysis validation of hub genes
The Kaplan-Meier plotter (http://kmplot.com/analysis/index.php?p=service&cancer=lung) for lung cancer was applied to assess the survival rate of more than 50 000 genes in patients with breast, ovarian, lung, and gastric cancer [13]. We investigated whether hub gene was associated with overall survival using Kaplan-Meier method and log-rank test. The criteria we selected were HR with 95% CI and log-rank P value <0.05 as a threshold.
TCGA verification of hub genes
GEPIA (http://gepia.cancer-pku.cn/index.html) is a customizable functionalities website for interactive analysis and visualization based on The Cancer Genome Atlas database [14]. To further verify the 9 hub genes identified from the PPI network, the GEPIA web server was used to plot a gene expression level heat map between lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUCS), and adjacent lung tissues in the TCGA database. The patient data were grouped according to the transcripts per million (TPM) value. Log2 (TPM+1) was used for log-scale, and four-way analysis of variance (ANOVA) was applied.
Results
Differentially expressed genes (DEGs) in NSCLC
Though filtering, analyzing, and sorting out the raw data by using GEO2R, 368 DEGs were extracted from GSE18842 as unique genes in NSCLC samples compared with control samples. The volcano plot of DEGs (Figure 1) consisted of 168 uDEGs and 200 dDEGs in NSCLC tissues compared with normal lung tissues.
Figure 1.
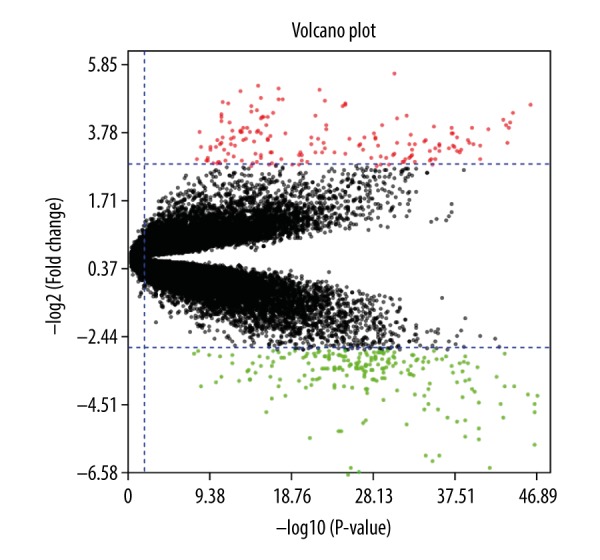
The volcano plot showing upregulated differentially expressed genes (uDEGs) and downregulated differentially expressed genes (dDEGs). The horizontal axis shows log10 (p value), the vertical axis indicates log2 (FC), red dots represent uDEGs, and green dots represent dDEGs.
Functional and pathway terms enrichment analysis
DAVID was utilized for gene ontology and Kyoto encyclopedia of genes and genomes analysis to investigate the functional and biological pathways of 168 uDEGs and 200 dDEGs. GO analysis showed that in the Biological process category, the uDEGs were considerably associated with keratinocyte differentiation, while the dDEGs were mainly involved in cell adhesion. Furthermore, CC analysis indicated that most of the uDEGs were located in the cytoplasm, and the dDEGs were mainly distributed in the plasma membrane. Additionally, according to the results of MF analysis, the uDEGs were significantly associated with structural molecule activity, while the dDEGs were associated with heparin binding (Table 1). Also, KEGG pathway analysis illustrated that the majority the uDEGs were involved in the Cell cycle, while the dDEGs were mainly involved in Malaria (Table 2).
Table 1.
GO function annotation of uDEGs and dDEGs associated with NSCLC (TOP 5).
| Category | Term | Involved in | Count | % | PValue |
|---|---|---|---|---|---|
| uDEGs | |||||
| GOTERM_BP_DIRECT | GO: 0030216 | Keratinocyte differentiation | 6 | 0.034227039 | 1.02E-04 |
| GOTERM_BP_DIRECT | GO: 0043066 | Negative regulation of apoptotic process | 6 | 0.034227039 | 0.029216325 |
| GOTERM_BP_DIRECT | GO: 0000281 | Mitotic cytokinesis | 5 | 0.028522533 | 3.52E-05 |
| GOTERM_BP_DIRECT | GO: 0018149 | Peptide cross-linking | 5 | 0.028522533 | 2.75E-04 |
| GOTERM_BP_DIRECT | GO: 0007267 | Cell-cell signaling | 5 | 0.028522533 | 0.001218331 |
| GOTERM_CC_DIRECT | GO: 0005737 | Cytoplasm | 38 | 0.216771249 | 1.22E-05 |
| GOTERM_CC_DIRECT | GO: 0005634 | Nucleus | 30 | 0.171135197 | 0.00966858 |
| GOTERM_CC_DIRECT | GO: 0016020 | Membrane | 15 | 0.085567598 | 0.002997772 |
| GOTERM_CC_DIRECT | GO: 0030496 | Midbody | 9 | 0.051340559 | 2.37E-07 |
| GOTERM_CC_DIRECT | GO: 0001533 | Cornified envelope | 5 | 0.028522533 | 9.17E-05 |
| GOTERM_MF_DIRECT | GO: 0005198 | Structural molecule activity | 11 | 0.062749572 | 4.91E-07 |
| GOTERM_MF_DIRECT | GO: 0005509 | Calcium ion binding | 13 | 0.074158585 | 0.001208322 |
| GOTERM_MF_DIRECT | GO: 0005524 | ATP binding | 18 | 0.102681118 | 0.005172406 |
| GOTERM_MF_DIRECT | GO: 0003777 | Microtubule motor activity | 4 | 0.022818026 | 0.006725855 |
| GOTERM_MF_DIRECT | GO: 0008201 | Heparin binding | 4 | 0.022818026 | 0.030940109 |
| dDEGs | |||||
| GOTERM_BP_DIRECT | GO: 0007155 | Cell adhesion | 10 | 7.633587786 | 0.00455356 |
| GOTERM_BP_DIRECT | GO: 0006898 | Receptor-mediated endocytosis | 9 | 6.870229008 | 4.47E-05 |
| GOTERM_BP_DIRECT | GO: 0006954 | Inflammatory response | 8 | 6.106870229 | 0.016151235 |
| GOTERM_BP_DIRECT | GO: 0001525 | Angiogenesis | 6 | 4.580152672 | 0.019308307 |
| GOTERM_BP_DIRECT | GO: 0007166 | Cell surface receptor signaling pathway | 6 | 4.580152672 | 0.041524321 |
| GOTERM_CC_DIRECT | GO: 0005886 | Plasma membrane | 45 | 34.35114504 | 0.001297306 |
| GOTERM_CC_DIRECT | GO: 0070062 | Extracellular exosome | 35 | 26.71755725 | 6.84E-04 |
| GOTERM_CC_DIRECT | GO: 0005576 | Extracellular region | 31 | 23.66412214 | 4.61E-07 |
| GOTERM_CC_DIRECT | GO: 0005615 | Extracellular space | 30 | 22.90076336 | 3.52E-08 |
| GOTERM_CC_DIRECT | GO: 0005887 | Integral component of plasma membrane | 23 | 17.55725191 | 2.95E-04 |
| GOTERM_MF_DIRECT | GO: 0008201 | Heparin binding | 6 | 4.580152672 | 0.003804314 |
| GOTERM_MF_DIRECT | GO: 0030246 | Carbohydrate binding | 5 | 3.816793893 | 0.038241324 |
| GOTERM_MF_DIRECT | GO: 0004888 | Transmembrane signaling receptor activity | 5 | 3.816793893 | 0.04997596 |
| GOTERM_MF_DIRECT | GO: 0044325 | Ion channel binding | 4 | 3.053435115 | 0.036771904 |
| GOTERM_MF_DIRECT | GO: 0005044 | Scavenger receptor activity | 3 | 2.290076336 | 0.038434779 |
Top 5 terms were selected depending on count and P-value. BP – biological process; CC – cellular component; GO – gene ontology; MF – molecular function.
Table 2.
KEGG pathway analysis of differentially expressed genes associated with NSCLC..
| Category | Term | Involved in | Count | % | P value |
|---|---|---|---|---|---|
| uDEGs | |||||
| KEGG_PATHWAY | hsa04110 | Cell cycle | 10 | 0.057045 | 5.05E-08 |
| KEGG_PATHWAY | hsa04114 | Oocyte meiosis | 8 | 0.045636 | 4.70E-06 |
| KEGG_PATHWAY | hsa04914 | Progesterone-mediated oocyte maturation | 6 | 0.034227 | 1.90E-04 |
| KEGG_PATHWAY | hsa04115 | p53 signaling pathway | 5 | 0.028523 | 7.62E-04 |
| KEGG_PATHWAY | hsa04512 | ECM-receptor interaction | 4 | 0.022818 | 0.016755 |
| dDEGs | |||||
| KEGG_PATHWAY | hsa05144 | Malaria | 5 | 3.816794 | 8.27E-04 |
| KEGG_PATHWAY | hsa03320 | PPAR signaling pathway | 5 | 3.816794 | 0.002661 |
| KEGG_PATHWAY | hsa04610 | Complement and coagulation cascades | 4 | 3.053435 | 0.022 |
| KEGG_PATHWAY | hsa05410 | Hypertrophic cardiomyopathy (HCM) | 4 | 3.053435 | 0.030239 |
| KEGG_PATHWAY | hsa05143 | African trypanosomiasis | 3 | 2.290076 | 0.033223 |
Top 5 terms were selected depending on count and P-value. KEGG – Kyoto Encyclopedia of Genes and Genomes.
Protein–protein interaction (PPI) network construction and module analysis
The identified DEGs PPI network (Figure 2) consists of 249 nodes and 1472 edges, including 168 uDEGs and 200 dDEGs. The top 10 highest-scoring nodes, including CDK1, UBE2C, AURKA, CCNA2, CDC20, CCNB1, TOP2A, ASPM, MAD2L1, KIF11 (Table 3), were selected from 12 algorithms in descending order according to the value of degree using the Cytohubba plug-in. Meanwhile, a significant module with 44 nodes and 913 edges, cluster score=42.465, was generated from the protein–protein interaction network calculated by the MCODE plug-in (Figure 3A). KEGG enrichment analysis demonstrated that the DEGs in the identified module were substantially associated with Cell cycle and Mitotic (Figure 3B). Other than the 10 genes mentioned above, the other nodes in the module were NUF2, CEP55, KIF4A, UHRF1, TPX2, KIF20A, UBE2T, PBK, TK1, FAM83D, ECT2, FOXM1, TRIP13, DLGAP5, KIAA0101, NUSAP1, ZWINT, CCNB2, PRC1, CDKN3, CENPF, BUB1, KIF2C, BUB1B, TTK, MELK, NEK2, CDCA7, GINS1, MCM2, ANLN, NDC80, BIRC5, and RRM2 (Figure 3A). All genes in the module were upregulated.
Figure 2.

PPI network construction for identified DEGs. Using the Search Tool for the Retrieval of Interacting Genes (STRING) online database, 368 differentially expressed genes (DEGs) were filtered into a PPI network complex. Yellow highlighted nodes represent the DEGs of degree>30 and the black line represents interaction among nodes.
Table 3.
Top 10 in network ranked by degree method.
| Rank | Name | Score |
|---|---|---|
| 1 | CDK1 | 49 |
| 2 | UBE2C | 48 |
| 3 | AURKA | 47 |
| 3 | CCNA2 | 47 |
| 5 | CDC20 | 46 |
| 5 | CCNB1 | 46 |
| 5 | TOP2A | 46 |
| 5 | ASPM | 46 |
| 9 | MAD2L1 | 45 |
| 9 | KIF11 | 45 |
Figure 3.

(A) The significant module generated from the PPI network; all differentially expressed genes in this module were upregulated. (B) Top 5 KEGG pathway terms enriched individually. KEGG pathway terms separated according to DEGs in this module. The horizontal axis represents the enriched KEGG pathway terms and the vertical axis represents the percentage of DEGs in this module.
Survival analysis of Hub genes
Eventually, the overall survival of 9 verified hub genes (Figure 4) was obtained by using the Kaplan-Meier plotter tool. The overall survival results demonstrated that overexpressed UBE2C [HR=1.77 (1.55–2.01), log-rank P=1e-16] was related to unsatisfactory overall survival for NSCLC patients, as were AURKA [HR=1.52 (1.33–1.72), log-rank P=1.2e-10]; CCNA2 [HR=1.57 (1.39–1.79), log-rank P=2.2×10e-12]; CDC20 [HR=1.82 (1.6–2.07), log-rank P=1e-16]; CCNB1 [HR=1. 63(1.38–1.92), log-rank P=7.3e-09]; TOP2A [HR=1.65 (1.45–1.87), log-rank P=1.9e-14]; ASPM [HR=1.76 (1.55–2.01), log-rank P=1e-16]; MAD2L1 [HR=1.55 (1.37–1.77), log-rank P=1.3e-11], and KIF11 [HR=1.52 (1.34–1.73), log-rank P=9e-11].
Figure 4.

Overall survival of 9 key genes in patients with NSCLC was evaluated by Kaplan-Meier curve with high and low expression of UBE2C (A), AURKA (B), CCNA2 (C), CDC20 (D), CCNB1 (E), TOP2A (F), ASPM (G), MAD2L1 (H), and KIF11 (I). The log-rank test was used to evaluate difference between the 2 curves.
Hub genes verified using GEPIA
To determine the reliability of DEGs identified from GSE18842, GEPIA was employed to evaluate the expression level of hub genes in the TCGA database in LUAD, LUCS, and normal lung tissues. Consistent with bioinformatics analysis results of GEO profiling, the expression level of each of the 9 genes identified in NSCLC tissues was significantly higher than that in normal tissues (Figure 5).
Figure 5.
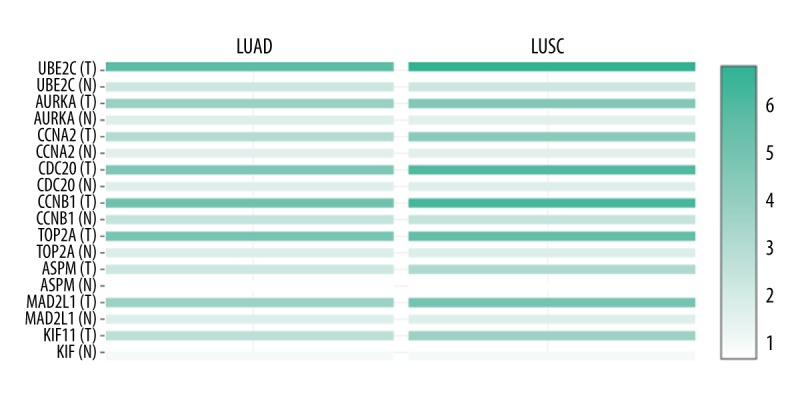
Heat map showing the expression level of 9 key genes (UBE2C, AURKA, CCNA2, CDC20, CCNB1, TOP2A, ASPM, MAD2L1, and KIF11) in LUAD, LUCS, and normal lung tissue based on TCGA database analyzed by GEPIA web server. The T represents LUAD or LUCS tumor tissues and the N represents normal lung tissue. LUAD – lung adenocarcinoma; LUCS – lung squamous cell carcinoma; TCGA – The Cancer Genome Atlas; GEIPA – gene expression profiling interactive analysis.
Discussion
Due to the high malignant degree of NSCLC and the low annual survival rate of patients, the exploration of effectual treatment has become the focus of attention in recent years. Although many unrealized pathogenic factors of NSCLC have been investigated, there are still many uncertainties regarding pathogenesis. A comprehensive understanding of acknowledged biomarkers and intrinsic molecular mechanism of NSCLC is elemental to diagnosis and therapy. In the present study, bioinformatics methods, especially gene expression analysis, were applied to reveal the possible dysregulated genes and pathways of NSCLC. A total of 368 DEGs were screened, including 205 uDEGs and 200 dDEGs. GO and KEGG pathway analyses of uDEGs and dDEGs were separately enriched. GO analysis revealed that the uDEGs were commonly involved in keratinocyte differentiation, and the dDEGs were mainly involved in cell adhesion. The uDEGs were substantially involved in Cell cycle, Oocyte meiosis, Progesterone-mediated oocyte maturation, and p53 signaling pathway, ECM-receptor interaction, and dDEGs were chiefly associated with Malaria, PPAR signaling pathway, Complement and coagulation cascades, Hypertrophic cardiomyopathy (HCM), and African trypanosomiasis. DEGs functional enrichment analysis provides major signaling pathways in the occurrence and development of NSCLC. Although it is unclear which is the pivotal culprit in the course of aggravation of the disease, these signaling pathways are closely associated with NSCLC [15–18]. After the PPI network was constructed, a critical network module was identified. The top 10 hub genes and 1 significant module extracted from the PPI network are all upregulated. In addition, survival analysis of hub genes demonstrated that 9 of these genes were markedly associated with the overall survival of patients with NSCLC. The module was sorted by KEGG analysis to be involved in Cell cycle and Mitotic.
UBE2C (Ubiquitin Conjugating Enzyme E2C) is an activated proto-oncogene in lung cancer, and its abnormal activation is associated with poor prognosis. UBE2C selectively inhibits autophagy in NSCLC, and the interruption of UBE2C-mediated autophagy inhibition can weaken the cell proliferation and invasive growth of NSCLC [19]. Guo J et al. reported that UBE2C was highly expressed in cisplatin-resistant NSCLC cells, which is involved in the induction of proliferation and invasion of cisplatin-resistant NSCLC cells [20]. UBE2C promotes the progression and metastasis of NSCLC by affecting the cell cycle and inhibiting apoptosis [21].
Previous studies of AURKA (Aurora Kinase A) have identified the relationship between the expression of AURKA and the progression of lung cancer. Katsha et al. reported that AURKA contributes to the activity of STAT3 by regulating the expression and phosphorylation of JAK2, and showed the importance of AURKA as a target in the treatment of gastric and esophageal cancer [22]. AURKA limits the ubiquitin degradation of survivin to promote drug resistance in gastric cancer, so the AURKA-Survivin axis can be used as a target to promote the curative effect [23]. Furthermore, Goos et al. revealed that the expression of AURKA in liver metastasis of colorectal cancer was positively correlated with its overexpression in the corresponding primary tumor. The expression of AURKA protein is not linked to the clinic pathological factors of colorectal cancer with liver metastasis, and it is a molecular biomarker with prognostic value [24].
Overexpression of CCNA2 (cyclin A) is associated with low recurrence-free survival in stage I NSCLC CCNA2 (cyclin A) [25]. There was a significant positive correlation between the expression of LINC00968, miR-9-3p, and CCNA2 in lung adenocarcinoma. The LINC00968/miR-9-3p/CCNA2 regulatory axis was a newly discovered regulatory mechanism in lung adenocarcinoma [26]. A previous study has shown that the expression of Cyclins A and B1 in thyroid papillary carcinoma may have specific immunostaining [27].
Cell Division Cycle 20 (CDC20) overexpression can be used as an independent predictor of biochemical recurrence in patients with clinically localized prostate cancer after laparoscopic radical prostatectomy without neoadjuvant therapy [28]. Studies have shown that CDC20 is an independent marker for predicting the clinical prognosis of patients with gastric and colon cancer, and its upregulation is associated with invasive progression and poor prognosis of gastric and colon cancer [29,30].
CCNB1 (cyclin B1) belongs to the highly conservative cyclin family and is significantly overexpressed in various types of cancer. Ding et al. reported that CCNB1 is a biomarker to prevent or even reverse hormone therapy resistance in ER+ breast cancer prognosis. The expression of CCNB1 may help to monitor hormone therapy and guide personalized treatment [31]. In addition, Cyniak-Magierska et al. revealed that the expression of cyclin B1 in papillary thyroid carcinoma may have a specific immunostaining pattern. If the results are confirmed in a larger patient population, the diagnostic panel constructed with antibodies to these proteins can improve diagnostic accuracy in papillary thyroid carcinoma cases [27]. Sabbaghi et al. reported that single-agent trastuzumab emtansine therapy induced by cyclin B1 was used as a pharmacodynamics predictive index for HER2-positive breast cancer [32].
TOP2A (DNA Topoisomerase II Alpha) amplification was associated with the characteristics of biologically invasive epithelial carcinoma of the urinary tract. Overexpression and/or amplification of TOP2A can help identify whether patients will benefit from targeted therapy [33]. de Resende et al. demonstrated that the evaluation of TOP2A protein is of prognostic importance, and because of its relationship with inferior prognosis, the evaluation of TOP2A immunohistochemistry in biopsies can be an meaningful tool for selecting the most appropriate surgical and clinical approaches for prostate cancer patients [34]. In addition, some studies have shown that TOP2A amplification is associated with the neoadjuvant chemotherapeutic sensitivity of anthracyclines, and TOP2A should be included as a predictive indicator in future studies of breast cancer [35].
ASPM (Abnormal Spindle Microtubule Assembly) is necessary for effective non-homologous terminal connections in mammalian cells. It can be used as a new target for combined radiotherapy or a functional biomarker for tumor prognosis [36]. Lin et al. showed that ASPM overexpression is a molecular marker for predicting the enhancement of invasion and metastasis risk of hepatic cell carcinoma; regardless of p53 mutation status and tumor stage, the risk of early tumor recurrence was higher and the prognosis was inferior [37]. Xie et al. indicated that the proportion of highly expressed ASPM cells in tumors was negatively correlated with the recurrence-free survival of prostate cancer patients [38].
MAD2L1 (Mitotic Arrest Deficient 2 Like 1) maintains spindle checkpoint function, and the genetic variation caused by the decrease of spindle checkpoint function due to the weakening of MAD2L1 function increases the susceptibility to lung cancer [39]. Wang et al. reported that reduction of MAD2L1 expression by siRNAs can reduce the growth of breast cancer cell lines MDA-MB-231 and MDA-MB-468, and inhibit cell migration and invasion [40]. Li et al. found that, as a promising therapeutic target and prognostic indicator of hepatocellular carcinoma, miR-200C-5p could inhibit the proliferation, migration, and invasion of hepatocellular carcinoma cells and induce apoptosis and cell cycle arrest by inhibiting MAD2L1 targets [41].
KIF11 (Kinesin Family Member 11) is a latent oncogene; its high expression may be a criterion for tumor invasiveness, and it can be used as a potential prognostic biomarker and therapeutic target in patients with prostate and oral cancer [42,43]. As a prevalent molecular regulator of heterogeneous cell growth and movement in tumors, KIF11 is an attractive therapeutic target for glioblastomas [44].
The diagnostic value and robustness of hub genes for predicting NSCLC was evaluated using the GEPIA web server based on the TCGA database. The function of these hub genes in NSCLC needed to be verified in vitro and in vivo by biological experiments in future research.
Conclusions
We attempted to identify DEGs through bioinformatics and to discover the regulatory mechanism of genes that can be actuated in clinical molecularly pathological diagnosis decision or antineoplastic protocols of NSCLC. However, in-depth research is needed to determine the exact mechanisms by which these genes are involved in NSCLC. We screened 372 DEGs. GO enrichment analysis indicated that in the Biological process (BP) category, the uDEGs were commonly enriched in keratinocyte differentiation, while the dDEGs were mainly involved in cell adhesion. These hub genes, with differential expression verified by GEPIA, significantly affect the survival rate of patients with lung cancer, and related research may improve our understanding of the etiology and pathogenesis, as well as improving diagnosis, treatment, and even prognostic assessment of NSCLC in years to come. We intend to verify the predicted results from bioinformatics analysis in further in vivo or in vitro experimental studies of these genes.
Footnotes
Source of support: This study was supported by the Natural Science Foundation Funding Scheme of Liaoning Province (No. 2019-MS-145), the Program for the Doctoral Scientific Research Foundation of Liaoning Province (No. 2019-BS-094), the Biological Anthropology Innovation Team Project of JZMU (No. JYLJ201702), and the Key Project of the Natural Science Foundation of Liaoning Province (No.20170540374)
References
- 1.Allemani C, Weir HK, Carreira H, et al. Global surveillance of cancer survival 1995-2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2) Lancet. 2015;385:977–1010. doi: 10.1016/S0140-6736(14)62038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–54. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 3.Tomaszek SC, Wigle DA. Surgical management of lung cancer. Semin Respir Crit Care Med. 2011;32:69–77. doi: 10.1055/s-0031-1272871. [DOI] [PubMed] [Google Scholar]
- 4.Caglevic C, Grassi M, Raez L, et al. Nintedanib in non-small cell lung cancer: From preclinical to approval. Ther Adv Respir Dis. 2015;9:164–72. doi: 10.1177/1753465815579608. [DOI] [PubMed] [Google Scholar]
- 5.Osmani L, Askin F, Gabrielson E, Li QK. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): Moving from targeted therapy to immunotherapy. Semin Cancer Biol. 2018;52:103–9. doi: 10.1016/j.semcancer.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: Current therapies and new targeted treatments. Lancet. 2017;389:299–311. doi: 10.1016/S0140-6736(16)30958-8. [DOI] [PubMed] [Google Scholar]
- 7.Prasad NB, Somervell H, Tufano RP, et al. Identification of genes differentially expressed in benign versus malignant thyroid tumors. Clin Cancer Res. 2008;14:3327–37. doi: 10.1158/1078-0432.CCR-07-4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: Archive for functional genomics data sets – update. Nucleic Acids Res. 2013;41:D991–95. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanchez-Palencia A, Gomez-Morales M, Gomez-Capilla JA, et al. Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. Int J Cancer. 2011;129:355–64. doi: 10.1002/ijc.25704. [DOI] [PubMed] [Google Scholar]
- 10.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 11.Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–68. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shannon P, Markiel A, Ozier O, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer. 2012;19:197–208. doi: 10.1530/ERC-11-0329. [DOI] [PubMed] [Google Scholar]
- 14.Tang Z, Li C, Kang B, et al. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu M, Zhang H, Li Y, et al. HOTAIR, a long noncoding RNA, is a marker of abnormal cell cycle regulation in lung cancer. Cancer Sci. 2018;109:2717–33. doi: 10.1111/cas.13745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, Liu J. Erroneous silencing of the mitotic checkpoint by aberrant spindle pole-kinetochore coordination. Biophys J. 2015;109:2418–35. doi: 10.1016/j.bpj.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong G, Chen X, Fang X, et al. Fra-1 is upregulated in lung cancer tissues and inhibits the apoptosis of lung cancer cells by the P53 signaling pathway. Oncol Rep. 2016;35:447–53. doi: 10.3892/or.2015.4395. [DOI] [PubMed] [Google Scholar]
- 18.Li D, Yang W, Zhang Y, et al. Genomic analyses based on pulmonary adenocarcinoma in situ reveal early lung cancer signature. BMC Med Genomics. 2018;11:106. doi: 10.1186/s12920-018-0413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo J, Wu Y, Du J, et al. Deregulation of UBE2C-mediated autophagy repression aggravates NSCLC progression. Oncogenesis. 2018;7:49. doi: 10.1038/s41389-018-0054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo J, Jin D, Wu Y, et al. The miR 495-UBE2C-ABCG2/ERCC1 axis reverses cisplatin resistance by downregulating drug resistance genes in cisplatin-resistant non-small cell lung cancer cells. EBioMedicine. 2018;35:204–21. doi: 10.1016/j.ebiom.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Jin D, Guo J, Wu Y, et al. UBE2C, directly targeted by miR-548e-5p, increases the cellular growth and invasive abilities of cancer cells interacting with the EMT marker protein zinc finger E-box binding Homeobox 1/2 in NSCLC. Theranostics. 2019;9:2036–55. doi: 10.7150/thno.32738. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Katsha A, Arras J, Soutto M, et al. AURKA regulates JAK2-STAT3 activity in human gastric and esophageal cancers. Mol Oncol. 2014;8:1419–28. doi: 10.1016/j.molonc.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamran M, Long ZJ, Xu D, et al. Aurora kinase A regulates Survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis. 2017;6:e298. doi: 10.1038/oncsis.2016.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goos JA, Coupe VM, Diosdado B, et al. Aurora kinase A (AURKA) expression in colorectal cancer liver metastasis is associated with poor prognosis. Br J Cancer. 2013;109:2445–52. doi: 10.1038/bjc.2013.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ko E, Kim Y, Cho EY, et al. Synergistic effect of Bcl-2 and cyclin A2 on adverse recurrence-free survival in stage I non-small cell lung cancer. Ann Surg Oncol. 2013;20:1005–12. doi: 10.1245/s10434-012-2727-2. [DOI] [PubMed] [Google Scholar]
- 26.Li DY, Chen WJ, Shang J, et al. Regulatory interactions between long noncoding RNA LINC00968 and miR-9-3p in non-small cell lung cancer: A bioinformatic analysis based on miRNA microarray, GEO and TCGA. Oncol Lett. 2018;15:9487–97. doi: 10.3892/ol.2018.8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cyniak-Magierska A, Stasiak M, Naze M, et al. Patterns of cyclin A and B1 immunostaining in papillary thyroid carcinoma. Ann Agric Environ Med. 2015;22:741–46. doi: 10.5604/12321966.1185787. [DOI] [PubMed] [Google Scholar]
- 28.Mao Y, Li K, Lu L, et al. Overexpression of Cdc20 in clinically localized prostate cancer: Relation to high Gleason score and biochemical recurrence after laparoscopic radical prostatectomy. Cancer Biomark. 2016;16:351–58. doi: 10.3233/CBM-160573. [DOI] [PubMed] [Google Scholar]
- 29.Wu WJ, Hu KS, Wang DS, et al. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J Transl Med. 2013;11:142. doi: 10.1186/1479-5876-11-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding ZY, Wu HR, Zhang JM, et al. Expression characteristics of CDC20 in gastric cancer and its correlation with poor prognosis. Int J Clin Exp Pathol. 2014;7:722–27. [PMC free article] [PubMed] [Google Scholar]
- 31.Ding K, Li W, Zou Z, et al. CCNB1 is a prognostic biomarker for ER+ breast cancer. Med Hypotheses. 2014;83:359–64. doi: 10.1016/j.mehy.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 32.Sabbaghi M, Gil-Gomez G, Guardia C, et al. Defective Cyclin B1 induction in trastuzumab-emtansine (T-DM1) acquired resistance in HER2-positive breast cancer. Clin Cancer Res. 2017;23:7006–19. doi: 10.1158/1078-0432.CCR-17-0696. [DOI] [PubMed] [Google Scholar]
- 33.Aumayr K, Klatte T, Neudert B, et al. HER2 and TOP2A gene amplification and protein expression in upper tract urothelial carcinomas. Pathol Oncol Res. 2018;24:575–81. doi: 10.1007/s12253-017-0260-0. [DOI] [PubMed] [Google Scholar]
- 34.de Resende MF, Vieira S, Chinen LT, et al. Prognostication of prostate cancer based on TOP2A protein and gene assessment: TOP2A in prostate cancer. J Transl Med. 2013;11:36. doi: 10.1186/1479-5876-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Xu B, Yuan P, et al. TOP2A amplification in breast cancer is a predictive marker of anthracycline-based neoadjuvant chemotherapy efficacy. Breast Cancer Res Treat. 2012;135:531–37. doi: 10.1007/s10549-012-2167-5. [DOI] [PubMed] [Google Scholar]
- 36.Kato TA, Okayasu R, Jeggo PA, Fujimori A. ASPM influences DNA double-strand break repair and represents a potential target for radiotherapy. Int J Radiat Biol. 2011;87:1189–95. doi: 10.3109/09553002.2011.624152. [DOI] [PubMed] [Google Scholar]
- 37.Lin SY, Pan HW, Liu SH, et al. ASPM is a novel marker for vascular invasion, early recurrence, and poor prognosis of hepatocellular carcinoma. Clin Cancer Res. 2008;14:4814–20. doi: 10.1158/1078-0432.CCR-07-5262. [DOI] [PubMed] [Google Scholar]
- 38.Xie JJ, Zhuo YJ, Zheng Y, et al. High expression of ASPM correlates with tumor progression and predicts poor outcome in patients with prostate cancer. Int Urol Nephrol. 2017;49:817–23. doi: 10.1007/s11255-017-1545-7. [DOI] [PubMed] [Google Scholar]
- 39.Guo Y, Zhang X, Yang M, et al. Functional evaluation of missense variations in the human MAD1L1 and MAD2L1 genes and their impact on susceptibility to lung cancer. J Med Genet. 2010;47:616–22. doi: 10.1136/jmg.2009.074252. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z, Katsaros D, Shen Y, et al. Biological and clinical significance of MAD2L1 and BUB1, genes frequently appearing in expression signatures for breast cancer prognosis. PLoS One. 2015;10:e0136246. doi: 10.1371/journal.pone.0136246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Bai W, Zhang J. miR-200c-5p suppresses proliferation and metastasis of human hepatocellular carcinoma (HCC) via suppressing MAD2L1. Biomed Pharmacother. 2017;92:1038–44. doi: 10.1016/j.biopha.2017.05.092. [DOI] [PubMed] [Google Scholar]
- 42.Piao XM, Byun YJ, Jeong P, et al. Kinesin family member 11 mRNA expression predicts prostate cancer aggressiveness. Clin Genitourin Cancer. 2017;15:450–54. doi: 10.1016/j.clgc.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Daigo K, Takano A, Thang PM, et al. Characterization of KIF11 as a novel prognostic biomarker and therapeutic target for oral cancer. Int J Oncol. 2018;52:155–65. doi: 10.3892/ijo.2017.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Venere M, Horbinski C, Crish JF, et al. The mitotic kinesin KIF11 is a driver of invasion, proliferation, and self-renewal in glioblastoma. Sci Transl Med. 2015;7:304ra143. doi: 10.1126/scitranslmed.aac6762. [DOI] [PMC free article] [PubMed] [Google Scholar]