

SUMMARY
Lipid metabolism is frequently perturbed in cancers, but the underlying mechanism is unclear. We present comprehensive evidence that oncogene MYC in collaboration with transcription factor Sterol Regulated Element-Binding Protein (SREBP1) regulates lipogenesis to promote tumorigenesis. We used human and mouse tumor-derived cell-lines, tumor xenografts, and four conditional transgenic mouse models of MYC-induced tumors to show that MYC regulates lipogenesis genes, enzymes, and metabolites. We found that MYC induces SREBP1 and they collaborate to activate fatty acid (FA) synthesis, and drive FA chain elongation from glucose and glutamine. Further, by employing desorption electrospray ionization mass spectrometry imaging (DESI-MSI), we observed in vivo lipidomic changes upon MYC induction across different cancers, for example, global increase in glycerophosphoglycerols. After inhibition of FA synthesis, tumorigenesis was blocked, and tumors regressed in both xenograft and primary transgenic mouse models, revealing the vulnerability of MYC-induced tumors to the inhibition of lipogenesis.
Graphical Abstract
eTOC blurb:
Gouw and Margulis et al. present comprehensive evidence that the oncogene MYC collaborates with the transcription factor SREBP1 in controlling lipogenesis to promote tumorigenesis. Utilizing multiple MYC-induced tumor models, they both identify key lipogenesis genes, enzymes and metabolites affected by MYC, and expose the vulnerability of MYC-cancers to lipogenesis inhibition.
INTRODUCTION
The MYC oncogene is often activated and/or overexpressed in cancers (Baudino and Cleveland, 2001; Cawley et al., 2004; Dang, 2012; Fernandez et al., 2003; Kalkat et al., 2017; Kolk et al., 2009; Wang et al., 1999). The MYC transcription factor dimerizes with MAX, binds to DNA, amplifies gene expression genome-wide, and regulates multiple cellular programs, including proliferation, metabolism, self-renewal, angiogenesis, and immune evasion (Blackwood et al., 1992; Casey et al., 2018; Dang et al., 2006; Grieb et al., 2016; McMahon, 2014). Experimentally, MYC inactivation regresses tumors (Casey et al., 2018; Gabay et al., 2014; Shroff et al., 2015); however, therapies that target MYC directly have yet to be accomplished.
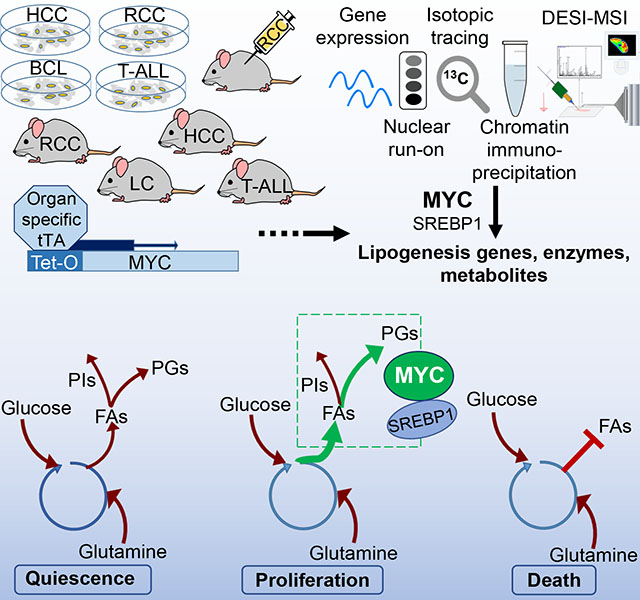
MYC has been shown to generally regulate glycolysis and glutaminolysis, which are required for energy production and macromolecular synthesis (Dang, 2012; Wahlstrom and Henriksson, 2015; Wang et al., 2011; Wise et al., 2008). MYC-stimulated growing cells need building blocks for new membranes, which can be assembled either from exogenous lipids or via endogenous de novo lipogenesis. Whether lipogenesis is regulated solely by MYC or collaboratively with other factors is not understood. Prior studies indicate that MYC may stimulate lipogenesis by enhancing incorporation of glucose and glutamine carbons into the backbones of fatty acids (FAs) through either forward or reverse tricarboxylic acid (TCA) cycling (Le et al., 2012; Metallo et al., 2012). Specifically, MYC regulates the conversion of glucose to acetyl-CoA and then to palmitate FA(16:0) (Morrish et al., 2010). In previous studies, we found that MYC overexpression correlates with changes in glycerophospholipids (GPs), but a direct mechanism was not established (Eberlin et al., 2014; Perry et al., 2013). Based on these results, we hypothesized that MYC may directly regulate lipogenesis, which is required for tumorigenesis. To test this, we used transgenic mouse models and human tumor-derived cell lines to examine three key pathways in lipogenesis: FA, cholesterol, and GP synthesis (Baenke et al., 2013; Swinnen et al., 2006). We employed desorption electrospray ionization mass spectrometry imaging (DESI-MSI) (Figure 1A) to monitor lipogenesis in situ without compromising metabolic homeostasis of the examined tissue (Bokhart and Muddiman, 2016; Eberlin et al., 2014; Perry et al., 2013; Shroff et al., 2015; Wiseman et al., 2006). Our study demonstrates that MYC regulates virtually all stages of lipogenesis and that this is required for the initiation and maintenance of tumor growth (Figure 1B).
Figure 1.

MYC as master regulator of proliferative lipid metabolic pathways: fatty acid synthesis, fatty acid elongation, and glycerophospholipid synthesis. (A) Schematic representation of DESI-MSI set-up used in this study. (B) Diagram showing that MYC and SREBP regulate multiple steps of the lipogenesis pathway uncovered in this study.
FA synthesis is required to generate cholesterol and complex lipids like GPs (Baenke et al., 2013; Swinnen et al., 2006), and includes multiple steps (Figure 2A). Initially, citrate from the TCA cycle is converted into acetyl-CoA by ATP citrate lyase (ACLY) (Morrish et al., 2010; Swinnen et al., 2006). The de novo FA synthesis starts with the production of malonyl-CoA from acetyl-CoA by acetyl-CoA carboxylase A (ACACA) (Morrish et al., 2010; Wang et al., 2009). Malonyl-CoA is then converted into palmitate [FA(16:0)] by sequential addition of 2 carbons via fatty acid synthase (FASN). Palmitate is subsequently converted to stearate [FA(18:0)] by elongase (ELOVL), and then desaturated to oleate [FA(18:1)] by stearoyl Co-A desaturase (SCD). The key FA synthesis regulators are known in normal cells but not in cancer. Here we identify the regulatory role of MYC in FA synthesis.
Figure 2.

Upregulation of lipid metabolic genes by MYC. (A) Diagram of MYC’s regulation of the fatty acid synthesis pathway. (B) qPCR shows fatty acid synthesis mRNA expression in high MYC (MYC ON, N=3 per cell line) compared to 24 hours of MYC inactivation (MYC OFF, N=3 per cell line) in BCL (P493–6) and RCC (E28). Expression of lipogenesis proteins for BCL (P493–6) and RCC (E28) cells with MYC ON vs. MYC OFF, by Western blot analysis. Statistical significance by t-test, ***P<0.001. (C) RNA-seq analysis of lipid metabolic pathways in fold-change of RPKM comparing MYC ON/MYC OFF in BCL (P493–6).
FA synthesis in normal cells is regulated by transcription factors called sterol regulatory element binding proteins (SREBPs) (Lewis et al., 2011; Shao and Espenshade, 2014), that consist of two (splice variants) of SREBP1 (SREBP1a and SREBP1c) and SREBP2. SREBPs are synthesized as precursors anchored in the endoplasmic reticulum (ER). Upon activation by diminished cholesterol, the SREBP cleavage-activating protein (SCAP) dissociates SREBPs from the ER into the nucleus (Shao and Espenshade, 2014; Swinnen et al., 2006), where SREBPs activate lipogenesis genes (Sun et al., 2015). SREBP1 activates fatty synthesis genes, whereas SREBP2 activates genes involved in mevalonate and cholesterol synthesis (Li et al., 2016).
In cancer metabolism, in addition to SREBPs, other regulators such as mTOR, YAP/TAZ, XBP, and MondoA control lipogenesis (Carroll et al., 2015; Lewis et al., 2011; Santinon et al., 2016; Xie et al., 2018a; Zhao et al., 2018), while SREBPs also regulate YAP/TAZ activity (Sorrentino et al., 2014). PI3K activation and mTOR signaling stimulate SREBP1 expression and nuclear localization (Fresno Vara et al., 2004; Huang and Chen, 2009; Lewis et al., 2011; Willems et al., 2012). Further, a gain-of-function p53 mutant can associate with SREBPs to activate mevalonate synthesis, thereby contributing to tumorigenesis (Freed-Pastor et al., 2012). Mevalonate synthesis activates YAP/TAZ to promote cellular growth through increased geranylation of Rho GTPases, which in turn might be regulated by MYC (Santinon et al., 2016; Sorrentino et al., 2014). Also, MYC was shown to regulate MondoA, which increases SREBP1 function and lipogenic gene expression (Carroll et al., 2015). Now, we demonstrate that SREBP1 and MYC collaborate to induce lipogenesis in cancer.
Cell division and growth requires glycerophospholipid synthesis (Baenke et al., 2013). GPs are long molecules comprised of two FA chains and a phosphate group ester-linked to glycerol and to a third substituent (Marsh, 2013). The chemical nature of this substituent divides GPs into several classes, whereas combinations of FA chain length and saturation give rise to different molecules within each class (Marsh, 2013). GPs promote biomass growth because they supply building blocks and provide suitable physical properties for membranes, act as cell signaling mediators, and are implicated in energy metabolism (Liu et al., 2014). Here, we show a causal role of MYC in GP synthesis.
We found that MYC with SREBP1 regulates lipogenesis in Tet-conditional transgenic mouse models of MYC-induced T-cell acute lymphoblastic leukemia (T-ALL), renal cell carcinoma (RCC), hepatocellular carcinoma (HCC), and lung carcinoma (LC) (Felsher, 2004, 2010; Felsher and Bishop, 1999; Gouw et al., 2017; Shroff et al., 2015), as well as in human and mouse tumor-derived cell lines and human tumor xenografts. We performed genome-wide expression profiling, nuclear run-on profiling, chromatin immunoprecipitation (ChIP) analysis, and DESI-MSI. DESI-MSI generates a detailed metabolomic map in situ without the use of matrices, labeling, or molecule pre-identification (Perry et al., 2013; Shroff et al., 2015). The inhibition of lipogenesis and FA synthesis diminished MYC induced tumorigenesis and elicited the regression of established tumors. We conclude that MYC activates SREBP1 and directly regulates FA synthesis and complex GP formation, and that the inhibition of lipogenesis both prevents and reverses MYC-induced tumorigenesis.
RESULTS
MYC Regulates Lipogenesis
We performed genome-wide expression profiling, nuclear run-on profiling, and ChIP analysis in the MYC Tet-On human P493–6 B-cell lymphoma line (BCL) (Pajic et al., 2000; Schuhmacher et al., 1999). MYC induction increased mRNA expression of FA synthesis genes, including ACLY, ACACA, FASN, and SCD1 and their corresponding proteins in BCL, and similarly in three in vivo transgenic mouse models of MYC-induced HCC, T-ALL, and RCC (Figures 2B, S1A). In contrast, catabolic FA oxidation genes were suppressed by MYC induction (Figure 2C). MYC was found bound to the promoters of these FA synthesis genes (shown by analyses of UCSC Genome Browser publicly available ChIP data (Lin et al., 2012), Figure S1B) and to promoters of mevalonate and cholesterol pathway genes, including HMGCR and DHCR7 (Figure S1C; for raw data: S1D). In fact, MYC bound to the promoters of most FA synthesis genes (Figure 3A), as measured by ChIP (Figure 3B; for raw data from UCSC: 3C) and initiated mRNA transcription, as validated by nuclear run-on (Figure 3D).
Figure 3.

MYC orchestrates glycolysis, glutaminolysis, and lipogenesis. (A) Diagram showing MYC direct binding and transcriptional activity on glycolysis, glutaminolysis, and lipogenesis genes. (B) ChIP-seq data of BCL (P493–6) showing MYC binding on glucose, glutamine, and fatty acid synthesis promoters upon MYC activation at 0, 1, and 24 hours following 24 hours of MYC inactivation. Y-axis shows 1 to 12 reads per million. (C) Binding of MYC on fatty acid synthesis genes by ChIP-Seq in BCL (P493–6) cells 0, 1 and 24 hours after tetracycline release. (D) Nuclear run-on of MYC transcriptional rate of lipogenesis genes in BCL(P493–6), N=3, Statistical significance by t-test, ***P<0.001. (E) UCSC genome browser display of publicly available MYC ChIP-Seq in BCL (P493–6) at 0, 1 and 24 hours after tetracycline release for glutaminolysis genes, and (F) glycolysis genes.
MYC Activates and Cooperates with SREBP1 to Induce Fatty Acid Synthesis
SREBP has a known role in regulating FA synthesis (Lewis et al., 2011; Shao and Espenshade, 2014). We therefore examined four possible interactions between MYC and SREBP1: First, careful ChIP and re-ChIP analysis of both proteins, using methods as described (Truax and Greer, 2012; Walz et al., 2014a), demonstrated that MYC and SREBP1 can both be found binding to the same DNA molecule in the promoters of FA synthesis genes (Figure S1E). Also, the knockdown of SREBP1 lowered expression of FA synthesis genes but not MYC gene expression, (Figure S1F).
Second, MYC by ChIP bound to the promoters and regulated the expression of not only FA synthesis genes, but also glycolysis and glutaminolysis genes (Figure 3E, F). Thus, MYC appears to regulate the genes involved sequentially in lipid synthesis: first inducing glucose and glutamine and then FA synthesis associated genes. By RNA-seq, increasing levels of MYC elicited the nonlinear induction of mRNA of glycolysis (Figure S2A), glutaminolysis (Figure S2B), and FA synthesis (Figure S2C).
Third, we found that MYC binds to the promoters of cholesterol biosynthesis genes (Figure S1C, S1D), induces key enzyme for cholesterol biosynthesis (Figure S2C), and regulates SREBP1 (Figure S2D). Further, MYC regulates both SREBP1 and SCAP in BCL and HCC, as seen by mRNA expression by RNA-seq (Figure S2D) and by protein analysis by Western blot (Figure S2E). This indicates that MYC’s regulation of lipogenesis is both through direct binding as well as through SREBP1.
Fourth, our results implicated that SREBP1 regulates FA synthesis genes in the presence of MYC (Figure S1E, F). To determine whether FA synthesis can be run by MYC independent of SREBP1, we knocked down SREBP1, through siRNA in a readily transfectable cell line, EC4, a MYC-induced HCC cell line (Beer et al., 2004). This enabled us to examine the immediate consequences of knockdown since SREBP1 knockout is lethal (Horton et al., 2002). The knockdown of Srebp1 decreased MYC-induced expression of Acaca, Fasn, and Scd1 (Figure 4A; for other genes: Figure S3A); whereas the inactivation of MYC suppressed SREBP1-induced expression of these FA synthesis genes. The knockdown of SREBP1 decreased FA synthesis genes without an effect MYC gene expression (Figure S1F). Importantly, MYC inactivation abolished SREBP1 binding to FA synthesis gene promoters, demonstrating that the function of SREBP1 depends on MYC (Figure 4B). In contrast, MYC binding to FA synthesis genes was not affected by SREBP1 knockdown (Figure 4B), even though mRNA levels of FA synthesis genes were downregulated after SREBP1 silencing (Figure 4A and S1F).
Figure 4.

MYC regulates SREBP and together they regulate lipogenesis. (A) mRNA expression of fatty acid synthesis genes: Acly, Acaca, Fasn, and Scd1 upon siRNA knockdown of SREBP1 or scramble RNA in HCC(EC4) under MYC ON and OFF conditions (N=3 per condition). Statistical significance by two-way ANOVA using Tukey’s multiple comparisons post tests, ***P<0.001. Overall, ***P<0.001 for SREBP1 effect, ***P<0.001 for MYC effect and ***P<0.001 for interaction between the two. (B) SREBP1 ChIP-qPCR on BCL P493–6 line shows decreased binding of SREBP1 upon MYC inactivation (top panel). MYC ChIP-qPCR on HCC EC4 line shows no change of MYC binding upon SREBP1 knockdown (bottom panel). (C) Glucose and glutamine incorporation into lipids by NMR in MYC ON (upper panel) and OFF (bottom panel) in BCL(P493–6). For close-up look see Figure S3B. (D) Incorporation of glucose and glutamine carbons to de novo synthesized fatty acids by mass spectrometry based on the fold change of 2C13 peak in BCL(P493–6) as measured upon 24 hours of MYC inactivation (N=5). Fold change in the de novo fatty acid synthesis when MYC is activated or inactivated (left panel); and glucose and glutamine incorporation into fatty acids when MYC is activated (middle panel), or inactivated (right panel). Statistical significance by t-test, **P<0.01, ***P<0.001.
Collectively, our results suggest that MYC can bind to the promoters of FA synthesis genes independent of SREBP1 (Figure 4B) but this binding is insufficient to promote expression of these genes. In addition, MYC drives the expression and promoter binding of SREBP1 (Figure S2D, S2E). MYC induces SREBP1 expression, and together MYC and SREBP1 bind to FA synthesis gene promoters to induce transcription (Figure S1E, 4A). Thus, MYC and SREBP1 cooperate to regulate FA synthesis in a feed-forward transcriptional loop.
Glucose and Glutamine Metabolism Regulated by MYC Contributes to Lipogenesis
MYC is known to regulate glucose and glutamine metabolism (Gao et al., 2009; Le et al., 2012; Wang et al., 2011; Wise et al., 2008). We found that MYC binds, transcribes, and increases the expression of glycolysis and glutaminolysis genes (Figure 3B, D, E, S2A, S2B). We investigated if carbons from glucose and glutamine are incorporated into de novo synthesized FAs. NMR tracing of 13C-glucose and 13C-glutamine 24 hours after MYC induction in BCL revealed that MYC drives the incorporation of carbons into lipogenesis (Figure 4C, S3B). Our analysis showed that in MYC-OFF cells, 84 ± 2% of the carbons incorporated in the de novo synthesized fatty acids originated from glucose and 16 ± 2 % were from glutamine as quantified by MS (Xie et al., 2018b) (Figure 4D). However, the ratio of glucose and glutamine carbons incorporated into de novo synthesized FAs with MYC-ON was similar to the ratio with MYC-OFF (Figure 4D). Thus, induction of fatty acid synthesis pathway by MYC is not associated with differential reprogramming of glutaminolysis versus glycolysis.
Increased FA synthesis in vivo after MYC activation was also detected by DESI-MSI through monitoring of unlabeled FA in transgenic mouse models of HCC, RCC, and LC, and in human BCL xenografts (Figure 5A). Specifically, we observed an increase in abundance of unsaturated FA oleate [FA(18:1)], m/z 281.248, across various MYC tumors (Figure S3C). However, 13C-oleate was not metabolized when administered to MYC- induced BCL cells (Figure S3D), and 13C-glucose increased labelled carbons in unsaturated FAs of these cells (Figure S3E), consistent with MYC’s induction of desaturase, SCD1 (Figures 2B, S1A). 3H-labeled palmitate oxidation rate in these cells was significantly higher when MYC was inactivated (Figure S3F). Oleate administrated to both MYC ON and MYC OFF BCL cells is not oxidized and remains as lipid droplets as shown by and oil-red-O staining (Figure S3G). Hence, MYC seems to suppress fatty acid oxidation while upregulating FA synthesis.
Figure 5.

Lipid signature of MYC-induced HCC, RCC, T-ALL, LC bronchiolar (b), and adenoma (a). All DESI-MSI analyses were performed in 6-week old transgenic mice (male and female, N=4 per disease), after either MYC induction for 2 weeks (MYC ON) or without MYC induction (MYC OFF). (A) PG and PI relative expression in MYC ON state compared to normal tissue for HCC, RCC, T-ALL, LC (b), and LC (a). (B) PG and PI relative expression in MYC OFF state compared to MYC ON in HCC, RCC, and T-ALL. Gray color represents undetected ion signal.
MYC Regulates Complex Lipid Synthesis In Vivo
To examine whether MYC controls GP biosynthesis in vivo, GPs were measured by DESI-MSI in four conditional transgenic mouse models: RCC, HCC, T-ALL, and LC; where we could regulate MYC expression (Eberlin et al., 2014; Felsher, 2004, 2010; Felsher and Bishop, 1999; Shroff et al., 2015; Taub et al., 1982). Note, DESI-MSI is a semi-quantitative analytical tool without an external standard and works best when comparing a change in relative MS peak abundances of similar sets of metabolites. We found that across different tumors MYC induction changed the abundances of glycerophosphoglycerols (PGs) (Figure 5A), which are glycerophospholipids with a glycerol phosphate group substituent. MYC increased most PG species across all tumors. Upon MYC inactivation, PG abundances returned to basal level (Figure 5B). PGs are formed from phosphatidic acid (PA) by the addition of two FAs to glycerol-3-phosphate (Athenstaedt et al., 1999; Hill et al., 1968) (Figure 6A). Therefore, PG induction could build upon the increased FA pool promoted by MYC as shown above. However, the relatively preferential synthesis of PGs over other GPs in vivo implies that MYC regulates lipogenesis not only through FA synthesis (Figure 6A).
Figure 6.

MYC induces PG synthesis and TOFA prevents MYC-induced tumorigenesis and tumor progression in vivo. (A) Diagram showing MYC’s regulation of PG synthesis genes uncovered in this study. (B) DESI-MSI shows the representative distribution of long acyl chain- and short acyl chain PGs in RCC following 15 days MYC activation, and 10 days MYC inactivation, N=3 per time point (for mass spectra, more PG species, tissue H&E, and for fold change observed in three different individual subjects see Figure S4 A–E). (C) qPCR shows mRNA expression of PG synthesis genes in RCC upon MYC activation at weeks 2, 3, 4 and subsequent inactivation at days 2, 3, 10, N=3 per time point. Statistical significance by t-test for MYC ON compared to control***P<0.001. (D) qPCR shows mRNA expression of fatty acid chain elongase genes in RCC upon MYC activation at weeks 2, 3, 4 and subsequent inactivation at days 2, 3, 10, N=3 per time point. Statistical significance by t-test for MYC ON compared to control, **P<0.01, ***P<0.001. (E) Tumor weight over 4 weeks in untreated transgenic RCC mice, TOFA prevention at week 0, and TOFA treatment at week 2 (top); pictures of treated and untreated kidneys (bottom), N=3 per treatment condition, 6-week old male and female mice, statistical significance by t-test, ***P<0.001. (F) DESI-MSI images show phospholipid (PG and PI) accumulation and reduction following MYC activation, inactivation, and TOFA treatment in mouse model of MYC-induced RCC. For H&E of tissues see Figure S6G, for other phospholipid species, see Figure S7A). (G) Tumor growth of human RCC 786–0 xenograft, human BCL xenograft (NSG mice) and in HCC primary transgenic mice in untreated vs. TOFA-treated (TOFA daily treatment starts at week 2 for one week). N=3 per treatment condition, 6-week old male and female mice, statistical significance by t-test, *P<0.05, **P<0.01. (H) DESI-MSI images show PG and PI levels following TOFA treatment in human RCC 786–0 xenograft (top panel, for other PG and PI species, see Figure S7B), and oleate levels following TOFA treatment in mouse model of MYC-induced RCC (bottom left panel) and mouse model of MYC-induced HCC (bottom right panel). For H&E of tissues see Figure S6G.
MYC Promotes In Vivo Differential Glycerophosphoglycerols Synthesis
To measure how MYC affects GPs in vivo, we performed DESI-MSI at different times after MYC induction in our transgenic model of RCC. Previously, we have shown in this mouse model that MYC expression can be induced synchronously throughout the kidney (Kolk et al., 2009; Shroff et al., 2015).
We detected significant differences in GP abundances (m/z range 700–1000) between normal kidneys and MYC-induced tumors (15 days MYC activation); notably an increase in m/z 865.500 and m/z 819.517 peaks identified by tandem MS (Table S1) as PG(22:6/22:6) and PG(18:1/22:6), respectively. These differences were largely reversible upon MYC inactivation (Figure S4A). DESI-MSI also confirmed increased abundances of PGs following MYC activation by measuring dynamic changes in time (Figure 6B, Figure S4B, for fold change: S4C, and Figure S4D, for fold change: S4E). However, there were differences in dynamics of induction between various PG species. PGs with shorter acyl side chains (18 carbons or less), increased initially (10 days of MYC activation), and then decreased following further MYC induction (Figure 6B; for other lipids and tissue H&E staining: Figure S4B; for fold change: Figure S4C). This was also observed for other PG species at m/z 745.501, 747.516, m/z 773.532, m/z 775.547, and m/z 771.517 (Figure S4B), identified as PG(18:2/16:0), PG(18:1/16:0), PG(18:1/18:1), PG(18:0/18:1), and PG(18:2/18:1), respectively (Table S1).
Following the short chain PG decrease at 15 days of MYC activation, there was an increase in PGs with at least one acyl chain longer than 20 carbons (Figure 6B; for tissue H&E staining: Figure S4B; for other lipids: Figure S4D; for fold change: Figure S4E). Molecular ions that followed this pattern were at m/z 865.500, m/z 819.517, m/z 867.519, m/z 821.534, m/z 841.500, and m/z 817.500 (Figure S4D), identified as PG(22:6/22:6), PG(18:1/22:6), PG(22:6/22:5), PG(18:1/22:5), PG(20:4/22:6), and PG(18:2/22:6), respectively (Table S1). These changes were reversed by MYC inactivation. Hence, MYC elicited precise changes in PG synthesis in vivo as measured by DESI-MSI.
MYC Induces PG Synthesis and Elongates FA Chains
MYC induction increased and MYC inactivation decreased the mRNA of PG synthesis genes (Figure 6A, C). This included the induction of cytidinediphosphate diacylglycerol (CDP-DAG) pathway genes: Pgs1 (phosphatidylglycerophosphate (PGP) synthase, converts CDP-DAG to PGP), and Ptpmt1 (PGP phosphatase, dephosphorylates PGP to generate PGs) (Chang et al., 1998; Nie et al., 2010; Shen and Dowhan, 1998; Visel et al., 2004; Yang et al., 2004; Zhang et al., 2011) (Figure 6C). Also, MYC upregulated Agpat1 and Cds1 (Figure 6A), which catalyze CDP-DAG conversion to PGs (Takeuchi and Reue, 2009; Yang et al., 2004). Therefore, MYC upregulates transcription of PG synthesis genes.
We observed that MYC activation preferentially elevated the abundances of longer acyl chain PGs. FA chains are constantly incorporated into de novo synthesized PGs through acyl chain remodeling (Nie et al., 2010; Yang et al., 2004). Hence, MYC may be promoting the elongation of existing shorter FA chains. Consistent with this idea, MYC induced mRNA expression of the FA elongases ELOVL2 and ELOVL6, which add two carbons to FA chains (Moon et al., 2014; Ofman et al., 2010; Pauter et al., 2014) (Figure 6D). This is consistent with the promoter binding data from ChIP-Seq in BCL (Figures S5A, B). Hence, MYC regulates genes that increase the production and incorporation of long unsaturated FAs.
MYC Alters Glycerophosphatidylinositol Metabolism in Tissue-Dependent Manner
Notably, MYC has tissue-specific effects on glycerophosphatidylinositol (PI) metabolism. Thus, in RCC and T-ALL, MYC reversibly decreased, but in LC and HCC increased PIs as measured by DESI-MSI (Figure 5A, Figure S5C; for tissue H&E staining: Figure S4B; for fold change Figure S5D). PIs have inositol as the phosphate group substituent, and they are competitively synthesized with PGs from the common CDP-DAG pool (Figure 6A) (Batenburg et al., 1985; Liu et al., 2014; Quirk et al., 1984). Significant PGS1 and PTPMT1 induction may preferentially promote CDP-DAG conversion to PGs, which could then shunt metabolites away from PI synthesis.
One explanation, is that MYC increased PIs in LC because of lung surfactants that are rich in both PGs and PIs (Agassandian and Mallampalli, 2013; Fickes et al., 2016; Griese et al., 1991). We infer that MYC generally increases PGs but differentially regulates other GPs, such as PIs in a tissue specific manner.
MYC Regulation of Lipogenesis is Required for Tumorigenesis
The role of lipogenesis in MYC-induced tumorigenesis was examined (Figure S6A). RCC (E28), HCC (EC4), and the human BCL (P493–6) proliferation was suppressed in vitro by FA synthesis inhibition (Figure S6). Cerulenin, an FASN inhibitor, decreased BCL proliferation in a dose-dependent manner (Figure S6B). Similarly, 5-tetradecyl-oxy-2-furoic acid (TOFA), an ACACA inhibitor (Luo et al., 2012; Wang et al., 2009), suppressed BCL (Figure S6C) and RCC (Figure S6D) proliferation, which was partially rescuable by administration of albumin-bound oleate at non-toxic dosages (Southam et al., 2015; Xie et al., 2018a) the downstream product of FA synthesis (Figure S6E). RCC tissue histology confirmed tumor regression (Figure S6F). Hence, inhibition of FA synthesis blocks MYC-induced cancer proliferation in vitro.
We examined the consequences of the inhibition of FA synthesis in vivo. We chose TOFA inhibition of ACACA rather than drug inhibition of FASN, because the latter leads to malonyl-CoA accumulation, which inhibits FA oxidation in addition to FA synthesis and is known to elicit ATP depletion and hence nonspecific effects (Shiragami et al., 2013; Thupari et al., 2001).
TOFA prevented tumorigenesis in multiple in vivo mouse and human tumor models. TOFA caused tumor regression in MYC-induced mouse and human RCC, human BCL, and mouse HCC (Figure 6E–H). By DESI-MSI, TOFA was found to block the formation of PGs and PIs during tumor initiation and regression in both transgenic MYC-induced RCC (Figures 6F; for tissue sections H&E staining: Figure S6G, for other ions: Figure S7A) and in human RCC xenografts (Figures 6H; for tissue sections H&E staining: Figure S6G; for other ions: Figure S7B). Histological analysis and measurement of phospho-histone 3 (PH3) and cleaved caspase 3 (CC3) confirmed strong antitumor activity of TOFA (Figure 7 A–C, S6F).
Figure 7.

TOFA treatment in various tumors. (A) Immunohistochemical staining for proliferation marker phosphor-histone 3 (PH3), apoptotic marker cleaved caspase 3 (CC3) in mouse model of MYC-induced RCC, (B) human xenograft BCL P493–6, and (C) mouse model of MYC-induced HCC. (D) Cell proliferation in three lymphoma cell lines driven by three different oncogenes (MYC (P493–6), KRAS (3588), and BCR-ABL (9662)) when inhibited by various TOFA concentrations vs untreated. N=3 per time point. Statistical significance by t-test, *P<0.05, **P<0.01, ***P<0.001 compared to untreated cells. (E) Tumor growth of transplanted MYC-induced lymphoma in 6-week old NSG mice (left panel), KRAS-induced lymphoma (middle panel) and BCR-ABL-induced lymphoma (right panel) allografts in 6-week old FVBN mice after 2 weeks of TOFA treatment vs no treatment. Statistical significance by t-test, **P<0.01, ***P<0.001. Western blots show levels of MYC in the three lymphoma cell lines, and they correlate with tumor sensitivity to ACACA inhibition. (F) Protein acetylation by Western blot analysis following TOFA addition to RCC (E28) cells. (G) Fold change of relative abundance of TCA cycle metabolites by DESI-MSI and schematic representation of TCA cycle following ACACA inhibition. (H) Etomoxir treatment of murine MYC-induced RCC in vivo does not significantly change tumor weight compared to control RCC injected with DMSO compared to normal kidney, N=4. Statistical significance by t-test, ***P<0.001. (I) siRNA knockdown of ACACA in HCC EC4 suppresses proliferation across 3 days in vitro, N=3. Statistical significance by t-test, ***P<0.001.
Next, we checked if TOFA mediated inhibition of tumor growth is dependent upon high levels of MYC expression. First, we found that TOFA’s ability to reduce proliferation of a tumor-derived cell line from a MYC-induced lymphoma in vitro correlated with higher levels of MYC expression (Figure S7C). We evaluated this by inhibiting doxycycline-regulated MYC system in our BCL model, where we could modulate MYC levels by administering different doxycycline concentrations. Here we found that at lower levels of MYC, proliferation inhibition by TOFA was not significant and the cells were still proliferating (Figure S7C). Second, TOFA’s effects varied both in vitro and in vivo in MYC versus KRAS or BCR-ABL driven lymphomas (Azimi et al., 2016; Felsher and Bishop, 1999; Rakhra et al., 2010) (Figure 7D, E). MYC-induced lymphomas were most sensitive to TOFA, followed by KRAS-induced then BCR-ABL-induced lymphomas (***P<0.001 difference between the three groups treated with 5 μg/mL of TOFA), which correlated with their levels of MYC expression (Figure 7E). Note that both activated KRAS and BCR-ABL are reported to depend on and increase MYC activity (Afar et al., 1994; Farrell and Sears, 2014; Kimmelman, 2015; Sawyers et al., 1992; Sears et al., 1999; Ying et al., 2012). Hence, the effect of ACACA inhibition by TOFA on tumor growth correlates with MYC expression.
We considered that TOFA inhibition of ACACA could be suppressing tumor growth through an epigenetic mechanism as shunting of accumulated acetyl-CoA as a substrate for histone acetylation can cause epigenetic changes that are able to induce cell death (Wellen et al., 2009). However, we found no significant changes in histone acetylation by Western blot analysis (Figure 7F). Instead, TOFA did induce changes in TCA cycle intermediates including increased succinate and malate and decreased pyruvate, as seen by DESI-MSI (Figure 7G).
We considered the possibility that fatty acid oxidation is involved in this process due to previous reports connecting MYC to fatty acid oxidation (Muller et al., 2014; Pacilli et al., 2013). To investigate the role of fatty acid oxidation, we inhibited carnitine palmitoyl transferase (CPT1) using Etomoxir in our transgenic MYC-induced RCC and found no effect on tumor weight (Figure 7H). On the other hand, ACACA knockdown in RCC-derived cell line had a significant effect on cell proliferation (Figure 7I) further confirming the significance of ACACA to MYC-induced tumor growth. To examine whether TOFA influenced the oxygen consumption rate (OCR), we measured OCR in two human cell lines sensitive to TOFA (Figure S7D), human RCC (786–0) and human glioblastoma (U87). We did not find a significant difference in OCR or ATP between treated and untreated cells (Figure S7E). Hence, the inhibition of FA synthesis blocks MYC-induced lipogenesis, prevents tumor initiation, and elicits tumor regression without increasing OCR or changing global histone acetylation profiles.
DISCUSSION
We report that MYC in coordination with SREBP1 regulates lipogenesis that is required for both the initiation and maintenance of tumorigenic growth in vitro and in vivo (Figure 1B). Our results suggest a therapeutic vulnerability of MYC-driven tumors to lipogenesis inhibition. These findings are consistent with several studies reporting that MYC regulates glucose, glutamine, other amino acid, and nucleotide metabolism (Dang, 2013; Gao et al., 2009; Li et al., 2005), and with others suggesting that MYC alters lipid metabolism (Carroll et al., 2015; Eberlin et al., 2014; Edmunds et al., 2015; Morrish et al., 2010; Perry et al., 2013; Shroff et al., 2015). However, the mechanism by which MYC regulates FA and GP synthesis was unclear prior to this study.
By using RNA-seq, ChIPseq, and nuclear run-on assays, we found that MYC amplifies gene expression nonlinearly and orderly, to induce specific groups of genes involved in metabolism and biomass accumulation. Upon MYC induction, FA synthesis is upregulated in human BCL and in transgenic mouse models of MYC-induced HCC, RCC, T-ALL, and LC. The induction of FA synthesis followed the induction of glycolytic and glutaminolytic genes, suggesting that MYC elicits the temporal activation of these metabolic programs.
Our observations reveal the complexity of MYC-dependent gene expression amplification (Lin et al., 2012; Nie et al., 2012). This behavior is likely to be determined by different transcription factors that partner with MYC to promote lipogenesis, as shown by some reports (Carroll et al., 2015; Collier et al., 2007; McPherson and Gauthier, 2004; Shao and Espenshade, 2014; Shimizu-Albergine et al., 2016; Zhang et al., 2010). Our results suggest that MYC induces SREBP1 and acts in conjunction with SREBP1 to synergistically induce FA synthesis genes. Our findings are consistent with prior reports of MYC activating and interacting with SREBP1 (McPherson and Gauthier, 2004; Shao and Espenshade, 2014; Shimizu-Albergine et al., 2016; Wu et al., 2016), but we show here that the relationship between MYC and SREBP1 is more complex. MYC’s binding to FAsee synthesis genes is not dependent on SREBP1, however, MYC requires SREBP1 to induce FA synthesis genes.
Our observations are also consistent with prior reports that MYC and associated proteins, including MAX, MXD, and MLX, collaborate with SREBP2, ChREBP, and MondoA to regulate FA and cholesterol biosynthesis (Carroll et al., 2015; Collier et al., 2007; Zhang et al., 2010). Based on our and aforementioned works, we infer that MYC orchestrates the induction of lipogenesis through interactions with discrete master regulators. Our results provide an example whereby MYC regulates the expression of genes not only by amplifying all genes expressed at the basal level (Lin et al., 2012; Nie et al., 2012; Sabo et al., 2014; Walz et al., 2014b).
DESI-MSI enabled us to visualize the in vivo spatiotemporal lipid modifications during tumorigenesis. MYC induction yields a distinct phospholipid signature across various human and mouse tumors manifested by a prominent PG increase. PGs are a minor component of intracellular membranes, accounting for <1% of phospholipids in non-neoplastic mammalian tissues (Morita and Terada, 2015). PGs are required for mitochondrial activity and membrane integrity (Dolinsky et al., 2016; Houtkooper and Vaz, 2008; Kiebish et al., 2008). The increase in PGs is consistent with induction of mitochondrial biogenesis orchestrated by MYC (Ji et al., 2011; Li et al., 2005).
We have defined the temporal sequence in the lipid changes associated with MYC-tumor initiation and progression. MYC increases long-chain PGs, by acyl chain remodeling and activation of FA elongases. Both MYC inactivation and FA synthesis inhibition were associated with PG depletion and resulted in tumor regression. While these observations are consistent with MYC-mediated de novo lipid synthesis, we recognize that the increase in PGs could be at least in part derived from fatty acids acquired from extracellular sources. Thus, by DESI-MSI, we have uncovered a lipid signature that should be useful to identify, diagnose, and prognosticate MYC-induced cancers, which can lead to the identification of novel therapeutics. Clinical biopsies identified with this signature may allow us to predict MYC involvement in examined tumors and suggest their therapeutic sensitivity to FA synthesis inhibition.
We conclude that MYC-induced tumors depend on lipogenesis, and specifically FA synthesis. This dependence can be attributed, at least in part, to MYC-driven lipogenesis. We observed that tumors driven by other oncogenes also were sensitive to the inhibition of lipogenesis, but this sensitivity correlated with MYC expression. Indeed, cancers driven by other oncogenes may also depend on FA synthesis, and that when there is MYC overexpression they appear to have higher sensitivity to inhibition of lipogenesis (Gouw et al., 2017; Svensson et al., 2016). TOFA mediated inhibition of ACACA prevented MYC tumorigenesis in vivo and regressed established tumors in autochthonous transgenic mice and human mouse xenografts. The inhibition of ACACA in vivo with TOFA inhibited tumorigenesis. The variety of more specific ACC inhibitors in development will be of future interest for their potential efficacy in the treatment of MYC-driven human cancers. The inhibition of FA synthesis may be therapeutically useful particularly in MYC-associated cancers.
We propose a general model (Figure 1B) by which MYC orchestrates the orderly activation of glycolysis, glutaminolysis as well as FA and PG synthesis, providing means for the balanced acquisition of nutrients and stoichiometric production of cellular biomass (Dang, 2013; Li et al., 2005). In cancer, MYC overexpression maximizes unrestrained growth, but at the expense of a remarkable vulnerability to the inhibition of lipogenesis that may be exploited to prevent and treat cancer. DESI-MSI enabled us to precisely map MYC-specific lipid metabolism in situ, and thus can be used to identify new anti-lipogenic drugs that will exploit this Achilles heel of cancer.
Limitations of Study
Fatty acid scavenging has been shown to contribute to RAS-driven cancer cell proliferation, but is likely insufficient to solely drive MYC-dependent cell growth. The amount of lipids present in the media during our experiments was too low to support proliferation through ‘fatty acid scavenging’ in response to treatment with TOFA or cerulenin, which inhibit ACACA and FASN respectively. Also, we observed a partial rescue by addition of oleate to TOFA treated cells. Further, subcellular membrane compositions are quite specific in terms of the relative amounts of unsaturated fatty acids, cholesterol or mitochondrial cardiolipin, and hence it is unlikely that fatty acid scavenging will supply the specific lipid composition needs of a growing cell without de novo lipogenesis, fatty acid elongation and desaturation. Future studies will need to address the intriguing possibility of an interplay between fatty acid scavenging and de novo lipogenesis to meet the needs of a MYC-mediated cell growth.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Lead Contact
Further information and request for resources and reagents used in this publication should be directed to Dean W. Felsher (dfelsher@stanford.edu).
Materials Availability
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Stanford University’s Research Compliance Office Administrative Panel on Laboratory Animal Care (APLAC) approved our animal protocols. MYC RCC mice were generated by crossing FVB/N-Tg(tetO-MYC)28aBop/J mice for MYC with Tg(Ggt1-tTA)#Agoc or (GGT-tTA) for kidney specificity. MYC HCC mice were generated by crossing FVB/N-Tg(tetO-MYC)28aBop/J mice for MYC with Tg(Cebpb-tTA)5Bjd or (LAP-tTA) for liver specificity. MYC LC mice were generated by crossing FVB/N-Tg(tetO-MYC)28aBop/J mice for MYC with B6. Cg-Tg(Scgb1a1-rtTA)1Jaw/J or (CCSP-tTA) for lung specificity. All parental lines had undergone at least 10 generations of backcrossing. MYC RCC and MYC HCC mice were bred on doxycycline until 4 weeks of age, when doxycycline is then replaced with water for 2–4 weeks for tumorigenesis depending on the experiment described in the manuscript. All mice were housed in Stanford Comparative Medicine Pavilion (CMP) under the auspices of the Veterinary Service Center (VSC) that are compliant with Asia Pacific Laboratory Animal Care (APLAC) regulations. Veterinarians in VSC monitored the health of every mouse daily. The Stanford VSC staff provided feeding & watering of animals; cleaning and/or changing animal cages; observing & reporting illness or death; maintaining facility at room temperature. VSC-approved staff and lab members performed basic breeding setups for experiments. Light cycle for the duration of the study was set to 12-hour light/12-hour darkness. Permissible temperature and relative humidity levels were 68F–79F and 30%–70%. VSC provided mice with Envigo Teklad Global 18% Protein Rodent Diet.
We performed all in vivo experiments using both male and female mice in equal proportion. All experiments were performed in a blinded fashion to avoid experimenter bias. In our study there was no statistically significant difference between male and female mouse models for experiments reported.
Cell Culture Conditions
BCL (P493–6) cells were cultured in Gibco’s® RPMI1640 media under normal culture conditions of 20% O2, 4% CO2, and 37°C. P493–6 cells were suspension cells, and they were kept at 0.2–1.8 million cells/mL density to avoid confluency. To turn off MYC, 1 μg/mL of tetracycline was applied to the media for 48 h to completely suppress MYC. Mouse-derived renal carcinoma line E28 and human RCC line 786–0 (ATCC CRL1932) were cultured for in vitro experiments under normal culture conditions of 20% O2, 4% CO2, and 37°C. 786–0 and E28 cells were maintained in Dulbecco’s modified eagle medium (DMEM), which was supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, 1% sodium pyruvate, 1% nonessential amino acids, and antibiotic-antimycotic. Trypsin-EDTA was used to passage E28 and 786–0 cells. All cell culture reagents were purchased from Gibco® (Thermo Fisher Scientific). Sex of cell lines used in this study was: 3588, 9662, EC4, 786–0, U87 - males. P493–6, E28 – unknown.
Cell Counting
A volume of cells was removed from culture medium and mixed with an equal volume of 0.4% Trypan blue stain. Then, 10 μL were taken out and placed into a hemocytometer for cell counting. Viable cell counts were used as a measure of cell proliferation.
RNA Extraction and cDNA Synthesis
RNA extraction from 2 × 107 cells was done using the Qiagen RNEasy extraction kit. RNA quality and concentration were assessed using the Thermo Scientific Nanodrop 3300 spectrophotometer. cDNA was then synthesized from 0.4 μg of the extracted RNA using Qiagen cDNA reverse transcription kit. The cDNA was then stored at −20°C.
METHOD DETAILS
Primers and qRT-PCR
Primers were designed to span the intron-exon junction using PrimerQuest online software, available from http://www.idt.com. Primer specificity was then tested using the Basic Local Alignment Search Tool (BLAST). Real-time PCR was performed in 96-well plates on an ABI Biosystems Thermo Cycler 7500. All primers were detected by using SYBR Green as a fluorophore. Reactions were carried out in 20 μL that contained 1.5 μL cDNA, 0.5 μM forward and reverse primers, 8 μL water, and 10 μL of 2x SYBR Green master mix (Applied Biosystems). The amplification cycle was as follows: 95°C for 3 min, 35 cycles of 95°C for 10 s, 63°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 5 min. At the end of amplification, a dissociation curve was obtained to verify nonspecific amplification. The cycler software yielded a threshold cycle number (Ct) for each gene, which was the number of cycles required to reach the threshold fluorescence. The Ct values were exported into Microsoft Excel for statistical analysis.
Nuclear Run-on Assay
Cells were harvested and washed with cold phosphate buffer saline (PBS). 10 mL of cold cell lysis buffer was added to resuspend each nuclei pellet with 100 μL of nuclei resuspension buffer to isolate the nuclei. Nuclear run-on buffer was added for 30 min then incubated at 30°C, and then the temperature reset at 37°C. 200 U of DNase I (10 U/μL, Roche Applied Sciences) was added to each reaction and incubated for 20 min at 37°C. 400 U of proteinase K (20 mg/mL, Ambion, premixed with 10% sodium dodecyl sulfate at 3:1) was added and then incubated for 15 min at 37°C. Extraction of total RNA was done using the Qiagen RNA extraction kit. Dynabeads and immobilized biotin-labeled nascent RNA were washed to prepare for first-strand and second-strand cDNA synthesis. This was then followed by cDNA purification, cRNA synthesis, cRNA purification, and array hybridization. The more detailed protocol is available (Fan et al., 2012).
Chromatin Immunoprecipitation (ChIP)
ChIP assays were carried out as previously described (for ChIP assay see (Walz et al., 2014a)), and for Re-ChIP assay see (Truax and Greer, 2012)). Briefly, cells were cross-linked with 1% formaldehyde for 10 min at room temperature and formaldehyde was then inactivated by the addition of 1M glycine for 5min at room temperature. Chromatin extracts containing DNA fragments with an average size of 200 bp were immunoprecipitated using anti-SREBP1 (2A4 mouse monoclonal home-made antibody), anti-c-Myc antibody (Y69- rabbit monoclonal antibody (ab32072)) or IgG (Sigma). ChIP data was analyzed using percent input method.
Small Molecule Inhibitors
The small molecule TOFA (Cayman Chemical) and Cerulenin (Sigma), diluted in dimethyl sulfoxide (DMSO) to the desired concentrations, was added to the cell culture medium to achieve inhibition of ACACA. TOFA is an allosteric inhibitor of ACACA, the enzyme that catalyzes the rate-limiting step of FA synthesis (Wang et al., 2009). Various concentrations of TOFA and Cerulenin were added to cells in culture over time to assess dose-response suppression of proliferation. Fatty acid oxidation measurements of labeled palmitate is performed upon TOFA treatment or MYC inactivation following the standard biochemical fatty acid oxidation protocol (Djouadi et al., 2003), where scintillation counter is used to measure labeled carbon dioxide release upon oxidation of labeled palmitate. For in vivo treatment of mice, 25 mg/kg TOFA was administered daily by intraperitoneal injections (IP). The stock TOFA was diluted in DMSO to 10 mg/mL, such that the final injected volume was around 50 μL because the mice weighed approximately 20 g. Etomoxir (Sigma) was dissolved in water. Mice received IP injections of 15 mg/kg starting on day 14 upon MYC induction in the MYC-induced RCC transgenic mouse models.
De Novo Lipogenesis Assay by NMR
For each NMR measurement, three T-75 flasks containing 18 mL of RPMI 1640 with 10% dialyzed FBS were inoculated with 2 × 107 P493–6 cells. The medium contained 2.0 mM [1-13C]glucose, 8.0 mM unenriched glucose, 3.0mM [U-13C]glutamine and no unenriched glutamine. After 14 hours of incubation in the 13C labeled medium, cells were harvested by centrifugation in 50-mL conical centrifuge tubes at 1000 rpm for 5 minutes. The supernatant was rapidly removed, the pellets were washed once with PBS containing 10% fatty acid free bovine serum albumin, then a second time with protein-free PBS before being frozen by submerging the tubes in liquid nitrogen.
A standard chloroform/methanol procedure was used to extract cellular lipids (Young et al., 2013). After suspension in a mixture of 2 mL methanol, 1 mL chloroform, and 1 mlLof double-distilled water (ddH2O) in a 15-mL glass centrifuge tube, the mixture was sonicated (probe sonicator) with the tube on ice for 1 min. An additional 1 mL of ddH2O and 1 mL of chloroform were added to the mixture before a second 1 min sonication was performed. The mixture was centrifuged at 8,000 rpm and 4 °C for 10 minutes to separate the hydrophobic and hydrophilic phases. The bottom hydrophobic phase was recovered and saved. The hydrophilic phase was extracted a second time with 1 mL of fresh chloroform and the mixture was recentrifuged. The second hydrophobic phase was added to the first and the combination was dried under a stream of nitrogen at room temperature. The dried residue was dissolved in 0.35 mL of CDCl3 and analyzed with a 400 MHz NMR spectrometer (Young et al., 2013).
Glucose and Oleate Tracing Assay by Mass Spectrometry
Media were aspirated, and cells were rinsed twice with 2 mL of PBS in room temperature. Then, 1 mL of methanol: water [1:1 (vol/vol)] solution was added with 0.1 M HCl at −20 °C. The resulting liquid and cell debris were scraped into a microfuge tube. 0.3 M KOH was then added, and the mix was incubated at 80 °C for 1 h to saponify FAs, acidified with 0.1 mL of formic acid, extracted twice with 1 mL of hexane, and N2-dried. Finally, it was reconstituted into 1:1:0.3 (vol) methanol: chloroform: water (1 mL of solvent per 2 μL of packed volume for cells, and 2 mL of solvent total for the medium samples) for liquid chromatography–mass spectrometry (LC-MS) analysis. Separation was done by reversed-phase ion-pairing chromatography on a C8 column coupled to negative-ion mode, full-scan LC-MS at 100,000 resolving power [stand-alone Orbitrap (Q-Exactive); Thermo Fischer Scientific].
Glucose and Glutamine Incorporation into De Novo Fatty Acid Synthesis by Mass Spectrometry
P493–6 cells were grown in RPMI1640 medium that contained 11 mM glucose and 3 mM glutamine. The medium was supplemented with 10% fetal bovine serum (Gemini Foundation) and 1% penicillin/streptomycin. For 13C labeling experiments, the medium contained either [U-13C6]glucose and unenriched glutamine or [U-13C5]glutamine and unenriched glucose. Twelve dishes were inoculated at an initial density of 3.2 × 105 cells/mL while the starting medium was not 13C enriched. Six dishes were allowed to grow without treatment, and six remaining dishes were treated with doxycycline 0.1 μg/mL (MYC OFF). After 24 hours, the cultures reached a density of approximately 6 × 105 cells/ml, the medium for the non-drug treated cultures was changed to 13C enriched medium. Three dishes were changed to medium with [U-13C6]glucose and unenriched glutamine. Three remaining dishes were changed to medium with [U-13C5]glutamine and unenriched glucose. After 24 hours, the medium was rapidly removed and the cultures were extracted with methanol and chloroform as described previously (Xie et al., 2018a). The doxycycline treated cultures were incubated in the presence of the drug for 42 hours to ensure a high level of MYC suppression. Subsequently, the medium was changed to RPMI1640 with doxycycline and either [U-13C6]glucose and unenriched glutamine (three dishes) or [U-13C5]glutamine and unenriched glucose (three dishes). After 24 hours of incubation in the enriched medium, during which no significant increase in cell density was observed, the medium was rapidly removed and the cultures were extracted with methanol and chloroform as described more in details previously (Xie et al., 2018a).
Desorption Electrospray Ionization Mass Spectrometry Imaging (DESI-MSI)
DESI-MSI was used for generating 2D chemical maps of tissue sections approximately 1cm in length to assess lipid and metabolite profiles of various organs. A detailed description of a standard protocol for this technique can be found in previously published papers (Eberlin et al., 2014; Perry et al., 2013; Shroff et al., 2015). Briefly, 16-μm thick tissue sections were imaged at a spatial resolution of 200 μm using a lab-built DESI-MSI source and a 2D moving stage coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific). The mass spectra were acquired in the negative ion mode using the Orbitrap as the mass analyzer at 60,000 resolving power, with the spray voltage set to −5 kV, the capillary voltage set to −65 V, and the tube lens voltage set to −120 V. These conditions were found to be optimal for the detection of free FAs, FA dimers, and complex GPs of different classes (Perry et al., 2013). At least three tissue samples for each disease model and state were imaged. We used tissue nondestructive solvent system dimethylformamide: acetonitrile [1:1 (vol:vol)] (Eberlin et al., 2011) at a flow rate of 0.8 μL/min assisted by a nebulizing gas (N2) at a pressure of 175 psi to desorb and ionize molecules of interest from the tissue. The same or adjacent specimen section was histologically stained after imaging using haematoxylin and eosin (H&E) staining protocol for unfixed tissue (Calligaris et al., 2013).
DESI-MSI Data Processing
Spatially accurate ion images were assembled using BioMap software (freeware, http://www.ms-imaging.org), and a rainbow color palette is used to visualize a variability in signal intensity. Each image was normalized by the total ion signal in BioMap to account for variations between experiments. All tissues were histopathologically evaluated after imaging using standard H&E staining to ensure precise tumor delineation. Regions of interest (ROIs) were then selected for each sample based on pathological evaluation, and the abundance of the exact m/z corresponding to each molecular species of interest was obtained from the data and reported as fold change values. Tandem MS analyses were performed using both the Orbitrap and the linear ion trap for mass analysis to determine the structural identity of the species. The lipid compositions were assigned based on the most prevalent tandem MS fragmentation pattern and had an absolute mass error of less than 4 ppm. The LipidMaps database (http://www.lipidmaps.org/) was employed to assist with lipid identification.
OCR measurement by Seahorse XFe96 Analyzer
The oxygen consumption rate (OCR) was analyzed with the Seahorse XF Cell Mito Stress Test (Part #103015–100; Agilent Technologies, Santa Clara, CA, USA) in a Seahorse XFe96 Analyzer (Agilent Technologies). The day before the experiment, 30,000 cells per well were plated in a 96-well Seahorse plate (pre-coated with D-lysine (Sigma)) in 80 μL DMEM 10% FCS medium. The Agilent Seahorse XFe96 Sensor Cartridge was hydrated with 200 μL/well of XF calibrant solution overnight in a non-CO2 incubator at 37 °C.
On the day of the experiment, 100 mL of Seahorse assay medium containing 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose was prepared. The pH of the pre-warmed (37 °C) medium was adjusted to 7.4 with 1 N NaOH. Cells were washed twice with 200 μL of the corresponding Seahorse medium and incubated with 175 μL of the Seahorse medium per well in a non-CO2 incubator at 37 °C for 1 h. Meanwhile, the Seahorse sensor cartridge ports were loaded with 25 μl of inhibitors to have a final concentration of 2 μM oligomycin (port A, Calbiochem), 1 μM FCCP (port B, Sigma-Aldrich), and 0.5 μM rotenone & antimycin A (port C, Sigma-Aldrich). The experimental design was setup using the WAVE software program, and measurements were performed in the Seahorse XFe96 Analyzer. After the measurement, supernatant from the cells was removed and the cells were fixed by addition of 100 μL 3.7% PFA (Merck) for 10 min at RT and air dried. Subsequently, the cells were stained using 0.1% crystal violet (Sigma) solution (in 20% Ethanol). For quantification, stained plates were incubated with 200 μl of 10% acetic acid per well with shaking for 15 min and the resulting solution was analyzed in a plate reader (Multiskan Ascent from Thermo labsystems) at 550 nm and the values were used for normalization of the seahorse data.
QUANTIFICATION AND STATISTICAL ANALYSIS
We performed power calculations based on our prior knowledge of variation in tumor latency, frequency, and progression. Thus, we used the minimal number of mice sufficient to detect the effect of interest and obtain reproducible, statistically significant, and biologically meaningful results. For example, we expected a two-fold difference in mean (M) tumor size in transgenic models between two groups, with a standard deviation (SD) of less than 50%. With Cohen’s effect size d=(M1−M2)/sqrt((SD1+SD2)/2), a type I error of 0.05, and a type II error of 0.9, the minimum sample size would be seven mice per group (R, packages: pwr, pwr2). Statistical analysis was carried out with Prism (GraphPad Software, Inc.). Unpaired 2-tailed Student’s t tests and a two-way Anova with a Tukey post hoc tests were used to analyze the data. Statistical significance values were set as *P<0.05, **P<0.01, ***P<0.001. A P-value less than 0.05 would be considered statistically significant. Unless otherwise indicated, results are presented as an arithmetic mean for each data set ± standard deviation. N represents repetition number or number of animals used for each condition point of experiment. Our data are consistent with the assumptions behind the statistical methods employed to analyze them. All statistical details regarding P-value and N can be found in main and supplementary figure legends.
ADDITIONAL RESOURCES
Publicly Available Data Accession Numbers
Gene Expression Omnibus (GEO) accession numbers for publicly available ChIP-Seq and RNA-seq data used in this study are GSE36354 and GSE120246 respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-SREBP1 (clone 2A4) [1:1000] | home-made antibody | N/A |
| Rabbit monoclonal anti-MYC (Y69) [1:1000] | Abcam | CAS# ab32072 |
| IgG from mouse serum [1:1000] | Sigma | CAS# I5006 |
| IgG from rabbit serum [1:1000] | Sigma | CAS# I5381 |
| MYC [1:1000] | Abcam | Ab32072 |
| SREBP1 [1:1000] | Novus Biologicals | NB600–582 |
| SCD1 [1:1000] | Cell Signaling Tech | CST 2794 |
| FASN [1:500] | Sigma-Aldrich | F9554 |
| ACLY [1:1000] | Cell Signaling Tech | CST 4332S |
| ACACA [1:1000] | Cell Signaling Tech | CST 3661 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Sigma-Aldrich | CAS# C2920 |
| Rotenone | Sigma-Aldrich | CAS# R8875 |
| Seahorse XFe96 FluxPak | Agilent Technologies | CAS# 102416–100 |
| XF Base Medium | Seahorse Bioscience | CAS# 102353–100 |
| D-Glucose-1-13C | Sigma | 297046 |
| L-Glutamine-13C5 | Sigma | 605166 |
| Critical Commercial Assays | ||
| ChIP-IT® Express Chromatin Immunoprecipitation Kits | Active Motif | 53008 |
| Qiagen RNA Extraction kit (RNEasy Plus Kit) | Qiagen | 74134 |
| Deposited Data | ||
| MYC Targeted Long Noncoding RNA DANCR Promotes Cancer in Part by Reducing p21 Levels. | GSE120246 | |
| Experimental Models: Cell Lines | ||
| Human: Glioblastoma U87 cells | Cell Services (CRUK-LRI) | N/A |
| Human: Renal cancer 786–0 cells | ATCC | CRL1932 |
| P493–6 Burkitt Lymphoma | (RRID:CVCL_6783) | |
| Experimental Models: Organisms/Strains | ||
| MYC transgenic mice also available from Jackson FVB/N-Tg(tetO-MYC)28aBop/J | Jackson Lab | 019376 |
| Liver-specific mice also available as MGI:Tg(Cebpb-tTA)5Bjd or (LAP-tTA) crossed with MYC transgenic mice to generate MYC HCC | MGI | MGI:3056818 |
| Kidney-specific mice also available at MGI: Tg(Ggt1-tTA)#Agoc or (GGT-tTA) crossed with MYC transgenic mice to generate MYC RCC | MGI | MGI:5705528 |
| Lung-specific mice also available from Jackson: B6.Cg-Tg(Scgb1a1-rtTA)1Jaw/J or (CCSP-tTA) crossed with MYC transgenic mice to generate MYC LC | Jackson Lab | 006232 |
| Oligonucleotides | ||
| ACLY/Acly | Harvard Primer Bank | GGTCTCGTTGGGGTCAACC / GAAGGGCTCGATCAGAAAGTTC |
| ACACA/Acaca | Harvard Primer Bank | ATGTCTGGCTTGCACCTAGTA / CCCCAAAGCGAGTAACAAATTCT |
| FASN/Fasn | Harvard Primer Bank | AAGGACCTGTCTAGGTTTGATGC / TGGCTTCATAGGTGACTTCCA |
| SCD1/Scd1 | Harvard Primer Bank | ACTGCCAGAGAACATCACACA / GGGGTAGGGTAAGCACAGTAG |
| SREBP1/Srebp1 | Harvard Primer Bank | ACAGTGACTTCCCTGGCCTAT / GCATGGACGGGTACATCTTCAA |
| HK2 | Harvard Primer Bank | GAGCCACCACTCACCCTACT / CCAGGCATTCGGCAATGTG |
| LDHA | Harvard Primer Bank | ATGGCAACTCTAAAGGATCAGC / CCAACCCCAACAACTGTAATCT |
| GLS | Harvard Primer Bank | AGGGTCTGTTACCTAGCTTGG / ACGTTCGCAATCCTGTAGATTT |
| SCAP | Harvard Primer Bank | TATCTCGGGCCTTCTACAACC / GGGGCGAGTAATCCTTCACA |
| CS | Harvard Primer Bank | TGCTTCCTCCACGAATTTGAAA / CCACCATACATCATGTCCACAG |
| Software and Algorithms | ||
| Seahorse Wave Controller Software | Instrument software by Agilent | https://www.agilent.com/en/products/cell-analysis/cell-analysis-software/instrument-software/wave-controller-2-4 |
| Bio Map software | Novartis | http://www.ms-imaging.org |
| Other | ||
| AGPAT1 | Harvard Primer Bank | AGGACGCAACGTCGAGAAC / GCAGTACCTCCATCATCCCAAG |
| PGS1 | Harvard Primer Bank | CTCCAGTTCTCACGTTAGGGT / CCTCTTGGCTACTCTTATCTGCC |
| PTPMT1 | Harvard Primer Bank | CAGAGGAGGCTGTAAGAGCCA / TGTGGATGTATGACCGGATCT |
Highlights.
MYC induces SREBP1, thereby regulating fatty acid synthesis and lipogenesis
Induction of MYC expression upregulates lipogenesis genes across MYC-cancers
Mass spectrometry imaging shows common lipid changes across different MYC-cancers
Inhibition of lipogenesis reverts MYC-tumors in both xenograft and mouse models
Context and Significance.
Understanding the metabolic requirements of cancer cells can lead to the development of new and effective therapies. In particular, metabolism that utilizes lipids (or fatty acids) is frequently perturbed in cancers but the underlying basis for the connection is unclear. Researchers at Stanford University and their collaborators have used a highly sensitive imaging approach called Desorption Electrospray Ionization Mass Spectrometry Imaging to monitor the tissue metabolism of various cancers that are driven by one of the most powerful oncogenes, MYC. The team employs MYC-cancer models as well as human cancer tissues to identify MYC-dependent metabolic signatures and then uses this information to decipher biological mechanisms of cancer generation. They present comprehensive evidence that the oncogene MYC, in collaboration with a DNA transcription factor known as SREBP1, regulates the production of lipids necessary for cancer formation. Their results point toward the promise of blocking lipid production as a general therapeutic strategy to combat common MYC-driven cancers.
ACKNOWLEDGEMENTS
We thank current and former members of the Dang, Felsher, Schulze, and Zare laboratories for their helpful suggestions during this project. We thank Livia Eberlin, Emelyn Shroff, Michael Wolfgang, Yunqi Lu, and Anne Le for their help.
AMG and KM are grateful to the Stanford Cancer Translational Nanotechnology Training (TNT) T32 training grant funded by the National Cancer Institute (T32 CA196585) and the Stanford Center of Molecular Analysis and Design, respectively. This work is supported by National Institutes of Health under R01 CA184384 (DWF and RNZ), R01 CA051497 (CVD) and R01 CA057341 (CVD) and the German Research Foundation (SCHU2670).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
International patent application No. PCT/US2019/019185 was issued based on results of this study. AMG, KM, RNZ and DWF are listed as inventors. The authors declare no other competing interests.
REFERENCES
- Afar DE, Goga A, McLaughlin J, Witte ON, and Sawyers CL (1994). Differential complementation of Bcr-Abl point mutants with c-Myc. Science 264, 424–426. [DOI] [PubMed] [Google Scholar]
- Agassandian M, and Mallampalli RK (2013). Surfactant phospholipid metabolism. Biochim Biophys Acta 1831, 612–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athenstaedt K, Weys S, Paltauf F, and Daum G (1999). Redundant systems of phosphatidic acid biosynthesis via acylation of glycerol-3-phosphate or dihydroxyacetone phosphate in the yeast Saccharomyces cerevisiae. Journal of bacteriology 181, 1458–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimi A, Kaufman KL, Ali M, Kossard S, and Fernandez-Penas P (2016). In Silico Analysis Validates Proteomic Findings of Formalin-fixed Paraffin Embedded Cutaneous Squamous Cell Carcinoma Tissue. Cancer genomics & proteomics 13, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenke F, Peck B, Miess H, and Schulze A (2013). Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 6, 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batenburg JJ, Klazinga W, and van Golde LM (1985). Regulation and location of phosphatidylglycerol and phosphatidylinositol synthesis in type II cells isolated from fetal rat lung. Biochim Biophys Acta 833, 17–24. [DOI] [PubMed] [Google Scholar]
- Baudino TA, and Cleveland JL (2001). The Max network gone mad. Molecular and cellular biology 21, 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer S, Zetterberg A, Ihrie RA, McTaggart RA, Yang Q, Bradon N, Arvanitis C, Attardi LD, Feng S, Ruebner B, et al. (2004). Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol 2, e332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood EM, Kretzner L, and Eisenman RN (1992). Myc and Max function as a nucleoprotein complex. Current opinion in genetics & development 2, 227–235. [DOI] [PubMed] [Google Scholar]
- Bokhart MT, and Muddiman DC (2016). Infrared matrix-assisted laser desorption electrospray ionization mass spectrometry imaging analysis of biospecimens. Analyst 141, 5236–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calligaris D, Norton I, Feldman DR, Ide JL, Dunn IF, Eberlin LS, Cooks RG, Jolesz FA, Golby AJ, Santagata S, et al. (2013). Mass spectrometry imaging as a tool for surgical decision-making. J Mass Spectrom 48, 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll PA, Diolaiti D, McFerrin L, Gu H, Djukovic D, Du J, Cheng PF, Anderson S, Ulrich M, Hurley JB, et al. (2015). Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer cell 27, 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey SC, Baylot V, and Felsher DW (2018). The MYC Oncogene is a Global Regulator of the Immune Response. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, et al. (2004). Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116, 499–509. [DOI] [PubMed] [Google Scholar]
- Chang SC, Heacock PN, Clancey CJ, and Dowhan W (1998). The PEL1 gene (renamed PGS1) encodes the phosphatidylglycero-phosphate synthase of Saccharomyces cerevisiae. The Journal of biological chemistry 273, 9829–9836. [DOI] [PubMed] [Google Scholar]
- Collier JJ, Zhang P, Pedersen KB, Burke SJ, Haycock JW, and Scott DK (2007). c-Myc and ChREBP regulate glucose-mediated expression of the L-type pyruvate kinase gene in INS-1-derived 832/13 cells. Am J Physiol Endocrinol Metab 293, E48–56. [DOI] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2013). MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, and Li F (2006). The c-Myc target gene network. Seminars in cancer biology 16, 253–264. [DOI] [PubMed] [Google Scholar]
- Djouadi F, Bonnefont JP, Munnich A, and Bastin J (2003). Characterization of fatty acid oxidation in human muscle mitochondria and myoblasts. Molecular genetics and metabolism 78, 112–118. [DOI] [PubMed] [Google Scholar]
- Dolinsky VW, Cole LK, Sparagna GC, and Hatch GM (2016). Cardiac mitochondrial energy metabolism in heart failure: Role of cardiolipin and sirtuins. Biochim Biophys Acta 1860, 1544–1554. [DOI] [PubMed] [Google Scholar]
- Eberlin LS, Ferreira CR, Dill AL, Ifa DR, Cheng L, and Cooks RG (2011). Nondestructive, histologically compatible tissue imaging by desorption electrospray ionization mass spectrometry. Chembiochem 12, 2129–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberlin LS, Gabay M, Fan AC, Gouw AM, Tibshirani RJ, Felsher DW, and Zare RN (2014). Alteration of the lipid profile in lymphomas induced by MYC overexpression. Proceedings of the National Academy of Sciences of the United States of America 111, 10450–10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds LR, Sharma L, Kang A, Lu J, Vockley J, Basu S, Uppala R, Goetzman ES, Beck ME, Scott D, et al. (2015). c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. The Journal of biological chemistry 290, 20100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Chen YC, Watkins T, Dang CV, Gorospe M, and Cheadle C (2012). Array-based nuclear run-on analysis. Methods Mol. Biol 809, 505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell AS, and Sears RC (2014). MYC degradation. Cold Spring Harbor perspectives in medicine 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsher DW (2004). Reversibility of oncogene-induced cancer. Current opinion in genetics & development 14, 37–42. [DOI] [PubMed] [Google Scholar]
- Felsher DW (2010). MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer 1, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsher DW, and Bishop JM (1999). Reversible tumorigenesis by MYC in hematopoietic lineages. Molecular cell 4, 199–207. [DOI] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, and Amati B (2003). Genomic targets of the human c-Myc protein. Genes & development 17, 1115–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickes R, Voelker DR, Berry KZ, and Murphy RC (2016). Tandem mass spectrometry of novel ether-linked phospholipid analogs of anionic pulmonary surfactant phospholipids. Rapid communications in mass spectrometry: RCM 30, 2601–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. (2012). Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, and Gonzalez-Baron M (2004). PI3K/Akt signalling pathway and cancer. Cancer treatment reviews 30, 193–204. [DOI] [PubMed] [Google Scholar]
- Gabay M, Li Y, and Felsher DW (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harbor perspectives in medicine 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouw AM, Eberlin LS, Margulis K, Sullivan DK, Toal GG, Tong L, Zare RN, and Felsher DW (2017). Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci U S A 114, 4300–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieb BC, Boyd K, Mitra R, and Eischen CM (2016). Haploinsufficiency of the Myc regulator Mtbp extends survival and delays tumor development in aging mice. Aging (Albany NY) 8, 2590–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griese M, Gobran LI, and Rooney SA (1991). ATP-stimulated inositol phospholipid metabolism and surfactant secretion in rat type II pneumocytes. The American journal of physiology 260, L586–593. [DOI] [PubMed] [Google Scholar]
- Hill EE, Husbands DR, and Lands WE (1968). The selective incorporation of 14C-glycerol into different species of phosphatidic acid, phosphatidylethanolamine, and phosphatidylcholine. The Journal of biological chemistry 243, 4440–4451. [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, and Brown MS (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, and Vaz FM (2008). Cardiolipin, the heart of mitochondrial metabolism. Cellular and molecular life sciences: CMLS 65, 2493–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XF, and Chen JZ (2009). Obesity, the PI3K/Akt signal pathway and colon cancer. Obesity reviews: an official journal of the International Association for the Study of Obesity 10, 610–616. [DOI] [PubMed] [Google Scholar]
- Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, Doan HM, Fan J, Cheadle C, Fallahi M, et al. (2011). Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PloS one 6, e26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, Tamachi A, Tu WB, and Penn LZ (2017). MYC Deregulation in Primary Human Cancers. Genes (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebish MA, Han X, Cheng H, Chuang JH, and Seyfried TN (2008). Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: lipidomic evidence supporting the Warburg theory of cancer. Journal of lipid research 49, 2545–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC (2015). Metabolic Dependencies in RAS-Driven Cancers. Clinical cancer research: an official journal of the American Association for Cancer Research 21, 1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolk MV, Meyberg D, Deuse T, Tang-Quan KR, Robbins RC, Reichenspurner H, and Schrepfer S (2009). LAD-ligation: a murine model of myocardial infarction. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell metabolism 15, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CA, Griffiths B, Santos CR, Pende M, and Schulze A (2011). Regulation of the SREBP transcription factors by mTORC1. Biochemical Society transactions 39, 495–499. [DOI] [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim JW, Yustein JT, Lee LA, and Dang CV (2005). Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Molecular and cellular biology 25, 6225–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wu JB, Li Q, Shigemura K, Chung LW, and Huang WC (2016). SREBP-2 promotes stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer. Oncotarget 7, 12869–12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang W, Shui G, and Huang X (2014). CDP-diacylglycerol synthetase coordinates cell growth and fat storage through phosphatidylinositol metabolism and the insulin pathway. PLoS genetics 10, e1004172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo DX, Tong DJ, Rajput S, Wang C, Liao DF, Cao D, and Maser E (2012). Targeting acetyl-CoA carboxylases: small molecular inhibitors and their therapeutic potential. Recent patents on anti-cancer drug discovery 7, 168–184. [DOI] [PubMed] [Google Scholar]
- Marsh D (2013). Handbook of Lipid Bilayers. (Boca Raton: CRC Press, Taylor & Francis Group; ). [Google Scholar]
- McMahon SB (2014). MYC and the control of apoptosis. Cold Spring Harb Perspect Med 4, a014407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson R, and Gauthier A (2004). Molecular regulation of SREBP function: the Insig-SCAP connection and isoform-specific modulation of lipid synthesis. Biochemistry and cell biology = Biochimie et biologie cellulaire 82, 201–211. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2012). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon YA, Ochoa CR, Mitsche MA, Hammer RE, and Horton JD (2014). Deletion of ELOVL6 blocks the synthesis of oleic acid but does not prevent the development of fatty liver or insulin resistance. Journal of lipid research 55, 2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita SY, and Terada T (2015). Enzymatic measurement of phosphatidylglycerol and cardiolipin in cultured cells and mitochondria. Scientific reports 5, 11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish F, Noonan J, Perez-Olsen C, Gafken PR, Fitzgibbon M, Kelleher J, VanGilst M, and Hockenbery D (2010). Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. The Journal of biological chemistry 285, 36267–36274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller I, Larsson K, Frenzel A, Oliynyk G, Zirath H, Prochownik EV, Westwood NJ, and Henriksson MA (2014). Targeting of the MYCN protein with small molecule c-MYC inhibitors. PLoS One 9, e97285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie J, Hao X, Chen D, Han X, Chang Z, and Shi Y (2010). A novel function of the human CLS1 in phosphatidylglycerol synthesis and remodeling. Biochim Biophys Acta 1801, 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. (2012). c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofman R, Dijkstra IM, van Roermund CW, Burger N, Turkenburg M, van Cruchten A, van Engen CE, Wanders RJ, and Kemp S (2010). The role of ELOVL1 in very long-chain fatty acid homeostasis and X-linked adrenoleukodystrophy. EMBO molecular medicine 2, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacilli A, Calienni M, Margarucci S, D’Apolito M, Petillo O, Rocchi L, Pasquinelli G, Nicolai R, Koverech A, Calvani M, et al. (2013). Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis. Journal of the National Cancer Institute 105, 489–498. [DOI] [PubMed] [Google Scholar]
- Pajic A, Spitkovsky D, Christoph B, Kempkes B, Schuhmacher M, Staege MS, Brielmeier M, Ellwart J, Kohlhuber F, Bornkamm GW, et al. (2000). Cell cycle activation by c-myc in a burkitt lymphoma model cell line. International journal of cancer 87, 787–793. [DOI] [PubMed] [Google Scholar]
- Pauter AM, Olsson P, Asadi A, Herslof B, Csikasz RI, Zadravec D, and Jacobsson A (2014). Elovl2 ablation demonstrates that systemic DHA is endogenously produced and is essential for lipid homeostasis in mice. Journal of lipid research 55, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RH, Bellovin DI, Shroff EH, Ismail AI, Zabuawala T, Felsher DW, and Zare RN (2013). Characterization of MYC-induced tumorigenesis by in situ lipid profiling. Analytical chemistry 85, 4259–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk JG Jr., Baumgarten B, and Bleasdale JE (1984). Effect of myo-inositol on the glycerophospholipid composition of adult and fetal rat lung tissue. Journal of perinatal medicine 12, 201–210. [DOI] [PubMed] [Google Scholar]
- Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, Fan AC, Yang Q, Braunstein L, Crosby E, et al. (2010). CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 18, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al. (2014). Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santinon G, Pocaterra A, and Dupont S (2016). Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends in cell biology 26, 289–299. [DOI] [PubMed] [Google Scholar]
- Sawyers CL, Callahan W, and Witte ON (1992). Dominant negative MYC blocks transformation by ABL oncogenes. Cell 70, 901–910. [DOI] [PubMed] [Google Scholar]
- Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, Eick D, and Kohlhuber F (1999). Control of cell growth by c-Myc in the absence of cell division. Current biology: CB 9, 1255–1258. [DOI] [PubMed] [Google Scholar]
- Sears R, Leone G, DeGregori J, and Nevins JR (1999). Ras enhances Myc protein stability. Molecular cell 3, 169–179. [DOI] [PubMed] [Google Scholar]
- Shao W, and Espenshade PJ (2014). Sterol regulatory element-binding protein (SREBP) cleavage regulates Golgi-to-endoplasmic reticulum recycling of SREBP cleavage-activating protein (SCAP). The Journal of biological chemistry 289, 7547–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, and Dowhan W (1998). Regulation of phosphatidylglycerophosphate synthase levels in Saccharomyces cerevisiae. The Journal of biological chemistry 273, 11638–11642. [DOI] [PubMed] [Google Scholar]
- Shimizu-Albergine M, Van Yserloo B, Golkowski MG, Ong SE, Beavo JA, and Bornfeldt KE (2016). SCAP/SREBP pathway is required for the full steroidogenic response to cyclic AMP. Proceedings of the National Academy of Sciences of the United States of America 113, E5685–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiragami R, Murata S, Kosugi C, Tezuka T, Yamazaki M, Hirano A, Yoshimura Y, Suzuki M, Shuto K, and Koda K (2013). Enhanced antitumor activity of cerulenin combined with oxaliplatin in human colon cancer cells. International journal of oncology 43, 431–438. [DOI] [PubMed] [Google Scholar]
- Shroff EH, Eberlin LS, Dang VM, Gouw AM, Gabay M, Adam SJ, Bellovin DI, Tran PT, Philbrick WM, Garcia-Ocana A, et al. (2015). MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proceedings of the National Academy of Sciences of the United States of America 112, 6539–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, et al. (2014). Metabolic control of YAP and TAZ by the mevalonate pathway. Nature cell biology 16, 357–366. [DOI] [PubMed] [Google Scholar]
- Southam AD, Khanim FL, Hayden RE, Constantinou JK, Koczula KM, Michell RH, Viant MR, Drayson MT, and Bunce CM (2015). Drug Redeployment to Kill Leukemia and Lymphoma Cells by Disrupting SCD1-Mediated Synthesis of Monounsaturated Fatty Acids. Cancer Res 75, 2530–2540. [DOI] [PubMed] [Google Scholar]
- Sun Y, He W, Luo M, Zhou Y, Chang G, Ren W, Wu K, Li X, Shen J, Zhao X, et al. (2015). SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. [DOI] [PubMed] [Google Scholar]
- Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, et al. (2016). Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nature medicine 22, 1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen JV, Brusselmans K, and Verhoeven G (2006). Increased lipogenesis in cancer cells: new players, novel targets. Current opinion in clinical nutrition and metabolic care 9, 358–365. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, and Reue K (2009). Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab 296, E1195–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, and Leder P (1982). Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proceedings of the National Academy of Sciences of the United States of America 79, 7837–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thupari JN, Pinn ML, and Kuhajda FP (2001). Fatty acid synthase inhibition in human breast cancer cells leads to malonyl-CoA-induced inhibition of fatty acid oxidation and cytotoxicity. Biochemical and biophysical research communications 285, 217–223. [DOI] [PubMed] [Google Scholar]
- Truax AD, and Greer SF (2012). ChIP and Re-ChIP assays: investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods in molecular biology (Clifton, N.J.) 809, 175–188. [DOI] [PubMed] [Google Scholar]
- Visel A, Thaller C, and Eichele G (2004). GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic acids research 32, D552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstrom T, and Henriksson MA (2015). Impact of MYC in regulation of tumor cell metabolism. Biochim Biophys Acta 1849, 563–569. [DOI] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al. (2014a). Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al. (2014b). Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Xu C, Sun M, Luo D, Liao DF, and Cao D (2009). Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochem. Biophys. Res. Commun 385, 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZR, Liu W, Smith ST, Parrish RS, and Young SR (1999). c-myc and chromosome 8 centromere studies of ovarian cancer by interphase FISH. Experimental and molecular pathology 66, 140–148. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems L, Tamburini J, Chapuis N, Lacombe C, Mayeux P, and Bouscary D (2012). PI3K and mTOR signaling pathways in cancer: new data on targeted therapies. Current oncology reports 14, 129–138. [DOI] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America 105, 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman JM, Ifa DR, Song Q, and Cooks RG (2006). Tissue imaging at atmospheric pressure using desorption electrospray ionization (DESI) mass spectrometry. Angew Chem Int Ed Engl 45, 7188–7192. [DOI] [PubMed] [Google Scholar]
- Wu Y, Chen K, Liu X, Huang L, Zhao D, Li L, Gao M, Pei D, Wang C, and Liu X (2016). Srebp-1 Interacts with c-Myc to Enhance Somatic Cell Reprogramming. Stem cells (Dayton, Ohio) 34, 83–92. [DOI] [PubMed] [Google Scholar]
- Xie H, Tang C-HA, Song JH, Mancuso A, Del Valle JR, Cao J, Xiang Y, Dang CV, Lan R, Sanchez DJ, et al. (2018a). IRE1α RNase–dependent lipid homeostasis promotes survival in Myc-transformed cancers. The Journal of Clinical Investigation 128, 1300–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Tang CH, Song JH, Mancuso A, Del Valle JR, Cao J, Xiang Y, Dang CV, Lan R, Sanchez DJ, et al. (2018b). IRE1alpha RNase-dependent lipid homeostasis promotes survival in Myc-transformed cancers. J Clin Invest 128, 1300–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Cao J, and Shi Y (2004). Identification and characterization of a gene encoding human LPGAT1, an endoplasmic reticulum-associated lysophosphatidylglycerol acyltransferase. The Journal of biological chemistry 279, 55866–55874. [DOI] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Ackerman D, Quinn ZL, Mancuso A, Gruber M, Liu L, Giannoukos DN, Bobrovnikova-Marjon E, Diehl JA, Keith B, et al. (2013). Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes & development 27, 1115–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Guan Z, Murphy AN, Wiley SE, Perkins GA, Worby CA, Engel JL, Heacock P, Nguyen OK, Wang JH, et al. (2011). Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell metabolism 13, 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Metukuri MR, Bindom SM, Prochownik EV, O’Doherty RM, and Scott DK (2010). c-Myc is required for the CHREBP-dependent activation of glucose-responsive genes. Mol Endocrinol 24, 1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Cao J, Xu L, Tang Q, Dobrolecki LE, Lv X, Talukdar M, Lu Y, Wang X, Hu DZ, et al. (2018). Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. The Journal of clinical investigation 128, 1283–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.