

Abstract
The regulators of mitochondrial cell death in cancer have remained elusive, hampering the development of new therapies. Here, we showed that protein isoforms of Mitochondrial Fission Factor (MFF1 and MFF2), a molecule that controls mitochondrial size and shape, i.e. mitochondrial dynamics, were overexpressed in patients with non-small cell lung cancer and formed homo- and heterodimeric complexes with the voltage-dependent anion channel-1 (VDAC1), a key regulator of mitochondrial outer membrane permeability. MFF inserted into the interior hole of the VDAC1 ring using Arg225, Arg236 and Gln241 as key contact sites. A cell-permeable MFF Ser223-Leu243 D-enantiomeric peptidomimetic disrupted the MFF-VDAC1 complex, acutely depolarized mitochondria and triggered cell death in heterogeneous tumor types, including drug-resistant melanoma, but had no effect on normal cells. In preclinical models, treatment with the MFF peptidomimetic was well-tolerated and demonstrated anticancer activity in patient-derived xenografts, primary breast and lung adenocarcinoma 3D organoids and glioblastoma neurospheres. These data identify the MFF-VDAC1 complex as a novel regulator of mitochondrial cell death and an actionable therapeutic target in cancer.
Keywords: Mitochondria, cell death, VDAC, cancer therapy
INTRODUCTION
As signaling hubs in the eukaryotic cell (1), mitochondria control multiple forms of regulated cell death (2), including apoptosis (3) and necrosis (4). These processes involve a sudden increase in the permeability of the mitochondrial outer membrane (2), which, through as yet unclear mechanisms, dissipates inner membrane potential, uncouples the electron transport chain and releases cell death-promoting proteins in the cytosol (5). The regulators of mitochondrial cell death have remained mostly elusive (6), and the composition of a putative permeability transition pore (PTP) is still much debated (7). However, there is agreement that isoforms of Voltage-Dependent Anion Channel (VDAC) (8) are key effectors of mitochondrial outer membrane permeability coupling to organelle cell death (9). These mechanisms are invariably subverted in cancer (10), where decreased mitochondrial permeability promotes tumor cell survival and confers treatment resistance (3).
A second process commonly exploited in cancer is mitochondrial dynamics (11), which controls the size, shape and subcellular position of mitochondria. Although important to titrate organelle fitness under stress conditions (12), deregulated mitochondrial dynamics, in particular fission has been implicated in MAPK- (13) and Ras-dependent tumorigenesis (14), as well as tumor cell invasion and metastatic spreading, in vivo (15,16). There is evidence that mitochondrial dynamics couples to apoptosis (17), but a direct, mechanistic link between mitochondrial fission and effectors of organelle cell death, especially in cancer, has not been demonstrated.
In this study, we uncovered a novel, therapeutically actionable interface between a regulator of mitochondrial fission, Mitochondrial Fission Factor (MFF) (18) and VDAC1 in the regulation of organelle cell death, selectively in cancer (19).
MATERIALS AND METHODS
Patient samples
A clinically-annotated series of 72 patients with histologically confirmed diagnosis of non-small cell lung cancer (NSCLC) was used in these studies (Supplementary Table S1). Archival tissues and clinical records were obtained from Fondazione IRCCS Caʹ Granda Hospital in Milan (Italy) as described (19) and written informed consent was waived for the retrospective nature of the study. NSCLC and normal bronchus tissue samples were arranged in tissue microarrays (TMA) blocks for immunohistochemical evaluations. For organotypic tissue cultures, one breast cancer and two NSCLC tissue samples (Supplementary Table S1) were subject to tissue slicing and ex-vivo culturing as described (20). Cultures were treated with cell-permeable (D) MFF peptidomimetic or cell-permeable scrambled peptide (50–100 μM) for 2 h or 24 h. At harvesting, tissues were formalin-fixed, paraffin-embedded and processed for immunohistochemical analyses.
Immunohistochemistry
Four μm-thick sections from each tissue block were stained with an antibody to MFF (Protein Tech#17090–1-AP), Ki67 (clone 30–9; Ventana Medical Systems, Roche Group, Tucson, AZ, USA) or cleaved caspase-3 (9661; Cell Signaling Technologies) using diaminobenzidine (DAB). Immunohistochemistry was performed as described (19). Two pathologists (V.V. and S.F.) blinded to clinical data evaluated and scored all slides. When discrepancies occurred, the case was further reviewed to reach an agreement score.
Cells and cell culture
Human prostate adenocarcinoma (LNCaP, C4–2, C4–2B, PC3, DU145), NSCLC (A549, H460), breast adenocarcinoma (MDA-231, MCF7), metastatic melanoma (A2058), normal prostate epithelial (RWPE-1) and human glioblastoma (LN229) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and maintained in culture according to the supplier’s specifications. Benign prostatic hyperplasia (BPH-1) cells were a gift from Dr. Simon Hayward (Vanderbilt University, Nashville, TN) and primary human foreskin fibroblasts (HFF) were established at The Wistar Institute. WM983B and WM35 human metastatic melanoma cell lines were established at The Wistar Institute as previously described (21). Resistant cell lines were maintained in the presence of PLX4720 (3 μM) plus PD0325901 (300 nM) as described previously (22). In all conditions, cell passaging was limited to <40 passages from receipt and cell lines were authenticated by STR profiling with AmpFlSTR Identifiler PCR Amplification Kit (Life Technologies) at the Wistar Institute’s Genomics Shared Resource (19). Mycoplasma-free cultures were confirmed at the beginning of the studies, and every 2 months afterwards, by direct polymerase chain reaction (PCR) of cultures using Bioo Scientific Mycoplasma Primer Sets (#375501) and Hot Start polymerase (QIAGEN).
Antibodies and reagents
The following antibodies to MFF (Protein Tech #17090–1-AP), Hexokinase-I (HK-I, Cell signaling #2024), Hexokinase-II (HK-II, Cell Signaling #2867), VDAC (Cell Signaling #4866), PARP (Cell Signaling #9532), Asp214-cleaved PARP (Cell Signaling #9541), Asp175-cleaved Caspase-3 (Cell signaling #9661), HMGB1 (Cell Signaling #6893), Cyclophilin A (Cell Signaling #2175), Flag (Sigma-Aldrich #F1804), β-tubulin (Sigma-Aldrich #T8328) or β-actin (Sigma-Aldrich #A5441) were used for Western blotting. Libraries of MFF peptides and their scrambled sequences used as control (purity >90%) were synthesized by Thermo Fisher Scientific or GenScript. Z-VAD-fmk and Cyclosporin A (CsA) were purchased from Selleckchem.
Plasmids and transfections
cDNA clones encoding human MFF1, MFF5 or control vector were purchased from GeneCopoeia (Cat. n. EX-Z4766, EX-Z0675). An MFF2 cDNA was obtained from Addgene. An MFF4 cDNA was constructed by mutagenesis of the MFF5 cDNA using QuikChange II XL Site-Directed Mutagenesis kit (Agilent Technologies) and confirmed by DNA sequencing. Mutations in the MFF sequence were also generated using QuikChange II and confirmed by DNA sequencing. An MFF2 cDNA deleted in the VDAC-binding region 223SARGILSLIQSSTRRAYQQILDVL246 was generated by site-specific mutagenesis using QuickChange II and confirmed by DNA sequencing. Transfection of plasmid DNA (1 μg) was carried out using 2 μl X-Treme gene HP (Roche) for 24 h.
Identification of MFF-associated proteins using 1D proteomics
PC3 cells were transfected with MFF1 cDNA, solubilized in lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA) in the presence of 1% CHAPS, EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich) and Phosphatase Inhibitor Cocktail PhosSTOP (Roche). After sonication, lysates were centrifuged at 15,000 × g for 30 min, and cell extracts were incubated with anti-Flag-conjugated agarose bead (Sigma-Aldrich) for 4 h at 4°C (19). After five washes in lysis buffer, MFF-associated proteins were separated by SDS gel electrophoresis. The entire gel region was excised and digested with trypsin, as described (23). Tryptic peptides were analyzed by LC-MS/MS on a Q Exactive HF mass spectrometer (ThermoFisher Scientific) coupled with a Nano-ACQUITY UPLC system (Waters). Samples were injected onto a UPLC Symmetry trap column (180 μm i.d. x 2 cm packed with 5 μm C18 resin; Waters), and tryptic peptides were separated by RP-HPLC on a BEH C18 nanocapillary analytical column (75 μm i.d. x 25 cm, 1.7 μm particle size; Waters) using a 95-min gradient. Eluted peptides were analyzed by the mass spectrometer set to repetitively scan m/z from 400 to 2000 in positive ion mode. The full MS scan was collected at 60,000-resolution followed by data-dependent MS/MS scans at 15,000-resolution on the 20 most abundant ions exceeding a minimum threshold of 20,000. Peptide match was set as preferred, the exclude isotopes option and charge-state screening were enabled to reject singly and unassigned charged ions. Peptide sequences were identified using MaxQuant 1.5.2.8 (24). MS/MS spectra were searched against the UniProt human protein database (August 2015) using full tryptic specificity with up to two missed cleavages, static carboxamidomethylation of Cys, and variable oxidation of Met and protein N-terminal acetylation. Consensus identification lists were generated with false discovery rates of 1% at protein and peptide levels.
Bioinformatics analysis
Proteomics intensity data were floored to the minimum detected signal (intensity of 105) and log 2-scaled. Proteins were then annotated as mitochondrial-related using MitoCarta 2.0 database (25). Proteins detected in both MFF experiments with at least 10 peptides as well as at least 10 MS/MS spectra counts and at least 10-fold higher intensity than controls were reported as significant interacting proteins (25). Ingenuity Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) was used to identify all known protein-protein interactions between proteins in the list of candidates and their involvement in mitochondrial functions.
Immunoprecipitation
Cells were harvested and lysed with IP buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA) containing 1% CHAPS, EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich) and Phosphatase Inhibitor Cocktail PhosSTOP (Roche). After sonication, lysates were centrifuged at 15,000 × g for 30 min, and cell extracts were incubated with anti-Flag-conjugated beads (Sigma-Aldrich). The precipitates were washed five times, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and processed for Western blotting, as described (19). For analysis of direct protein-protein interaction, increasing concentrations (0–250 ng) of bead-coupled Flag-MFF1 proteins affinity-purified from PC3 cells were rinsed with reaction buffer (25 mM Hepes, pH 7.5) and mixed with recombinant VDAC1 in 500 μl of 25 mM Hepes (pH 7.5) for 2 h at 4°C. Upon completion of the binding reaction, 50 μl of supernatant from each sample (unbound material) and 10 μl out of 100 μl eluate pool obtained from the beads were separated by SDS gel electrophoresis followed by Western blotting with antibodies to MFF or VDAC.
Expression and purification of human VDAC1
A full-length human VDAC1 cDNA (hVDAC1) was cloned into pET28b vector containing an N-terminal hexahistidine-SUMO fusion tag, with a flexible linker cleavable by tobacco etch virus (TEV) protease. The hVDAC1 was expressed in E.coli ScarabXpress T7 lac competent cells (Scarab genomics) for 16 h at 16°C using 1 mM IPTG (Denville Scientific Inc.). The cells were harvested by centrifugation and lysed on ice via sonication in buffer containing 25 mM Tris-HCl (pH 7.5), 1 M Urea, 1 M KCl, 5% glycerol, 1 mM benzamidine and 1 mM PMSF (Ni Buffer A). After centrifugation at 18,000 rpm for 20 min at 4°C, the cell pellet was washed extensively in Ni Buffer A with 1% Triton X-100 and then solubilized in buffer containing 20 mM Tris-HCl (pH 7.9), 500 mM NaCl, 4 M guanidine-HCl and 10% glycerol for 45 min with gentle stirring. The supernatant was collected following centrifugation at 20,000 rpm for 10 min at 4°C. The protein was purified over nickel-nitrilotriacetic acid (Ni-NTA - Qiagen) column, buffer-exchanged to 25 mM Tris-HCl (pH 7.5), 500 mM KCl, 5% glycerol, 1 mM benzamidine and 1 mM PMSF (Ni buffer C) with 2% n-Octylglucoside (Research Products International), eluted with 300 mM imidazole and treated overnight with TEV at 4°C to cleave the His-SUMO tag. The protein was then buffer exchanged to buffer C with 100 mM salt and loaded onto tandem HS(poros)-HQ(poros) column to remove the TEV and the His-SUMO fusion tag. The cleaved, full-length hVDAC1 was collected from the HS-HQ flow through, concentrated using amicon ultra filter (10 kDa cut off) and used for further experiments.
Isothermal titration calorimetry (ITC)
ITC experiments were performed using MIcroCal iTC200 (Malvern). Purified full-length hVDAC1 was buffer-exchanged into 25 mM HEPES-KOH (pH 7.5), 0.1 M KCl, 5% glycerol, 2% n-Octylglucoside, and 1 mM TCEP (ITC buffer). Wild type (WT) MFF peptide 8#11 corresponding to the minimal VDAC binding site, 223SARGILSLIQSSTRRAYQQILDVL246, and its scrambled control, SSQLRYLARSQRITIQLIAGS (see below) were also prepared in ITC buffer. The ITC binding experiments were carried out at 20°C. Peptides at a concentration of 100 μM were added by 2.47 μl injections to 10 μM hVDAC1. The data collected was processed in MicroCal Origin software (Malvern).
hVDAC1-MFF model generation
The hVDAC1-MFF model was generated using the CABS-dock server, which uses an efficient protocol for the flexible docking of proteins and peptides (26,27). The coordinates of hVDAC1 (PDB ID: 2JK4 (28)) and the WT MFF peptide sequence (SARGILSLIQSSTRRAYQQIL) were provided for the modeling. The MFF peptide docking into hVDAC1 structure was carried out in three steps as described (26,27). In this study, we use the best binding mode of the peptide from the 10-top scored.
Peptidyl mimicry of MFF recognition
A library of partially overlapping synthetic peptides duplicating the entire MFF1 sequence is presented in Supplementary Table S1. A library of deletion mutant peptides based on MFF peptide #8 sequence 217DGANLSSARGILSLIQSSTRRAYQQILDVL246 was also synthesized (Supplementary Table S2). The minimal MFF interacting sequence with VDAC, designated peptide 8#11 with the sequence 223SARGILSLIQSSTRRAYQQIL243 and its corresponding scrambled version, SSQLRYLARSQRITIQLIAGS were also synthesized. To target the MFF-VDAC complex in tumor cells, the MFF peptide 8#11 was made cell permeable with the addition of an NH2-terminus biotin-Ahx linker and HIV Tat cell-penetrating sequence RQIKIWFQNRRMK. A cell-permeable control scrambled peptide #8–11 and an MFF peptide #8–11 mutant containing the double mutation Arg225Asp/Arg236Asp (DD) were also synthesized. To generate a clinical candidate of the MFF-VDAC pathway, in vivo, a retro-inverso D-enantiomer peptidomimetic of MFF #8–11 sequence was synthesized containing all D-amino acid in the reverse orientation, as described (29). A scrambled D-enantiomer peptide was also synthesized as control. All peptides were synthesized with >95% purity. For analysis of intramitochondrial accumulation using the Colorimetric Biotin Assay kit (Sigma #MAK171), PC3 cells were incubated with biotin-conjugated, cell-permeable MFF (D) 8–11 peptidomimetic (10 μM) or cell-permeable scrambled peptide (10 μM) for 30 min at 22°C. Isolated mitochondrial extracts were then treated with HABA (2-(4-Hydroxyphenylazo) benzoic acid)/avidin assay mixture for 5 min at 22°C and absorbance was quantified at 500 nm. In this assay, accumulation of the biotinylated peptidomimetic in mitochondrial samples displaces HABA from HABA/Avidin complex, thus leading to reduction in absorbance.
Analysis of mitochondrial membrane potential
PC3 cells were treated with cell-permeable scrambled peptide or cell-permeable MFF #8–11 peptide, washed three times in PBS, pH 7.4, and analyzed on a FACSCalibur flow cytometer, with the TMRE signal as FL1. Intact cells were gated in the FSC/SSC plot to exclude small debris. Cells treated with 20 μM FCCP 10 min prior to TMRE staining were used as a negative control. The resulting FL1 data were plotted on a histogram. For quantification of membrane potential of purified mitochondria, mitochondria isolated from PC3 cells (50 μg) were suspended in 400 μl of SB buffer (0.2 M sucrose, 10 mM Tris-MOPS, pH 7.4, 5 mM succinate, 1 mM sodium phosphate, 10 μM EGTA-Tris, 2 μM rotenone). Samples were treated with control scrambled peptide or the various MFF peptides for 20 min at 22°C, and incubated with 100 nM TMRE for 30 min at 22°C. After two washes with SB buffer, mitochondria were recovered by centrifugation at 6000 x g for 10 min, suspended in 100 μl of SB buffer and mitochondrial membrane potential was quantified on a florescence microplate reader (Ex/Em = 535/595 nm, Beckman Coulter). Samples treated with 2 mM CaCl2 were used as control for the lowest membrane potential (fully depolarized state).
Glioblastoma (GBM) neurospheres
Primary, patient-derived GBM neurospheres were isolated and maintained in culture as described (30). Tissue samples were obtained from chemotherapy- or radiotherapy-naïve patients with confirmed histological diagnosis of WHO grade IV glioma (GBM) undergoing treatment at the Neurosurgery Division of Fondazione IRCCS Caʹ Granda Ospedale Maggiore Policlinco (Milan, Italy). Written informed consent was obtained from all patients. Neurospheres were cyto-spinned on charged slides (Thermo Scientific, Waltham, MA) and treated with cell-permeable scrambled peptide (50 μM) or cell-permeable retro-inverso D-enantiomer MFF peptide (10–50 μM) in PBS, pH 7.4, for up to 24 h. Control-normalized changes in cell viability under the various conditions were determined by the ratio of Calcein (live cells) and To-Pro (dead cells) staining and fluorescence microscopy (Eclipse Ti-E, Nikon Instruments, Florence, Italy).
Animal studies
Studies involving vertebrate animals (rodents) were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011). Protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of The Wistar Institute (protocol #112625 and 112610). Groups of 4–6 weeks-old male athymic nude mice (Crl:NU(NCr)-Foxn1nu, Charles River Laboratory) were injected s.c. with PC3 cells (5×106 cells in 50% Matrigel) and superficial tumors were allowed to grow to ~50–75 mm3 volume. Tumor-bearing animals were randomized into two groups receiving cell-permeable scrambled peptide (50 mg/kg) or cell-permeable retro-inverso D-enantiomer MFF peptide (10 mg/kg or 50 mg/kg) as daily i.p. injections in PBS, pH 7.2. Tumor growth in the animal groups was measured with a caliper during a two-week time interval and quantified. For in vivo studies using patient-derived xenograft (PDX) melanoma models, NOD SCID γ (NSG) mice were implanted with WM3960 tumor tissue. The WM3960 model was derived from a patient with metastatic (stage IV) melanoma who progressed while treated with the standard-of-care combination of the BRAF inhibitor Dabrafenib and the MEK inhibitor Trametinib. When implanted tumors reached a palpable volume (100–200 mm3), tumor-bearing animals were randomized into three groups receiving cell-permeable scrambled peptide (50 mg/kg), Dabrafenib/Trametinib chow (150 mg/kg and 1.5 mg/kg, respectively) or cell-permeable retro-inverso D-enantiomer MFF peptide (50 mg/kg) plus Dabrafenib/Trametinib chow as daily i.p. injections in PBS, pH 7.2.
Statistical analysis
Data were analyzed using the two-sided unpaired t, chi-square or Kruskal-Wallis (with p value correction for multiple testing) tests using a GraphPad software package (Prism 8.1) for Windows. Data are expressed as mean±SD of replicates from a representative experiment out of at least two or three independent determinations or as mean±SD of three individual experiments. A p value of <0.05 was considered as statistically significant.
RESULTS
Identification of an MFF-VDAC1 complex in cancer
We began this study by examining the expression of MFF, a mitochondrial outer membrane receptor for the fission effector, Dynamin-Related Protein-1 (Drp1) (18), in a clinically-annotated cohort of non-small cell lung cancer (NSCLC) patients (Supplementary Table S3). We found that MFF was differentially overexpressed in NSCLC patients, including cases of adenocarcinoma (AdCa) and squamous cell carcinoma (SCC), compared to normal bronchus (Fig. 1A and B). In this cohort, MFF expression did not correlate with tumor grade (Fig. 1C), nodal status (Supplementary Fig. S1A) or tumor size (Supplementary Fig. S1B). In independent confirmatory studies, MFF was also differentially overexpressed in patient samples of prostatic adenocarcinoma, compared to normal prostate (19). Similarly, MFF was highly expressed in multiple tumor cell lines, including prostate cancer (Supplementary Fig. S1C).
Figure 1.

MFF-VDAC complex in cancer. A, MFF expression in non-small cell lung cancer (NSLC) patients by immunohistochemistry. N, normal; AdCa, adenocarcinoma; SCC, squamous cell carcinoma. Insets, image magnification of indicated regions. Scale bars (top), 300 μm; (bottom), 200 μm. B and C, Quantification of MFF expression in NSCLC by immunohistochemistry (IHC) compared to normal lung (B) or according to tumor grade (C). Mean±SD. ***, p<0.0001; ns, not significant, by Kruskal-Wallis test. D, Ingenuity pathway analysis of MFF-associated proteins identified by a 1D proteomics screening in PC3 cells. E, PC3 cells were transfected with vector, Flag-MFF1 or Flag-MFF2 cDNA, immunoprecipitated (IP) with an antibody to Flag and analyzed by Western blotting. The position of endogenous or Flag-MFF isoforms is indicated. Sup, supernatant; Unb, unbound. F, PC3 extracts were immunoprecipitated with IgG or an antibody to MFF and endogenous co-associated proteins were identified by Western blotting. G, Increasing concentrations of recombinant VDAC1 were mixed with affinity-purified Flag-MFF1 followed by pull-down and Western blotting. H and I, PC3 cells were transfected with vector or Flag-tagged MFF1 (H) or MFF2 (I) cDNA, immunoprecipitated (IP) with an antibody to Flag and analyzed by Western blotting. TCE, total cell extracts.
To uncover potential function(s) of MFF in cancer, we next carried out a proteomics screen for MFF-associated proteins in prostate adenocarcinoma PC3 cells. We identified 42 proteins that associate with MFF (Supplementary Fig. S2A), including regulators of mitochondrial cell death (2), such as VDAC1, −2 and −3 (8), metaxin-1 and −2 (MTX1, MTX2), and hexokinase-I and -II (HK-I, HK-II) (Supplementary Fig. S2A, Fig. 1D). Other MFF-associated proteins comprised mitochondrial receptors for protein import (TOMM70, TOMM40), protein sorting (SAM50), and cristae remodeling (DNAJC11, APOOL-MIC27, CHCHD3) (Supplementary Fig. S2A, Fig. 1D). Therefore, we focused on VDAC1 as the most abundant regulator of mitochondrial outer membrane permeability in cancer (8).
Processing of the human MFF locus is complex and predicted to generate at least five protein isoforms by alternative splicing (Supplementary Fig. S2B). Of these, MFF1 and MFF2 were the most prominently expressed in PC3 and DU145 prostate cancer cells (Supplementary Fig. S2C). Consistent with the results of the proteomics screen, Flag-MFF1 or Flag-MFF2 bound VDAC1 in co-immunoprecipitation experiments, in vivo (Fig. 1E) (19). HK-II, which associates with VDAC at the mitochondrial outer membrane and is important for cell survival (6), was also present in MFF-VDAC1 complexes (Fig. 1E). Similar results were obtained in analysis of endogenous proteins, as endogenous MFF associated with endogenous HK-I, HK-II and VDAC1 in co-immunoprecipitation experiments from PC3 cells (Fig. 1F). We next asked if this interaction was direct, and we mixed increasing concentrations of recombinant VDAC1 with affinity-purified Flag-MFF1. In these experiments, Flag-MFF1 directly bound recombinant VDAC1 in a concentration-dependent manner, whereas uncoupled beads had no effect (Fig. 1G). Next, we looked at the role of other MFF isoforms (Supplementary Fig. S2B) in VDAC1 binding. In co-immunoprecipitation experiments of differentially tagged recombinant proteins, we found that MFF isoforms assembled in homodimers, such as MFF1-MFF1 (Fig. 1H), as well as heterodimers, including MFF1-MFF2 (Fig. 1I, Supplementary Fig. S2D) and MFF1-MFF4 (Supplementary Fig. S2E and F) in the VDAC1 complex.
Structural requirements of an MFF-VDAC1 complex
Based on these results, we next asked if MFF affected the function of VDAC1 in mitochondrial outer membrane permeability (9). To test this hypothesis, we examined partially overlapping synthetic peptides duplicating the entire MFF1 sequence (Supplementary Table S1) in a high-throughput assay that measures depolarization of isolated tumor (PC3) mitochondria (Fig. 2A). We found that peptide #8 corresponding to the MFF sequence Asp217-Leu246 depolarized tumor mitochondria, whereas none of the other MFF peptides had an effect (Fig. 2B). This response was selective because MFF peptide #8 depolarized isolated mitochondria from PC3 cells in a concentration-dependent manner, whereas BPH-1 cells were not affected (Supplementary Fig. S3A). Among MFF isoforms (Supplementary Fig. S2B), only MFF1 and MFF2 contained the peptide #8 region (Supplementary Fig. S3B), consistent with the ability of these isoforms to directly bind VDAC1, in vivo (Fig. 1E and F) and in vitro (Fig. 1G).
Figure 2.

Peptidyl mimicry of MFF-VDAC1 complex. A, Schematic diagram of high-throughput screening of MFF-derived peptides for inhibition of mitochondrial inner membrane potential. MTE, mitochondrial extracts. B, The conditions are as in (A) and MFF-derived peptides (20 μM for 40 min) were screened for modulation of mitochondrial membrane potential by TMRE staining. Mean±SD (n=2). C, Isothermal titration calorimetry binding data of WT (MFF, left) or scrambled (Scramb, right) MFF peptide to human recombinant VDAC1. Representative experiment (n=4). D, PC3 cell extracts were fractionated on beads-coupled MFF peptide #8 by affinity chromatography and bound proteins were identified by Western blotting. Sup, supernatant. E, Structural model of VDAC1-MFF peptide #8 complex generated with the CABS-Dock server. The MFF peptide (green stick) binds the interior cavity of VDAC1 (blue carton/stick) in an extensive network of interactions blocking access to solutes. F, Predicted contact sites of MFF residues R225, R236 or Q241 with VDAC1. G, DU145 or PC3 cells transfected with vector (Vect) or Flag-MFF1 cDNA were immunoprecipitated (IP) with an antibody to Flag, treated with MFF peptide #8 and analyzed by Western blotting. IgL, Ig light chain. H and I, PC3 cells transfected with WT Flag-MFF or Flag-MFF R225D, R236D or Q241A mutant cDNA were immunoprecipitated (IP) with an antibody to Flag and immune complexes were analyzed by Western blotting (H) and quantified by densitometry (I). AU, arbitrary units.
To quantify the interaction between MFF peptide #8 and VDAC1, we next performed isothermal titration calorimetry (ITC) studies. In these experiments, MFF peptide #8 bound human recombinant VDAC1 with a Kd of ~12 μM, whereas a control peptide with the scrambled sequence had no effect (Fig. 2C). Next, we asked if this MFF sequence bound VDAC1, in vivo. Accordingly, bead-immobilized MFF peptide #8 affinity-purified HK-I, HK-II and VDAC1 from PC3 cell extracts, in vivo (Fig. 2D). Molecular modeling studies using the CABS dock server (26,27) placed the MFF peptide #8 sequence in the interior hole of the VDAC1 ring (Fig. 2E), with an average root-mean-square deviation (RMSD) of 4.1, cluster density of 36 and number of contact elements of 148. This model identified three MFF residues that coordinate the binding interface with VDAC1: MFF Arg225 is predicted to contact VDAC1 Glu40 and Glu59, MFF Arg236 contacts the side chains of VDAC1 Thr6, Gln154 and Asp128 and MFF Gln241 is within coordinating distance of VDAC1 Asn185 and Glu189 (Fig. 2F).
Based on these data, we next asked if MFF peptide #8 acted as a competitive inhibitor of the MFF-VDAC1 complex. Consistent with this, addition of MFF peptide #8 to DU145 (Fig. 2G, top) or PC3 (Fig. 2G, bottom) cell extracts inhibited the association of MFF with VDAC1 or HK-I, in co-immunoprecipitation experiments, in vivo (Fig. 2G). To independently test our molecular modeling predictions, we next generated variants of the MFF1 protein containing single amino acid substitutions, Arg225Asp, Arg236Asp or Gln241Ala predicted to disrupt the contact sites with VDAC1 (Fig. 2F). In co-immunoprecipitation experiments, each of the three MFF1 mutants showed significantly reduced binding to VDAC1 and HK-II, compared to wild type (WT) MFF1 (Fig. 2H and I).
Next, we wished to further narrow the MFF-VDAC1 binding interface, and we synthesized variants of peptide #8 with amino- or carboxy-terminus deletions (Supplementary Table S2). Using our high-throughput assay of mitochondrial depolarization (Fig. 2A), we found that variants #1, #2 and #6 of peptide #8 depolarized isolated tumor mitochondria (Supplementary Fig. S3C). None of the other MFF peptide #8-derived sequences was effective (Supplementary Fig. S3C). Therefore, we synthesized a new MFF peptide #8–11 containing the sequence Ser223-Leu243, corresponding to a minimal binding site for VDAC1 (Supplementary Fig. S3B).
An MFF-VDAC1 complex is required for tumor cell survival
To understand the function of an MFF-VDAC1 complex in tumor cells, we next made the MFF peptide #8–11 cell-permeable via the addition of an NH2-terminus RQIKIWFQNRRMK HIV-Tat cell-penetrating sequence (31). The cell-permeable MFF peptide, but not a cell-permeable scrambled sequence, induced sudden loss of mitochondrial membrane potential, by single-cell time-lapse videomicroscopy (Fig. 3A and B), as well as analysis of whole tumor cell populations, by flow cytometry (Supplementary Fig. S4A and B). Consistent with increased mitochondrial outer membrane permeability (6), treatment with the cell-permeable MFF peptide induced cytochrome c release from PC3 cells (Fig. 3C), whereas BPH-1 cells were minimally affected (Fig. 3D). An unrelated MFF #1 peptide did not induce cytochrome c release from PC3 cells (Fig. 3C). In line with acute loss of mitochondrial integrity, treatment with the cell-permeable MFF peptide #8–11 killed (IC50 ~10 μM) multiple prostate cancer cell types in a concentration- and time-dependent manner (Fig. 3E). Normal prostatic epithelial cells or fibroblasts were not affected, and a cell-permeable scrambled peptide did not kill normal or prostate cancer cells (Fig. 3E). In addition, treatment with the cell-permeable MFF peptide #8–11 comparably killed isogenic pairs of drug-sensitive and drug-resistant melanoma cells (Fig. 3F, Supplementary Fig. S4C). To validate the specificity of these findings, we next introduced the Arg225Asp/Arg236Asp double mutation (DD) that prevents VDAC1 binding in the cell-permeable MFF peptide. This mutant peptide entirely lost the ability to induce tumor (PC3) cell death, whereas WT MFF peptide #8–11 efficiently killed tumor cells in a time- and concentration-dependent manner (Fig. 3G).
Figure 3.

Regulation of tumor cell death by MFF-VDAC1 complex. A, PC3 cells were incubated with vehicle, cell-permeable scrambled peptide (scrambled) or cell-permeable MFF peptide #8–11 (10 μM), and MitoTracker (MT) and TMRE fluorescence reactivity was imaged continuously by time-lapse videomicroscopy. Representative images at t=0 min and t=120 min. B, The conditions are as in (A) and changes in TMRE and MitoTracker labeling were quantified at the indicated time intervals. The decrease in MitoTracker signal after treatment with MFF peptide #8–11 may reflect activation of mitophagy. C and D, PC3 (C) or BPH-1 (D) cells were treated with the indicated MFF peptides or CaCl2 and supernatants (Sup) or mitochondrial extracts (Mito) were analyzed by Western blotting. E, The indicated tumor (LNCaP, C4–2B, PC3, DU145, PC3) or normal (BPH-1, HFF) cell types were treated with increasing concentrations (0–40 μM) of cell-permeable scrambled peptide (Scr) or MFF peptide #8–11 (MFF) and analyzed for cell death after 2 h by CellTox reactivity. Mean±SD (n=3). F, Isogenic drug-sensitive (2058_S) or drug-resistant (2058_R) melanoma cells were incubated with increasing concentrations of cell-permeable MFF peptide #8–11 or cell-permeable scrambled peptide (50 μM) and analyzed for cell death at the indicated time intervals by CellTox reactivity. Mean±SD (n=2). G, PC3 cells were treated (0–50 μM) with cell-permeable scrambled peptide, WT MFF peptide #8–11 or MFF peptide #8–11 containing the double mutation Arg225Asp/Arg236Asp (DD) and analyzed for cell death at the indicated time intervals. Mean±SD (n=3). H, PC3 cells were incubated with cell-permeable scrambled peptide or cell-permeable MFF peptide #8–11 (0–50 μM), mixed with the indicated concentrations of cyclosporine A (CsA, 0–10 μM for 75 min), and analyzed for cell death at the indicated time intervals. Mean±SD (n=2). I, Immortalized wild type (WT) mouse embryonic fibroblasts (MEF) or CypD−/− MEF were incubated with cell-permeable scrambled peptide (Scr) or MFF peptide #8–11 (25 μM) and analyzed for cell death. Mean±SD (n=2). J, Immortalized WT or VDAC1−/− MEF were incubated with the indicated increasing concentrations of cell permeable MFF peptide #8–11 (10–50 μM) or scrambled peptide (50 μM) and analyzed for cell death at the indicated time intervals. Mean±SD (n=2).
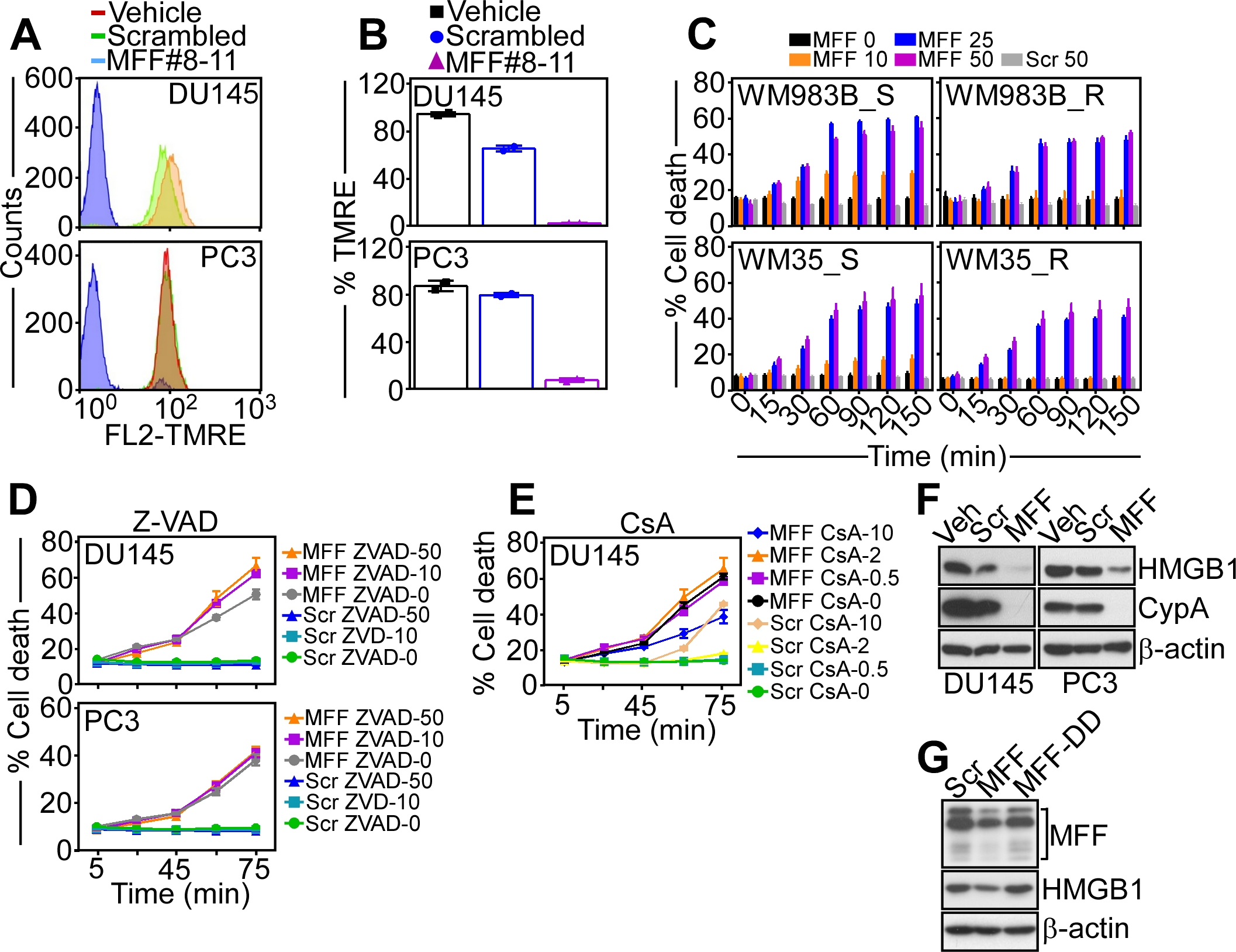
Next, we used these reagents to characterize more in detail the type of mitochondrial cell death induced by MFF-VDAC1 targeting. In these experiments, a pan-caspase inhibitor, Z-VAD-fmk did not significantly reduce tumor cell death induced by the cell-permeable MFF peptide #8–11 (Supplementary Fig. S4D). Instead, an inhibitor of regulated necrosis (4), cyclosporine A (CsA), partially reversed tumor cell killing induced by MFF peptide #8–11 (Fig. 3H, Supplementary Fig. S4E). Biochemically, tumor cell death in these settings was accompanied by loss of plasma membrane integrity, another marker of regulated necrosis (6), as judged from the release of HMGB1 and cyclophilin A (CypA) from tumor cells (Supplementary Fig. S4F). Conversely, a cell-permeable scrambled peptide or MFF-DD mutant peptide #8–11 did not cause HMGB1 release (Supplementary Fig. S4F and G). The mitochondrial matrix protein, Cyclophilin D (CypD) is an essential effector of regulated necrosis (32), and its role in this pathway was next investigated. Treatment with the cell-permeable MFF peptide #8–11 killed immortalized wild type (WT) mouse embryonic fibroblasts (MEF) in a concentration-dependent manner (Fig. 3I). In contrast, CypD−/− MEFs were resistant to cell death induced by the MFF peptide (Fig. 3I). Finally, we tested the effect of MFF targeting on immortalized VDAC1−/− MEF. Consistent with the data above, treatment with MFF peptide #8–11 efficiently killed WT MEF in a time- and concentration-dependent response (Fig. 3J, left). Conversely, VDAC1−/− immortalized MEF were insensitive to cell death in these settings (Fig. 3J, right).
Targeting the MFF-VDAC1 complex for cancer therapy
From these proof-of-concept studies, we next generated a clinical candidate to target the MFF-VDAC1 complex for cancer therapy, in vivo. To accomplish this, we synthesized a cell-permeable retro-inverso peptidomimetic of the Ser223-Leu243 MFF sequence, containing all D-amino acids in the reverse orientation (Fig. 4A) to improve stability during systemic administration (33). This retro-inverso D-enantiomeric MFF peptidomimetic, designated MFF (D) 8–11 rapidly (<20 min) accumulated intracellularly (Fig. 4B) and became associated with isolated mitochondria (Supplementary Fig. S5A). Functionally, the MFF (D) 8–11 peptidomimetic was more active than the L-enantiomeric (L) MFF peptide #8–11 at depolarizing tumor mitochondria (Fig. 4C) and killed tumor cells in a time- and concentration-dependent manner also more efficiently than the MFF (L) peptide #8–11 (Fig. 4D). By time-lapse videomicroscopy, the MFF (D) 8–11 peptidomimetic killed tumor cells within 15 min of exposure with morphologic features of regulated necrosis (Supplementary Movie S1). A cell-permeable scrambled peptidomimetic had no effect (Supplementary Movie S2).
Figure 4.

Preclinical targeting of MFF-VDAC1 complex for cancer therapy. A, Predicted structure of MFF peptide #8–11 with L-amino acids (top) or retro-inverso D-enantiomer (bottom). B, PC3 cells were treated with biotin-labeled MFF (D) 8–11 peptidomimetic (10 μM), incubated with streptavidin-FITC (TRITC) and intracellular peptidomimetic accumulation was analyzed after 20 min by fluorescence microscopy. Nuclei were stained with DAPI. Representative images (n=2). C and D, PC3 cells were incubated with the indicated concentrations (10–25 μM) of cell-permeable scrambled peptide (Scr), MFF peptide #8–11 (L) or MFF (D) 8–11 peptidomimetic and analyzed for mitochondrial membrane potential by TMRE labeling and flow cytometry (C) or cell death (D) at the indicated time intervals. Mean±SD (n=2). E, PC3 cells (5×106 in 50% Matrigel) were engrafted onto the flanks of immunocompromised athymic mice, and animals randomized in two groups were treated with cell-permeable scrambled peptide or MFF (D) 8–11 peptidomimetic (50 mg/kg, daily i.p.) with quantification of tumor growth. Mean±SD (n=8–10). **, p=0.008 by unpaired two-tailed t test (tumor measurements at d. 13). F, Patient-derived melanoma xenografts resistant to the combination of Dabrafenib (Dab) plus Trametinib (Tra) were treated with vehicle (Veh) or MFF (D) 8–11 peptidomimetic (50 mg/kg) and tumor growth was quantified at the indicated time intervals. Mean±SD (n=5) *, p=0.02 by unpaired two-tailed t test. G, Primary, patient-derived NSCLC 3D organoids were treated with cell-permeable scrambled peptide (Scramb, 100 μM) or MFF (D) 8–11 peptidomimetic (100 μM) and analyzed after 24 h by hematoxylin-eosin (H&E) staining or immunohistochemistry for Ki-67 or cleaved caspase-3 (Cl. Casp.3) expression. Scale bar, 100 μm. H and I, The conditions are as in (G) and the percentage of cells stained for Ki-67 or cleaved caspase-3 in patient-derived NSCLC (H) or breast adenocarcinoma (AdCa, I) 3D organoids was quantified. Mean±SD (average of 2–3 independent fields). *, p=0.02; ***, p=0.0004 for 100 μM MFF (D) 8–11 peptidomimetic compared to scrambled peptide, by unpaired two-tailed t test. J and K, Patient-derived human glioblastoma (GBM) neurospheres in culture were treated with cell-permeable scrambled peptide (50 μM) or the indicated increasing concentrations of MFF (D) 8–11 peptidomimetic (μM) for 1, 3 or 24 h (J, representative GBM neurospheres after 24-h treatment are shown), stained with calcein (live cells) or To-Pro (dead cells), and normalized fluorescence units (FU) were quantified (K). BF, bright field. Mean±SD, two individual patients analyzed. The statistical analysis for each time point is as follows: 1-h, Scrambled peptide (Scr) vs. MFF 10, p=0.002; Scr vs. MFF 25, p=0.03; Scr vs. MFF 50, ns; 3-h, Scr vs. MFF 10, p=0.001; Scr vs. MFF 25, p=0.01; Scr vs. MFF 50, p=0.01; 24-h, Scr vs. MFF 10, p=0.03; Scr vs. MFF 25, p=0.02; Scr vs. MFF 50, p=0.01, all by unpaired two-tailed t test.
Although xenograft models are suboptimal due to the lack of immune competence and superficial, as opposed to visceral tumor engraftment, daily systemic administration of the MFF (D) 8–11 peptidomimetic (10–50 mg/kg i.p.) inhibited PC3 tumor growth in immunocompromised mice in a concentration-dependent manner (Supplementary Fig. S5B, Fig. 4E). Systemic administration of a cell-permeable scrambled peptidomimetic had no effect (Supplementary Fig. S5B, Fig. 4E). Next, we examined the effect of MFF-VDAC1 targeting in patient-derived xenograft (PDX) models of melanoma resistant to the combination therapy of Dabrafenib (mutant BRAF inhibitor) plus Trametinib (MEK inhibitor). When exposed to vehicle or the combination Dabrafenib/Trametinib, these PDX tumors grew exponentially in immunocompromised mice (Fig. 4F). Conversely, treatment with the MFF (D) 8–11 peptidomimetic significantly inhibited the growth of drug-resistant PDX melanoma (Fig. 4F), without overt signs of toxicity or animal weight loss (Supplementary Fig. S5C).
In a third preclinical model, treatment with the MFF (D) 8–11 peptidomimetic inhibited cell proliferation (loss of Ki-67+ cells) and induced markers of cell death (increased cleaved caspase-3-positive cells) in patient-derived NSCLC (Fig. 4G and H) and breast adenocarcinoma (Supplementary Fig. S5D, Fig. 4I) 3D organoids in culture. A cell-permeable scrambled peptidomimetic had no effect on 3D tumor organoids (Fig. 4G–I, Supplementary Fig. S5D). Consistent with these results, matched tumor cell lines, NSCLC A549 and H460 or breast adenocarcinoma MDA-231 and MCF-7 cells expressed MFF1, MFF2 and MFF4 (Supplementary Fig. S5E), and exhibited mitochondrial depolarization and nearly complete killing after treatment with the MFF (D) 8–11 peptidomimetic, but not a scrambled sequence (Supplementary Fig. S5F). Finally, we looked at patient-derived glioblastoma (GBM) neurospheres as a preclinical model enriched in cancer stem cells. In validation experiments, the cell-permeable MFF (D) 8–11 peptidomimetic efficiently killed GBM LN229 cells in a time- and concentration-dependent manner (Supplementary Fig. S6A). Normal HFF were not affected and a control scrambled sequence did not cause LN229 cell killing (Supplementary Fig. S6A). Consistent with these data, treatment with the MFF (D) 8–11 peptidomimetic, but not a scrambled peptide, was highly cytotoxic for patient-derived GBM neurospheres (Fig. 4J, Supplementary Fig. S6B), resulting in time- and concentration-dependent loss of viability, as early as one hour after drug exposure (Fig. 4K).
DISCUSSION
In this study, we have shown that a regulator of mitochondrial fission, MFF is overexpressed in NSCLC and assembles in isoform-specific (MFF1 and MFF2) homo- and heterodimeric complexes with VDAC1 at the mitochondrial outer membrane. Peptidyl mimicry of the MFF-VDAC1 interaction is feasible and a cell-permeable MFF Ser223-Leu243 D-enantiomeric peptidomimetic disrupted MFF-VDAC1 complex(es), in vivo, triggered mitochondrial outer membrane permeability and killed genetically disparate tumor types, including drug-resistant melanoma, predominantly by regulated necrosis. Treatment with the MFF peptidomimetic was well tolerated and delivered potent anticancer activity in multiple preclinical, patient-derived tumor models.
The molecular details of how MFF regulates VDAC1-directed mitochondrial outer membrane permeability (2), including potential post-translational modifications, remain to be fully elucidated (19). Although only MFF1 and MFF2 contain the sequence that directly binds VDAC1, structure-function studies demonstrated that other MFF isoforms are recruited in a multimeric complex with VDAC1, in vivo. NMR and reconstitution studies with isolated mitochondria have shown that also VDAC1 forms oligomers that assemble in “open” or “closed” configurations (34) to regulate channel conductance (35). In this context, the predicted insertion of MFF multimers in the interior hole of the VDAC1 ring appears ideally suited to shut off oligomeric channel conductance and oppose cell death initiation in tumors (2). A similar scenario of “channel closure” has been proposed for other VDAC binding-proteins with pro-survival functions, including HK-I (36) and HK-II (37), which are also recruited to MFF-VDAC1 complex(es).
Although the role of VDAC in channel conductance is established (35), how this pathway connects to downstream steps of mitochondrial cell death remains controversial (6). In fact, earlier mouse knockout studies suggested that none of the three VDAC isoforms may be required for cell death, in vivo (38). On the other hand, this pathway may be differentially exploited to preserve mitochondrial integrity in transformed cells. Accordingly, VDAC1 is a recognized regulator of tumor cell death (8), and actionable therapeutic target (39), VDAC2 participates in Bax-mediated apoptosis during tumor progression (40), and cysteine oxidation of VDAC3 may control pore conductance and ROS sensing (41), including in cancer. A similar context-specific rewiring may apply to MFF. Although MFF knockout mice showed better tissue protection and reduced mitochondrial apoptosis after ischemia-reperfusion injury (42), MFF was recently characterized as a critical survival factor in prostate cancer, preserving the cancer stem cell pool (43), and in line with the findings presented here. In addition, a pro-survival function of MFF via VDAC1 binding as described here may be important to oppose the propensity to apoptosis associated with mitochondrial fragmentation (17), and fully enable the role of deregulated fission in primary (13,14) and metastatic (15,16) tumor growth.
Targeting protein-protein interactions at the mitochondrial outer membrane is a validated therapeutic strategy, and has produced the first apoptosis modifier clinically approved for cancer therapy (44). Here, comprehensive studies of peptidyl mimicry identified a cell-permeable MFF Ser223-Leu243 D-enantiomeric peptidomimetic as a potential clinical candidate of this pathway. Treatment with this agent induced hallmarks of acute mitochondrial dysfunction, with sudden depolarization of the organelle inner membrane, release of cytochrome c in the cytosol (6) and massive cell death in disparate tumor types, including drug-resistant melanoma. This cell death response, which was dependent on VDAC1, had hallmarks of both apoptosis, with cytochrome release and caspase activation, in vivo, as well as regulated necrosis (4), as characterized by loss of plasma membrane integrity, insensitivity to caspase inhibition, and requirement of CypD (32). The coexistence of both apoptosis and necrosis is common after acute mitochondrial dysfunction (45) and reflects mechanistic overlap of organelle cell death pathways (46).
In our preclinical evaluation, treatment with the cell-permeable MFF D-enantiomeric peptidomimetic was well tolerated, and delivered potent anticancer activity in multiple, patient-derived tumor models representative of acquired drug resistance (Dabrafenib/Trametinib-resistant melanoma), 3D tumor architecture (breast adenocarcinoma and NSCLC organoids) and cancer stem cells (GBM neurospheres). Although the long-term safety of this regimen remains to be established, the encouraging therapeutic index observed here may reflect the differential distribution of MFF in cancer, compared to normal tissues, but also a general dependence on this pathway to preserve mitochondrial integrity in transformed cells. This is consistent with the importance of mitochondrial fitness to sustain bioenergetics (47), buffer ROS production (48), and elevate the threshold of “mitochondrial priming” in tumors (49). Compared to approved therapies that target Bcl2 family proteins at the mitochondrial outer membrane (3), disruption of the MFF-VDAC1 complex may offer broader efficacy in multiple, genetically heterogenous malignancies and bypass drug resistance mechanisms maintained by pro-survival Bcl2 proteins.
Supplementary Material
{kind=link}
Significance.
Findings describe mitochondrial fission regulation using a peptidomimetic agent that disturbs the MFF-VDAC complex and displays anticancer activity in multiple tumor models
ACKNOWLEDGMENTS
We thank James Hayden and Frederick Keeney for assistance with time-lapse videomicroscopy in the Imaging Core Facility as well as the Proteomics and Metabolomics Core Facility for LC-MS/MS analysis. This work was supported by National Institutes of Health (NIH) grants P01 CA140043 (D.C.A., L.R.L. and D.W.S.), R35 CA220446 (D.C.A.), R50 CA221838 (H.-Y.T) and R50 CA211199 (A.V.K.), by Fondazione Cariplo (2014–1148 to V.V.), by the Italian Minister of Health- Ricerca Corrente program 2017 (to S.F.), and by the National Research Foundation of Korea funded by the Ministry of Education (2018R1D1A1B07048104, 2018R1A6A1A03025810) and the Ministry of Science and ICT (2014M3A9D8034459). Y.C.C is the recipient of the Research Fund (1.170074.01) of Ulsan National Institute of Science and Technology (UNIST) and the National Research Foundation of Korea (2014M3A9D8034459) funded by the Ministry of Science and ICT. A.M.S is supported by a fellowship from the Doctorate School in Molecular and Translational Medicine at the University of Milan. Support for Core Facilities utilized in this study was provided by Cancer Center Support Grant (CCSG) CA010815 to The Wistar Institute.
Footnotes
Conflict of interest: The authors declare no competing financial interest
REFERENCES
- 1.Chandel NS. Evolution of Mitochondria as Signaling Organelles. Cell Metab 2015;22(2):204–6. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018;25(3):486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams JM, Cory S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ 2018;25(1):27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2016;15(5):348–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol 2015;16(6):329–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol 2016;26(9):655–67. [DOI] [PubMed] [Google Scholar]
- 7.Karch J, Molkentin JD. Identifying the components of the elusive mitochondrial permeability transition pore. Proc Natl Acad Sci U S A 2014;111(29):10396–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazure NM. VDAC in cancer. Biochim Biophys Acta 2017;1858(8):665–73. [DOI] [PubMed] [Google Scholar]
- 9.Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol Cell 2015;60(1):47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 11.Chen H, Chan DC. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab 2017;26(1):39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eisner V, Picard M, Hajnoczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 2018;20(7):755–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 2015;57(3):537–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell 2015;57(3):521–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caino MC, Seo JH, Aguinaldo A, Wait E, Bryant KG, Kossenkov AV, et al. A neuronal network of mitochondrial dynamics regulates metastasis. Nat Commun 2016;7:13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013;32(40):4814–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev 2008;22(12):1577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 2010;191(6):1141–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seo JH, Agarwal E, Chae YC, Lee YG, Garlick DS, Storaci AM, et al. Mitochondrial fission factor is a novel Myc-dependent regulator of mitochondrial permeability in cancer. EBioMedicine 2019:pii: S2352–3964(19)30615–2. doi: 10.1016/j.ebiom.2019.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faversani A, Vaira V, Moro GP, Tosi D, Lopergolo A, Schultz DC, et al. Survivin family proteins as novel molecular determinants of doxorubicin resistance in organotypic human breast tumors. Breast Cancer Res 2014;16(3):R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest 2016;126(5):1834–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu H, Liu S, Zhang G, Bin W, Zhu Y, Frederick DT, et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 2017;550(7674):133–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chae YC, Angelin A, Lisanti S, Kossenkov AV, Speicher KD, Wang H, et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat Commun 2013;4:2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26(12):1367–72. [DOI] [PubMed] [Google Scholar]
- 25.Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 2016;44(D1):D1251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaszczyk M, Kurcinski M, Kouza M, Wieteska L, Debinski A, Kolinski A, et al. Modeling of protein-peptide interactions using the CABS-dock web server for binding site search and flexible docking. Methods 2016;93:72–83. [DOI] [PubMed] [Google Scholar]
- 27.Kurcinski M, Jamroz M, Blaszczyk M, Kolinski A, Kmiecik S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res 2015;43(W1):W419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, Villinger S, et al. Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci U S A 2008;105(40):15370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plescia J, Salz W, Xia F, Pennati M, Zaffaroni N, Daidone MG, et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005;7(5):457–68. [DOI] [PubMed] [Google Scholar]
- 30.Di Cristofori A, Ferrero S, Bertolini I, Gaudioso G, Russo MV, Berno V, et al. The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma. Oncotarget 2015;6(19):17514–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raucher D, Ryu JS. Cell-penetrating peptides: strategies for anticancer treatment. Trends Mol Med 2015;21(9):560–70. [DOI] [PubMed] [Google Scholar]
- 32.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434(7033):658–62. [DOI] [PubMed] [Google Scholar]
- 33.de la Fuente-Nunez C, Reffuveille F, Mansour SC, Reckseidler-Zenteno SL, Hernandez D, Brackman G, et al. D-enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem Biol 2015;22(2):196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Betaneli V, Petrov EP, Schwille P. The role of lipids in VDAC oligomerization. Biophys J 2012;102(3):523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colombini M VDAC structure, selectivity, and dynamics. Biochim Biophys Acta 2012;1818(6):1457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villinger S, Briones R, Giller K, Zachariae U, Lange A, de Groot BL, et al. Functional dynamics in the voltage-dependent anion channel. Proc Natl Acad Sci U S A 2010;107(52):22546–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem 2002;277(9):7610–8. [DOI] [PubMed] [Google Scholar]
- 38.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 2007;9(5):550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prezma T, Shteinfer A, Admoni L, Raviv Z, Sela I, Levi I, et al. VDAC1-based peptides: novel pro-apoptotic agents and potential therapeutics for B-cell chronic lymphocytic leukemia. Cell Death Dis 2013;4:e809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chin HS, Li MX, Tan IKL, Ninnis RL, Reljic B, Scicluna K, et al. VDAC2 enables BAX to mediate apoptosis and limit tumor development. Nat Commun 2018;9(1):4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okazaki M, Kurabayashi K, Asanuma M, Saito Y, Dodo K, Sodeoka M. VDAC3 gating is activated by suppression of disulfide-bond formation between the N-terminal region and the bottom of the pore. Biochim Biophys Acta 2015;1848(12):3188–96. [DOI] [PubMed] [Google Scholar]
- 42.Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P, et al. Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved mPTP Opening. J Am Heart Assoc 2017;6(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Civenni G, Bosotti R, Timpanaro A, Vazquez R, Merulla J, Pandit S, et al. Epigenetic Control of Mitochondrial Fission Enables Self-Renewal of Stem-like Tumor Cells in Human Prostate Cancer. Cell Metab 2019. [DOI] [PubMed] [Google Scholar]
- 44.Croce CM, Reed JC. Finally, An Apoptosis-Targeting Therapeutic for Cancer. Cancer Res 2016;76(20):5914–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Declercq W, Takahashi N, Vandenabeele P. Dual face apoptotic machinery: from initiator of apoptosis to guardian of necroptosis. Immunity 2011;35(4):493–5. [DOI] [PubMed] [Google Scholar]
- 46.Lemasters JJ. V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am J Physiol 1999;276(1):G1–6. [DOI] [PubMed] [Google Scholar]
- 47.Anderson RG, Ghiraldeli LP, Pardee TS. Mitochondria in cancer metabolism, an organelle whose time has come? Biochim Biophys Acta Rev Cancer 2018;1870(1):96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 2014;14(11):709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011;334(6059):1129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.