

Abstract
Each of the four acute hepatic porphyrias is due to mutation of an enzyme in the heme biosynthetic pathway. The accumulation of pathway intermediates that occur most notably when these diseases are active is the basis for screening and establishing a biochemical diagnosis of these rare disorders. Measurement of enzyme activities and especially DNA testing also are important for diagnosis. Suspicion of the diagnosis and specific testing, particularly measurement of urinary porphobilinogen, are often delayed because the symptoms are nonspecific, even when severe. Urinary porphyrins are also measured, but their elevation is much less specific. If porphobilinogen is elevated, second line testing will establish the type of acute porphyria. DNA testing identifies the familial mutation and enables screening of family members. Management includes removal of triggering factors whenever possible. Intravenous hemin is the most effective treatment for acute attacks. Carbohydrate loading is sometimes used for mild attacks. Cyclic attacks, if frequent, can be prevented by a GnRH analogue. Frequent noncyclic attacks are sometime preventable by scheduled (e.g. weekly) hemin infusions. Long term complications may include chronic pain, renal impairment and liver cancer. Other treatments, including RNA interference, are under development.
Keywords: Porphyrias, porphyrins, porphobilinogen, hemin, GnRH analogues, RNA interference
Introduction
This review summarizes preferred approaches to diagnosis and short and long term management of the acute porphyrias, as related to the current understanding of the pathophysiology of these disorders. Because these diseases are rare and their neurovisceral symptoms and signs mimic those of other more common diseases, their diagnosis is often delayed. This is unfortunate, because specific and sensitive diagnostic testing is available that can confidently diagnose these disorders, and effective treatment is available particularly to resolve acute attacks of neurovisceral symptoms. Therefore, there are needs for increased physician awareness of these disorders and to make testing for acute porphyrias part of the differential diagnosis and laboratory workup of patients who present with nonspecific symptoms that are difficult to diagnose.
Overview of genetic and clinical features
The acute hepatic porphyrias are four types of porphyria that are due to inherited deficiencies of four different enzymes in the heme biosynthetic pathway (Figure 1)(1, 2). In contrast to other porphyrias in which only porphyrins accumulate and skin photosensitivity develops, they are associated with accumulation of porphyrin precursors - δ-aminolevulinic acid (ALA – an amino acid) and porphobilinogen (PBG – a pyrrole), and by symptoms and signs resulting from effects on the nervous system at many levels. ALA and PBG are the substrate and product of ALA dehydratase (ALAD, Figure 2), the second enzyme in the pathway. Patients usually experience acute attacks with a combination of disease manifestations lasting for days or weeks with intervening intervals of good health. But it is increasingly recognized that symptoms can become chronic especially in patients who have experienced repeated exacerbations (2, 3).
Figure 1.
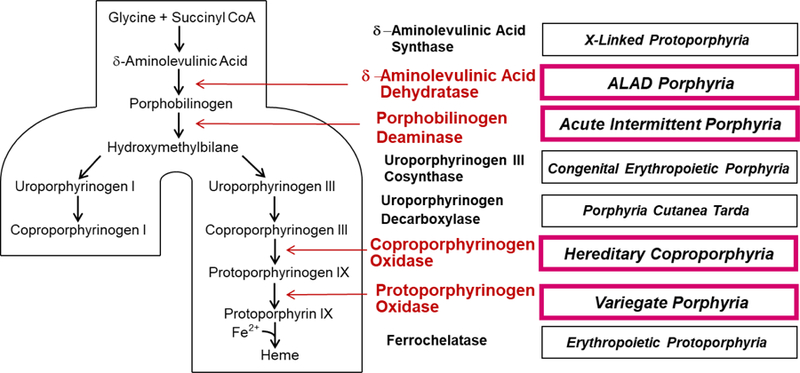
Intermediates and enzymes of the heme biosynthetic pathway, the types of porphyria associated with altered activity of each enzyme. The four acute porphyrias are highlighted. Abbreviation: ALAD, δ-aminolevulinic acid dehydratase.
Figure 2.
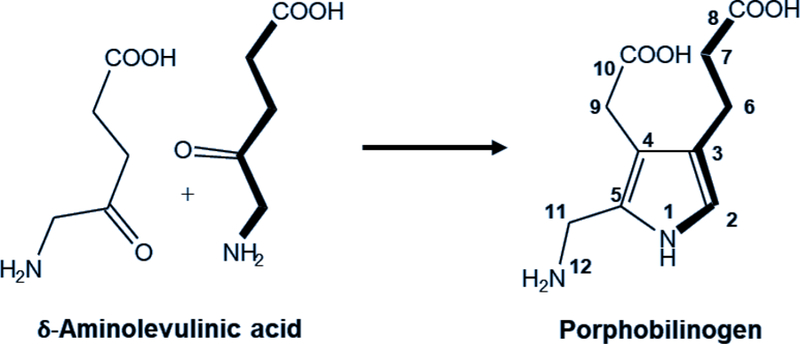
Chemical structures of the porphyrin precursors, which are increased in the acute porphyrias. δ-Aminolevulinic acid is the substrate and porphobilinogen the product of δ-aminolevulinic acid dehydratase, the second enzyme in the heme biosynthetic pathway.
Acute intermittent porphyria (AIP), the most common acute porphyria and the third most common human porphyria, results from mutation of the third enzyme in the heme biosynthetic pathway, known as PBG deaminase (PBGD) or hydroxymethylbilane synthase (HMBS). Hereditary coproporphyria (HCP) and variegate porphyria (VP) result from mutation of the sixth and seventh enzymes in the pathway [coproporphyrinogen oxidase (CPOX) and protoporphyrinogen oxidase (PPOX), respectively]. The substrates for these enzymes accumulate in the liver in HCP and VP and are believed to inhibit PBGD (4), thereby increasing ALA and PBG and causing neurological symptoms. These porphyrinogens (i.e. reduced porphyrinogens) also become oxidized to the corresponding porphyrins, which are photosensitizing and may cause blistering skin lesions. Elevations of ALA and PBG can be less prominent in HCP and VP than in AIP. Nonetheless, attacks of HCP and VP can be severe and life-threatening. ALAD porphyria (ADP) is due to mutation of ALAD, the second enzyme in the pathway, and is very rare. Biochemical findings are marked elevations in urinary ALA and coproporphyrin III and erythrocyte zinc protoporphyrin.
AIP, HCP and VP are autosomal dominant inherited disorders with low penetrance. Individuals who have latent or active disease are heterozygous for a mutant allele, and have approximately half-normal activity of the affected enzyme. In contrast, ADP is autosomal recessive and results from a severe deficiency of ALAD; parents are unaffected carriers with half-normal ALAD activity. Only 8 cases have been documented in the literature (5, 6), so it remains the least understood type of acute porphyria.
Most individuals who inherit mutations that can cause AIP, HCP and VP never develop symptoms – i.e. penetrance is low (7). These porphyrias become active when hepatic δ-aminolevulinic acid synthase (ALAS1 – the ubiquitous form of the first and rate-limiting enzyme of this pathway) becomes induced by environmental and metabolic factors. Synthesis of hepatic ALAS1 is under negative feedback control by a regulatory heme pool, which becomes depleted when heme availability is low. When heme availability is adequate, synthesis of ALAS1 and its import into mitochondria are repressed to a level needed to maintain heme homeostasis. The inherited enzyme deficiencies in the AIP, HCP and VP only reduce enzyme activities to approximately 50% of normal. Although not sufficient by itself to cause substantial heme deficiency in hepatocytes, these partial deficiencies can lead to enhanced induction of ALAS1 when heme synthesis is stimulated by certain drugs, hormones or metabolic factors. Under these circumstances, ALA and PBG accumulate due to marked induction of ALAS1 and decreased activity of PBGD. Evidence suggests, although not conclusively, that ALA is neuropathic and may account for the neuropathic symptoms of the acute hepatic porphyrias (8).
Why AIP, HCP and VP remain “latent” (defined often as without PBG elevation or symptoms) in the great majority of individuals who inherit them is not known. Unknown modifying genetic traits are likely important. Symptoms develop after puberty and more commonly in females. This suggests that female hormones (especially progesterone) play a role. Indeed, increases in progesterone levels are associated with attacks during the luteal phase of the menstrual cycle in some women (9). Environmental, nutritional and metabolic factors such as drugs (e.g. barbiturates, phenytoin, rifampin) and hormonal products (e.g. progesterone and progestins), smoking and dietary restriction are recognized as triggers of acute attacks. Such factors act by inducing hepatic ALAS1 and cytochrome P450 enzymes (CYPs); hepatic ALAS1 induction resulting from dietary restriction is mediated by peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1-δ) (10).
Diagnosis: clinical presentations that suggest acute porphyria
The clinical manifestations of the acute porphyrias are nonspecific, even when severe, and thus, many patients remain undiagnosed or are misdiagnosed as having other medical or surgical conditions (2). The diagnosis can be suspected based on combinations of nonspecific symptoms, but laboratory confirmation is always required to establish a diagnosis (3).
A combination of nonspecific clinical manifestations in the following three broad categories – sometimes referred to as a “classic triad” – may occur in the acute porphyrias and suggest the diagnosis:
Severe abdominal pain is the most common and often the initial symptom of an attack. It is typically generalized rather than localized, and is often accompanied by nausea, vomiting, distension, constipation or diarrhea. Pain is typically severe, with few of the physical findings found in inflammatory conditions such as appendicitis.
Peripheral neuropathy can be manifested as pain in multiple areas such as the back, buttocks, chest or limbs. Paresis may develop and progress, especially with an advanced attack. Early detection of paresis, which often begins proximally in the upper extremities, requires a careful neurological examination.
Central and autonomic nervous system involvement may cause mental status changes, seizures, psychosis, insomnia and anxiety. Hypothalamic involvement can cause the syndrome of inappropriate antidiuretic hormone production (SIADH), leading to hyponatremia. Autonomic nervous system manifestations include tachycardia, hypertension and bladder dysfunction with urinary retention, incontinence, and dysuria)(2, 3).
Other clues may include reddish or brown urine due to excess porphyrins or porphobilin, respectively, unless misdiagnosed as hematuria. Renal impairment and abnormal liver function tests may occur during attacks. The importance of chronic symptoms and impact on quality of life has gained emphasis in the recent literature (2, 3, 11, 12).
A “classic triad” or other combination of nonspecific symptoms cannot by itself be diagnostic of acute porphyria. In clinical practice, the diagnosis is often ruled out when patients with “classic symptoms” are properly tested. MacAlpine and Hunter (13) pointed to the “classic triad” of symptoms displayed by King George III of England as justifying a retrospective diagnosis of acute porphyria, a claim that is now largely discounted (14).
Abdominal pain is warranted as a particular focus in considering a diagnosis of acute porphyria because this is the most common presenting symptom. Abdominal pain has always been a difficult diagnostic problem in medicine. Acute porphyria should be considered in the differential diagnosis of abdominal pain when:
Other symptoms are present that suggest central, peripheral, or autonomic nervous system involvement.
Associated factors that are often associated with attacks are present, such as female sex, the luteal phase of the menstrual cycle, or certain medications reported to precipitate attacks of porphyria (e.g. phenytoin, sulfonamide antibiotics, rifampin, progestins), keeping in mind that such features are not always present (12).
An initial diagnostic workup does not suggest a cause of abdominal pain. Initial evaluation of this presentation in an emergency department often includes a history and physical examination, complete blood cell count, urinalysis, blood chemistries including lipase and amylase and imaging (plain films, ultrasound or CT). Negative results at that point require that the differential diagnosis be expanded, and screening for acute porphyrias would be appropriate at this point rather than at some later time in the diagnostic workup.
Primary care and emergency physicians are especially likely to see patients who should be screened for acute porphyria. Specialist consultants should also be aware that acute porphyrias can involve multiple areas and organ systems in addition to the gastrointestinal tract, particularly the nervous system, skin, liver and kidneys. Undiagnosed patients with porphyria may be regarded as “drug seekers” when asking for effective relief of severe pain, and may be referred to pain specialists or not considered worthy of further follow up.
Any physician seeing a patient with suggestive symptoms should be able to screen for acute porphyrias, which are multisystem diseases that do not fall exclusively within the purview of a single specialty. Screening for acute porphyrias is relatively simple using tests that are widely available, and does not require referral to a specialist, often having little more training and experience with these disorders than do primary care physicians.
Acute porphyrias are not considered often enough in the differential diagnosis of commonly seen symptoms such as abdominal pain, so screening for elevated PBG is not part of the standard workup for such symptoms. Therefore, screening for acute porphyrias does not occur as often as it should, resulting in diagnostic delay. Screening for acute porphyrias should be considered even if the level of clinical suspicion is not particularly high, given that the presenting clinical picture is nonspecific and often not highly suggestive of the diagnosis.
Diagnosis: Currently recommended biochemical and molecular approaches
Many laboratory tests are available for diagnosis of porphyrias. These include porphyrin precursors in urine, plasma, feces and erythrocytes, cytosolic enzyme activities in erythrocytes and DNA testing. The recommended approach is to regard some tests as first-line tests that can be used frequently for screening, and others as second-line tests to be used much less frequently, i.e. only when a first-line test is positive. This enables one to consider and screen for these conditions more frequently and without undue cost.
It is a mistake to order all available testing, as an expensive “shotgun” approach, whenever acute porphyrias are suspected, or to request a “urine porphyrin screen” or “porphyrin panel” when there is uncertainty about what test to order. There is no single screening test, or a panel of tests, for all types of porphyria. The primary screening test for acute porphyrias is a measurement of urine PBG, but an order for urine porphyrins may or may not include PBG. Some other porphyrias do not increase urine porphyrins or porphyrin precursors (e.g. diagnosis of erythropoietic protoporphyria requires measurement of erythrocyte protoporphyrin).
The choice of tests is also governed by the clinical circumstances and is different (and simpler) for a patient with acute or recent abdominal pain than in one with past symptoms. The workup can be narrowed if it is known that a relative had a specific type of porphyria and the documentation for that diagnosis is available for review. As will be discussed, DNA testing has become important, but does not replace biochemical testing for initial diagnosis of patients presenting with symptoms that suggest acute porphyria.
Testing for acute porphyrias in patients with symptoms:
First-line testing (screening).
This should consist only of measurement of urine PBG, total porphyrins and creatinine using a spot (random, single void) urine sample. If available, PBG should be measured in house using a rapid method. (Unfortunately, a kit that was useful for rapid semi-quantitative assessment of urine PBG is no longer available (15)). If necessary, urine samples can be frozen for several months without loss of PBG. To allow for a possible dilute urine sample (e.g. after administration of large volumes of IV fluids), which can yield a falsely negative result if expressed per L, final results should be expressed per gram or mmol of creatinine (16). An example of misleading results expressed per L rather than per gram of creatinine is provided in Table 1. If the patient’s condition is life-threatening, the testing laboratory should be contacted and asked to expedite the result, and to confirm that creatinine will also be measured. PBG can be measured in plasma or serum in patients with advanced renal disease, but plasma levels are less elevated than in urine in patients with normal renal function.
Table 1.
An example of patient with an attack of acute intermittent porphyria and a urine porphobilinogen level expressed as mg/L that appears normal on Day 1 because the urine sample was dilute, as indicated by an unusually low creatinine concentration. PBG was elevated when normalized to creatinine on Day 1, and is also clearly elevated on Day 2 with a urine sample that is not dilute.
| Day 1 | Day 2 | Units | Range | |
|---|---|---|---|---|
| Porphobilinogen (PBG) | 0.7 | 83.4 | mg/L | - |
| 23.3 | 35.5 | mg/g creatinine | 0~4 | |
| Creatinine | 0.03 | 2.35 | g/L | 0.7~2 |
PBG results expressed per L should be interpreted with caution, with consideration of how dilute the urine sample might be; results expressed per g creatinine are more reliably interpreted, assuming kidney function and creatinine output are normal. Dilute urine samples (creatinine < 0.2 g/L) should be considered unreliable for PBG testing if results are expressed per L and not normalized for creatinine.
Results of first-line testing may include the following.
-
Urine PBG is substantially elevated. Urine PBG should exceed 10 mg/g creatinine (5 umol/mmol creatinine) and be ~5-fold above the upper limit of normal. This is a diagnostic finding for AIP, HCP or VP, which does not occur in any other medical condition. Treatment with hemin, if clinically indicated, can be started immediately, after obtaining samples for second-line testing (see below) to differentiate these three porphyrias.
PBG excretion is generally 20 to 200 mg/day (normal range: 0 to ~4 mg/day) during acute attacks of AIP. Excretion of ALA is usually about half that of PBG (expressed as mg/day). Increases in ALA and PBG can persist for prolonged periods between attacks, especially in AIP. Increases in ALA and PBG are less striking during acute attacks of HCP and VP and often decrease more rapidly.
Increases in urinary porphyrins always accompany elevations in porphyrin precursors. These account for reddish urine, since ALA and PBG are colorless. Uroporphyrin can form nonenzymatically from PBG prior to excretion in urine. However, there is evidence that the excess porphyrins in this condition are predominantly isomer III, which suggests they are formed enzymatically, perhaps from ALA in nonhepatic tissues after this porphyrin precursor is transported from the liver (17–19).
- Urine PBG and porphyrins are both normal. This effectively excludes all four acute porphyrias as a cause of concurrent or recent symptoms.
- If some clinical suspicion of acute porphyria remains, the same testing can be repeated at another time if symptoms reappear. Even though the likelihood of a positive result is small, the initial screening results will be confirmed. Second-line testing is not recommended if first-line testing is negative in a symptomatic patient.
- Because hemin treatment reduces levels of porphyrin precursors and porphyrins, a negative first-line result on a sample obtained after starting hemin is not definitive. Therefore, a spot urine should be obtained before hemin is started.
-
Urine PBG is modestly elevated. Modest PBG elevations, after normalization by the creatinine concentration, may be seen with especially sensitive methods of measurement, such as mass spectroscopy, and may be difficult to interpret (20). Concurrent normal porphyrin levels may suggest that PBG is not significantly elevated, and that any severe symptoms are due to another cause. If there is still concern, this testing can be repeated at another time if symptoms recur.
Since PBG may be less elevated and normalize more rapidly in HCP and VP than in AIP, fecal and plasma porphyrins may be measured, since these are the most sensitive biochemical tests for active and even quiescent stages of these porphyrias (3).
Urine PBG is normal and total porphyrins are elevated. Particularly if porphyrin elevation is substantial, this may suggest ADP (in which ALA but not PBG is substantially elevated) or either HCP or VP (in which PBG may become normal more quickly than in AIP). But unfortunately, elevation of urine porphyrins is a nonspecific finding that can occur in many medical conditions. Therefore, second line testing is usually indicated, as described below.
Second-line testing (for confirmation or exclusion of porphyrias as causing first-line abnormal results and to define the type of acute porphyria).
Comprehensive testing using the same urine sample with substantial PBG elevation, plus blood collected before hemin is administered and a fecal sample(3). Because the rate of fecal flow is slow, a fecal sample collected within 1–2 days of starting hemin is likely to be suitable.
Urine porphyrin separation by high performance liquid chromatography (HPLC) to ascertain the pattern of individual porphyrins. Substantial elevation in total urinary porphyrins is expected during acute attacks. Uroporphyrin elevation often parallels elevation in PBG. Coproporphyrin elevation may persist longer in HCP and VP than in AIP. However, porphyrinuria and especially coproporphyrinuria as isolated findings are nonspecific and by themselves do not support a diagnosis of porphyria.
Plasma porphyrins. These are usually normal or only slightly elevated in AIP and HCP. Plasma porphyrins are elevated in the few HCP patients with skin lesions and are commonly elevated in VP. Fluorescence scanning of diluted plasma at neutral pH shows a distinctive peak at 626nm that is specific for VP, and is a particularly useful finding for diagnosis of this porphyria (21–24).
Fecal porphyrins. These are markedly elevated in active cases of HCP and VP, and normal or only modestly elevated in AIP. The porphyrin pattern by HPLC is predominantly coproporphyrin III in HCP, and shows roughly equal amounts of coproporphyrin III and protoporphyrin in VP. The ratio of coproporphyrin III to coproporphyrin I is a particularly sensitive marker for HCP, and exceeds 1.5 in that disorder (19, 25).
Erythrocyte PBG deaminase activity. The disease-causing mutant allele in AIP generally expresses little or no enzyme, so the normal allele accounts for virtually all of the remaining activity of this cytosolic enzyme in erythrocytes and other cells. As a result, this enzyme activity is approximately half-normal in erythrocytes of most patients with AIP, helping to confirm a diagnosis of AIP. But there is an overlap between the ranges of this enzyme activity in AIP patients and normal individuals (26), which reduces the value of this assay. This is in part because distinct erythroid-specific and housekeeping isoforms of this enzyme are produced through alternative splicing of two distinct primary mRNA transcripts, such that some AIP mutations can affect the housekeeping (hepatic) but not the erythroid form of the enzyme (27). Also, the enzyme activity in circulating erythrocytes declines considerably as the cells age in the circulation, and changes in the proportion of younger red cells, which have higher enzyme activity, can substantially affect the results of this assay (28). Also, enzyme activity may be falsely lower if conditions for processing and shipping samples are not optimal.
Urine ALA is often measured with PBG, or after PBG elevation is found, but is less sensitive for diagnosis of AIP, HCP and VP. Urine ALA and coproporphyrin III, but not PBG, are markedly elevated in ADP.
As proposed by Whatley et al (22), second line testing in a patient with elevated PBG can be completed efficiently first by a plasma fluorescence scan to establish a diagnosis of VP; if this excludes VP a fecal porphyrin analysis including the coproporphyrin III/I ratio follows to differentiate HCP from AIP.
Diagnosis of an acute attack.
First-line testing as described above aims to establish with certainty whether or not a patient with compatible nonspecific symptoms has one of the acute porphyrias. But because biochemical abnormalities, including PBG and porphyrin elevations, may persist or recur in the absence of symptoms, these laboratory findings alone do not establish that an acute attack is in progress. For example, patients with acute porphyrias may have elevations in these levels when they develop other conditions causing abdominal pain. Therefore, the diagnosis of an acute attack in a patient already proven to have acute porphyria is largely clinical, and is not based on a specific level of ALA or PBG, or a certain degree of increase above baseline (3). Levels of ALA and PBG between attacks may become normal, but often remain elevated and fluctuate considerably, making it difficult to establish a true attack-free baseline. Also, acute attack results are seldom available in a timely manner to support a diagnosis of an attack. It is important to measure ALA and PBG before treatment with intravenous hemin, which can cause dramatic, rapid but often transient decreases in these levels.
The role of DNA testing.
Confirmation of a diagnosis is acute porphyria by identifying the pathogenic familial mutation is now considered standard of care (3, 12, 22). Once the familial mutation is identified, other family members can undergo reliable targeted mutation analysis. Several caveats regarding DNA testing must be kept in mind:
Finding a pathogenic mutation does not necessarily explain symptoms. The great majority of those in a family who inherit the familial mutation remain asymptomatic for all or most of their lifetimes. An individual with a pathogenic mutation and normal levels of PBG and porphyrins is regarded as having latent disease, so any concurrent symptoms would not be explained by porphyria. Therefore, biochemical testing remains essential in order to diagnose active disease as a cause of symptoms.
Not all sequence variants that are identified by gene sequencing are pathogenic, and at it may not be known immediately whether or not an identified variant is pathogenic. Some variants originally reported as pathogenic were found later to be benign (7).
A small percentage of pathogenic mutations are not detected by gene sequencing and require other methods such as gene dosage analysis for identification (22, 29). Such analysis may not be readily available, so results may be considerably delayed. Therefore, biochemical diagnosis remains especially important in such cases for documenting acute porphyria.
As with all DNA test results, it is important to maintain confidentiality of genetic information, which might be misused by employers and others.
Testing for acute porphyrias in patients without symptoms:
Individuals with past symptoms that suggest acute porphyria.
Elevations in PBG and porphyrins may persist in patients with acute porphyrias, so it is useful to measure urine PBG and porphyrins, plasma porphyrins and fecal porphyrins in patients with past symptoms. If these are negative, there are two options. Firstly, DNA testing and sequencing of 3 genes – HMBS, COPX and PPOX, keeping in mind that a small percentage of mutations are not detected by sequencing. Sequencing ALAD is probably not necessary because all patients with ADP reported to date have had persistent biochemical abnormalities. Secondly, urine ALA and porphyrins can be repeated at a future time when symptoms recur.
Relatives of patients with known porphyria.
Screening of all first-degree relatives of patients with proven acute porphyrias is recommended. For this to be done accurately, it is important as a first step to define the type of acute porphyria and identify the pathogenic mutation in an index case. First degree relatives are then screened for that mutation – a targeted process that is much less costly than gene sequencing. Targeted gene testing is much more dependable for testing relatives than measuring activities of enzymes, such as erythrocyte PBGD.
An example of this approach in a patient with AIP and her family is shown in Table 2. This 17-year-old year old female with autism developed grand mal seizures 3 months after menarche at age 13. She was started on carbamazepine, which was increased to 800 mg TID to control seizures. At age 17 she developed severe abdominal pain, constipation and extremity pain elevated BP and tachycardia. Over the next several years she had multiple ER visits and hospitalizations, with recurrent episodes of abdominal pain, ileus, hyponatremia, seizures and weight loss of 20 pounds. Acute porphyria was suspected and demonstrated by measurement of elevated urinary PBG, decreased erythrocyte PBG deaminase activity and a HMBS mutation previously associated with AIP. Her mother had been previously hospitalized for unexplained abdominal pain, and was also found to have AIP (elevated urine PBG, reduced erythrocyte PBGD activity and the same familial HMBS mutation), and the patient’s dizygotic twin sister was found to have latent AIP (normal PBG, reduced erythrocyte PBGD activity and the familial HMBS mutation).
Table 2.
Example of the current approach to biochemical and molecular diagnosis of acute intermittent porphyria first in an affected index case and then in two family members. See text for details.
| Testing | Ref | Patient | Sister | Mother | Father |
|---|---|---|---|---|---|
| Urine PBG (mg/g creatinine) | <4 mg/24 h | 27.5 | 1.7 | 19.4 | 1.2 |
| Erythrocyte PBG deaminase (nmol/mL/h) | 20–50 | 13 | 13 | 15 | 29 |
| HMBS mutation analysis | None | c.1084delT | c.1084delT | c.1084delT | ND |
| Diagnosis | - | AIP | Latent AIP | AIP | - |
Population screening.
It is currently not feasible to screen healthy individuals who are not relatives of known patients or those in the general population for these genetic disorders. Biochemical methods are not useful because penetrance of mutations that can cause acute porphyrias is very low. Interestingly, screening of large genomic databases has shown that heterozygous mutations that can cause AIP are unexpectedly common in healthy populations, and penetrance may be as low as 1%, which is much lower than previously thought, suggesting that unknown modifying genes or environmental influences are especially important for disease expression (7).
Biomarkers of disease activity
Levels of ALA, PBG and porphyrins and urine and plasma are important for diagnosis of acute porphyrias, but also serve as biomarkers of active disease. An important recently developed biomarker is ALAS1 mRNA, which can be measured in exosomes - small membrane vesicles of that are secreted by hepatocytes, as well as other cells, and are found in plasma and urine. Exosomal ALAS1 mRNA measured in plasma and urine serves as a noninvasive assessment of the degree of upregulation of ALAS1 mRNA in hepatocytes. This biomarker, along with ALA and PBG, responds to treatment with hemin as well as to givosiran, a new agent that down-regulates hepatic ALAS1 by RNA interference (12, 30, 31).
Current treatment and management
Effective treatment and management can be launched once a diagnosis of acute porphyria is established. Phases of treatment and management include treatment of the acute attack, prevention of attacks and monitoring for long term complications.
Treatment of the acute attack
Hospitalization is usually required for treatment of attacks. Cases with frequently recurring attacks are sometimes managed in outpatient settings if the patient is well-characterized and the attacks are always similar and respond rapidly to treatment. Hospitalization facilitates treatment of severe symptoms (e.g. pain requiring opioids, nausea and vomiting requiring promethazine or another phenothiazine, or ondansetron), agitation and other central nervous system manifestations, intravenous therapies and monitoring of respiration, electrolytes and nutritional status. Indeed, admission to intensive care is warranted if the vital capacity is impaired (3, 12).
Drugs known or likely to be unsafe in acute porphyrias should be discontinued whenever possible. Low doses of short-acting benzodiazepines are probably safe for anxiety and insomnia. β-Adrenergic blocking agents can be used for hypertension or for symptomatic tachycardia and hypertension, keeping in mind that risk from these drugs may be increased in patients with hypovolemia or incipient cardiac failure (32). Hyponatremia is carefully corrected, if present. Many anticonvulsant drugs can exacerbate acute porphyrias, although clonazepam may be less harmful than phenytoin, barbiturates, or valproic acid (33–35). Gabapentin, vigabatrin, levetiracetam, lamotrigine and bromides (used previously as an anticonvulsant) are considered safe.
Specific therapies are directed at down-regulating hepatic ALAS1 and thereby decreasing amounts of ALA and PBG originating in the liver and found in plasma and urine.
Carbohydrate loading can repress hepatic ALAS1 and reduce porphyrin precursor excretion. This effect is mediated in the liver by PGC-1α (peroxisome proliferator-activated receptor-γ coactivator-1α), and may be most effective in patients who are malnourished or when dietary restriction has contributed to the attack. However, the effects of this treatment are relatively weak, and high quality evidence for efficacy is lacking. Carbohydrate loading should be relied upon only for mild attacks (not requiring opioids for pain and without seizures, other severe central nervous system manifestations or complications such as hyponatremia or motor neuropathy). It is also used if hemin has been ordered but is not yet available. Carbohydrate may be administered as oral glucose polymer solutions if tolerated, or more often as intravenous treatment with 300 to 500 g of glucose, usually administered as a 10 percent solution in water or half-normal saline. Close monitoring of electrolytes is required because dilutional effects of large volumes of free water may contribute to hyponatremia. A more complete parenteral nutrition regimen is sometimes needed for prolonged attacks that impair oral intake (3, 12).
Intravenous hemin is much more potent than glucose in reducing ALA and PBG and is currently considered the most effective treatment for acute attacks, albeit in the absence of high quality clinical trial evidence (3, 12, 36, 37). Hemin is available in the United States as a lyophilized hematin preparation (Panhematin™, Recordati Rare Diseases, Northfield, IL) (38). It was the first drug approved under the U.S. Orphan Drug Act. The hemin preparation available in Europe and South Africa is heme arginate (Normosang™, Orphan Europe, Paris, France), a preparation of heme and arginine that is stable in solution (36, 39–41). When either of these products is infused intravenously, hemin becomes bound to circulating hemopexin and albumin and is then taken up primarily by hepatocytes. In hepatocytes, it reconstitutes the regulatory heme pool, and this leads to down-regulation of hepatic ALAS1 and a prompt reduction, usually within several days, in porphyrin precursor excretion.
For treatment of the acute attack the standard regimen is 3 to 4 mg/kg of hemin daily for 4 days, but this may be extended if a response is not observed within this time. Response may be slow particularly if treatment is not started early in an attack. Based on experience at many centers, hemin can be given safely during pregnancy (3, 12, 36, 42). Product labeling recommends reconstitution of hematin with sterile water, but it was discovered later that it then degrades rapidly, and degradation products bind to endothelial cells, clotting factors and platelets, causing infusion site phlebitis (especially with use of small peripheral veins) and a transient coagulopathy (43, 44). Repeated treatment can lead to significant loss of venous access and require insertion of an intravenous port. Bonkovsky and coworkers found that stabilization of hematin by reconstitution with 25 percent human albumin can prevent these adverse effects (45). This off-label approach has become widely used (46). Other less common side effects of hemin include fever, aching, malaise, hemolysis, anaphylaxis, and circulatory collapse (47, 48). Excessive dosing in one case caused reversible acute renal tubular damage (49). Heme arginate is also reconstituted with variable amounts of albumin at some centers.
There is a consensus that hemin is an effective and sometimes life-saving treatment for acute attacks. It is also used for prevention of attacks by providing infusions weekly or even biweekly, although this experience is more limited (50). A recent concern is that repeated dosing with hemin may itself have adverse effects on the liver in acute porphyrias and perpetuate a pattern of chronicity and increase the frequency of attacks (51). However, further studies are needed that distinguish between long term and progressive adverse hepatic effects of these diseases themselves from any effects that may result from treatment with hemin. Controlled trials are in progress that aim to provide better evidence for efficacy of hemin for treating and preventing attacks.
Cimetidine is a well-known inhibitor of hepatic CYPs, and was found to lessen the severity of porphyria and the degree of ALAS1 induction in rodents after administration of agents such as allylisopropylacetamide (52). This finding is not surprising, because this chemical is known to require metabolic activation by hepatic CYPs in order to cause chemically-induced porphyria. Cimetidine was then recommended for attacks of acute porphyrias based on uncontrolled observations in small numbers of patients. The drug was postulated to directly inhibit ALAS1 (53, 54). However, cimetidine could not be demonstrated to be an inhibitor of ALAS1, and there is no rationale to suggest that inhibition of CYP activities would be beneficial in human acute porphyrias. Therefore, cimetidine should not be recommended as an inexpensive alternative to hemin (3, 12).
Prevention of acute attacks
Management of patients with an ongoing pattern of repeated acute attacks is challenging (12). These patients generally have severely impaired quality of life and require considerable medical and family support. Efforts should be made to eliminate or reduce any factors that can trigger attacks, keeping in mind that attacks are most often due to a number of different factors that are additive. These efforts should include:
Review of current medications, including hormonal contraceptives, to eliminate any that might be harmful in acute porphyrias.
Search for concurrent infections or other underlying conditions that might trigger attacks.
Dietary history and nutritional assessment. This may be best done by a dietitian experienced in nutritional assessment and identifying dietary indiscretions.
Patients should be advised to maintain a well-balanced diet somewhat high in carbohydrate (60 to 70 percent of total calories). There is little evidence that additional dietary carbohydrate helps further in preventing attacks, and efforts to increase carbohydrate intake with the aim of preventing attacks may only lead to undesired weight gain. Patients who wish to lose excess weight should do so gradually and when they are clinically stable. Iron deficiency, if present, should be corrected (3, 12).
Smoking cessation is advised.
Menstrual history with attention to identifying whether onset of attacks occurs during the luteal phase of the cycle, which is a common association in women with acute porphyrias. Other factors, such as a harmful drug or dieting may combine with high levels of progesterone during the luteal phase to cause an attack.
Gonadotropin-releasing hormone analogues can be considered to prevent repeated attacks that are confined to the luteal phase of the cycle and occur at least 3–4 times per year (9, 55–57). Gynecological consultation should be obtained for a detailed baseline evaluation before this approach is considered. A GnRH analogue can be tried but is less effective in patients with attacks partially associated with the cycle. There is little risk from a trial of GnRH analogue treatment started in the first 1–3 days of a menstrual cycle and continue for a short period of time, such as several months. This will answer the question, using an agent that will not induce an attack, whether interruption of ovulation by any method will be effective in preventing attacks in that patient. This treatment can then be stopped if not effective. If effective, continuing a GnRH analogue beyond 6 months entails risk for irreversible bone loss. So if such treatment is continued, low-dose estradiol, preferably by the transdermal route, or a bisphosphonate may be added to prevent bone loss and other side effects, or treatment changed to a low-dose oral contraceptive. Unopposed estrogen effects on the endometrium, which can predispose to endometrial carcinoma, is a concern if a GnRH analogue with add-back estradiol is continued long term, and careful gynecological follow up is needed. It is reasonable to stop treatment after 1 year to see if it is still needed, since a pattern of frequent premenstrual attacks may continue for some years but seldom persists throughout the reproductive life in women with acute porphyrias.
Hemin prophylaxis may be effective in preventing attacks, and is appropriate especially for preventing noncyclic attacks. When used for this purpose, hemin is administered once or twice weekly in the absence of symptoms (50, 58). “On demand” treatment with hemin is a related approach that is not truly preventive or prophylactic, whereby arrangements are made in advance for patients to have access to hemin treatment as soon as possible after attack symptoms begin. Patients on a prophylactic regimen may at times delay a prophylactic dose for a few days and await the onset of symptoms, so are at times combining the prophylactic and on demand approaches. This is understandable, because hemin infusions are expensive, inconvenient to administer and sometimes have side effects (noted above). Patients on prolonged prophylaxis often have intravenous ports to facilitate venous access and sometimes require phlebotomies for iron overload. Ferritin levels may increase acutely after hemin infusion, so are not always reliable indicators of iron overload in this setting. Hemin is a short-acting drug, since it is rapidly metabolized in hepatocytes by heme oxygenase to biliverdin, which is then reduced by biliverdin reductase to bilirubin. Therefore, prophylactic administration of hemin is less likely to be effective if administered less frequently than weekly. Stopping prophylactic infusions or changing to on demand treatment is advisable after 1–2 years to see if this treatment is still needed.
RNA interference is a new and innovative approach, in the form of givosiran, which is currently in clinical trials. Givosiran is a small interfering RNA (siRNA) directed against ALAS1, and is derivatized with N-acetylgalactosamine for targeting to hepatocytes (30, 59). Because it is long-acting after subcutaneous injection to patients with AIP, it can reduce levels of ALA and PBG to normal or near-normal levels for a month or longer. For this reason, it is being developed for prevention of attacks, particularly in patients with 3–4 or more attacks per year (12).
Gene therapy, in which a viral vector was used to deliver a normal HMBS gene to hepatocytes, was promising in preclinical studies. Although a Phase 1 clinical trial did not show efficacy in preventing frequent attacks of AIP at the doses given, work is continuing to optimize vector design and increase delivered dose and transduction (60–63).
Liver transplantation has been highly effective as a last resort in patients who are disabled by repeated attacks and repeated hospitalizations (56, 64–66). This major surgical approach is clearly an option for severely affected patients, but advanced motor neuropathy increases the risk of postoperative complications. An increased risk of hepatic artery thrombosis is reported after transplantation for AIP (66). Therefore, careful patient selection and timing are important.
Cimetidine is also used to attempt to prevent attacks of porphyria. But as discussed above, there is no convincing rationale or supporting evidence for its use for treating acute porphyrias at any stage.
PBGD mRNA is another new and innovative treatment approach that is promising in early preclinical studies. A single strand of PBGD mRNA is packaged into biodegradable lipid nanoparticles, which are taken up by hepatocytes. In AIP mice, more than 90% of hepatocytes over-expressed human PBGD protein for 10 days, and levels of porphyrin precursors were reduced (67, 68). Clinical trials in AIP are anticipated.
Long-Term Monitoring
Patients with acute porphyrias are at risk for hypertension, renal damage, primary liver cancer (hepatocellular carcinoma or cholangiocarcinoma), depression and suicide (69). Hypertension can contribute to progression of renal disease, and should be controlled with drugs that are considered safe for acute porphyrias. Patients with acute porphyrias may develop end stage renal disease, possibly due to tubular and interstitial damage induced by ALA. It was found recently that a high-affinity variant of the human peptide transporter 2 (PEPT2), which is expressed by proximal tubular cells mediates the reabsorption of ALA, predisposed to development of renal damage in AIP (70). Renal function should be monitored in these patients and nephrotoxic drugs avoided. Losartan or other inhibitors of PEPT2 may have potential for preventing progression of renal disease associated with the acute porphyrias (70). Patients older than age 50 years, and especially those with continued elevations of ALA and PBG, have a 60–110-fold increased risk of primary liver cancer, and should be screened at least annually by ultrasound or an alternative imaging method to detect such lesions at an early stage (3, 12, 16, 37, 56)[Barelli]. Attention should also be given to long term treatment of depression and chronic pain.
Conclusions
The acute hepatic porphyrias are difficult to recognize because they are rare, and their nonspecific symptoms, even when life-threatening, resemble those of many other more common diseases. However, accurate and timely diagnosis is important because specific and effective treatment is available. Diagnostic screening should rely on first line testing, especially urine PBG as well as porphyrins. Second line testing establishes the type of acute porphyria, and DNA testing provides further confirmation and enables screening of family members for the identified pathogenic mutation. Specific treatment includes hemin and carbohydrate loading. Newer therapies, including RNA interference, are under development. These porphyrias deserve more attention in medical practice, and testing for elevated PBG and porphyrins should more often be part of the workup for unexplained symptoms such as abdominal pain. Once a diagnosis is established, effective treatment is available and new therapeutic approaches are under development.
Acknowledgments
Supported in part by a grant for the Porphyrias Consortium (1 U54 DK083909) of the NIH Rare Diseases Clinical Research Network from the National Center for Advancing Translational Sciences and the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, by the Institute for Translational Sciences at the University of Texas Medical Branch, supported in part by a Clinical and Translational Science Award (UL1TR001439) from the National Center for Advancing Translational Sciences, National Institutes of Health, and by the American Porphyria Foundation.
Abbreviations:
- ADP
ALA dehydratase porphyria
- AIP
acute intermittent porphyria
- ALAD
ALA dehydratase
- ALA
δ-aminolevulinic acid
- ALAS1
ubiquitous form of ALA synthase
- ALAS2
erythroid specific form of ALA synthase
- CPOX
coproporphyrinogen oxidase
- CYP
cytochrome P450 enzymes
- GnRH
gonadotropin releasing hormone
- HCP
hereditary coproporphyria
- HMBS
hydroxymethylbilane synthase
- PBG
porphobilinogen
- PBGD
PBG deaminase
- PGC1
δ peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PPOX
protoporphyrinogen oxidase
- SIADH
syndrome of inappropriate antidiuretic hormone production
- siRNA
small interfering RNA
- VP
variegate porphyria
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References cited
- 1.Phillips JD, Anderson KE. The porphyrias (Chapter 58). In: Kaushansky K, Lichtman MA, Prchal JT, et al. , eds. Williams Hematology, 9th edition. New York: McGraw-Hill; 2016:839–63. [Google Scholar]
- 2.Bonkovsky HL, Maddukuri VC, Yazici C, Anderson KE, Bissell DM, Bloomer JR, et al. Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. Am J Med 2014;127(12):1233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 2005;142(6):439–50. [DOI] [PubMed] [Google Scholar]
- 4.Meissner P, Adams P, Kirsch R. Allosteric inhibition of human lymphoblast and purified porphobilinogen deaminase by protoporphyrinogen and coproporphyrinogen. A possible mechanism for the acute attack of variegate porphyria. J Clin Invest 1993;91(4):1436–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akagi R, Kato N, Inoue R, Anderson KE, Jaffe EK, Sassa S. δ-Aminolevulinate dehydratase (ALAD) porphyria: The first case in North America with two novel ALAD mutations. Mol Genet Metab 2006;87:329–36. [DOI] [PubMed] [Google Scholar]
- 6.Neeleman RA, van Beers EJ, Friesema EC, Koole-Lesuis R, van der Pol WL, Wilson JHP, et al. Clinical remission of delta-aminolevulinic acid dehydratase deficiency through suppression of erythroid heme synthesis. Hepatology 2019. [DOI] [PMC free article] [PubMed]
- 7.Chen B, Solis-Villa C, Hakenberg J, Qiao W, Srinivasan RR, Yasuda M, et al. Acute Intermittent Porphyria: Predicted Pathogenicity of HMBS Variants Indicates Extremely Low Penetrance of the Autosomal Dominant Disease. Hum Mutat 2016;37(11):1215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bissell DM, Lai JC, Meister RK, Blanc PD. Role of delta-aminolevulinic acid in the symptoms of acute porphyria. Am J Med 2015;128(3):313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson KE, Spitz IM, Bardin CW, Kappas A. A GnRH analogue prevents cyclical attacks of porphyria. Arch Int Med 1990;150:1469–74. [PubMed] [Google Scholar]
- 10.Handschin C, Lin J, Rhee J, Peyer AK, Chin S, Wu PH, et al. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell 2005;122(4):505–15. [DOI] [PubMed] [Google Scholar]
- 11.Naik H, Stoecker M, Sanderson SC, Balwani M, Desnick RJ. Experiences and concerns of patients with recurrent attacks of acute hepatic porphyria: A qualitative study. Mol Genet Metab 2016;119(3):278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balwani M, Wang B, Anderson KE, Bloomer JR, Bissell DM, Bonkovsky HL, et al. Acute hepatic porphyrias: Recommendations for evaluation and long-term management. Hepatology 2017;66(4):1314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macalpine I, Hunter R, Rimington C, Brooke J, Goldberg A. Porphyria - A Royal Malady London: British Medical Association; 1968. [Google Scholar]
- 14.Peters T King George III, bipolar disorder, porphyria and lessons for historians. Clin Med 2011;11(3):261–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deacon AC, Peters TJ. Identification of acute porphyria: evaluation of a commercial screening test for urinary porphobilinogen. Ann Clin Biochem 1998;35(Pt 6):726–32. [DOI] [PubMed] [Google Scholar]
- 16.Stein PE, Badminton MN, Rees DC. Update review of the acute porphyrias. Br J Haematol 2017;176(4):527–38. [DOI] [PubMed] [Google Scholar]
- 17.Anderson KE, Drummond GS, Freddara U, Sardana MK, Sassa S. Porphyrogenic effects and induction of heme oxygenase in vivo by d-aminolevulinic acid. Biochim Biophys Acta 1981;676:289–99. [DOI] [PubMed] [Google Scholar]
- 18.Jacob K, Egeler E, Gross U, Doss MO. Investigations on the formation of urinary coproporphyrin isomers I-IV in 5-aminolevulinic acid dehydratase deficiency porphyria, acute lead intoxication and after oral 5-aminolevulinic acid loading. Clin Biochem 1999;32(2):119–23. [DOI] [PubMed] [Google Scholar]
- 19.Kuhnel A, Gross U, Jacob K, Doss MO. Studies on coproporphyrin isomers in urine and feces in the porphyrias. Clin Chim Acta 1999;282(1–2):45–58. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Yasuda M, Desnick RJ, Balwani M, Bishop D, Yu C. A LC-MS/MS method for the specific, sensitive, and simultaneous quantification of 5-aminolevulinic acid and porphobilinogen. J Chromatogr B Analyt Technol Biomed Life Sci 2011;879(24):2389–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poh-Fitzpatrick MB. A plasma porphyrin fluorescence marker for variegate porphyria. Arch Dermatol 1980;116:543–7. [PubMed] [Google Scholar]
- 22.Whatley SD, Mason NG, Woolf JR, Newcombe RG, Elder GH, Badminton MN. Diagnostic strategies for autosomal dominant acute porphyrias: retrospective analysis of 467 unrelated patients referred for mutational analysis of the HMBS, CPOX, or PPOX gene. Clin Chem 2009;55(7):1406–14. [DOI] [PubMed] [Google Scholar]
- 23.Hift RJ, Davidson BP, van der Hooft C, Meissner DM, Meissner PN. Plasma fluorescence scanning and fecal porphyrin analysis for the diagnosis of variegate porphyria: precise determination of sensitivity and specificity with detection of protoporphyrinogen oxidase mutations as a reference standard. Clin Chem 2004;50(5):915–23. [DOI] [PubMed] [Google Scholar]
- 24.Enriquez de Salamanca R, Sepulveda P, Moran MJ, Santos JL, Fontanellas A, Hernandez A. Clinical utility of fluorometric scanning of plasma porphyrins for the diagnosis and typing of porphyrias. Clin Exp Dermatol 1993;18(2):128–30. [DOI] [PubMed] [Google Scholar]
- 25.Blake D, McManus J, Cronin V, Ratnaike S. Fecal coproporphyrin isomers in hereditary coproporphyria. Clin Chem 1992;38:96–100. [PubMed] [Google Scholar]
- 26.Sassa S, Granick S, Bickers DR, Bradlow HL, Kappas A. A microassay for uroporphyrinogen I synthase, one of three abnormal enzyme activities in acute intermittent porphyria, and its application to the study of the genetics of this disease. Proc Natl Acad Sci USA 1974;71:732–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C-H, Astrin KH, Lee G, Anderson KE, Desnick RJ. Acute intermittent porphyria: identification and expression of exonic mutations in the hydroxymethylbilane synthase gene: an initiation codon missense mutation in the housekeeping transcript causes “variant acute intermittent porphyria” with normal expression of the erythroid-specific enzyme. J Clin Invest 1994;94:1927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson KE, Sassa S, Peterson CM, Kappas A. Increased erythrocyte uroporphyrinogen-I-synthetase, δ-aminolevulinic acid dehydratase and protoporphyrin in hemolytic anemias. Am J Med 1977;63:359–64. [DOI] [PubMed] [Google Scholar]
- 29.Whatley SD, Badminton MN. Role of genetic testing in the management of patients with inherited porphyria and their families. Ann Clin Biochem 2013. [DOI] [PubMed]
- 30.Chan A, Liebow A, Yasuda M, Gan L, Racie T, Maier M, et al. Preclinical Development of a Subcutaneous ALAS1 RNAi Therapeutic for Treatment of Hepatic Porphyrias Using Circulating RNA Quantification. Mol Ther Nucleic Acids 2015;4:e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manceau H, Gouya L, Puy H. Acute hepatic and erythropoietic porphyrias: from ALA synthases 1 and 2 to new molecular bases and treatments. Curr Opin Hematol 2017;24(3):198–207. [DOI] [PubMed] [Google Scholar]
- 32.Bonkowsky HL, Tschudy DP. Hazard of propranolol in treatment of acute porphyria (letter). Br Med J 1974;4:47–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki A, Aso K, Ariyoshi C, Ishimaru M. Acute intermittent porphyria and epilepsy: safety of clonazepam. Epilepsia 1992;33:108–11. [DOI] [PubMed] [Google Scholar]
- 34.Bonkowsky HL, Sinclair PR, Emery S, Sinclair JF. Seizure management in acute hepatic porphyria: risks of valproate and clonazepam. Neurology 1980;30:588–92. [DOI] [PubMed] [Google Scholar]
- 35.Larson AW, Wasserstrom WR, Felsher BF, Shih JC. Posttraumatic epilepsy and acute intermittent porphyria: effects of phenytoin, carbamazepine, and clonazepam. Neurology 1978;28:824–8. [DOI] [PubMed] [Google Scholar]
- 36.Mustajoki P, Nordmann Y. Early administration of heme arginate for acute porphyric attacks. Arch Int Med 1993;153:2004–8. [PubMed] [Google Scholar]
- 37.Harper P, Sardh E. Management of acute intermittent porphyria. Expert Opin Orphan Drugs 2014;2(4):349–68. [Google Scholar]
- 38.Siegert SW, Holt RJ. Physicochemical properties, pharmacokinetics, and pharmacodynamics of intravenous hematin: a literature review. Adv Ther 2008;25(9):842–57. [DOI] [PubMed] [Google Scholar]
- 39.Timonen TTT, Kauma H. Therapeutic effect of heme arginate in myelodysplastic syndromes. Eur J Haematol 1992;49:234–8. [DOI] [PubMed] [Google Scholar]
- 40.Tenhunen R Heme arginate: its characterization, metabolism and clinical implications. In: Mustajoki P, ed. New Therapeutic Approaches to Hepatic Porphyrias Vammala: Vammalan Kirjapaino Oy; 1987:36–43. [Google Scholar]
- 41.Tenhunen R, Tokola O, Lindén IB. Haem arginate: a new stable haem compound. J Pharm Pharmacol 1987;39:780–6. [DOI] [PubMed] [Google Scholar]
- 42.Badminton MN, Deybach JC. Treatment of an acute attack of porphyria during pregnancy. Eur J Neurol 2006;13(6):668–9. [DOI] [PubMed] [Google Scholar]
- 43.Green D, Tsao CH. Hematin: effects on hemostasis. J Lab Clin Med 1990;115:144–7. [PubMed] [Google Scholar]
- 44.Jones RL. Hematin-derived anticoagulant. Generation in vitro and in vivo. J Exp Med 1986;163:724–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonkovsky HL, Healey JF, Lourie AN, Gerron GG. Intravenous heme-albumin in acute intermittent porphyria: evidence for repletion of hepatic hemoproteins and regulatory heme pools. Am J Gastroenterol 1991;86:1050–6. [PubMed] [Google Scholar]
- 46.Anderson KE, Bonkovsky HL, Bloomer JR, Shedlofsky SI. Reconstitution of hematin for intravenous infusion. Ann Intern Med 2006;144(7):537–8. [DOI] [PubMed] [Google Scholar]
- 47.Daimon M, Susa S, Igarashi M, Kato T, Kameda W. Administration of heme arginate, but not hematin, caused anaphylactic shock. Am J Med 2001;110(3):240. [DOI] [PubMed] [Google Scholar]
- 48.Khanderia U Circulatory collapse associated with hemin therapy for acute intermittent porphyria. Clin Pharmacy 1986;5:690–2. [PubMed] [Google Scholar]
- 49.Jeelani Dhar G, Bossenmaier I, Cardinal R, Petryka ZJ, Watson CJ. Transitory renal failure following rapid administration of a relatively large amount of hematin in a patient with acute intermittent porphyria in clinical remission. Acta Med Scand 1978;203:437–43. [DOI] [PubMed] [Google Scholar]
- 50.Marsden JT, Guppy S, Stein P, Cox TM, Badminton M, Gardiner T, et al. Audit of the Use of Regular Haem Arginate Infusions in Patients with Acute Porphyria to Prevent Recurrent Symptoms. JIMD Rep 2015;22:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmitt C, Lenglet H, Yu A, Delaby C, Benecke A, Lefebvre T, et al. Recurrent attacks of acute hepatic porphyria: major role of the chronic inflammatory response in the liver. J Intern Med 2018. [DOI] [PubMed]
- 52.Marcus DL, Nadel H, Lew G, Freedman ML. Cimetidine Suppresses Chemically Induced Experimental Hepatic Porphyria. Am J Med Sci 1990;300:214–7. [DOI] [PubMed] [Google Scholar]
- 53.Cherem JH, Malagon J, Nellen H. Cimetidine and acute intermittent porphyria. Ann Intern Med 2005;143(9):694–5. [DOI] [PubMed] [Google Scholar]
- 54.Horie Y, Norimoto M, Tajima F, Sasaki H, Nanba E, Kawasaki H. Clinical usefulness of cimetidine treatment for acute relapse in intermittent porphyria. Clin Chim Acta 1995;234(1–2):171–5. [DOI] [PubMed] [Google Scholar]
- 55.Yamamori I, Asai M, Tanaka F, Muramoto A, Hasegawa H. Prevention of premenstrual exacerbation of hereditary coproporphyria by gonadotropin-releasing hormone analogue. Intern Med 1999;38(4):365–8. [DOI] [PubMed] [Google Scholar]
- 56.Stein P, Badminton M, Barth J, Rees D, Stewart MF. Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann Clin Biochem 2013:217–23. [DOI] [PubMed]
- 57.Schulenburg-Brand D, Gardiner T, Guppy S, Rees DC, Stein P, Barth J, et al. An Audit of the Use of Gonadorelin Analogues to Prevent Recurrent Acute Symptoms in Patients with Acute Porphyria in the United Kingdom. JIMD Rep 2017;36:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anderson KE, Egger NG, Goeger DE. Heme arginate for prevention of acute porphyric attacks (abstract). Acta Haematologica 1997;98, Supplement 1:120. [Google Scholar]
- 59.Yasuda M, Gan L, Chen B, Kadirvel S, Yu C, Phillips JD, et al. RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc Natl Acad Sci U S A 2014;111(21):7777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.D’Avola D, Lopez-Franco E, Sangro B, Paneda A, Grossios N, Gil-Farina I, et al. Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria. J Hepatol 2016;65(4):776–83. [DOI] [PubMed] [Google Scholar]
- 61.Brunetti-Pierri N, Newsome PN. AAV-mediated liver-directed gene therapy for acute intermittent porphyria: It is safe but is it effective? J Hepatol 2016;65(4):666–7. [DOI] [PubMed] [Google Scholar]
- 62.Serrano-Mendioroz I, Sampedro A, Serna N, de Salamanca RE, Sanz-Parra A, Corrales F, et al. Bioengineered PBGD variant improves the therapeutic index of gene therapy vectors for acute intermittent porphyria. Hum Mol Genet 2018;27(21):3688–96. [DOI] [PubMed] [Google Scholar]
- 63.Serrano-Mendioroz I, Sampedro A, Alegre M, Enriquez de Salamanca R, Berraondo P, Fontanellas A. An Inducible Promoter Responsive to Different Porphyrinogenic Stimuli Improves Gene Therapy Vectors for Acute Intermittent Porphyria. Hum Gene Ther 2018;29(4):480–91. [DOI] [PubMed] [Google Scholar]
- 64.Yasuda M, Erwin AL, Liu LU, Balwani M, Chen B, Kadirvel S, et al. Liver transplantation for acute intermittent porphyria: biochemical and pathologic studies of the explanted liver. Mol Med 2015;21:487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singal AK, Parker C, Bowden C, Thapar M, Liu L, McGuire BM. Liver transplantation in the management of porphyria. Hepatology 2014;60(3):1082–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dowman JK, Gunson BK, Mirza DF, Bramhall SR, Badminton MN, Newsome PN, et al. Liver transplantation for acute intermittent porphyria is complicated by a high rate of hepatic artery thrombosis. Liver Transpl 2012;18(2):195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang L, Berraondo P, Jerico D, Guey LT, Sampedro A, Frassetto A, et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat Med 2018;24(12):1899–909. [DOI] [PubMed] [Google Scholar]
- 68.Fontanellas A, Avila MA, Berraondo P. Emerging therapies for acute intermittent porphyria. Expert Rev Mol Med 2016;18:e17. [DOI] [PubMed] [Google Scholar]
- 69.Jeans JB, Savik K, Gross CR, Weimer MK, Bossenmaier IC, Pierach CA, et al. Mortality in patients with acute intermittent porphyria requiring hospitalization: a United States case series. Am J Med Genet 1996;11:269–73. [DOI] [PubMed] [Google Scholar]
- 70.Tchernitchko D, Tavernier Q, Lamoril J, Schmitt C, Talbi N, Lyoumi S, et al. A Variant of Peptide Transporter 2 Predicts the Severity of Porphyria-Associated Kidney Disease. J Am Soc Nephrol 2017;28(6):1924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]