

Abstract
This review describes the effects of FLT3 mutations that alter its intracellular localization and modify its glycosylation, leading to differences in downstream signaling pathways. The most common type of FLT3 mutation, internal tandem duplication (FLT3-ITD), leads to localization in the endoplasmic reticulum and constitutive strong activation of STAT5. In contrast, the ligand-activated FLT3-wild type is mainly expressed on the cell surface and activates MAP kinases. Based on these backgrounds, several reports have demonstrated that glycosylation inhibitors are effective for inhibition of FLT3-ITD expression and intracellular localization. The general subcellular localization regulatory mechanisms for receptor tyrosine kinases are also discussed.
Keywords: FLT3, Internal tandem duplication, Glycosylation, Intracellular localization, Downstream signaling
1. Introduction
FLT3 (Fms-like tyrosine kinase 3) is a member of the class III receptor tyrosine kinase family. Notably, approximately one-third of acute myeloid leukemia (AML) patients have mutations of this gene, and such mutations are one of the most frequently identified types of genetic alterations in AML [1,2]. The majority of the mutations involve an internal tandem duplication (ITD) in the juxtamembrane domain of FLT3 that is specifically found in AML [3]. The second most common type of FLT3 mutation in AML are mutations in the activation loop of the tyrosine kinase domain (TKD). Almost all of these mutations involve an aspartate-to-tyrosine substitution at codon 835, although other substitutions have also been identified [4,5]. These mutations cause a conformational change of the molecule and disrupt its autoinhibitory function, thereby rendering the receptor constitutively active [6], [7], [8].
Differences in the downstream pathways of FLT3-ITD and FLT3-TKD have been extensively characterized using the cytokine-dependent 32D and Ba/F3 cell lines. Several studies have demonstrated that the most unique downstream signaling effect of FLT3-ITD is STAT5 activation [9], [10], [11]. FLT3-ITD, FLT3-TKD, and ligand-activated FLT3-wild-type (WT) downstream signaling pathways commonly activate MAP kinase, Akt, and Shc [11]. The differences in these signaling pathways are an attractive target focus for research, and can be partially explained by the location changes observed after FLT3 mutations that affect the maturation of the surface glycoproteins.
As there are many excellent reviews on the roles of FLT3 in hematological malignancies [1,2,[6], [7], [8],12], this review particularly focuses upon on the differences in intracellular localization of wild-type and mutant FLT3 receptors that affect the modification status for glycosylation and influence the downstream signaling pathways. The author also summarizes recent knowledge on the maturation and localization changes of mutated FLT3 receptors and other receptor tyrosine kinases and the signaling differences arising from these regulations.
2. Glycosylation status is affected by mutational status of FLT3 receptor
It was reported that two species of FLT3 receptor can be detected by western blotting [13]: a 150-kDa species representing the complex glycosylated mature form and a 130-kDa species representing the under-glycosylated immature form with mannose-rich structures. In cells expressing FLT3-ITD, the 130-kDa species was predominantly detectable, while cells expressing FLT3-WT had proportionally more of the 150-kDa species [13]. Galanthus nivalis lectin, which is selective for mannose-rich structures, was shown to interact specifically with the 130-kDa species expressed as FLT3-ITD [14]. Furthermore, the 130-kDa form was selectively deglycosylated by digestion with endo-H, which is highly specific for immature mannose-rich carbohydrates [14]. Because mannose-rich glycoproteins are a hallmark of endoplasmic reticulum (ER) proteins, it was concluded that the FLT3 130-kDa species resides in an ER compartment [13]. In addition, it was also demonstrated that FLT3-ITD is localized in a perinuclear region [15]. Collectively, attenuated glycoprotein maturation and intracellular localization are probably cause and effect, and the mutational state of FLT3 may regulate these statuses.
3. FLT3-ITD affects the maturation and intracellular localization, thereby regulating the downstream pathways
Accumulating evidence suggests that dynamic changes in the intracellular localization of FLT3 and maturation of the glycoprotein affect the control of its downstream signaling pathways (Fig. 1(A)). A very important paper by Choudhary et al. [16] described that FLT3-ITD localized at the ER aberrantly activates STAT5, while FLT3-ITD localized at the membrane strongly activates the MAPK and PI3K pathways with diminished phosphorylation of STAT5. Köthe et al. [17] further confirmed that FLT3-ITD localized at the plasma membrane leads to constitutive activation of K-Ras. It was recently reported that FLT3-ITD and FLT3-D835Y (FLT3-TKD) are retained in the perinuclear ER, while FLT3-WT is expressed in the plasma membrane [18]. After addition of the tyrosine kinase inhibitor (TKI) AC220, the intracellular localization of FLT3-ITD, as well as FLT3-TKD, changes to a plasma membrane localization, similar to FLT3-WT or FLT3-N676K, another type of FLT3-TKD (Fig. 1(B)) [18]. This phenomenon was also reported by Schmidt-Arras et al. [13], who noted that inhibition of FLT3-ITD kinase by small molecules, inactivating point mutations, or co-expression with protein-tyrosine phosphatases promotes complex glycosylation and surface expression (Fig. 1(B)) [13]. Based on these observations, Reiter et al. [18] demonstrated that TKI treatment boosts FLT3 × CD3 antibody-mediated cytotoxicity against FLT3-ITD-positive AML cells.
Fig. 1.

Subcellular localizations of FLT3-ITD and FLT3-TKD, their maturation statuses, and their effects on downstream pathways. (A) FLT3-ITD localized at the ER activates STAT5, while FLT3-ITD localized at the membrane strongly activates the MAPK and PI3K pathways [16]. (B) Addition of TKI, inactivating point mutations, or co-expression with protein-tyrosine phosphatases (PTP) promotes complex glycosylation and surface expression of FLT3-ITD and FLT3-TKD, similar to FLT3-WT [13,17,18].
The mechanisms, which leads to changes in subcellular localization of FLT3 as consequence of activating mutations, remained unclear. However, Rudorf et al. [19] recently revealed one of these mechanisms, that FLT3-TKD is able to activate the downstream effector molecule STAT5 in the presence of mutated Nucleophosmin (NPM), NPM1c (Fig. 2). They showed that NPM1c alters the cellular localization of FLT3-TKD from the cell surface to ER, which may lead to the aberrant activation of STAT5. They revealed that co-immunoprecipitation of FLT3 shows interaction with NPM1c in OCI-AML3 (NPM1c) FLT3-TKD (D835Y) cells, low interaction in OCI-AML3 (NPM1c) FLT3-ITD cells, but not in HL-60 (NPM wild type)-FLT3-TKD cells. Phosphorylation of FLT3 at amino acid 835 is crucial for NPM1c interaction. In addition, they clearly demonstrated that aberrant STAT5 activation occurs not only in primary murine cells but also in patients with AML with combined FLT3-TKD and NPM1c mutations. These findings may provide potential mechanisms leading to intracellular retention or altered trafficking of mutated FLT3 receptors.
Fig. 2.

Mechanisms of the change of subcellular localization of FLT3-TKD and effect for downstream pathways. Mutations of NPM1 result in cytosolic form of NPM1, called NPM1c, alters the cellular localization of FLT3-TKD from the cell surface to ER, which leads to the aberrant activation of STAT5 [19].
Moloney et al. [20] demonstrated that FLT3-ITD at the plasma membrane is responsible for activation and phosphorylation of the AKT signaling pathway and production of p22phox-generated H2O2. Inhibition of FLT3-ITD-generated ROS at the plasma membrane leads to NOX4 de-glycosylation and p22phox proteasomal degradation (Fig. 3) [20]. The same group also demonstrated that not only AKT but also ERK1/2, GSK3β and STAT5 result in activation and production of DNA-damaging NOX4D-generated H2O2 at the nuclear membrane (Fig. 2) [21]. Collectively, these findings suggest that the change in localization with surface modification of the glycoprotein alter the effects of FLT3 downstream pathways.
Fig. 3.
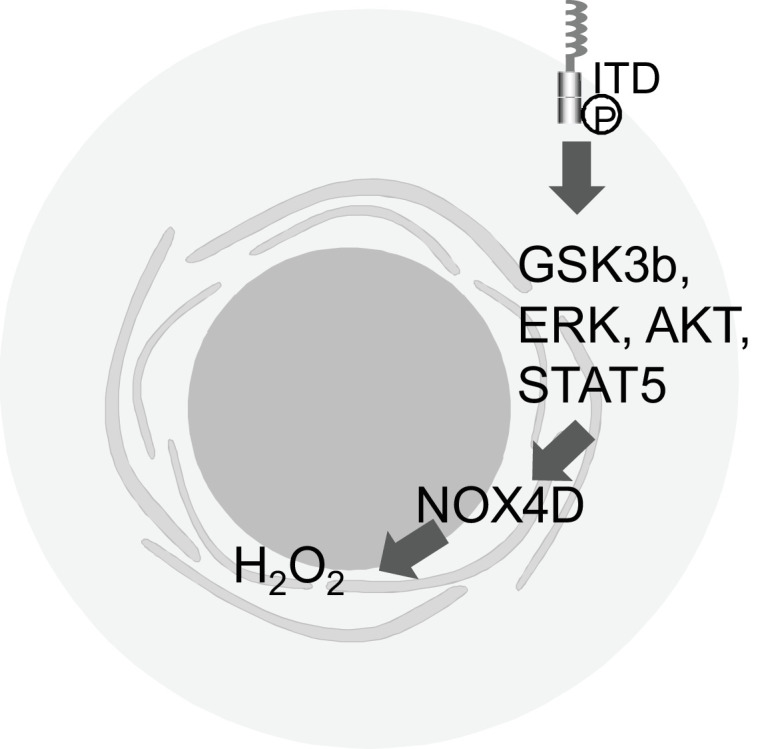
Downstream signaling of FLT3-ITD at the plasma membrane. FLT3-ITD at the plasma membrane is responsible for activation and phosphorylation of AKT, ERK1/2, GSK3β and STAT5, resulting in activation and production of DNA-damaging NOX4D-generated H2O2 [20, 21].
4. Effects of glycosylation inhibitors for FLT3 function
Fluvastatin, a statin, was reported to attenuate mutant FLT3 kinase activity by preventing complex glycosylation of the receptor [22]. This effect induces altered localization and signaling and ultimately leads to induction of apoptosis [22].
Statins were developed to lower cholesterol and triglyceride. They act by blocking 3‑hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase, a rate-limiting step in the mevalonate pathway [23]. The mevalonate pathway produces dolichol, which is responsible for co-translational transfer of oligosaccharides to nascent polypeptides that undergo N-linked glycosylation [24]. Williams et al. [22] clearly demonstrated that fluvastatin inhibits FLT3 glycosylation and prolongs survival of mice with FLT3-ITD leukemia. The anticancer drug 2-deoxy-d-glucose (2-DG), which also inhibits N-linked glycosylation, has been extensively investigated in solid tumors [25]. However, there have been few reports [26] on the effects of 2-DG in leukemia cells. Larrue et al. [27] recently described that 2-DG efficiently inhibits the activity of FLT3-ITD and c-KIT mutants in AML. They clearly showed that 2-DG acts through N-linked glycosylation inhibition to inhibit the cell-surface expression and cellular signaling of FLT3-ITD and c-KIT mutant, and induce apoptotic cell death [27]. Recently, Tsitsipatis et al. [28] demonstrated that abrogation of FLT3-ITD glycoprotein maturation by low doses of the N-glycosylation inhibitor tunicamycin has antiproliferative and pro-apoptotic effects. These effects are partly mediated by arresting FLT3-ITD in an under-glycosylated state and thereby attenuating FLT3-ITD-driven AKT and ERK signaling [28]. In combination with FLT3 kinase inhibitors, tunicamycin exhibits a strong and specific synergy in killing of FLT3-ITD-expressing cell lines and primary AML cells [28]. Although tunicamycin is currently not a clinically approved drug, it was reported to be well-tolerated in vivo using mouse models [29,30]. Therefore, possible use of tunicamycin as a clinical drug combined with FLT3 inhibitors can be expected in the future. Collectively, the findings suggest that this glycosylation inhibitor can disturb the cell-surface expression and cellular signaling of FLT3 and c-KIT mutants.
Although FLT3 inhibitors have shown promising efficacies in AML, their duration of clinical response is short because of the rapid development of resistance [31]. Because there are many good examples of combination therapy with FLT3 inhibitors [32], [33], [34], FLT3 inhibition with glycosylation inhibition may be a new attractive combination therapy for AML.
5. Subcellular localization of receptor tyrosine kinases may generally affect the activation of downstream kinase pathways
ER or Golgi compartment retention is not FLT3-specific, but rather a general mechanism for activation of receptor tyrosine kinases (RTKs) and downstream pathways. Cleavage at the transmembrane domain of RTK, which yields an intracellular domain may occur by various mechanisms in response to various stimuli [35]. RTK intracellular domain fragments are stabilized and intracellularly transported into subcellular compartments, such as the nucleus, by binding to chaperones or transcription factors, while membrane-bound RTKs (full-length or truncated) are transported from the plasma membrane to the ER through association with cargo proteins, such as Rab- or clathrin-binding adaptor proteins, through retrograde trafficking pathways [36,37]. Rab functions in internalization and transport to degradation, as well as recycling to the plasma membrane and the Golgi [36,37].
Cellular receptors for growth factors and cytokines are post-translationally modified with N-linked branched carbohydrate chains [13]. Newly synthesized polypeptide chains initially become glycosylated with mannose-rich branched oligosaccharides in the ER. The glycoproteins are then subjected to partial de-glycosylation, and transferred to the Golgi compartment for more complex glycosylation [13,38]. As already described in several reviews, irreversible RTK modifications, such as proteolytic cleavage events, are related to changes in intracellular localization [36,39,40].
Abnormal maturation and trafficking have been observed for many tyrosine kinase receptors, including platelet-derived growth factor (PDGF), KIT, and colony stimulating factor (CSF) 1 receptors, which are type III RTKs like FLT3. Intracellular activation of PDGF receptors by the v-sis protein is related to sis-mediated transformation [41]. Like FLT3 receptor, normal cells synthesize the receptor as a 160-kDa precursor that contains core, endo-H-sensitive, and asparagine-linked sugars. The precursor is converted to a 180-kDa mature form that contains complex endo-H-resistant oligosaccharides and is expressed at the cell surface [41,42]. In v-sis-transformed cells, a 140-kDa incompletely-processed form of the receptor is observed in addition to the 160-kDa precursor. Autocrine activation of PDGF receptor occurs in intracellular compartments [41]. Tabone-Eglinger et al. [43] reported that mutations in KIT, also known as c-KIT, induce intracellular retention and activation of an immature form of KIT protein in gastrointestinal stromal tumors (GISTs). They showed that GIST-type KIT mutations induce activation-dependent alterations in normal maturation and trafficking, resulting in intracellular retention of the activated kinase within the cell. Kim et al. [44] reported that mutant KIT proteins are intrinsically less stable than wild type KIT due to proteasome mediated degradation. They and others have shown that mutant KIT, similar to FLT3, abnormally localized to the ER or Golgi complex [43], [44], [45], [46]. Importantly, by screening a mutant KIT stabilizing factor, Kim et al. [44] have found that protein kinase (PKC)-θ is strongly expressed in GISTs. They also found that PKC-θ interacts with intracellular mutant KIT to promote its stabilization by increased retention in the Golgi complex. Another example is CSF1 receptor, which exhibits activating mutations that retard transport to the cell surface and lead to tyrosine phosphorylation in the absence of a ligand, resulting in CSF1-independent signals for cell growth and transformation [47].
In the maturation of fibroblast growth factor receptor 3 (FGFR3), an RTK, an inhibitory role of tyrosine phosphorylation has been suggested. Lievens et al. [48] demonstrated that amino acid substitutions at the Lys-650 codon within the activation loop kinase domain of FGFR3 result in constitutive phosphorylation of the receptor. Highly activated tyrosine-phosphorylated SADDAN (severe achondroplasia with developmental delay and acanthosis nigricans) mutants lead to accumulation of the immature and phosphorylated form in the ER, which fails to be degraded. The ER-retained constitutively-active FGFR3 activates the Janus kinase (JAK)/STAT pathway by recruiting Jak1, while wild-type FGFR3 is unable to activate STAT1 [48]. A similar mechanism is observed for anaplastic lymphoma kinase (ALK). When common mutations of ALK, a membrane-associated RTK, occur, the ALK mutants are essentially intracellular and largely retained in ER/Golgi compartments, and this localization is correlated with a defect of N-linked glycosylation [49]. Furthermore, Mazot et al. [49] reported that constitutive activity of ALK impairs receptor trafficking.
Not only point mutations but also fusions of RTKs play a role in the aberrant intracellular localization and downstream signaling of receptors. Neel et al. [50] recently reported that clinically relevant ROS1 RTK fusion oncoproteins show differential subcellular localizations, which impart distinct cell signaling and oncogenic properties. When ROS1 fusion proteins are localized to endosomes, the strongest activations of MAPK signaling are observed. A list of the RTKs described in this review is provided in Table 1.
Table 1.
List of RTKs whose functions are affected by intracellular localizations.
| Name of RTK | Description | Refs. |
|---|---|---|
| FLT3-ITD | In ER, FLT3-ITD activates STAT5, while in membrane FLT3-ITD strongly activates MAPK and PI3K | [16,17] |
| FLT3-ITD, FLT3-TKD, FLT3-N676K | After the addition of tyrosine kinase inhibitor (TKI) AC220, a intracellular localization of FLT3-ITD, as well as FLT3-TKD was changed to plasma membrane, which is similar to FLT3-WT or FLT3-N676K, another type of FLT3 TKD | [18] |
| FLT3-TKD | NPM1c alters the cellular localization of FLT3-TKD from the cell surface to ER, which may lead to the aberrant activation of STAT5. | [19] |
| FLT3-ITD | In plasma membrane, FLT3ITD activates AKT signaling pathway and produces of p22phox-generated H2O2 | [20] |
| PDGFR | The precursor of PDGF receptor is converted to a 180-kD mature form. Intracellular activation of PDGFR by the v-Sis protein has been related to sis-mediated transformation. | [41,42] |
| KIT mutation | KIT mutations induce intracellular retention and activation of an immature form of the KIT protein in gastrointestinal stromal tumors | [43], [44], [45], [46] |
| CSF1 mutation | CSF1 activating mutation retarded in transport to the cell surface and were phosphorylated on tyrosine in the absence of ligand, resulting in CSF-1-independent signals for cell growth and transformation. | [47] |
| FGFR3 mutation | The highly activated tyrosine phosphorylated SADDAN mutants accumulates its immature and phosphorylated from in the ER, which fails to be degraded. ER retained constitutively active FGFR3 activates JAK/STAT pathway. | [48] |
| ALK mutation | Constitutive active form of ALK impairs receptor trafficking. Mutated ALK variants were essentially intracellular and were largely retained in the reticulum/Golgi compartments, and this is corroborated with a defect of N-linked glycosylation | [49] |
| ROS1 RTK fusion proteins | SDC4-ROS1 and SLC34A2-ROS1 fusion oncoproteins resided on endosomes and activated the MAPK pathway. | [50] |
In summary, not only FLT3 and other type III RTKs, but also many additional RTKs have similar characteristics for the effects of their mutation, maturation, intracellular localization, and activation on downstream signaling.
6. Conclusion
Collectively, subcellular localization and surface glycoprotein modification impact the functions of FLT3 and many other RTKs and lead to in changes in downstream pathways. Based on these mechanisms, several promising drugs have been attracting attention, including fluvastatin [22], 2-DG [27], and tunicamycin [28]. In particular, statins are a class of drugs already approved by the US Food and Drug Administration and may be repurposed for the management of AML cases with FLT3-ITD. Further clarification of the mechanisms of these regulations may lead to the development of effective therapies for RTK-activated leukemia.
Funding
This work was supported in part by a Grant-in-Aid for Scientific Research (No. 17K09019) from the Ministry of Education, Science and Culture, Japan, Astellas Research Support (Astellas Pharma Inc.) and Pfizer Academic Contribution (Pfizer Inc.).
Declaration of Competing Interest
The author declares that they have no competing interests.
CRediT authorship contribution statement
Shinichiro Takahashi: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing.
Acknowledgments
The authors thank Alison Sherwin, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
References
- 1.Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J. Hematol. Oncol. 2011;4:13. doi: 10.1186/1756-8722-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takahashi S. Current findings for recurring mutations in acute myeloid leukemia. J. Hematol. Oncol. 2011;4:36. doi: 10.1186/1756-8722-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yokota S., Kiyoi H., Nakao M., Iwai T., Misawa S., Okuda T., Sonoda Y., Abe T., Kahsima K., Matsuo Y., Naoe T. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia. 1997;11(10):1605–1609. doi: 10.1038/sj.leu.2400812. [DOI] [PubMed] [Google Scholar]
- 4.Taketani T., Taki T., Sugita K., Furuichi Y., Ishii E., Hanada R., Tsuchida M., Ida K., Hayashi Y. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood. 2004;103(3):1085–1088. doi: 10.1182/blood-2003-02-0418. Epub 2003 Sep 22. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto Y., Kiyoi H., Nakano Y., Suzuki R., Kodera Y., Miyawaki S., Asou N., Kuriyama K., Yagasaki F., Shimazaki C., Akiyama H., Saito K., Nishimura M., Motoji T., Shinagawa K., Takeshita A., Saito H., Ueda R., Ohno R., Naoe T. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
- 6.Gilliland D.G., Griffin J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 7.Markovic A., MacKenzie K.L., Lock R.B. FLT-3: a new focus in the understanding of acute leukemia. Int. J. Biochem. Cell Biol. 2005;37(6):1168–1172. doi: 10.1016/j.biocel.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Stirewalt D.L., Radich J.P. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer. 2003;3(9):650–665. doi: 10.1038/nrc1169. [DOI] [PubMed] [Google Scholar]
- 9.Mizuki M., Fenski R., Halfter H., Matsumura I., Schmidt R., Muller C., Gruning W., Kratz-Albers K., Serve S., Steur C., Buchner T., Kienast J., Kanakura Y., Berdel W.E., Serve H. FLT3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the RAS and STAT5 pathways. Blood. 2000;96(12):3907–3914. [PubMed] [Google Scholar]
- 10.Spiekermann K., Bagrintseva K., Schwab R., Schmieja K., Hiddemann W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin. Cancer Res. 2003;9(6):2140–2150. [PubMed] [Google Scholar]
- 11.Choudhary C., Schwable J., Brandts C., Tickenbrock L., Sargin B., Kindler T., Fischer T., Berdel W.E., Muller-Tidow C., Serve H. AML-associated FLT3 kinase domain mutations show signal transduction differences compared with FLT3 ITD mutations. Blood. 2005;106(1):265–273. doi: 10.1182/blood-2004-07-2942. [DOI] [PubMed] [Google Scholar]
- 12.Daver N., Schlenk R.F., Russell N.H., Levis M.J. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312. doi: 10.1038/s41375-018-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt-Arras D.E., Bohmer A., Markova B., Choudhary C., Serve H., Bohmer F.D. Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol. Cell Biol. 2005;25(9):3690–3703. doi: 10.1128/MCB.25.9.3690-3703.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibuya N., Goldstein I.J., Van Damme E.J., Peumans W.J. Binding properties of a mannose-specific lectin from the snowdrop (Galanthus nivalis) bulb. J. Biol. Chem. 1988;263(2):728–734. [PubMed] [Google Scholar]
- 15.Koch S., Jacobi A., Ryser M., Ehninger G., Thiede C. Abnormal localization and accumulation of FLT3-ITD, a mutant receptor tyrosine kinase involved in leukemogenesis. Cells Tissues Organs. 2008;188(1–2):225–235. doi: 10.1159/000118788. [DOI] [PubMed] [Google Scholar]
- 16.Choudhary C., Olsen J.V., Brandts C., Cox J., Reddy P.N., Bohmer F.D., Gerke V., Schmidt-Arras D.E., Berdel W.E., Muller-Tidow C., Mann M., Serve H. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell. 2009;36(2):326–339. doi: 10.1016/j.molcel.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 17.Kothe S., Muller J.P., Bohmer S.A., Tschongov T., Fricke M., Koch S., Thiede C., Requardt R.P., Rubio I., Bohmer F.D. Features of RAS activation by a mislocalized oncogenic tyrosine kinase: FLT3 ITD signals through K-Ras at the plasma membrane of acute myeloid leukemia cells. J. Cell Sci. 2013;126(Pt 20):4746–4755. doi: 10.1242/jcs.131789. [DOI] [PubMed] [Google Scholar]
- 18.Reiter K., Polzer H., Krupka C., Maiser A., Vick B., Rothenberg-Thurley M., Metzeler K.H., Dorfel D., Salih H.R., Jung G., Nossner E., Jeremias I., Hiddemann W., Leonhardt H., Spiekermann K., Subklewe M., Greif P.A. Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia. 2018;32(2):313–322. doi: 10.1038/leu.2017.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudorf A., Muller T.A., Klingeberg C., Kreutmair S., Poggio T., Gorantla S.P., Ruckert T., Schmitt-Graeff A., Gengenbacher A., Paschka P., Baldus C., Zeiser R., Vassiliou G.S., Bradley A., Duyster J., Illert A.L. NPM1c alters FLT3-D835Y localization and signaling in acute myeloid leukemia. Blood. 2019;134(4):383–388. doi: 10.1182/blood.2018883140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moloney J.N., Stanicka J., Cotter T.G. Subcellular localization of the FLT3-ITD oncogene plays a significant role in the production of NOX- and p22(phox)-derived reactive oxygen species in acute myeloid leukemia. Leuk Res. 2017;52:34–42. doi: 10.1016/j.leukres.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Moloney J.N., Jayavelu A.K., Stanicka J., Roche S.L., O'Brien R.L., Scholl S., Bohmer F.D., Cotter T.G. Nuclear membrane-localised NOX4D generates pro-survival ROS in FLT3-ITD-expressing AML. Oncotarget. 2017;8(62):105440–105457. doi: 10.18632/oncotarget.22241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams A.B., Li L., Nguyen B., Brown P., Levis M., Small D. Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood. 2012;120(15):3069–3079. doi: 10.1182/blood-2012-01-403493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corsini A., Maggi F.M., Catapano A.L. Pharmacology of competitive inhibitors of HMG-COA reductase. Pharmacol. Res. 1995;31(1):9–27. doi: 10.1016/1043-6618(95)80042-5. [DOI] [PubMed] [Google Scholar]
- 24.Mo H., Elson C.E. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp. Biol. Med. (Maywood) 2004;229(7):567–585. doi: 10.1177/153537020422900701. [DOI] [PubMed] [Google Scholar]
- 25.Pelicano H., Martin D.S., Xu R.H., Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 26.Tsunekawa-Imai N., Miwa H., Shikami M., Suganuma K., Goto M., Mizuno S., Takahashi M., Mizutani M., Horio T., Komatsubara H., Gotou M., Yamamoto H., Wakabayashi M., Watarai M., Hanamura I., Imamura A., Mihara H., Nitta M. Growth of xenotransplanted leukemia cells is influenced by diet nutrients and is attenuated with 2-deoxyglucose. Leuk Res. 2013;37(9):1132–1136. doi: 10.1016/j.leukres.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 27.Larrue C., Saland E., Vergez F., Serhan N., Delabesse E., Mansat-De Mas V., Hospital M.A., Tamburini J., Manenti S., Sarry J.E., Recher C. Antileukemic activity of 2-Deoxy-D-glucose through inhibition of N-linked glycosylation in acute myeloid leukemia with FLT3-ITD or c-KIT mutations. Mol. Cancer Ther. 2015;14(10):2364–2373. doi: 10.1158/1535-7163.MCT-15-0163. [DOI] [PubMed] [Google Scholar]
- 28.Tsitsipatis D., Jayavelu A.K., Muller J.P., Bauer R., Schmidt-Arras D., Mahboobi S., Schnoder T.M., Heidel F., Bohmer F.D. Synergistic killing of FLT3ITD-positive AML cells by combined inhibition of tyrosine-kinase activity and N-glycosylation. Oncotarget. 2017;8(16):26613–26624. doi: 10.18632/oncotarget.15772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Contessa J.N., Bhojani M.S., Freeze H.H., Ross B.D., Rehemtulla A., Lawrence T.S. Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin. Cancer Res. 2010;16(12):3205–3214. doi: 10.1158/1078-0432.CCR-09-3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hou H., Sun H., Lu P., Ge C., Zhang L., Li H., Zhao F., Tian H., Zhang L., Chen T., Yao M., Li J. Tunicamycin potentiates cisplatin anticancer efficacy through the DPAGT1/Akt/ABCG2 pathway in mouse xenograft models of human hepatocellular carcinoma. Mol. Cancer Ther. 2013;12(12):2874–2884. doi: 10.1158/1535-7163.MCT-13-0201. [DOI] [PubMed] [Google Scholar]
- 31.Wu M., Li C., Zhu X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018;11(1):133. doi: 10.1186/s13045-018-0675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi S., Harigae H., Yokoyama H., Ishikawa I., Abe S., Imaizumi M., Sasaki T., Kaku M. Synergistic effect of arsenic trioxide and flt3 inhibition on cells with flt3 internal tandem duplication. Int. J. Hematol. 2006;84(3):256–261. doi: 10.1532/IJH97.06076. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi S. Combination therapy with arsenic trioxide for hematological malignancies. Anticancer Agents Med. Chem. 2010;10(6):504–510. doi: 10.2174/1871520611009060504. [DOI] [PubMed] [Google Scholar]
- 34.Nagai K., Hou L., Li L., Nguyen B., Seale T., Shirley C., Ma H., Levis M., Ghiaur G., Duffield A., Small D. Combination of ATO with FLT3 TKIs eliminates FLT3/ITD+ leukemia cells through reduced expression of FLT3. Oncotarget. 2018;9(68):32885–32899. doi: 10.18632/oncotarget.25972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carpenter G., Liao H.J. Receptor tyrosine kinases in the nucleus. Cold Spring Harb. Perspect. Biol. 2013;5(10) doi: 10.1101/cshperspect.a008979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen M.K., Hung M.C. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015;282(19):3693–3721. doi: 10.1111/febs.13342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wandinger-Ness A., Zerial M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 2014;6(11) doi: 10.1101/cshperspect.a022616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helenius A., Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291(5512):2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 39.Kreitman M., Noronha A., Yarden Y. Irreversible modifications of receptor tyrosine kinases. FEBS Lett. 2018;592(13):2199–2212. doi: 10.1002/1873-3468.13095. [DOI] [PubMed] [Google Scholar]
- 40.Miaczynska M. Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2013;5(11) doi: 10.1101/cshperspect.a009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keating M.T., Williams L.T. Autocrine stimulation of intracellular PDGF receptors in v-sis-transformed cells. Science. 1988;239(4842):914–916. doi: 10.1126/science.2829358. [DOI] [PubMed] [Google Scholar]
- 42.Keating M.T., Williams L.T. Processing of the platelet-derived growth factor receptor. Biosynthetic and degradation studies using anti-receptor antibodies. J. Biol. Chem. 1987;262(16):7932–7937. [PubMed] [Google Scholar]
- 43.Tabone-Eglinger S., Subra F., El Sayadi H., Alberti L., Tabone E., Michot J.P., Theou-Anton N., Lemoine A., Blay J.Y., Emile J.F. KIT mutations induce intracellular retention and activation of an immature form of the KIT protein in gastrointestinal stromal tumors. Clin. Cancer Res. 2008;14(8):2285–2294. doi: 10.1158/1078-0432.CCR-07-4102. [DOI] [PubMed] [Google Scholar]
- 44.Kim W.K., Yun S., Park C.K., Bauer S., Kim J., Lee M.G., Kim H. Sustained mutant kit activation in the Golgi complex is mediated by PKC-theta in gastrointestinal stromal tumors. Clin. Cancer Res. 2017;23(3):845–856. doi: 10.1158/1078-0432.CCR-16-0521. [DOI] [PubMed] [Google Scholar]
- 45.Obata Y., Horikawa K., Takahashi T., Akieda Y., Tsujimoto M., Fletcher J.A., Esumi H., Nishida T., Abe R. Oncogenic signaling by KIT tyrosine kinase occurs selectively on the Golgi apparatus in gastrointestinal stromal tumors. Oncogene. 2017;36(26):3661–3672. doi: 10.1038/onc.2016.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obata Y., Toyoshima S., Wakamatsu E., Suzuki S., Ogawa S., Esumi H., Abe R. Oncogenic KIT signals on endolysosomes and endoplasmic reticulum are essential for neoplastic mast cell proliferation. Nat Commun. 2014;5:5715. doi: 10.1038/ncomms6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roussel M.F., Downing J.R., Rettenmier C.W., Sherr C.J. A point mutation in the extracellular domain of the human CSF-1 receptor (c-fms proto-oncogene product) activates its transforming potential. Cell. 1988;55(6):979–988. doi: 10.1016/0092-8674(88)90243-7. [DOI] [PubMed] [Google Scholar]
- 48.Lievens P.M., Mutinelli C., Baynes D., Liboi E. The kinase activity of fibroblast growth factor receptor 3 with activation loop mutations affects receptor trafficking and signaling. J. Biol. Chem. 2004;279(41):43254–43260. doi: 10.1074/jbc.M405247200. [DOI] [PubMed] [Google Scholar]
- 49.Mazot P., Cazes A., Boutterin M.C., Figueiredo A., Raynal V., Combaret V., Hallberg B., Palmer R.H., Delattre O., Janoueix-Lerosey I., Vigny M. The constitutive activity of the ALK mutated at positions F1174 or R1275 impairs receptor trafficking. Oncogene. 2011;30(17):2017–2025. doi: 10.1038/onc.2010.595. [DOI] [PubMed] [Google Scholar]
- 50.Neel D.S., Allegakoen D.V., Olivas V., Mayekar M.K., Hemmati G., Chatterjee N., Blakely C.M., McCoach C.E., Rotow J.K., Le A., Karachaliou N., Rosell R., Riess J.W., Nichols R., Doebele R.C., Bivona T.G. Differential subcellular localization regulates oncogenic signaling by ROS1 kinase fusion proteins. Cancer Res. 2019;79(3):546–556. doi: 10.1158/0008-5472.CAN-18-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]