

Abstract
Exercise stimulates cellular and physiological adaptations that are associated with widespread health benefits. To uncover conserved protein phosphorylation events underlying this adaptive response, we performed mass spectrometry‐based phosphoproteomic analyses of skeletal muscle from two widely used rodent models: treadmill running in mice and in situ muscle contraction in rats. We overlaid these phosphoproteomic signatures with cycling in humans to identify common cross‐species phosphosite responses, as well as unique model‐specific regulation. We identified > 22,000 phosphosites, revealing orthologous protein phosphorylation and overlapping signaling pathways regulated by exercise. This included two conserved phosphosites on stromal interaction molecule 1 (STIM1), which we validate as AMPK substrates. Furthermore, we demonstrate that AMPK‐mediated phosphorylation of STIM1 negatively regulates store‐operated calcium entry, and this is beneficial for exercise in Drosophila. This integrated cross‐species resource of exercise‐regulated signaling in human, mouse, and rat skeletal muscle has uncovered conserved networks and unraveled crosstalk between AMPK and intracellular calcium flux.
Keywords: AMPK, calcium, exercise, phosphorylation, STIM1
Subject Categories: Metabolism; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Global analysis of contraction‐regulated protein phosphorylation in skeletal muscle uncovers signalling nodes conserved between mouse, rat and human.

Introduction
Exercise remains the most effective treatment for a variety of chronic diseases such as type 2 diabetes, dementia, sarcopenia, and heart disease (Berlin & Colditz, 1990; Boulé et al, 2001; Pitkälä et al, 2013; Landi et al, 2014). Such health benefits are linked to activation of an extensive range of coordinated biochemical processes in the exercising muscle to restore homeostasis, and resulting adaptations confer the systemic and tissue‐specific health benefits of exercise. Many of the long‐term beneficial effects of exercise arise from the contracting muscle itself, and these likely have benefit within the muscle as well as have the potential to trigger beneficial effects in other tissues. These include increased energy expenditure and clearance of ectopic lipid stores, improved insulin sensitivity and reduced circulating insulin levels, and increased secretion of exercise‐regulated myokines, such as IL‐6 or irisin (Hawley et al, 2014) and extracellular vesicles (Whitham et al, 2018). The breadth of molecular machinery involved in eliciting these integrative exercise adaptations is unknown and is currently the focus of extensive research utilizing models of exercise and muscle contraction in rodents and humans. Filling this knowledge gap requires the development of highly rigorous methods capable of capturing the diverse biological changes that occur in muscle acutely during exercise. We suggest that protein phosphorylation regulates many of these biological processes, and with current developments in mass spectrometry, it is now feasible to quantify such changes on a global scale (Needham et al, 2019).
We have previously used a phosphoproteomics approach to quantify and map > 1,000 phosphosites significantly regulated by a single high‐intensity cycling bout in humans (Hoffman et al, 2015). Of these exercise‐regulated sites, more than 900 have not been functionally validated. Such studies in humans have yielded invaluable information, but the ability to validate exercise‐associated pathways is restricted, as genome‐wide association studies have poor phenotype predictability power (Marigorta et al, 2018). Therefore, the use of rodent models of exercise and muscle contraction combined with genetic or pharmacological manipulations is essential to comprehensively explore signal transduction and mechanisms underlying the benefits of exercise. Two commonly used models to simulate the human exercise response are rodent treadmill running and in situ electrical stimulation of skeletal muscle with an intact nerve and blood supply (Gehrig et al, 2012; Sylow et al, 2017). For example, phosphoproteomic analysis of mouse skeletal muscle has been utilized to investigate the role of AMP‐activated protein kinase (AMPK) in muscle contraction‐stimulated fatty acid oxidation (Dzamko et al, 2008), the role of mTORC2 in treadmill exercise‐induced signal transduction (Kleinert et al, 2017), and signal transduction during the recovery phase following muscle contraction (Potts et al, 2017).
Herein, we mapped the phosphoproteomic responses to acute in situ contraction in rats and a single bout of treadmill running in mice. We then compared these datasets across species with our previously published human cycling dataset (Hoffman et al, 2015). Our goals were to pinpoint common and unique signaling events across commonly used experimental models of exercise, and to uncover exercise‐regulated nodes that were robustly activated across exercise models and conserved across species. Among the most highly enriched signaling pathways across all three exercise models were AMPK and Ca2+. In view of the key roles of AMPK (Winder & Hardie, 1996; Mu et al, 2001; Narkar et al, 2008; O'Neill et al, 2011; Toyama et al, 2016) and Ca2+ (Ebashi & Endo, 1968; Melzer et al, 1995; Rose et al, 2006; Kwong et al, 2018) in the exercise response in skeletal muscle, we further interrogated a potential point of crosstalk between these two pathways. We found stromal interaction molecule 1 (STIM1), a protein critical for store‐operated Ca2+ entry (SOCE), to be phosphorylated by AMPK across all three datasets at two sites (S257 and S521). We showed AMPK‐mediated phosphorylation of STIM1 at S257 to play a key role in regulating STIM1 conformation and SOCE in L6 myoblasts, as well as exercise capacity in a Drosophila model of exercise. Through integrated, cross‐species phosphoproteomic analysis of human, rat, and mouse skeletal muscle, we have uncovered conserved exercise‐regulated signaling networks and unraveled a novel mechanism whereby AMPK regulates skeletal muscle Ca2+ dynamics.
Results
Rodent exercise models stimulate key exercise signaling pathways
We have recently used phosphoproteomics analysis to construct a global map of exercise signaling, comprising more than 2,400 regulated phosphosites (1,004 changing by more than 50% after exercise) in human muscle from volunteers who underwent acute intense cycling exercise (Hoffman et al, 2015). Here we compared the human dataset to changes induced by in situ muscle contraction in rats or by treadmill running in mice (Fig 1A). For in situ contraction, intact rat tibialis anterior (TA) skeletal muscle was contracted by electrical stimulation of the sciatic nerve (100 Hz for 5 min; 1 s on followed by 3 s off). The contracted muscles were compared to the contralateral non‐contracted muscles connected to the force transducer at resting tension (sham). This contraction protocol induced skeletal muscle fatigue over the 5‐min period (Appendix Fig S1). For treadmill exercise, gastrocnemius muscle was collected from mice subjected to a single bout of treadmill running at 20 m/min over 30 min at a 10° incline (65% of maximal running capacity determined by a maximal running test as in Sylow et al (2017) and compared to mice that were placed on the treadmill but did not undergo the running protocol (rest). Western blotting confirmed that in situ contraction and treadmill running both induced activation of key signaling pathways that were observed in human muscle after cycling (Hoffman et al, 2015), including regulatory site phosphorylation of AMPK (T172) and the AMPK substrate acetyl‐CoA carboxylase (ACC; S79) (Fig 1B).
Figure 1. Skeletal muscle exercise‐regulated phosphoproteome.
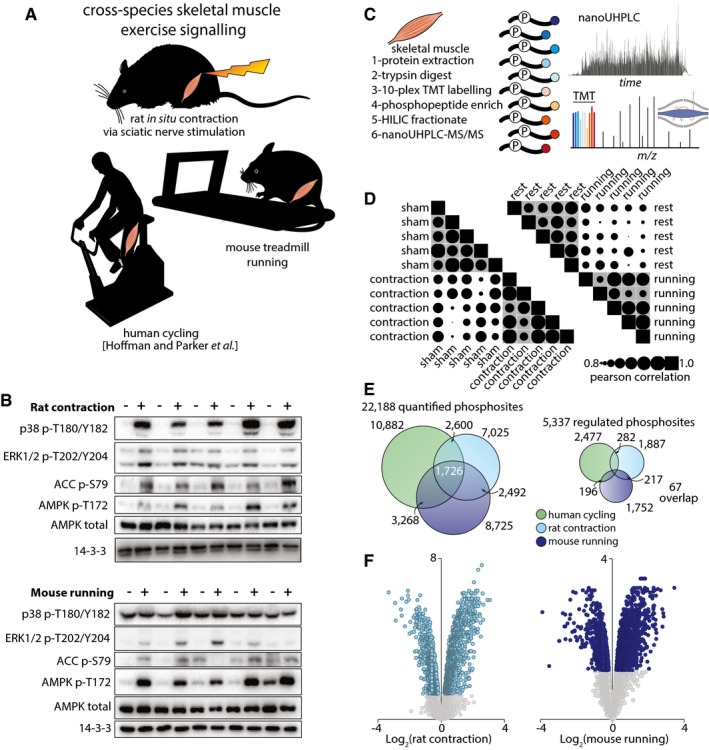
- Illustration of the three exercise models analyzed in this study. Rat tibialis skeletal muscles were subjected to either in situ contraction or sham surgery. Protein was extracted and digested with Lys‐C/trypsin, and peptides were isobarically labeled with TMT tags. Phosphopeptides were enriched by titanium dioxide chromatography and sequential elution from immobilized metal ion affinity chromatography (SIMAC). The unbound non‐phosphorylated fraction and phosphorylated fraction were further separated by hydrophilic interaction liquid chromatography (HILIC). Each fraction was analyzed by nano‐ultra high‐pressure liquid chromatography coupled to tandem MS (nanoUHPLC‐MS/MS).
- Western blots for exercise‐associated phosphorylated proteins in (top) contracted (+) or sham (−) rat muscle and (bottom) muscle from mice that underwent a treadmill running protocol (+) or remained at rest (−). Total AMPK and 14‐3‐3 were used as loading controls.
- Schematic representation of the workflow for mass spectrometry from skeletal muscle.
- Pearson correlation plot representing the reproducibility of phosphoprotein protein profiles between samples analyzed via mass spectrometry.
- Venn diagrams representing the number of (left) total or (right) significantly regulated phosphopeptides detected in human, rat, and mouse models of exercise.
- Volcano plots of phosphopeptide median Log2 fold‐change (contraction/sham surgery) plotted against the −Log10 p‐value in rat contraction (left) or mouse running (right). Colored dots represent significantly regulated phosphopeptides (P < 0.05, n = 5, moderated t‐test). Gray dots represent phosphopeptides that were detected but not significantly altered in exercise.
Rodent exercise‐ and contraction‐regulated phosphoproteomes reveal coordinate kinase activity and robust regulation of calcium machinery
Quantitative phosphoproteomic analyses of skeletal muscle signaling induced by contraction in rats and treadmill running in mice was performed using multiplexed tandem mass tag (TMT) isobaric labeling and phosphopeptide enrichment coupled to LC‐MS/MS (Fig 1C). The phosphorylation profiles in the biological replicates were more highly correlated in the respective groups (gray) compared to between the groups, highlighting reproducible quantification (Fig 1D). A total of 8,989 unique phosphopeptides (7,035 phosphosites with > 90% localization probability) were quantified in all 10 rat skeletal muscle samples subjected to sham or contraction (n = 5), while 9,722 unique phosphopeptides (8,725 phosphosites with > 90% localization probability) were quantified in all 10 mouse skeletal muscles samples subject to rest or treadmill running (n = 5) (Fig 1E; Table EV1). Of the phosphosites quantified, 1,887 and 1,752 were significantly regulated by rat contraction and mouse running, respectively (Fig 1E and F; q‐value < 0.05 Benjamini–Hochberg FDR). Although the observed global fold‐changes and number of significant sites were similar between the two exercise models, the overall P‐values were lower for rat contraction, suggesting this model displays higher reproducibility compared to treadmill running (Fig 1F). We next mapped orthologous phosphosites between human, rat, and mouse muscles using PhosphOrtholog (Chaudhuri et al, 2015) and integrated these data with our previously published phosphoproteome of exercised human muscle (Hoffman et al, 2015). Here, our aim was to identify the regulation of highly conserved phosphorylation events, which are more likely to have important biological roles and represent top priorities for functional validation (Beltrao et al, 2012). Proteins were retrieved from orthologous reference databases followed by global sequence alignment and generation of a statistical significance score for the identification of exact orthologous phosphorylated amino acids. These integrated data contained > 22,000 unique phosphosites, of which 1,726 were orthologous across all three datasets. Sixty‐seven orthologous phosphosites were significantly regulated across all three datasets and, of these, 78% were regulated in the same direction (Fig 1E; Appendix Table S1). Despite a relatively low degree of overlap between the models, Appendix Table S1 provides a high‐confidence compendium of what likely constitute conserved exercise regulators, many of which have not previously been studied in this context. Notably, 19% of these sites represent proteins that are dually‐phosphorylated (HSPB1, MLLT4, NFIX, C18orf25, TNS1, VAPA, SYNPO2, and NDRG2) or even triply‐phosphorylated (STIM1, LMOD2, XIRP1, and ALPK3). Among the proteins listed in Appendix Table S2, there is a high degree of convergence on protein kinases (AMPK, PHKA1, SPEG, CAMK, S6K, ALPK3, and mTOR), protein phosphatases (PPP2R5A, PPP1R14A, and PPP1R3C) and transcription factors (NFIX and TFEB) that play a role in the regulation of metabolism and gene expression to allow muscle adaptation. Also observed were scaffolding and actin/myosin regulatory proteins (SYNPO2, LMOD2, AKAP13, RCSD1, CLASP1, FLNC, PLEC, SYNPO2, and TNS1); vesicle transport regulatory proteins (RABGEF2, TBC1D1, LNPEP, VIPAS39, VAPA, and GPHN); and proteins involved in proteostasis including regulators of protein synthesis (RPTOR, EIF4B, and EIF4G1) and chaperone proteins (TFEB, CRYAB, HSPB1, and MAPT). Many of these exercise‐regulated proteins represent high‐priority targets for further mechanistic interrogation.
A relatively low number of conserved exercise‐regulated phosphosites overlapped between the different models; however, we observed several examples of regulated phosphorylation of neighboring amino acids between human, rat, and mouse. That is, although the exact orthologous sites were not phosphorylated, we observed regulated phosphorylation in local protein regions conserved between the different species. Phosphorylation of a protein anywhere in a local structural region may serve a similar function via modulation of charge density (Holt et al, 2009; Beltrao et al, 2012). To investigate this, we created “sequence windows” consisting of ± 5% of the protein length around the phosphosites and searched for regulated phosphosites with the same directionality anywhere within the sequence window between the species. Like PhosphoSitePlus (PSP) and PhosphOrtholog, the sequence window filters out phosphosites in structural protein domains such as I‐set, leaving a higher proportion of phosphosites in domains that are frequently of interest, including protein kinase and transcription factor domains (Appendix Fig S2A–D). As expected from expanding the stringency from PSP and PhosphOrtholog, the functional prediction score (preprint: Ochoa et al, 2019) was intermediate compared to all phosphosites identified in human muscle and high confidence mapped orthologs (Appendix Fig S2E). Mapping potential orthologs by sequence window resulted in the identification of 190 phosphosites regulated in all three models (114 up‐ and 76 down‐regulated phosphosites), representing 80 proteins (Table EV2). For example, all three datasets identified increased phosphorylation of genethonin‐1 (starch‐binding domain‐containing protein 1; STBD1), a glycogen binding protein associated with transport of glycogen to lysosomes and glycophagy (Jiang et al, 2010) (Appendix Fig S3). A cluster of four to five phosphosites was observed in all three datasets directly adjacent to or within the GABARAPL1 binding site in the Atg8 interacting motif (AIM; Jiang et al, 2011). However, an INDEL mutation and series of point mutations resulted in no single orthologous phosphosite identified in all three species, but rather adjacent phosphosites were regulated. A rodent‐specific phosphorylation site (S199 and S195 in mouse and rat, respectively) is orthologous to D214 in human directly in the AIM suggesting evolutionary selection of local negative charge. It is worth noting that, although several phosphosites were co‐regulated in sequence windows and may serve common functions, they are not necessarily within the same kinase consensus motifs. Therefore, the sites may be regulated by different, possibly redundant, kinases.
To investigate kinase activity between the models, we mapped the phosphosites to the PSP database to retrieve kinase: substrate relationships (KSRs) and performed an enrichment analysis based on the direction of substrate phosphorylation changes (Fig 2A; q‐value < 0.05). A total of 553 low‐throughput or high‐throughput KSRs were annotated in PSP and mapped to our combined phosphoproteomics data (204 mapped across all three species). Kinase enrichment analysis revealed consistent directionality of kinases across the rodent models and human cycling (Fig 2A). However, AMPK and PKA were the only kinases to be significantly enriched in all three exercise models using this directional kinase enrichment method (Fig 2A). To investigate similarities and differences in kinase activity in greater detail, we visualized individual substrates of these kinases (Fig 2B). Notably, substrates of these various kinases were not observed in all forms of exercise. Often, this was because the cognate site was not detected, rather than detected and not regulated. However, differences in regulated sites may reflect systemic versus local biochemical responses to exercise. For example, in the case of both human cycling and mouse running, we observed decreased Akt activity based on substrate enrichment analysis. However, in the case of rat contraction, AKT2 S474 was increased, whereas several downstream substrates were either not detected or not regulated. The observed inhibition of the PI3K/Akt/mTORC2 pathway in human cycling and mouse running but not rat contraction likely reflects a known role of catecholamines to inhibit insulin secretion during exercise, which likely does not occur in the contraction model.
Figure 2. Phosphorylation landscapes of exercise models.
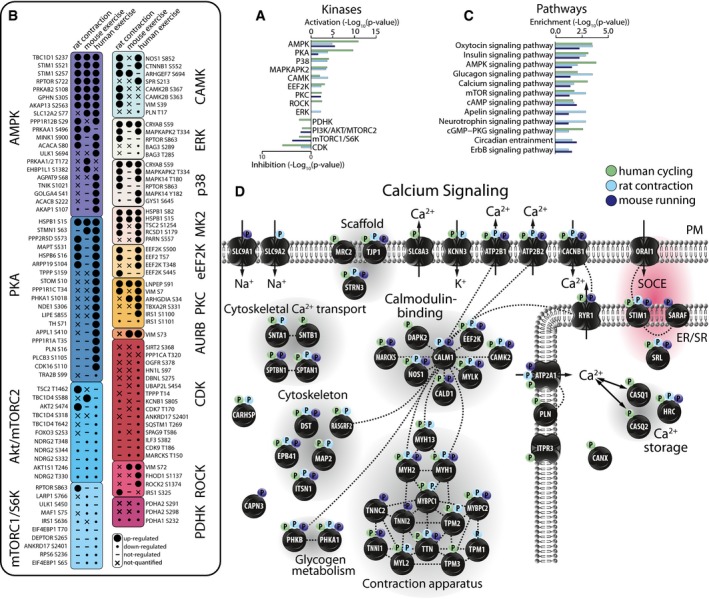
-
A–D(A) Kinase activation enrichment analysis, (B) phosphorylation status of specific phosphosites associated with known or predicted upstream kinases, (C) biological pathway enrichment analysis, and (D) phosphorylation status of proteins involved in Ca2+ signaling in human, rat, and mouse models of exercise.
We next interrogated differences in the downstream pathways by mapping the proteins containing regulated phosphosites to KEGG and performing an enrichment analysis. This analysis does not accommodate for directionality, as some proteins contain both up‐ and down‐regulated phosphosites. This analysis revealed excellent concordance of the regulated pathways between models (Fig 2C; Table EV3). Two hundred and ten proteins contained at least one regulated phosphorylation site across all three datasets (Table EV3). Taken together, these data highlight differences in biological pathway activation between the models, such as those associated with catecholamine signaling, but also reveal potential conserved regulation at the protein and pathway levels.
Exercise‐ and contraction‐regulated phosphorylation of STIM1 by AMPK
Our analyses of orthologous and local protein region phosphorylation regulated by exercise and contraction, as well as of kinase and pathway enrichment, identified AMPK and Ca2+ signaling as major points of convergence between the exercise models. We manually curated a comprehensive map of Ca2+ signaling and overlaid the regulated phosphosites from the models to investigate this regulation in greater detail (Fig 2D). Several of these proteins including ATP2B1, CAM2K, and MYH1 contained regulated phosphorylation sites in close proximity to sequence windows, whereas only PHKA1 (regulates glycogen metabolism downstream of the Ca2+‐responsive protein CALM1) and STIM1 contained orthologous phosphosites regulated in all three datasets. STIM1 was of particular interest because it shared three orthologous phosphosites that were regulated with the same directionality across all three datasets (Appendix Table S1) and two of these sites (S257 and S521) are predicted AMPK phosphosites (Hoffman et al, 2015) (Fig 3A). STIM1 is a sarcoplasmic reticulum (SR) transmembrane protein that functions as an ER Ca2+ sensor critical for store‐operated Ca2+ entry (SOCE; Roos et al, 2005). In response to reduced SR Ca2+ levels, STIM1 undergoes conformational changes that trigger its dimerization and interaction with the Ca2+ release activated Ca2+ (CRAC) channel Orai1 at the plasma membrane (PM) to facilitate SOCE (Feske & Prakriya, 2013). Given the central roles of Ca2+ and AMPK in the skeletal muscle exercise response, we further explored this potential crosstalk between AMPK and Ca2+ regulation. Based on structural data (Park et al, 2009; Jha et al, 2013; Zhou et al, 2013), the position of these STIM1 phosphosites suggested that AMPK regulates STIM1 intra‐ and/or inter‐molecular interactions, potentially impacting STIM1‐Orai1 complex activation and SOCE. AMPK has not been previously implicated in regulating SOCE during exercise in muscle, and this provided further impetus to rigorously characterize these phosphosites. One predicted AMPK phosphosite on STIM1, S257, is within a region of STIM1 critical for its activation by switching between inter‐molecular interaction and dimerization (Muik et al, 2011). The other predicted AMPK site, S521, is within a region of STIM1 thought to regulate the interaction of STIM1 with the PM and/or Orai1 (Baba et al, 2006; Jha et al, 2013).
Figure 3. STIM1 is a novel exercise‐regulated AMPK substrate.
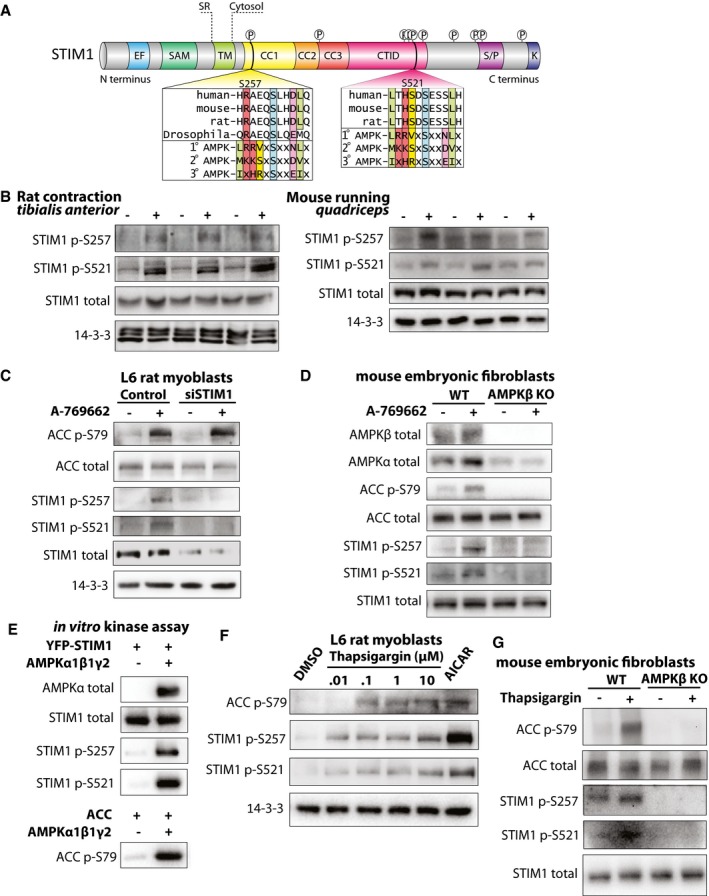
- Schematic of STIM1 domain architecture is shown, with the AMPK consensus motifs for S257 and S521 (Gwinn et al, 2008).
- Western blots for STIM1 phosphorylated at S257 or S521 in (left) contracted (+) or sham (−) rat muscle and (right) muscle from mice that underwent a treadmill running protocol (+) or remained at rest (−). Total STIM1 and 14‐3‐3 were used as loading controls. 14‐3‐3 data for mouse running were re‐used from Fig 1B.
- Western blots for phosphorylated ACC (S79) and STIM1 (S257 or S521) in L6 rat myoblasts with siRNA‐mediated knockdown of endogenous STIM1 (siSTIM1) or transfected with scramble control siRNA (Control). Cells were treated with the AMPK activator A‐769662 (+) or vehicle (−) for 30 min. Total STIM1 was used to demonstrate STIM1 knockdown efficiency. Total ACC and 14‐3‐3 were used as loading controls.
- Western blots for phosphorylated ACC (S79) and STIM1 (S257 or S521) in wild‐type (WT) or AMPK β1/β2 knockout (AMPKβ KO) mouse endothelial fibroblasts were isolated and treated with A‐769662 (+) or vehicle (−) for 30 min. AMPKβ was used to demonstrate knockout, and AMPKɑ was used to demonstrate specificity for AMPKβ. Total ACC and STIM1 were used as loading controls.
- Western blots for total ACC and STIM1 and phosphorylated STIM1 (S257 and S521) and ACC (S79) in samples from an in vitro kinase assay to assess the ability of AMPK to phosphorylate YFP‐STIM1.
- Representative Western blots for phosphorylated ACC (S79) and STIM1 (S257 and S521) in response to a 15‐min treatment with increasing doses of thapsigargin (0.01, 0.1, 1, or 10 μM) or 0.5 mM AICAR. DMSO was used as a vehicle control. 14‐3‐3 was used as a loading control. n = 3 independent experiments.
- Western blots for phosphorylated ACC (S79) and STIM1 (S257 or S521) in wild‐type (WT) or AMPK KO mouse endothelial fibroblasts were isolated and treated with 2 μM thapsigargin (+) or vehicle (−) for 15 min.
Source data are available online for this figure.
STIM1 is an AMPK substrate
To further characterize these predicted AMPK phosphosites on STIM1, we produced antibodies against each phosphosite. To validate these antibodies, we expressed human wild‐type (WT) STIM1‐mRuby3 in HEK cells, or STIM1‐mRuby3 with either serine 257 or 521 mutated to alanine (S257A and S521A, respectively). As expected, these antibodies recognized a band of ~120 kDa. Both antibodies detected an increase in phosphorylation of WT STIM1‐mRuby3 when cells were stimulated with the AMPK agonist A‐769662, and they were specific to their respective phosphosites, as no phosphorylation was detected in cells expressing the respective alanine mutants in the presence of A‐769662 (Appendix Fig S4A). Using these phosphosite‐specific antibodies, we showed that phosphorylation of STIM1 S257 and S521 was increased in contracted rat muscle and exercised mouse muscle compared to sham and rested controls, respectively (Fig 3B; Appendix Fig S4B and C). Consistent with these sites being AMPK substrates, we observed increased phosphorylation of both sites in L6 myoblasts treated with A‐769662, and knockdown of STIM1 using siRNA (siSTIM) resulted in loss of A‐769662‐induced phosphorylation at both sites compared to control cells transfected with scrambled siRNA (Control) (Fig 3C). Genetic deletion of AMPK β1/β2 subunits (AMPKβ KO) in mouse embryonic fibroblasts (MEFs) resulted in a complete loss of STIM1 phosphorylation (Fig 3D), confirming that A‐769662 is acting through AMPK to cause STIM1 phosphorylation. Finally, we performed in vitro kinase assays to test whether STIM1 S257 and S521 are directly phosphorylated by AMPK. We observed an increase in phosphorylation at STIM1 S257 and S521, as well as the positive control, ACC S79, in the presence of AMPKα1β1γ2 (Fig 3E). Collectively, these findings indicate that both S257 and S521 are bona fide AMPK substrate phosphosites.
It is established that elevated cytosolic Ca2+ activates AMPK (Jensen et al, 2007). We therefore investigated whether pharmacologically elevated cytosolic Ca2+ is sufficient to cause STIM1 phosphorylation using the SERCA pump inhibitor thapsigargin, which, by inhibiting Ca2+ re‐entry into the SR, results in elevated cytosolic Ca2+. We used the AMPK agonist AICAR as a positive control. Treatment of L6 myoblasts with thapsigargin dose‐dependently induced phosphorylation of STIM1 at both S257 and S521 (Fig 3F and Appendix Fig S4D). Phosphorylation of ACC S79 was also induced in response to thapsigargin treatment, confirming that cytosolic Ca2+ elevation upon thapsigargin treatment leads to activation of AMPK (Fig 3F and Appendix Fig S4D). Moreover, thapsigargin did not cause STIM1 phosphorylation in AMPKβ KO MEFs, confirming that AMPK is required for STIM1 phosphorylation in this context (Fig 3G).
Phosphorylation by AMPK regulates STIM1 conformation and activity
Considering the critical function of STIM1 in SOCE, we next investigated the potential role of AMPK‐mediated STIM1 phosphorylation, focusing on STIM1 conformation and intracellular Ca2+ levels. Interestingly, the S257 site lies within the Orai1‐activating small fragment (OASF) of STIM1 (amino acids 233–474), which is sufficient to interact with and activate Orai1 (Muik et al, 2009). The OASF comprises three coiled‐coil domains (CC1‐CC3) that form intra‐ and inter‐molecular interactions depending on the activation state of STIM1 (Muik et al, 2011; Fahrner et al, 2014). S257 lies within CC1α1 and disruption of this domain via mutation of leucines (including L251S) results in a conformational change of the OASF and constitutive activation of Orai1 and SOCE (Muik et al, 2011). We constructed a STIM1 OASF Förster resonance energy transfer (FRET) sensor similar to that described by Romanin and colleagues (Muik et al, 2011) to measure STIM1 conformation via fluorescence lifetime imaging microscopy (FLIM). This sensor comprises the OASF flanked by two FRET‐paired fluorescent proteins, one at the N‐terminus and one at the C terminus (Clover2‐OASF‐TagRFP‐T). When STIM1 is in its inactive conformation, it forms a tight fold so that the N and C termini are in close proximity, allowing high rates of energy transfer between the FRET pair. When STIM1 is activated, the protein unfolds and extends, preventing energy transfer between the FRET pair. The mClover2: tagRFPt FRET pair used in this study was validated by measuring the amplitude weighted lifetime of mClover2 linked by either a short flexible linker or a long rigid linker expressed in HEK‐E cells. The long linker (minimal FRET) had a median lifetime of 2.4 ± 0.06 ns, similar to the lifetime of mClover2 alone (2.56 ± 0.01 ns). This decreased to 1.91 ± 0.07 ns in the presence of a short flexible linker (high FRET), suggesting efficient energy transfer can occur between mClover2 and tagRFPt (Fig 4A). To investigate the role of AMPK‐induced phosphorylation on the conformation of the OASF sensor, we performed FLIM in L6 myoblasts expressing the sensor before and after treatment with A‐769662. A‐769662 treatment resulted in a decrease in fluorescence lifetime, indicating a shift to the inactive, folded conformation (Fig 4B). To specifically assess the involvement of S257 phosphorylation in changes to STIM1 conformation, we mutated serine 257 of the OASF sensor to alanine (S257A), which is a non‐phosphorylatable (phospho‐dead) mutant, or to glutamic acid (S257E), to mimic the presence of a phosphate group at this position (phospho‐mimetic). Compared to cells expressing the phospho‐dead S257A OASF, cells expressing the phospho‐mimetic S257E OASF displayed decreased fluorescence lifetime (Fig 4C). These data are consistent with a model whereby phosphorylation at S257 favors the folded, inactive STIM1 conformation.
Figure 4. Phosphorylation of STIM1 affects its conformation.
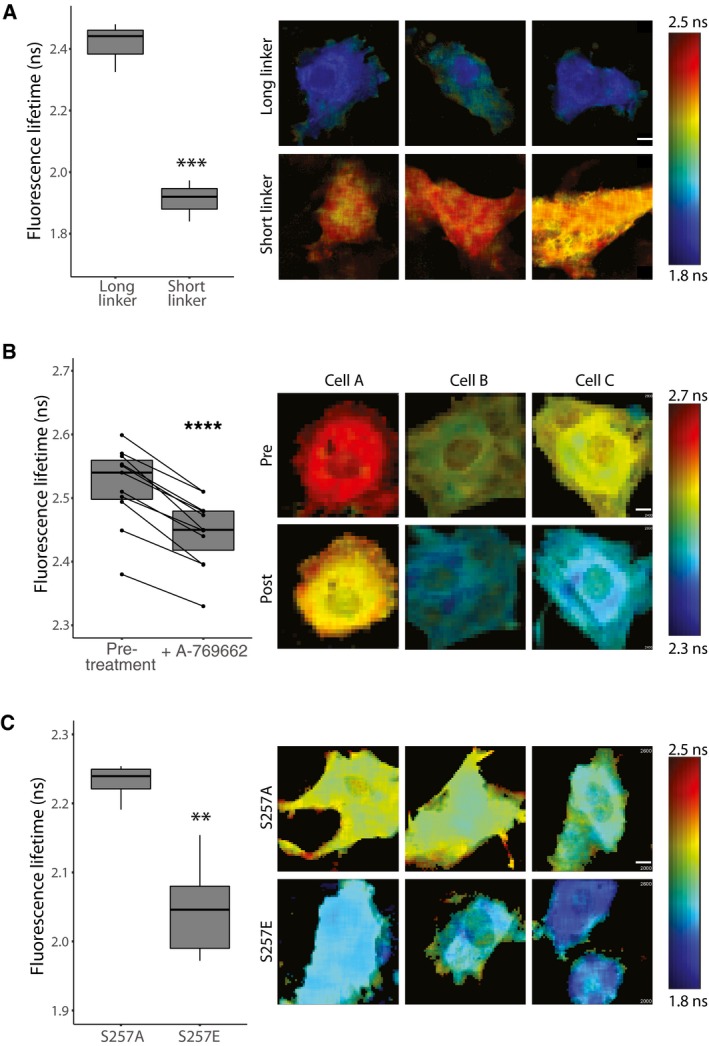
- The mClover2:tagRFP‐T fret pair was validated in HEK‐E cells with the fluorophores joined by either a long rigid or short flexible linker.
- STIM1 OASF expressed in live L6 myoblasts before (−) or after (+) treatment with A‐769662.
- L6 myoblasts with S257 mutated to A or E.
We then tested whether this phosphorylation‐induced change in STIM1 conformation translates to a change in STIM1 activity in terms of SOCE. We measured cytosolic Ca2+ in L6 myoblasts under a range of conditions, including STIM1 knockdown and re‐expression of full‐length human STIM1 WT or mutants including phospho‐dead or phospho‐mimetic. We also utilized the constitutively active L251S mutant (Muik et al, 2011) as an additional experimental control. STIM1‐mRuby3 was localized to the SR as expected (Appendix Fig S5A). Cytosolic Ca2+ was measured by co‐expressing the genetically encoded cytosolic Ca2+ sensor GCaMP6s (Chen et al, 2013). Cells were switched from buffer comprising 1 mM Ca2+ to Ca2+‐free buffer (with equimolar Mg2+ replacing Ca2+ and EGTA to chelate residual extracellular Ca2+; 0 mM Ca2+ + EGTA) then treated with thapsigargin to deplete SR Ca2+ stores and activate STIM1. To induce SOCE, we switched cells to 1 mM Ca2+ buffer and in the continued presence of thapsigargin (Fig 5A–C; Appendix Fig S5B and C). Store‐operated Ca2+ entry was blunted in siSTIM1 cells compared to control cells and re‐expression of WT STIM1 rescued, or even enhanced, SOCE (Fig 5A–C). Compared to STIM1 WT, SOCE was significantly attenuated in cells re‐expressing STIM1 S257E, whereas SOCE was unaltered in cells expressing STIM1 S257A (Fig 5A–C) or S521E (Appendix Fig S5B and C). As expected, the constitutively active L251S mutant did not respond to thapsigargin (Fig 5A–C). There was no significant difference in mRuby3 expression levels of the various STIM1 constructs used in these experiments as determined by fluorescence microscopy (Appendix Fig S5D). Additionally, the response to thapsigargin in the absence of Ca2+ represents the size of SR Ca2+ stores (Fig 5B). These stores appear to be severely depleted in cells expressing either the L215S or S257E mutant, consistent with impaired STIM1 function and a reduced ability to refill SR Ca2+. We note that this is not observed with STIM1 KD alone. This suggests that the residual STIM1 activity in the KD cells is sufficient for the maintenance of SR Ca2+ stores, and depletion with the mutants occurs as a result of the mutants exerting a dominant negative effect. Taken together, these data show that phosphorylation of STIM1 at S257 causes a decrease in STIM1 SOCE activity.
Figure 5. Phosphorylation of STIM1 by AMPK suppresses SOCE .
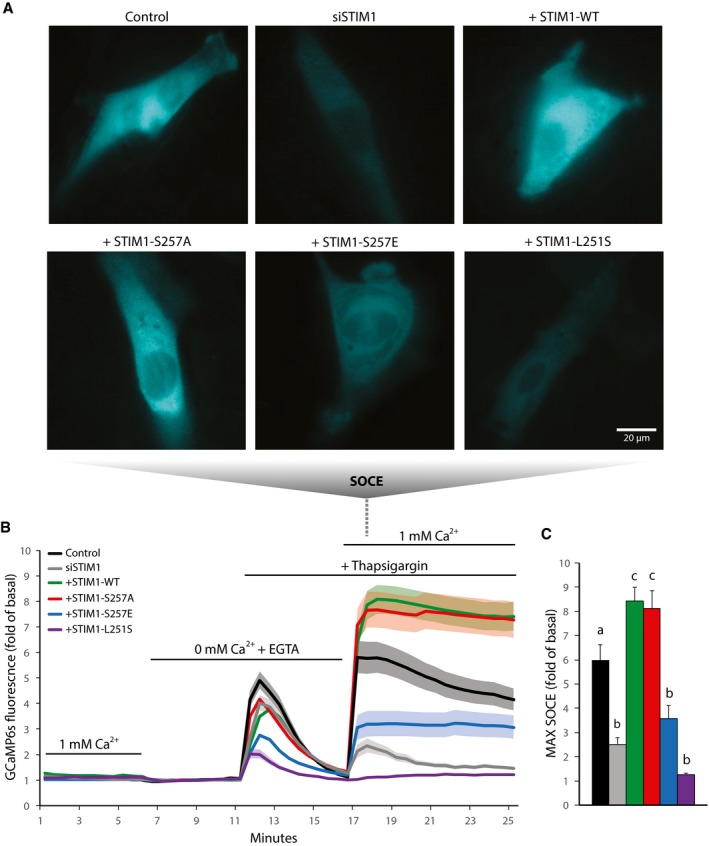
-
A–CL6 myoblasts were transfected with the genetic cytosolic Ca2+ sensor GCaMP6s. Cells were also transfected with non‐targeted siRNA (Control) or siRNA directed at endogenous STIM1 (siSTIM1). Some cells with endogenous STIM1 knockdown were also transfected with full‐length wild‐type (WT) or mutant (S257A, S257E, or L251S) STIM1‐mRuby3. (A) Representative microscopy images of GCaMP6s signal indicating SOCE in L6 myoblasts, (B) Ca2+ levels over time (fold of 0 mM Ca2+ + EGTA basal), and (C) quantification of SOCE in L6 myoblasts incubated in buffer containing 1 or 0 mM Ca2+ and treated with 2 μM thapsigargin during the indicated times. For (C), “b” is significantly different from “a” and “c”, and “c” is significantly different from “a” and “b”, P < 0.05 by one‐way ANOVA with Tukey's post‐hoc test. For (B‐C), data are represented as mean ± SEM, n = 5 independent experiments (n = 3 for L251S).
Source data are available online for this figure.
Finally, we investigated whether phosphorylation of STIM1 at S257 has functional role in exercise. For these experiments, we used Drosophila (flies) because STIM1 is conserved (STIM; 57% sequence similarity compared to human), including S257 and the surrounding AMPK consensus sequence (Fig 3A). First, we made whole‐body STIM knockdown flies using the GAL4‐UAS system (Brand & Perrimon, 1993) by crossing three independent STIM RNAi lines (RNAi26‐1 and RNAi8‐4 on the yw background, and RNAiGD16187 on the w1118 background) with a tubulin‐GAL4 driver and found that this cross was lethal during development (data not shown). We then made muscle‐specific STIM knockdown flies by crossing these STIM RNAi lines with the mef2‐GAL4 driver line. These flies were viable, so we proceeded to test their exercise ability via the negative geotaxis climbing assay (Ganetzky & Flanagan, 1978). When flies were in the rested state, there was no detectable difference in climbing speeds during the negative geotaxis climbing assay between the STIM knockdown flies and the respective controls (Fig 6A and B). Flies were then exercised for 2.5 h by continuous climbing, and then, the negative geotaxis climbing assay was repeated. This revealed a significant increase in the time required to climb up the vial wall in control flies compared to pre‐exercise, indicating fatigue. Strikingly, the STIM1 RNAi knockdown lines did not fatigue, as there was no change in climbing speed post‐exercise compared to pre‐exercise (Fig 6A and B). To specifically test a role for STIM1 S257 in exercise, we crossed STIM CRISPR whole‐body knockout flies with flies expressing WT, S257A phospho‐dead or S257E phospho‐mimetic human STIM1‐mRuby3. Western blotting of STIM1 in these flies showed equivalent levels of overexpression between WT, S257A, and S257E (Appendix Fig S6A and B). Overexpression of human STIM1‐mRuby3 WT, S257A, or S257E rescued lethality; however, there was not a complete rescue in climbing ability compared to normal flies. Only 59% of WT‐, 50% of S257A‐, and 77% of S257E‐expressing flies were able to climb when stimulated in the negative geotaxis assay. Of the flies able to climb, the S257E‐expressing flies were able to climb for longer and did not fatigue as quickly as the S257A‐expressing flies, whereas WT‐expressing flies showed an intermediate phenotype (Fig 6C). These exercise experiments demonstrate an advantage to limiting SOCE during exercise, either by STIM knockdown or by STIM1 inhibitory phosphorylation.
Figure 6. STIM1 inhibition prevents exercise‐induced fatigue.
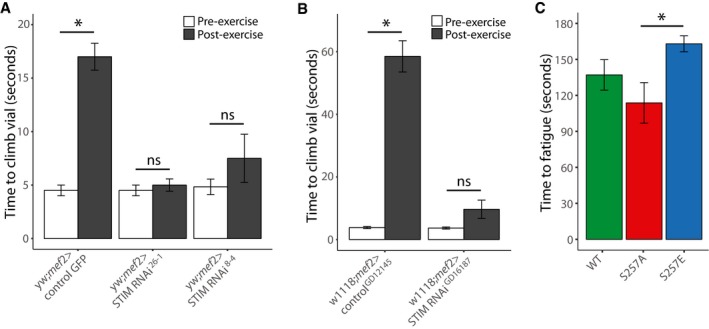
-
A, BNegative geotaxis climbing assays in Drosophila (flies). Time required for 50% of flies to climb to the target line before or after 2.5 h of continuous climbing exercise (A) in the yw strain with muscle‐specific STIM knockdown using two independent RNAi sequences (RNAi26‐1 or RNAi8‐4) compared to GFP‐expressing controls, or (B) in the w1118 strain with muscle‐specific STIM knockdown using RNAiGD16187 compared to non‐targeted RNAiGD12145 controls.
-
CTime to fatigue, when flies were no longer able to climb, during a 3‐min period after negative geotaxis stimulation in STIM whole‐body knockout flies overexpressing wild‐type (WT) or mutant (S257A or S257E) human STIM1‐mRuby3.
Discussion
Understanding the phosphoproteomic landscape across various species and exercise modalities
In this study, we have utilized unbiased global phosphoproteomics of skeletal muscle to provide novel insights into the biological responses to exercise. A small number of large abundant contractile proteins, which account for over 50% of skeletal muscle mass, presents a considerable challenge for biochemical analysis of this tissue (Geiger et al, 2013). Here we combined optimized phosphopeptide enrichment strategies with multiplexed isobaric labeling and high‐resolution separation coupled to tandem MS to identify > 18,000 unique phosphosites across rat in situ contraction and mouse treadmill running, ~20% of which were significantly regulated with exercise. To gain insight into the overlap of phosphoproteomes of commonly used exercise models in different species, we integrated the rat contraction and mouse running datasets with our previously published human cycling dataset (Hoffman et al, 2015). At the level of individual orthologous phosphosites, there was relatively low overlap between the three datasets (67 sites). This is not necessarily surprising as different modes of exercise, protocols, and muscle groups were used, and phosphorylated amino acids are only marginally conserved across species owing to their enriched localization in disordered and faster‐evolving protein regions under low selection pressure (Landry et al, 2009). We created “sequence windows” to identify regulated phosphosites within the same region of the protein that may have a similar function. This revealed greater overlap (190 sites) in phosphorylation of protein regions between datasets. Although these phosphosites are not necessarily orthologous, we speculate that these phosphorylation clusters may share a similar role in regulating protein function. Furthermore, biological pathway enrichment analysis using proteins containing regulated phosphosites showed excellent overlap between the three datasets, suggesting, although differences in substrate phosphorylation are observed between the models, there may be functional convergence at the higher pathway level. Taken together, these analyses highlight robust phosphorylation events and biological processes that are common to the exercise models used here. We believe that these studies provide a major advance in our endeavors to map key exercise‐regulated pathways in skeletal muscle.
Kinase activity across exercise models
Kinase: substrate relationships enrichment analysis revealed increased activity of stress‐sensing kinases such as AMPK, PKA, P38, MAPKAPK2, and CAMK in at least one of the three datasets. However, in many cases kinases were not significantly regulated in one or more of the models, suggesting either disparate activation/inhibition, differential regulation of phosphatases/negative feedback, or skewed enrichment statistics arising from limited KSR annotation in the database. AMPK was the only kinase with significantly enriched activity across all three datasets. The major upstream kinase for AMPK activation is LKB1, which is believed to be constitutively active in skeletal muscle. AMP, which is elevated during metabolic stress such as exercise, stabilizes LKB1‐mediated AMPK phosphorylation by preventing dephosphorylation of AMPK by phosphatases (Richter & Ruderman, 2009; Kjøbsted et al, 2018). AMPK has been shown to be activated by exercise at intensities greater than 60% of maximal aerobic capacity or prolonged low‐intensity exercise (Richter & Ruderman, 2009). Activity of Akt/mTORC2 and mTORC1/S6K signaling was decreased in human cycling and mouse running, while in rat contraction, phosphorylation of some targets of these kinases including TSC2, AKT2, and RPTOR was increased. These data agree with previous findings that acute high‐intensity in situ contraction increases Akt (all isoforms 1–3) activity in multiple rat muscle types, dependent on an unidentified circulating factor(s) (Sakamoto et al, 2002). Our observation of only a subset of mTOR targets may reflect temporal dynamics, as this kinase is fully activated one hour post‐contraction (O'Neil et al, 2009), whereas we collected muscle immediately after the completion of the contraction protocol. Other divergences between species and exercise modes may be attributed to differences in central catecholamine release or delivery of circulating factors to the exercising muscle and muscle fiber type composition resulting in potential alterations in cellular composition (Hoffman, 2017). In situ contraction of isolated muscle has several advantages as an exercise model including the ability to override cognitive and neural factors to produce controlled, regular contractions to the point of muscle fatigue. However, there are limitations to this model that should be considered. Electrical stimulation causes recruitment of large, fast motor units before smaller ones, as opposed to the voluntary exercise, which follows a pattern of recruitment of smaller slow motor units followed by increasingly larger, fast motor units. Additionally, this in situ contraction recruitment pattern causes more rapid onset of fatigue and muscle cytoskeletal damage (Gregory & Bickel, 2005; Crameri et al, 2007; Doucet et al, 2012). Our kinase activity analyses identify points of divergence between models that should guide selection of appropriate models for future investigation of exercise‐induced skeletal muscle adaptations.
STIM1 phosphorylation by AMPK
AMP‐activated protein kinase and Ca2+ signaling were among the top five biological pathways enriched in all three exercise models. Furthermore, consensus sequence analysis revealed potential crosstalk between AMPK signaling and Ca2+ regulation at STIM1, which is critical for SOCE through its interaction with the Ca2+ channel Orai1. In all three exercise models, phosphorylation of STIM1 was enriched at two distinct predicted AMPK consensus sites. Herein, we validated STIM1 as a direct substrate of AMPK and demonstrated that phosphorylation of STIM1 by AMPK alters STIM1 conformation and decreases SOCE activity. These data are consistent with a previous study in endothelial cells showing that AMPK activation stimulates phosphorylation of STIM1 on unidentified residues, and this resulted in decreased SOCE (Sundivakkam et al, 2013).
STIM1 activation and points of regulation
Stromal interaction molecule 1 activation is multifactorial and has been shown by our group and others to be regulated by phosphorylation at multiple distinct phosphosites. STIM1 contains a single transmembrane domain, which spans the SR membrane. When SR Ca2+ levels are depleted, hydrophobic regions of STIM1 located in the EF‐SAM domain (AA 63–127) are exposed, causing the SR region of STIM1 to homodimerize. This forces the cytosolic region of STIM1 into an extended conformation and promotes inter‐molecular association between STIM1 CC1 domains (AA 238–344) (Stathopulos et al, 2006; Covington et al, 2010). A single mutation of two individual leucines within CC1α1 to serines (including L251) is sufficient to induce STIM1 extension independent of SR Ca2+ sensing, emphasizing that this coiled‐coil domain is critical for STIM1 intra‐ and inter‐molecular interaction (Muik et al, 2011). One of the AMPK‐regulated phosphosites we identified, S257, lies within the CC1α1 domain. Our data show that phosphorylation of S257 inhibits extension of the STIM1 coiled‐coil region, and we propose this is either due to electrostatic hindrance between phosphorylated residues of opposing STIM1 molecules or the negatively charged phosphorylation in CC1α1 might enhance intra‐molecular interaction with a basic segment in CC2. STIM1 dimers span ER‐PM junctions, associate with PIP2 at the PM, and accumulate to form puncta. Orai1 is trapped within these puncta (Liou et al, 2007) and STIM1 binds to Orai1 via the OASF (AA 233–474), which consists of the CC1 domain (CC1α1, CC1α2, and CC1α3) and two additional coiled‐coil domains (CC2 and CC3). Phosphorylation of STIM1 in its CC2 domain, at Y361 or T389, promotes STIM1 interaction with Orai1 and SOCE (Yazbeck et al, 2017; Thompson et al, 2018). Additionally, phosphorylation of a cluster of sites close to or within the serine/proline‐rich domain near the PM interaction region (S572, S575, S608, and S621) promotes STIM1 interaction with Orai1 (Pozo‐Guisado et al, 2010; Casas‐Rua et al, 2015). The CTID (AA 448–530) of STIM1 prevents its constitutive association with Orai1 and confers dependency of SOCE on ER Ca2+ stores (Baba et al, 2006; Jha et al, 2013). The other AMPK‐regulated phosphosite we identified, S521, is located within the CTID. Although we did not find evidence from our experiments for a regulatory role of this phosphosite in SOCE, we cannot rule out that in certain settings this site may influence CTID interaction with Orai1. The CTID contains a cluster of serine residues that we found to be regulated with exercise, so we speculate that these phosphosites act in a coordinated fashion and/or have overlapping function, which is likely not observable by studying only one site in isolation.
Biological implications for AMPK‐mediated regulation of STIM1 activity in skeletal muscle
AMP‐activated protein kinase is robustly activated during exercise, and mice lacking both the AMPK catalytic α and regulatory β subunits in skeletal muscle have defects in whole‐body exercise capacity and adaptations (O'Neill et al, 2011; Lantier et al, 2014). Our findings that STIM1 activity is decreased by AMPK during exercise are complex from the perspective that SOCE is proposed to be an important mechanism for refilling of the SR to allow repeated muscle contraction (Stiber et al, 2008; Wei‐Lapierre et al, 2013). However, while these studies have clearly demonstrated a requirement for STIM1 in muscle development, a robust requirement for SOCE in SR refilling has not been demonstrated (Cully & Launikonis, 2013). Furthermore, even during high‐intensity muscle contraction, SOCE‐mediated Ca2+ entry is four orders of magnitude slower than the rate of SR Ca2+ release and, so, is likely to contribute minimally to SR Ca2+ refilling in this scenario (Launikonis et al, 2010). Notably, SR Ca2+ reuptake is diminished over repeated contractions (Ward et al, 1998; Leppik et al, 2004; Cheng et al, 2018) and the resulting accumulation of cytosolic Ca2+ is associated with muscle fatigue characterized by loss of force generation, diminished excitation–contraction coupling, and proteolysis (Lamb et al, 1995; Cheng et al, 2018). Even beyond the exercise scenario, excessive SOCE is associated with muscular dystrophy and mitochondrial Ca2+ overload (Vandebrouck et al, 2006; Goonasekera et al, 2014). Therefore, we and others (Launikonis et al, 2010) suggest a primary role for STIM1 is to fine‐tune cytosolic Ca2+ levels in order to regulate diverse signaling and transcriptional processes (Berridge et al, 2003; Ivarsson et al, 2019), and that its activity is appropriately dampened during exercise (we propose via AMPK) to prolong the time to cytosolic Ca2+ overload and fatigue. Notably, SERCA pumps consume considerable amounts of ATP to pump Ca2+ from the cytosol into the SR (Norris et al, 2010) and so the dampening of SOCE by AMPK is consistent with one of the major roles of this kinase to conserve energy when ATP:AMP ratios drop, such as during exercise (Hardie, 2003).
Broader implications for the AMPK‐STIM1 axis in health and disease
In this study, we have discovered a crosstalk between AMPK and Ca2+ signaling via STIM1 in skeletal muscle. Intriguingly, STIM1 is ubiquitously expressed in human tissues and Ca2+ overload, as well as deficiencies in AMPK signaling, have been proposed to underlie numerous pathologies in diverse tissues. For example, in insulin‐responsive tissues such as skeletal muscle and adipose tissue, decreased AMPK signaling and Ca2+ overload have been proposed to trigger the development of insulin resistance (Levy et al, 1994; Gauthier et al, 2011). Ca2+ overload and alterations in AMPK signaling have also been shown to impair insulin secretion and induce pancreatic beta cell death in the etiology of type 2 diabetes (Choi et al, 2007; Fu et al, 2013). Elevated cytosolic Ca2+ levels and dampened AMPK activity have been observed in autoimmune disease, and inhibition of SOCE suppresses inflammatory cytokine release from immune cells (Nath et al, 2009; Lin et al, 2013). Cytosolic Ca2+ overload can induce hepatocyte death leading to liver fibrosis, whereas reduction of intracellular Ca2+ or AMPK activation protects from hepatocyte injury and cell death (Da Silva Morais et al, 2010; Vasques et al, 2016). Neuropathic pain commonly features elevated cytosolic Ca2+, and blocking Ca2+ influx or activating AMPK has been shown to alleviate pain in various models of nerve injury (Dray, 2008; Fernyhough & Calcutt, 2010; Melemedjian et al, 2011). Excessive SOCE is associated with tubular aggregate myopathy in muscle (Böhm et al, 2017; Böhm & Laporte, 2018). Finally, AMPK activation has been associated with decreased SOCE in platelets and dendritic cells (Lang et al, 2012; Nurbaeva et al, 2012). Thus, the functional role of the AMPK‐STIM1 regulatory axis in exercise and various tissues and disease settings remains to be explored and warrants further investigation.
Summary
Exercise has long been recognized to be among the most effective means of protecting against lifestyle‐related diseases, largely attributable to adaptations in skeletal muscle. However, the precise signaling pathways activated in response to exercise across widely used experimental models have not been fully elucidated. Recent technical advances in MS have allowed us to capture a global, unbiased snapshot of the exercise landscape in skeletal muscle with high resolution. These integrated analyses of the exercise phosphoproteome in skeletal muscle across exercise models and species have revealed thousands of phosphorylation events, many of which have never been previously identified or implicated in the exercise response. Using this approach, we identified orthologous phosphosites, as well as protein regions, that were consistently phosphorylated across the three models and these likely represent highly conserved exercise‐regulated processes and thus central to exercise‐regulated adaptive responses. We also identified high consistency at the level of biological pathways across the three models. Among the most enriched pathways in all models were AMPK and Ca2+ signaling. STIM1 represented a point of convergence between AMPK and Ca2+ signaling, so we further explored this regulatory axis. We found that STIM1 was phosphorylated by AMPK at two independent sites (S257 and S521), and that phosphorylation of STIM1 S257 affects STIM1 conformation and decreases its SOCE activity. Furthermore, we have shown a role for SOCE in muscle fatigue and demonstrated that phosphorylation of STIM1 may be an important mechanism by which AMPK signaling allows muscle to adapt to exercise and delay fatigue. This study expands the known role of AMPK to regulating intracellular Ca2+ dynamics and SOCE, sheds light on the vast exercise‐regulated signaling network, and opens doors for further exploration of protein phosphorylation and its breadth of roles in regulating tissue exercise adaptations. This cross‐species reference will inform the development and use of exercise models and ideally lead to identification of new therapeutic targets to help achieve health benefits associated with exercise.
Materials and Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents may be directed to and will be fulfilled by the Lead Contact, Professor David James (david.james@sydney.edu.au).
Experimental model and subject details
Animal husbandry
All animals were group housed at 21–23°C in a humidity‐controlled environment on a 12:12 h light:dark cycle in filter‐top cages with ad libitum access to standard rodent chow and water. All animal husbandry and experimentation was conducted in accordance with the Australian code of practice for the care and use of animals for scientific purposes as stipulated by the National Health and Medical Research Council (Australia). Rat and mouse exercise experiments were approved by the Animal Ethics Committee of The University of Melbourne and the University of Copenhagen (permit 2015‐15‐0201‐00477 from the Danish Animal Experiments Inspectorate), respectively. AMPK β1/β2 KO MEFs were generated from wild‐type or AMPK‐β1 floxed/β2 knockout embryos as previously described (Davies et al, 2014) in accordance with Austin Health and St. Vincent's Hospital animal ethics committees, and these MEFs were immortalized as previously described (Dite et al, 2017).
Models of exercise
Human cycling exercise samples were generated as previously described (Hoffman et al, 2015). For in situ muscle contraction experiments, 3‐month‐old male Wistar rats were provided with ~50% of overnight food intake determined from pilot experiments then anaesthetized the following day by intraperitoneal administration of sodium pentobarbitone (Nembutal, Sigma‐Aldrich, NSW, Australia; 60 mg/kg i.p.) such that they were unresponsive to tactile stimuli. Tibialis anterior muscles (n = 10) were isolated and subjected to either sham surgery or a sciatic nerve stimulated high‐intensity in situ contraction protocol with the distal tendon connected to a custom‐built force transducer. Nerve and blood supply remained intact throughout. The muscle was stimulated at a frequency of 100 Hz for 1 s, followed by 3‐s rest. This protocol was repeated for 5 min with the animal resting on a warm water‐jacketed platform and temperature maintained by a heat lamp with warmed mineral oil (37°C) applied periodically to the nerve. Maximum force (mN) was monitored over the duration of the contraction protocol to assess muscle fatigue. Immediately after 5 min at resting tension (sham) or the contraction protocol, muscles were quickly removed from the force transducer, excised, rinsed in ice‐cold saline, blotted on gauze, and snap‐frozen in liquid nitrogen. For treadmill running experiments, fed 3‐month‐old female mice [wild‐type littermate mice from inducible Rac1 muscle‐specific KO (mKO) mice breeding (Sylow et al, 2013)], extensively backcrossed to C57BL/6 background) were acclimatized to the treadmill 3 × 5 min at 10 m/min and 2 × 5 min at 16 m/min at a 0° incline during the week prior to the maximal running capacity test. The test was performed at a 10° incline beginning with a 5‐min warm up at 10 m/min, after which the speed was increased by ∼1.2 m/min every minute until exhaustion. Exhaustion was defined as the speed at which the mouse was unable to keep up with the treadmill. The test was performed blinded. On experimental day, each mouse was exercised at a relative workload corresponding to ∼65% of its maximal running speed (∼20 m/min for 30 min at a 10° incline, or left resting on a still treadmill. Immediately after the exercise bout, mice were killed by cervical dislocation and quadriceps muscles were quickly removed and stored at −80°C until further processing. Rat and mouse muscles were ground under liquid nitrogen and stored at −80°C until further processing for MS or Western blotting.
Method details
Skeletal muscle lysis, isobaric labeling, and phosphopeptide enrichment
Muscles were ground under liquid nitrogen and approximately 40 mg of tissue lysed in 6 M urea, 2 M thiourea, 25 mM triethylammonium bicarbonate (TEAB), pH 7.9 containing phosphatase and protease inhibitor cocktails (Roche) by tip‐probe sonication (2 × 15 s) on ice. The lysates were centrifuged at 17,000 g, 15 min, 4°C and the supernatant precipitated with 6 volumes of acetone, overnight, −20°C. Protein pellets were resuspended in 6 M urea, 2 M thiourea, 25 mM TEAB, pH 7.9 and quantified by Qubit fluorescence (Invitrogen). Concentrations were normalized and 1 mg of protein reduced with 10 mM dithiothreitol for 60 min at 25°C followed by alkylation with 25 mM iodoacetamide for 30 min at 25°C in the dark. The reaction was quenched to a final concentration of 20 mM dithiothreitol and digested with Lys‐C (Wako) at 1:50 enzyme to substrate ratio for 2 h at 25°C. The mixture was diluted 5‐fold with 25 mM TEAB and digested with trypsin at 1:50 enzyme to substrate ratio for 12 h at 30°C. The peptide mixture was acidified to a final concentration of 2% formic acid, 0.1% trifluoroacetic acid (TFA) and centrifuged at 16,000 g for 15 min. Peptides were desalted using hydrophilic lipophilic balance—solid‐phase extraction (HLB‐SPE) cartridges (Waters) followed by elution with 50% acetonitrile, 0.1% TFA and dried by vacuum centrifugation. Peptides were resuspended in 30 μl of 100 mM TEAB, quantified by Qubit fluorescence, and normalized to 250 μg/30 μl (pH 7.5). Peptides were labeled with 10‐plex tandem mass tags (TMT) in 50% acetonitrile for 90 min at room temperature and quenched with 0.5% hydroxylamine. The labeled peptides were pooled within the respective experiments and dried to approximately 50 μl by vacuum centrifugation. Phosphopeptides were enriched essentially as described previously and consisted of titanium dioxide followed by sequential elution from immobilized metal ion affinity chromatography and fractionation by hydrophilic interaction liquid chromatography (TiSH; Engholm‐Keller et al, 2012). Briefly, peptides were resuspended in 1 ml of titanium dioxide loading buffer (1 M glycolic acid, 80% acetonitrile, 5% TFA), and a 20 μg aliquot was saved for total proteomic analysis. Titanium dioxide beads (15 mg in 150 μl acetonitrile) (GL Science, Japan) were added to the peptide mixture and rotated at room temperature for 20 min. The mixture was centrifuged at 10,000 g, 1 min, and the supernatant was applied to a second aliquot of titanium dioxide (7.5 mg in 75 μl acetonitrile) and rotated at room temperature for 20 min. The mixture was centrifuged at 10,000 g, 1 min, and the supernatant was applied to a third aliquot of titanium dioxide (4 mg in 40 μl acetonitrile) and rotated at room temperature for 20 min. The beads were washed with 100 μl titanium dioxide loading buffer followed by 80% acetonitrile, 2% TFA and finally 16% acetonitrile, 0.4% TFA. The beads were dried briefly by vacuum centrifugation and eluted with 50 μl of 1% ammonium hydroxide by shaking at room temperature for 15 min. The titanium dioxide elution slurry was loaded onto a C8‐plugged microcolumn and eluted with gentle air pressure to trap beads. The beads were eluted with an additional 50 μl 1% ammonium hydroxide, and the elution was pooled. Enriched phosphopeptides were acidified to a final concentration of 10% formic acid and dried by vacuum centrifugation. The enriched phosphopeptides were resuspended in 500 μl of 50% acetonitrile, 0.2% TFA and rotated with 50 μl of Fe(III)‐IMAC beads (Sigma‐Aldrich) at room temperature for 45 min. The IMAC slurry was loaded onto a crushed GeLoader microcolumn and eluted with gentle air pressure to trap beads. Mono‐phosphorylated peptides were eluted with 50% acetonitrile, 0.1% TFA followed by 20% acetonitrile, 1% TFA; pooled with the flow‐through; and dried by vacuum centrifugation. Multi‐phosphorylated peptides were eluted with 1% ammonium hydroxide, acidified to a final concentration of 10% formic acid, 0.1% TFA, and desalted with C18 microcolumns. Enriched mono‐phosphorylated peptides were subjected to another round of titanium dioxide as described above. The enriched mono‐phosphorylated peptides and non‐phosphorylated peptides were fractionated on in‐house packed TSK‐amide HILIC column as described previously (Palmisano et al, 2010). All isobarically labeled peptides were analyzed by data‐dependent acquisition (DDA) as described below.
Mass spectrometry
Peptides were resuspended in 2% acetonitrile, 0.1% formic acid and loaded onto a 50 cm × 75 μm inner diameter column packed in‐house with 1.9 μm C18AQ particles (Dr Maisch GmbH HPLC) using an Easy nLC‐1000 UHPLC operated in single‐column mode with intelligent flow control loading at 950 bar. Peptides were separated using a linear gradient of 5–30% Buffer B over 100 min at 250 nl/min (Buffer A = 0.5% acetic acid; Buffer B = 80% acetonitrile, 0.5% acetic acid). The column was maintained at 50°C using a PRSO‐V1 ion‐source (Sonation) coupled directly to a Q‐Exactive mass spectrometer (MS). A full‐scan MS1 was measured at 70,000 resolution at 200 m/z (300–1,750 m/z; 100 ms injection time; 3e6 AGC target) followed by isolation of up to 20 most abundant precursor ions for MS/MS (2 m/z isolation; 8.3e5 intensity threshold; 30.0 normalized collision energy; 17,500 resolution at 200 m/z; 60‐ms injection time; 5e5 AGC target). Charge state reduction of isobarically labeled peptides was achieved with a 10% ammonium hydroxide vapor underneath the ESI source (Thingholm et al, 2010).
Bioinformatics
Mass spectrometry data were process with MaxQuant (v1.5.2.10) and searched with Andromeda against either the mouse or rat UniProt databases (Cox & Mann, 2008; Cox et al, 2011). All settings were default and searched with the following variable modifications; methionine oxidation; serine, threonine, and tyrosine phosphorylation; and N‐terminus and lysine TMT labeling. Carbamidomethylation of cysteine was set as a fixed modification. Bioinformatic analysis was performed primarily in the R programming environment (R Development Core Team, 2008). All data were normalized to the median of each muscle. Significantly regulated phosphopeptides were determined using a moderated t‐test from LIMMA package in R (Smyth, 2004). Linear models were fit to the data and empirical Bayes was used for variance shrinkage in order to determine regulated sites, and P‐values were corrected for multiple testing controlling for 5% FDR using Benjamini and Hochberg method (Benjamini & Yekutieli, 2001). Kinase and pathway enrichment were performed using a mean‐rank gene set test. The gene sets for kinase enrichment were known substrates from PhosphoSitePlus (Hornbeck et al, 2012) and manual curation. Pathway enrichment gene sets were obtained from KEGG (Ogata et al, 1999) and Reactome (Fabregat et al, 2018), and filtered to present only the 12 most enriched pathways.
For comparison of human, mouse, and rat phosphosites, an algorithm and automated tool was developed in‐house (Chaudhuri et al, 2015). Based on global pairwise sequence alignment of orthologous proteins from human and rat, sites quantified from independent cross‐species experiments can be easily mapped to each other using this tool. Global pairwise sequence alignment between each orthologous protein pair was performed using the BLOSUM62 (Styczynski et al, 2008) substitution matrix, gapOpening of 10, gapExtension of 0.5 using Biostrings package in R (Pages et al, n.d.). We scanned this sequence alignment using the human phosphosite numbers from the MS‐based phosphoproteomics experiment as the reference to identify the aligned amino acid residues and their positions in the rat and mouse MS‐phosphoproteomics data. If there was a match in a phosphorylated residue and if the corresponding aligned position existed in the rat and/or mouse phosphoproteomics data, we considered this as a “match” and the Uniprot protein accession name, residue, and site number match were retained as a modified site that could be mapped between species. To match phosphosites within 5% of the protein length, protein orthologs were compiled from NCBI and biomaRt (Smedley et al, 2015). Protein lengths were computed from Uniprot sequences. If multiple sites were within the 5% threshold, only the closest site to the human phosphosite was selected.
Plasmids
Human STIM1 YFP was a gift from Anjana Rao (Addgene plasmid #19754). pKanCMV‐mClover3‐mRuby3 was a gift from Michael Lin (Addgene plasmid #74252). mClover2‐C1 was a gift from Michael Davidson (Addgene plasmid #54577). pGP‐CMV‐GCaMP6s was a gift from Douglas Kim (Addgene plasmid #40753; Chen et al, 2013).
Cloning
The fluorescent tag on Human STIM1 YFP was switched to mRuby3 to minimize aggregation, improve localization, and improve the signal. PCR‐based site‐directed mutagenesis was used to introduce the S257A, S257E, S521A, and S521E mutations to the plasmid DNA.
FRET controls
The FRET construct was generated by amplifying and inserting TagRFP‐1 into pClover2‐C1 (Addgene) via EcoRI and BamHI sites. STIM1 OASF (aa 233–474) was amplified from human STIM1 cDNA (Addgene) and inserted into BglII and HindIII sites of the Clover2‐TagRFP‐t plasmid to generate the OASF‐FRET construct (Clover2‐OASF‐TagRFP‐T). The long and short linker FRET control constructs were synthesized as a GeneBlock by IDT (Integrated DNA Technologies Pte. Ltd. Baulkham Hills, Australia). The short flexible linker construct was made by annealing oligos encoding the amino acid sequence AGGGGS into the BspEI and EcoRI sites of Clover2‐TagRFP‐T. The long rigid linker construct contained amino acid sequence GA[EAAAK]4ELEA[EAAAK]4A between the BspEI and EcoRI sites of Clover2‐TagRFP‐T. For the STIM1 S257A and S257E FRET constructs, the S257 residue was mutated to either alanine (S257A) or glutamic acid (S257E) via PCR‐based site‐directed mutagenesis.
Muscle cell culture, transfection, and drug treatment
L6 rat myoblasts were cultured in α‐Minimum essential medium (α‐MEM; Gibco) (5 mM d‐glucose, 10% fetal bovine serum (FBS; Hyclone Laboratories), 1x GlutaMAX, and 1 mM Na‐pyruvate). All cells were kept in a 10% CO2 humidified incubator at 37°C. L6 myoblasts were transiently transfected with the FRET constructs using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions. For Ca2+ imaging experiments, three independent siRNA oligonucleotides directed at rat STIM1 (sequence 1: 5′–3′ GCUGCUGGUUUGCCUAUAUTT, sequence 2: 5′–3′ GGAUCUCAGAGGGAUUUGATT, sequence 3: 5′–3′ GUAGCCGAAACACACGAAUTT) were used to achieve knockdown of endogenous STIM1 in L6 myoblasts, or a non‐targeted oligonucleotide (5′–3′ UUCUCCGAACGUGUCACGUTT) was used for the Control condition. All siRNA oligonucleotides were synthesized and provided by Shanghai GenePharma Co., Ltd. For these experiments, the STIM1‐mRuby3 and GCaMP6s DNA and the siRNA oligonucleotides were combined, then mixed with Lipofectamine and added to cells simultaneously according to the manufacturer's instructions. All transfections were performed in OptiMEM without FBS. Media was changed to normal culture media 16 h after transfection. 48 h after transfection, cells were re‐seeded into imaging wells, and cells were imaged or drug‐treated for 30 min and used for Western blotting 72 h after transfection. Thapsigargin (Sigma‐Aldrich), A‐769662 (Tocris Bioscience), and AICAR (Tocris Bioscience) were made up to 1,000× solutions in DMSO and used at final concentrations of 2 μM, 100 μM, and 0.5 mM, respectively, unless otherwise noted.
Time‐domain fluorescence lifetime imaging
Cells were seeded into Ibidi 8‐well glass‐bottom μ‐slides. Fixed samples were imaged in PBS with 2.5% DABCO. Live cells were imaged in KRP as indicated in Calcium imaging. Data were acquired on a Nikon A1 laser scanning confocal microscope (Coherent Scientific Pty Ltd) with a Picoquant FCS/FCCS/FLIM upgrade kit using a Nikon CFI Plan Apo IR 60XWI NA1.27 WD 0.17 objective. Lifetime measurements were obtained using a TimeHarp 260 TCSPC module controlled by Symphotime64. mClover2 was excited by a pulsed 485 nm diode laser (40 Hz), and photon emission was detected with a PMA hybrid 40 detector (Coherent Scientific Pty Ltd) after passing through a 525/50 emission filter. The data were analyzed with the FLIMfit software tool developed at Imperial College London (Warren et al, 2013) using a single exponential decay to fit the TCSPC data.
Calcium imaging
L6 myoblasts were seeded at a concentration of 1 × 106/ml into Matrigel‐coated CellAsic M04S microfluidic plates (Merck) 1 day prior to imaging using gravity with the plate angled to ~30° to enhance even cell loading. One hour prior to imaging, cells were switched to 1 mM Ca2+ KRP‐based buffer: 155 mM NaCl, 4.5 mM KCl, 5 mM HEPES, 5 mM d‐glucose, 0.2% BSA, MEM essential vitamins, 1× GlutaMAX and 1 mM sodium pyruvate, pH 7.4, 1 mM CaCl2, and 2.5 mM MgCl2. The Ca2+‐free buffer had the following modifications: 0 mM CaCl2, 3.5 mM MgCl2, and 1 mM EGTA. The relevant buffers were loaded into the individual wells of the CellAsic plate. The plate was attached to the manifold of a CellAsic Onix2 microfluidic platform (Merck) and placed on the stage of a Nikon TiE‐inverted microscope equipped with an OKOlab microscope enclosure maintained at 37°C. G6CaMP6s was stimulated with a 470 nm LED (Lumencor), and emission was captured on an Andor 888 emCCD camera after passing through a 525‐/50‐nm filter. Buffer switching was performed using the CellAsic Onix 2 software. Processing and analysis of the microscopy images was performed in Fiji (ImageJ; Schindelin et al, 2012).
Drosophila negative geotaxis exercise experiments
Flies were maintained under standard conditions of 25°C, 65% humidity, and a 12‐h light/dark cycle, and raised on standard food containing yeast, cornmeal, and dextrose. For the STIM1 knockdown models, tubulin‐GAL4 was crossed to UAS‐STIM.RNAi.E 26‐1, UAS‐STIM.RNAi.E 8‐4, or UAS‐STIM.RNAi GD16187 and it was determined that the whole‐body STIM.RNAi lines were lethal. mef2‐GAL4 was then crossed with the above UAS‐STIM.RNAi or the appropriate UAS‐control lines, and these muscle‐specific STIM.RNAi lines were viable. 3‐ to 5‐day‐old male progeny were collected for negative geotaxis exercise experiments. Researchers were blinded to genotype, and flies were acclimated for 30 min prior to climbing assay. The vials containing flies were tapped three times, then climbing speed was observed (time required for 50% of the flies in a vial to climb 50% of the way up the wall of the tube). Flies were then exercised by tapping to the bottom of the vial every 30 s for 2.5 h using an automated rotator. Flies were removed from the rotator, then climbing speed during a negative geotaxis assay was immediately observed to obtain post‐exercise data. Three vials per condition were used for each experiment, and the experiment was repeated on two separate days in independent batches of flies. For the STIM1 transgenic models, human STIM1‐mRuby3 WT, S257A, and S257E cDNA were excised using BglII and NOTI restriction enzymes and directionally cloned into the Drosophila expression vector pJFRC‐MUH. pJFRC‐MUH was a gift from Gerald Rubin (Janelia Research Campus, Howard Hughes Medical Institute, Ashburn, VA, USA; Addgene plasmid #26213). Plasmids were sent for injection (Bestgene, Chino Hills, CA, USA). FM7/Y; UAS‐mRuby3‐hSTIM1 WT, FM7/Y; UAS‐mRuby3‐STIM1 S257A and FM7/Y; UAS‐mRuby3‐hSTIM1 S257E males were crossed to STIM KO/FM7; tubulin‐GAL4/TM3,Sb females. 3‐ to 5‐day‐old STIM KO/Y; UAS‐mRuby3‐hSTIM1 WT; tubulin‐GAL4 (STIM KO; UAS‐hSTIM1 WT), STIM KO/Y; UAS‐mRuby3‐hSTIM1 S257A; tubulin‐GAL4 (STIM KO; UAS‐hSTIM1 S257A) and STIM KO/Y; UAS‐mRuby3‐hSTIM1 S257E; tubulin‐GAL4 (STIM KO; UAS‐hSTIM1 S257E) were used for negative geotaxis exercise experiments. Researchers were blinded to genotype, and flies were acclimated for 30 min prior to climbing assay. The vials containing flies were tapped three times and flies were then allowed to climb for 3 min. This process was repeated four times to allow the flies to acclimate to the assay, and, at the 5th repeat, the time the flies climbed during the 3‐minute period was measured. This experiment was repeated on three different days with independent batches of flies. Fly stocks for tubulin‐GAL4 (BL #5138), mef2‐GAL4 (BL #27390), UAS‐mCD8GFP (BL# 32186), UAS‐STIM1 RNAi (BL #41758 and #41759), and UAS‐STIM1 FL (BL #41756) were obtained from Bloomington Stock Center (Bloomington, Indiana, USA). The UAS‐STIM1 RNAi GD16187 and control GD3003 was obtained from VDRC (Vienna, Austria; stock #47073 and #12145), and the STIM1 CRISPR KO was a gift from Gaiti Hasan (Bangalore, India).
Western blotting
Frozen muscles were homogenized with stainless steel pellets using the Tissuelyser II (Qiagen) at 2 × 45 s at 30 Hz and ice‐cold homogenization buffer (10% Glycerol, 20 mM Na‐pyrophosphate, 150 mM NaCl, 50 mM HEPES (pH 7.5), 1% NP‐40, 20 mM ß‐glycerophosphate, 10 mM NaF, 2 mM PMSF, 1 mM EDTA (pH 8.0), 1 mM, EGTA (pH 8.0), 10 μg/ml Aprotinin, 10 μg/mL Leupeptin, 2 mM Na3VO4, 3 mM Benzamidine, 5 mM Nicotinamide). Homogenates were rotated end‐over‐end for 1 h at 4°C, and supernatant was obtained by centrifugation at 13,000 g for 20 min at 4°C. Supernatant was removed to a new tube, protein concentration was measured by BCA method, and lysates were diluted to the same protein concentration (1 μg/μl). Cells were collected for Western blotting by washing 3× with ice‐cold PBS, placing cells on ice, then adding NP‐40 lysis buffer containing protease and phosphatase inhibitors. Cells were incubated in lysis buffer on ice for 10 min with scraping, and then, lysate was homogenized by passing lysate through an 18‐gauge needle 10×. Flies were homogenized in 2% SDS lysis buffer. Supernatant was obtained by centrifugation at 13,000 g for 15 min at 4°C. Protein concentration of the supernatant was measured by BCA method. 4× Laemmli buffer (277.8 mM Tris–HCl pH 6.8, 40% (v/v) glycerol, 4% SDS, 0.02% bromophenol blue, 10% 2‐mercaptoethanol) was added to samples, and samples were heated at 65°C for 15 min. 15 μg protein was separated by SDS–PAGE. The resolved proteins were transferred to PVDF membrane. Membranes were blocked in 5% skim milk in TBS‐Tween for 1 h at room temperature then incubated in primary antibody (1:1000) in TBS with 5% BSA overnight at 4°C. The phosphor‐specific STIM1 p‐S257 and p‐S521 antibodies were generated by 21st Century Biochemicals. All other phospho‐specific and total antibodies were purchased from Cell Signaling Technology. Membranes were washed in TBS‐Tween then incubated in HRP‐conjugated secondary antibody (1:10,000) in TBS for 1 h at room temperature, then washed in TBS‐Tween. Membranes were analyzed via autoradiography using the Bio‐Rad ChemiDoc Gel Imager system. Western blot quantification was performed in ImageJ 1.51s.
AMPK in vitro kinase assay
HEK293 cells were cultured in Dulbecco's modified essential medium (DMEM; Gibco) (25 mM d‐glucose, 10% fetal bovine FBS). HEK293 cells expressing YFP‐STIM1 were serum starved for 2 h, washed 3× with ice‐cold PBS, and lysed in IP Buffer (1% NP‐40, 10% glycerol, 137 mM NaCl, 25 mM Tris–HCl, pH 7.5 containing protease, and phosphatase inhibitors). Cell debris was removed by centrifugation at 16,000 g, 10 min at 4°C, and the supernatant incubated with anti‐GFP antibody with rotation for 2 h at 4°C (1:200; Roche). The supernatant:antibody mixture was added to 30 μl of Protein‐G‐agarose beads pre‐equilibrated with IP Buffer and rotated for 45 min at 4°C. The beads were washed 3× with IP Buffer followed by 2× with 25 mM Tris, pH 7.5. A separate batch of non‐transfected HEK293 cells was prepared as described above except the lysate was incubated with 30 μl of streptavidin‐agarose beads to enrich endogenously biotinylated ACC1/2 as a positive control. A third batch of HEK293 cells expressing FLAG‐AMPKα1β1γ2 was serum starved for 2 h and stimulated with 5 mM AICAR and 100 μM of A‐769662 for 30 min. Cells were lysed and immunoprecipitation performed as described above except with the use of an anti‐FLAG primary antibody (1:200; Sigma). The FLAG‐AMPKα1β1γ2 was eluted from the beads with 3xFLAG peptide (16 μg/30 μl; Sigma) in Kinase Buffer (1 mM ATP, 10 mM MgCl2, 25 mM Tris–HCl, pH 7.5). Kinase reactions were performed by adding purified FLAG‐AMPKα1β1γ2 directly to the agarose beads containing enriched ACC1/2 or YFP‐STIM1 and incubated for 30 min at 37°C with shaking. Proteins were eluted with 2× Laemmli buffer and analyzed by Western blotting.
Statistical analysis
Data visualization was performed in R Studio and Adobe Illustrator. Statistical analyses for Figs 1 and 2 are described elsewhere in the methods section. Statistical analyses for Figs 3 and 4 were performed in GraphPad Prism V7. Experiment numbers are detailed in the figure legends. Error bars represent mean ± standard error of the mean (SEM) unless otherwise noted.
Author contributions
DEJ, JS, GSL, and EAR conceptualized and designed the study, oversaw experiments, provided expert guidance, and wrote the manuscript. MEN, BLP, JGB, and NJH designed experiments and wrote the manuscript. MEN performed transfections, cell sample preparation, Western blotting of muscle samples and cells, preparation of cells and buffers for microscopy experiments, and analysis of the Ca2+ microscopy data. BLP and NJH prepared muscle samples for phosphoproteomics and Western blotting. BLP performed the phosphoproteomics and in vitro kinase assays. JGB, MEN, and DMN performed the microscopy experiments. JGB performed and analyzed the TCSPC. KCC, JS, and MEN performed the cloning. TN and NJH performed the rat muscle contraction experiments. LS performed the mouse treadmill exercise experiments. DF developed Drosophila STIM1 models and performed fly genetics. EJN and MEN developed and performed climbing protocols. NXYL and JSO generated and performed experiments in AMPK β 1/2 knockout mouse embryonic fibroblasts. BLP, EJN, and RC performed analyses of the phosphoproteomics data. EJN developed and performed the 5% sequence window analysis. All authors approved the final version of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Table EV2
Table EV3
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We are grateful to Fiona Colarossi for assistance with the animal ethics application and Jørgen Jensen for helpful discussions regarding skeletal muscle contraction models. We thank Dr. Anjana Rao (La Jolla Institute for Allergy and Immunology) for providing the STIM1 and ORAI1 plasmids. This work was supported by National Health and Medical Research Council (NHMRC) project grants GNT1120201 and GNT1086850 (D.E.J.), a grant from the Diabetes Australia Research Trust (D.E.J.), and a grant from the Danish Council for Independent Research/Medicine 6108‐00203 (E.A.R.). D.E.J. is an NHMRC Senior Principal Research Fellow. B.L.P is supported by an NHMRC Early Career fellowship. Lykke Sylow is supported by a Novo Nordisk Foundation Excellence grant (32082). The contents of the published material are solely the responsibility of the individual authors and the Administering Institution and do not reflect the views of the NHMRC.
The EMBO Journal (2019) 38: e102578
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD010452 (http://www.ebi.ac.uk/pride).
References
- Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, Numaga T, Mori Y, Iino M, Hikida M et al (2006) Coupling of STIM1 to store‐operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Natl Acad Sci USA 103: 16704–16709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, Lim WA, Fraser JS, Frydman J, Krogan NJ (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150: 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29: 1165–1188 [Google Scholar]
- Berlin JA, Colditz GA (1990) A meta‐analysis of physical activity in the prevention of coronary heart disease. Am J Epidemiol 132: 612–628 [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517 [DOI] [PubMed] [Google Scholar]
- Böhm J, Bulla M, Urquhart JE, Malfatti E, Williams SG, O'Sullivan J, Szlauer A, Koch C, Baranello G, Mora M et al (2017) ORAI1 mutations with distinct channel gating defects in tubular aggregate myopathy. Hum Mutat 38: 426–438 [DOI] [PubMed] [Google Scholar]
- Böhm J, Laporte J (2018) Gain‐of‐function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium 76: 1–9 [DOI] [PubMed] [Google Scholar]
- Boulé NG, Haddad E, Kenny GP, Wells GA, Sigal RJ (2001) Effects of exercise on glycemic control and body mass in type 2 diabetes mellitus: a meta‐analysis of controlled clinical trials. JAMA 286: 1218–1227 [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415 [DOI] [PubMed] [Google Scholar]
- Casas‐Rua V, Tomas‐Martin P, Lopez‐Guerrero AM, Alvarez IS, Pozo‐Guisado E, Martin‐Romero FJ (2015) STIM1 phosphorylation triggered by epidermal growth factor mediates cell migration. Biochim Biophys Acta 1853: 233–243 [DOI] [PubMed] [Google Scholar]
- Chaudhuri R, Sadrieh A, Hoffman NJ, Parker BL, Humphrey SJ, Stöckli J, Hill AP, James DE, Yang JYH (2015) PhosphOrtholog: a web‐based tool for cross‐species mapping of orthologous protein post‐translational modifications. BMC Genom 16: 617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T‐W, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V et al (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499: 295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AJ, Place N, Westerblad H (2018) Molecular basis for exercise‐induced fatigue: the importance of strictly controlled cellular Ca2+ handling. Cold Spring Harb Perspect Med 8: a029710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S‐E, Kim H‐E, Shin H‐C, Jang H‐J, Lee K‐W, Kim Y, Kang SS, Chun J, Kang Y (2007) Involvement of Ca2+‐mediated apoptotic signals in palmitate‐induced MIN6N8a beta cell death. Mol Cell Endocrinol 272: 50–62 [DOI] [PubMed] [Google Scholar]
- Covington ED, Wu MM, Lewis RS (2010) Essential role for the CRAC activation domain in store‐dependent oligomerization of STIM1. Mol Biol Cell 21: 1897–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Crameri RM, Aagaard P, Qvortrup K, Langberg H, Olesen J, Kjaer M (2007) Myofibre damage in human skeletal muscle: effects of electrical stimulation versus voluntary contraction. J Physiol 583: 365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully TR, Launikonis BS (2013) Store‐operated Ca2+ entry is not required for store refilling in skeletal muscle. Clin Exp Pharmacol Physiol 40: 338–344 [DOI] [PubMed] [Google Scholar]
- Da Silva Morais A, Abarca‐Quinones J, Guigas B, Viollet B, Stärkel P, Horsmans Y, Leclercq IA (2010) Development of hepatic fibrosis occurs normally in AMPK‐deficient mice. Clin Sci 118: 411–420 [DOI] [PubMed] [Google Scholar]
- Davies M, Fraser SA, Galic S, Choy S‐W, Katerelos M, Gleich K, Kemp BE, Mount PF, Power DA (2014) Novel mechanisms of Na+ retention in obesity: phosphorylation of NKCC2 and regulation of SPAK/OSR1 by AMPK. Am J Physiol Renal Physiol 307: F96–F106 [DOI] [PubMed] [Google Scholar]
- Dite TA, Ling NXY, Scott JW, Hoque A, Galic S, Parker BL, Ngoei KRW, Langendorf CG, O'Brien MT, Kundu M et al (2017) The autophagy initiator ULK1 sensitizes AMPK to allosteric drugs. Nat Commun 8: 571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet BM, Lam A, Griffin L (2012) Neuromuscular electrical stimulation for skeletal muscle function. Yale J Biol Med 85: 201–215 [PMC free article] [PubMed] [Google Scholar]
- Dray A (2008) Neuropathic pain: emerging treatments. Br J Anaesth 101: 48–58 [DOI] [PubMed] [Google Scholar]
- Dzamko N, Schertzer JD, Ryall JG, Steel R, Macaulay SL, Wee S, Chen Z‐P, Michell BJ, Oakhill JS, Watt MJ et al (2008) AMPK‐independent pathways regulate skeletal muscle fatty acid oxidation. J Physiol 586: 5819–5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebashi S, Endo M (1968) Calcium ion and muscle contraction. Prog Biophys Mol Biol 18: 123–183 [DOI] [PubMed] [Google Scholar]
- Engholm‐Keller K, Birck P, Størling J, Pociot F, Mandrup‐Poulsen T, Larsen MR (2012) TiSH–a robust and sensitive global phosphoproteomics strategy employing a combination of TiO2, SIMAC, and HILIC. J Proteomics 75: 5749–5761 [DOI] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B et al (2018) The reactome pathway knowledgebase. Nucleic Acids Res 46: D649–D655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner M, Muik M, Schindl R, Butorac C, Stathopulos P, Zheng L, Jardin I, Ikura M, Romanin C (2014) A coiled‐coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J Biol Chem 289: 33231–33244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernyhough P, Calcutt NA (2010) Abnormal calcium homeostasis in peripheral neuropathies. Cell Calcium 47: 130–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Prakriya M (2013) Conformational dynamics of STIM1 activation. Nat Struct Mol Biol 20: 918–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu A, Eberhard CE, Screaton RA (2013) Role of AMPK in pancreatic beta cell function. Mol Cell Endocrinol 366: 127–134 [DOI] [PubMed] [Google Scholar]
- Ganetzky B, Flanagan JR (1978) On the relationship between senescence and age‐related changes in two wild‐type strains of Drosophila melanogaster. Exp Gerontol 13: 189–196 [DOI] [PubMed] [Google Scholar]
- Gauthier M‐S, O'Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, Gokce N, Apovian C, Ruderman N (2011) Decreased AMP‐activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole‐body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun 404: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrig SM, van der Poel C, Sayer TA, Schertzer JD, Henstridge DC, Church JE, Lamon S, Russell AP, Davies KE, Febbraio MA et al (2012) Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 484: 394–398 [DOI] [PubMed] [Google Scholar]
- Geiger T, Velic A, Macek B, Lundberg E, Kampf C, Nagaraj N, Uhlen M, Cox J, Mann M (2013) Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol Cell Proteomics 12: 1709–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonasekera SA, Davis J, Kwong JQ, Accornero F, Wei‐LaPierre L, Sargent MA, Dirksen RT, Molkentin JD (2014) Enhanced Ca2+ influx from STIM1–Orai1 induces muscle pathology in mouse models of muscular dystrophy. Hum Mol Genet 23: 3706–3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory CM, Bickel CS (2005) Recruitment patterns in human skeletal muscle during electrical stimulation. Phys Ther 85: 358–364 [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30: 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2003) Minireview: the AMP‐activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144: 5179–5183 [DOI] [PubMed] [Google Scholar]
- Hawley JA, Hargreaves M, Joyner MJ, Zierath JR (2014) Integrative biology of exercise. Cell 159: 738–749 [DOI] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher‐Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ et al (2015) Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise‐regulated kinases and AMPK substrates. Cell Metab 22: 922–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NJ (2017) Omics and exercise: global approaches for mapping exercise biological networks. Cold Spring Harb Perspect Med 7: a029884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325: 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post‐translational modifications in man and mouse. Nucleic Acids Res 40: D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivarsson N, Mattsson CM, Cheng AJ, Bruton JD, Ekblom B, Lanner JT, Westerblad H (2019) SR Ca2+ leak in skeletal muscle fibers acts as an intracellular signal to increase fatigue resistance. J Gen Physiol 151: 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TE, Rose AJ, Hellsten Y, Wojtaszewski JFP, Richter EA (2007) Caffeine‐induced Ca2+ release increases AMPK‐dependent glucose uptake in rodent soleus muscle. Am J Physiol‐Endocrinol Metab 293: E286–E292 [DOI] [PubMed] [Google Scholar]
- Jha A, Ahuja M, Maléth J, Moreno CM, Yuan JP, Kim MS, Muallem S (2013) The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J Cell Biol 202: 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Heller B, Tagliabracci VS, Zhai L, Irimia JM, DePaoli‐Roach AA, Wells CD, Skurat AV, Roach PJ (2010) Starch binding domain‐containing protein 1/genethonin 1 is a novel participant in glycogen metabolism. J Biol Chem 285: 34960–34971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Wells CD, Roach PJ (2011) Starch‐binding domain‐containing protein 1 (Stbd1) and glycogen metabolism: identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem Biophys Res Commun 413: 420–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjøbsted R, Hingst JR, Fentz J, Foretz M, Sanz M‐N, Pehmøller C, Shum M, Marette A, Mounier R, Treebak JT et al (2018) AMPK in skeletal muscle function and metabolism. FASEB J 32: 1741–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinert M, Parker BL, Fritzen AM, Knudsen JR, Jensen TE, Kjøbsted R, Sylow L, Ruegg M, James DE, Richter EA (2017) Mammalian target of rapamycin complex 2 regulates muscle glucose uptake during exercise in mice. J Physiol 595: 4845–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Huo J, Bround MJ, Boyer JG, Schwanekamp JA, Ghazal N, Maxwell JT, Jang YC, Khuchua Z, Shi K et al (2018) The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight 3: 121689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb GD, Junankar PR, Stephenson DG (1995) Raised intracellular [Ca2+] abolishes excitation‐contraction coupling in skeletal muscle fibres of rat and toad. J Physiol 489(Pt 2): 349–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi F, Marzetti E, Martone AM, Bernabei R, Onder G (2014) Exercise as a remedy for sarcopenia. Curr Opin Clin Nutr Metab Care 17: 25–31 [DOI] [PubMed] [Google Scholar]
- Landry CR, Levy ED, Michnick SW (2009) Weak functional constraints on phosphoproteomes. Trends Genet 25: 193–197 [DOI] [PubMed] [Google Scholar]
- Lang F, Eylenstein A, Shumilina E (2012) Regulation of Orai1/STIM1 by the kinases SGK1 and AMPK. Cell Calcium 52: 347–354 [DOI] [PubMed] [Google Scholar]
- Lantier L, Fentz J, Mounier R, Leclerc J, Treebak JT, Pehmøller C, Sanz N, Sakakibara I, Saint‐Amand E, Rimbaud S et al (2014) AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J 28: 3211–3224 [DOI] [PubMed] [Google Scholar]
- Launikonis BS, Murphy RM, Edwards JN (2010) Toward the roles of store‐operated Ca2+ entry in skeletal muscle. Pflugers Arch 460: 813–823 [DOI] [PubMed] [Google Scholar]
- Leppik JA, Aughey RJ, Medved I, Fairweather I, Carey MF, McKenna MJ (2004) Prolonged exercise to fatigue in humans impairs skeletal muscle Na + ‐K + ‐ATPase activity, sarcoplasmic reticulum Ca2+ release, and Ca2+ uptake. J Appl Physiol 97: 1414–1423 [DOI] [PubMed] [Google Scholar]
- Levy J, Gavin JR III, Sowers JR (1994) Diabetes mellitus: a disease of abnormal cellular calcium metabolism? Am J Med 96: 260–273 [DOI] [PubMed] [Google Scholar]
- Lin F‐F, Elliott R, Colombero A, Gaida K, Kelley L, Moksa A, Ho S‐Y, Bykova E, Wong M, Rathanaswami P et al (2013) Generation and characterization of fully human monoclonal antibodies against human Orai1 for autoimmune disease. J Pharmacol Exp Ther 345: 225–238 [DOI] [PubMed] [Google Scholar]
- Liou J, Fivaz M, Inoue T, Meyer T (2007) Live‐cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA 104: 9301–9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigorta UM, Rodríguez JA, Gibson G, Navarro A (2018) Replicability and prediction: lessons and challenges from GWAS. Trends Genet 34: 504–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J, Peebles KA, Lepow T et al (2011) Targeting adenosine monophosphate‐activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain 7: 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer W, Herrmann‐Frank A, Lüttgau HC (1995) The role of Ca2+ ions in excitation‐contraction coupling of skeletal muscle fibres. Biochim Biophys Acta 1241: 59–116 [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT Jr, Valladares O, Bucan M, Birnbaum MJ (2001) A role for AMP‐activated protein kinase in contraction‐ and hypoxia‐regulated glucose transport in skeletal muscle. Mol Cell 7: 1085–1094 [DOI] [PubMed] [Google Scholar]
- Muik M, Fahrner M, Derler I, Schindl R, Bergsmann J, Frischauf I, Groschner K, Romanin C (2009) A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J Biol Chem 284: 8421–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik M, Fahrner M, Schindl R, Stathopulos P, Frischauf I, Derler I, Plenk P, Lackner B, Groschner K, Ikura M et al (2011) STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J 30: 1678–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang Y‐X, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H et al (2008) AMPK and PPARδ agonists are exercise mimetics. Cell 134: 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath N, Khan M, Rattan R, Mangalam A, Makkar RS, de Meester C, Bertrand L, Singh I, Chen Y, Viollet B et al (2009) Loss of AMPK exacerbates experimental autoimmune encephalomyelitis disease severity. Biochem Biophys Res Commun 386: 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham EJ, Parker BL, Burykin T, James DE, Humphrey SJ (2019) Illuminating the dark phosphoproteome. Sci Signal 12: eaau8645 [DOI] [PubMed] [Google Scholar]
- Norris SM, Bombardier E, Smith IC, Vigna C, Tupling AR (2010) ATP consumption by sarcoplasmic reticulum Ca2+ pumps accounts for 50% of resting metabolic rate in mouse fast and slow twitch skeletal muscle. Am J Physiol Cell Physiol 298: C521–C529 [DOI] [PubMed] [Google Scholar]
- Nurbaeva MK, Schmid E, Szteyn K, Yang W, Viollet B, Shumilina E, Lang F (2012) Enhanced Ca2+ entry and Na+/Ca2+ exchanger activity in dendritic cells from AMP‐activated protein kinase‐deficient mice. FASEB J 26: 3049–3058 [DOI] [PubMed] [Google Scholar]
- Ochoa D, Jarnuczak AF, Gehre M, Soucheray M (2019) The functional landscape of the human phosphoproteome. BioRxiv https://www.biorxiv.org/content/10.1101/541656v1.abstract [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (1999) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 27: 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil TK, Duffy LR, Frey JW, Hornberger TA (2009) The role of phosphoinositide 3‐kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J Physiol 587: 3691–3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA et al (2011) AMP‐activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA 108: 16092–16097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages H, Aboyoun P, Gentleman R, DebRoy S (n.d.) Biostrings: string objects representing biological sequences, and matching algorithms. R package version 2.26.3
- Palmisano G, Lendal SE, Engholm‐Keller K, Leth‐Larsen R, Parker BL, Larsen MR (2010) Selective enrichment of sialic acid–containing glycopeptides using titanium dioxide chromatography with analysis by HILIC and mass spectrometry. Nat Protoc 5: 1974 [DOI] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136: 876–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkälä KH, Pöysti MM, Laakkonen M‐L, Tilvis RS, Savikko N, Kautiainen H, Strandberg TE (2013) Effects of the Finnish Alzheimer disease exercise trial (FINALEX): a randomized controlled trial. JAMA Intern Med 173: 894–901 [DOI] [PubMed] [Google Scholar]
- Potts GK, McNally RM, Blanco R, You J‐S, Hebert AS, Westphall MS, Coon JJ, Hornberger TA (2017) A map of the phosphoproteomic alterations that occur after a bout of maximal‐intensity contractions. J Physiol 595: 5209–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo‐Guisado E, Campbell DG, Deak M, Alvarez‐Barrientos A, Morrice NA, Alvarez IS, Alessi DR, Martín‐Romero FJ (2010) Phosphorylation of STIM1 at ERK1/2 target sites modulates store‐operated calcium entry. J Cell Sci 123: 3084–3093 [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2008) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; ISBN3‐900051‐07‐0. http://www.R-project.org [Google Scholar]
- Richter EA, Ruderman NB (2009) AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J 418: 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Ashot Kozak J, Wagner SL, Cahalan MD et al (2005) STIM1, an essential and conserved component of store‐operated Ca2+ channel function. J Cell Biol 169: 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ, Kiens B, Richter EA (2006) Ca2+–calmodulin‐dependent protein kinase expression and signalling in skeletal muscle during exercise. J Physiol 574 (Pt 3): 889–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Hirshman MF, Aschenbach WG, Goodyear LJ (2002) Contraction regulation of Akt in rat skeletal muscle. J Biol Chem 277: 11910–11917 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh MH, Baldock R, Barbiera G et al (2015) The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res 43: W589–W598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article 3 [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Li G‐Y, Plevin MJ, Ames JB, Ikura M (2006) Stored Ca2+ depletion‐induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF‐SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem 281: 35855–35862 [DOI] [PubMed] [Google Scholar]
- Stiber J, Hawkins A, Zhang Z‐S, Wang S, Burch J, Graham V, Ward CC, Seth M, Finch E, Malouf N et al (2008) STIM1 signalling controls store‐operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol 10: 688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styczynski MP, Jensen KL, Rigoutsos I, Stephanopoulos G (2008) BLOSUM62 miscalculations improve search performance. Nat Biotechnol 26: 274–275 [DOI] [PubMed] [Google Scholar]
- Sundivakkam PC, Natarajan V, Malik AB, Tiruppathi C (2013) Store‐operated Ca2+ entry (SOCE) induced by protease‐activated receptor‐1 mediates STIM1 protein phosphorylation to inhibit SOCE in endothelial cells through AMP‐activated protein kinase and p38β mitogen‐activated protein kinase. J Biol Chem 288: 17030–17041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylow L, Jensen TE, Kleinert M, Mouatt JR, Maarbjerg SJ, Jeppesen J, Prats C, Chiu TT, Boguslavsky S, Klip A et al (2013) Rac1 is a novel regulator of contraction‐stimulated glucose uptake in skeletal muscle. Diabetes 62: 1139–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylow L, Møller LLV, Kleinert M, D'Hulst G, De Groote E, Schjerling P, Steinberg GR, Jensen TE, Richter EA (2017) Rac1 and AMPK account for the majority of muscle glucose uptake stimulated by ex vivo contraction but not in vivo exercise. Diabetes 66: 1548–1559 [DOI] [PubMed] [Google Scholar]
- Thingholm TE, Palmisano G, Kjeldsen F, Larsen MR (2010) Undesirable charge‐enhancement of isobaric tagged phosphopeptides leads to reduced identification efficiency. J Proteome Res 9: 4045–4052 [DOI] [PubMed] [Google Scholar]
- Thompson JL, Zhao Y, Stathopulos PB, Grossfield A, Shuttleworth TJ (2018) Phosphorylation‐mediated structural changes within the SOAR domain of STIM1 enable specific activation of distinct Orai channels. J Biol Chem 293: 3145–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC et al (2016) Metabolism. AMP‐activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351: 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandebrouck A, Ducret T, Basset O, Sebille S, Raymond G, Ruegg U, Gailly P, Cognard C, Constantin B (2006) Regulation of store‐operated calcium entries and mitochondrial uptake by minidystrophin expression in cultured myotubes. FASEB J 20: 136–138 [DOI] [PubMed] [Google Scholar]
- Vasques ER, Cunha JEM, Coelho AMM, Sampietre SN, Patzina RA, Abdo EE, Nader HB, Tersariol ILS, Lima MA, Godoy CMG et al (2016) Trisulfate disaccharide decreases calcium overload and protects liver injury secondary to liver ischemia/reperfusion. PLoS One 11: e0149630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CW, Spangenburg EE, Diss LM, Williams JH (1998) Effects of varied fatigue protocols on sarcoplasmic reticulum calcium uptake and release rates. Am J Physiol 275: R99–R104 [DOI] [PubMed] [Google Scholar]
- Warren SC, Margineanu A, Alibhai D, Kelly DJ, Talbot C, Alexandrov Y, Munro I, Katan M, Dunsby C, French PMW (2013) Rapid global fitting of large fluorescence lifetime imaging microscopy datasets. PLoS One 8: e70687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei‐Lapierre L, Carrell EM, Boncompagni S, Protasi F, Dirksen RT (2013) Orai1‐dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat Commun 4: 2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham M, Parker BL, Friedrichsen M, Hingst JR, Hjorth M, Hughes WE, Egan CL, Cron L, Watt KI, Kuchel RP et al (2018) Extracellular vesicles provide a means for tissue crosstalk during exercise. Cell Metab 27: 237–251.e4 [DOI] [PubMed] [Google Scholar]
- Winder WW, Hardie DG (1996) Inactivation of acetyl‐CoA carboxylase and activation of AMP‐activated protein kinase in muscle during exercise. Am J Physiol 270: E299–E304 [DOI] [PubMed] [Google Scholar]
- Yazbeck P, Tauseef M, Kruse K, Amin M‐R, Sheikh R, Feske S, Komarova Y, Mehta D (2017) STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca2+ Entry. Sci Rep 7: 42758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Srinivasan P, Razavi S, Seymour S, Meraner P, Gudlur A, Stathopulos PB, Ikura M, Rao A, Hogan PG (2013) Initial activation of STIM1, the regulator of store‐operated calcium entry. Nat Struct Mol Biol 20: 973–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Table EV2
Table EV3
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD010452 (http://www.ebi.ac.uk/pride).