

Abstract
Repetitive DNA sequences are often associated with chromosomal rearrangements in cancers. Conventionally, single‐strand annealing (SSA) is thought to mediate homology‐directed repair of double‐strand breaks (DSBs) between two repeats, causing repeat‐mediated deletion (RMD). In this report, we demonstrate that break‐induced replication (BIR) is used predominantly over SSA in mammalian cells for mediating RMD, especially when repeats are far apart. We show that SSA becomes inefficient in mammalian cells when the distance between the DSBs and the repeats is increased to the 1–2 kb range, while BIR‐mediated RMD (BIR/RMD) can act over a long distance (e.g., ~ 100–200 kb) when the DSB is close to one repeat. Importantly, oncogene expression potentiates BIR/RMD but not SSA, and BIR/RMD is used more frequently at single‐ended DSBs formed at collapsed replication forks than at double‐ended DSBs. In contrast to short‐range SSA, H2AX is required for long‐range BIR/RMD, and sequence divergence strongly suppresses BIR/RMD in a manner partially dependent on MSH2. Our finding that BIR/RMD has a more important role than SSA in mammalian cells has a significant impact on the understanding of repeat‐mediated rearrangements associated with oncogenesis.
Keywords: break‐induced replication, H2AX, oncogenic stress, repeat‐mediated deletion, single‐strand annealing
Subject Categories: DNA Replication, Repair & Recombination
Mammalian cells preferentially use BIR rather than single‐strand annealing for long‐distance recombination between repetitive DNA sequences associated with oncogenic chromosomal rearrangements.
Introduction
Repetitive DNA sequences are abundant and comprise approximately half of the human genome (Lander et al, 2001). Alu sequences, which are short interspersed elements about 300 bp in size, are present at more than 1 million copies (Batzer & Deininger, 2002; Belancio et al, 2010). These repetitive sequences are a major source of chromosomal rearrangements, thereby contributing to genome instability (Deininger et al, 2003).
Chromosomal structural changes can arise when DNA breaks, mainly double‐strand breaks (DSBs), are not correctly repaired. Nonhomologous end joining (NHEJ), which re‐ligates the broken ends without a template, and homologous recombination (HR), which uses a homologous donor for repair, are two major DSB repair pathways (Paques & Haber, 1999; Jasin & Rothstein, 2013; Chang et al, 2017). There are several different HR pathways, including gene conversion (GC), break‐induced replication (BIR), and single‐strand annealing (SSA; Anand et al, 2013; Mehta & Haber, 2014; Bhargava et al, 2016). All pathways are initiated by extensive 5′–3′ resection of DSB ends, generating long 3′ single‐strand DNA (ssDNA) for recombination repair. When both DSB ends share homology with the donor sequences, the break is usually repaired by GC, which can be mediated by synthesis‐dependent strand annealing (SDSA) or by formation of a double‐Holliday junction (dHJ; Jasin & Rothstein, 2013; Mehta & Haber, 2014). When homology with a donor is present for only one end of a DSB, BIR is used for repair. This often occurs at collapsed replication forks and eroding telomeres where single‐ended DSBs are generated (Davis & Symington, 2004; McEachern & Haber, 2006; Anand et al, 2013; Malkova & Ira, 2013). A major BIR mechanism is RAD51‐dependent, requiring invasion of the RAD51 filament into the homologous template in the same manner as in GC. However, BIR is different from GC in that BIR replication can extend for a long distance and requires the Pol32 subunit (POLD3 in mammalian cells) of Polδ (Lydeard et al, 2007; Costantino et al, 2014). Recent studies have also revealed an important role of RAD52 in BIR and BIR‐related mechanisms in mammalian cells (Bhowmick et al, 2016; Sotiriou et al, 2016), although its function is dispensable for GC (Feng et al, 2011; Wang et al, 2018a). When a DSB is flanked by direct repeats, SSA is used for repair, resulting in the deletion of one repeat as well as the intervening sequence (Bhargava et al, 2016). SSA also requires end resection to generate ssDNA within the repeats, which enables the annealing of flanking repeats. SSA is suppressed by RAD51 but dependent on RAD52 (Rudin & Haber, 1988; Fishman‐Lobell et al, 1992; Sugawara & Haber, 1992; Stark et al, 2004).
Repeat‐mediated deletions (RMDs) have been described as contributing to the etiology of inherited diseases and cancers (Kolomietz et al, 2002; Pavlicek et al, 2004; Belancio et al, 2010). The BRCA1 gene is highly enriched in Alu elements, and recombination between Alu elements has been implicated in the generation of pathogenic BRCA1 mutants (Petrij‐Bosch et al, 1997; Puget et al, 1997; Pavlicek et al, 2004). In the conventional model, SSA is thought to mediate RMDs. However, we found that SSA becomes inefficient when the repeats are separated at a range of ~ 1–2 kb, while BIR can be efficiently used to mediate RMDs when the repeats are located over a long distance, even when they are ~ 100–200 kb apart. Our study suggests that BIR plays a more significant role than SSA in long‐range RMD in mammalian cells, which is different from the prevalent thought that SSA is the dominant way to mediate recombination of two identical repeats, resulting in RMD. Our findings also provide new evidence to support the notion that replicative mechanisms are attributed to repeat‐mediated rearrangement.
Results
BIR, not SSA, is used for recombination between two repeats when the DSB is close to one repeat
SSA is believed to be a major pathway mediating recombination between two repeats to generate RMD. We established a new EGFP‐based SSA reporter (EGFP‐SSA) in which the 5′EGFP and 3′EGFP fragments share 315 bp homology (repeat) and are separated by a 2,035 bp DNA sequence (Fig 1A). We obtained clones carrying a single copy of the EGFP‐SSA reporter stably integrated into the genome. Cleavage by I‐SceI 9 bp away from the 3′ repeat readily induced EGFP signals in multiple clones carrying an integrated single copy of the EGFP‐SSA reporter (Appendix Fig S1A). PCR analysis of sorted green cells confirmed the presence of the SSA or RMD product with the deletion of one repeat along with the intervening sequence (Appendix Fig S1B).
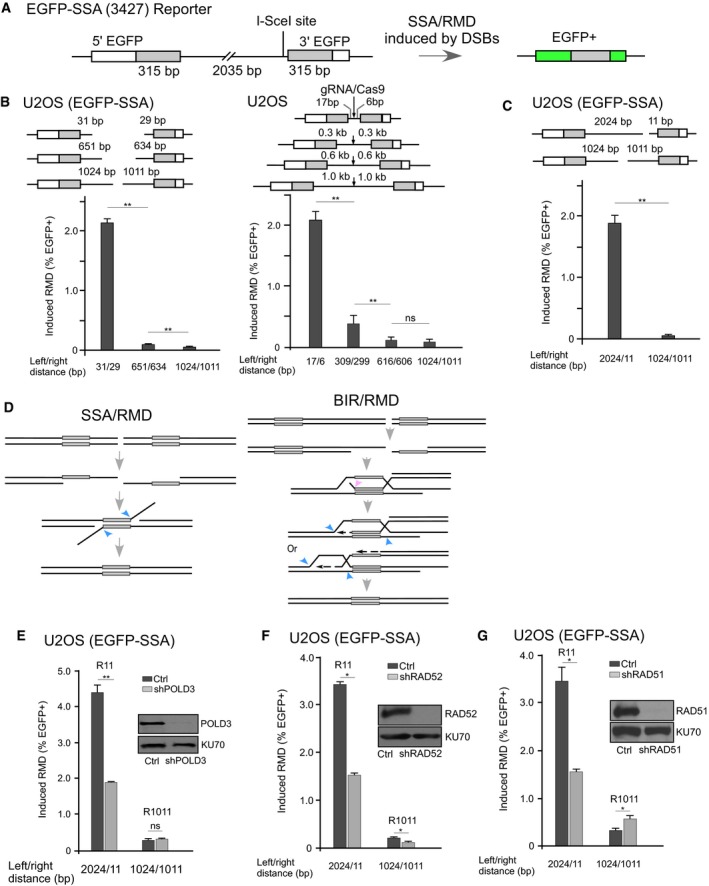
Figure 1. BIR is involved in RMD when a DSB is close to one repeat.
-
ASchematic drawing of the EGFP‐based SSA reporter, EGFP‐SSA. The EGFP coding sequence is split into two fragments, which share a 315 bp overlapping homology (repeat) and are separated by a 2,035 bp DNA sequence. An I‐SceI cleavage sequence is inserted in the intervening sequence, 9 bp away from the 3′ repeat. The EGFP open reading frame is restored after DSB‐induced RMD of the two EGFP fragments.
-
BRMD assays performed after DSBs have been generated by sgRNAs/Cas9. The distance between the sgRNA/Cas9 cleavage sites and the repeat sequences is indicated. U2OS cells carrying a single copy of the EGFP‐SSA reporter (3427‐17) were transfected with sgRNA/Cas9 plasmids, and the percentage of GFP‐positive cells was determined (left). U2OS cells transfected with the indicated reporters EGFP‐SSA‐17/6 bp, EGFP‐SSA‐0.3 kb, EGFP‐SSA‐0.6 kb, and EGFP‐SSA‐1.0 kb were pooled as a population after selection of the drug marker on the reporters, followed by sgRNA/Cas9 cleavage (right).
-
CRMD is strongly induced when a DSB is generated close to one repeat. The U2OS (EGFP‐SSA‐3427‐17) cell line carrying a single copy of the reporter was transfected with sgRNA/Cas9 plasmids as indicated, and the percentage of GFP‐positive cells was determined.
-
DModels of RMD mediated by SSA (RMD/SSA) or BIR (RMD/BIR). When a DSB is created in the middle of two repeats, the DNA ends of both sides are resected, generating 3′ ssDNA overhangs, until the repeat sequences on opposite sides are both exposed. Then, ssDNA of the repeat sequences anneals, resulting in a product with internal sequences deleted. When a DSB is generated close to one repeat, ssDNA formed in one repeat first can invade the other repeat still in duplex using BIR, which can also give rise to a deletion product identical to the SSA product after cleavage of D‐loop. Pink arrow: putative structure‐specific endonucleases to cleave nonhomologous tails; blue arrow: putative structure‐specific endonucleases to cleave the D‐loop.
-
E–GDetermination of RMD frequency in U2OS (EGFP‐SSA‐3427‐17) cells when POLD3 (E), RAD52 (F), or RAD51 (G) is depleted by lentiviral expression of shRNA. Cells infected with a lentiviral empty vector were used as control. The knockdown level of the indicated proteins was shown by Western blot, using KU70 as the loading control.
To study the effect of the distance of DSBs from the repeats, we used Cas9 along with single or double single‐guide RNAs (sgRNAs) to generate DSBs in the intervening sequence. In support of the requirement of end resection to reach the repeats, SSA/RMD frequency is much higher when the DSBs are ~ 30 bp away compared to when they are ~ 0.6 kb away from the two repeats, with even lower frequency when the distance is increased to ~ 1 kb (Fig 1B, left). We also generated EGFP‐SSA reporters containing the intervening sequences of varying lengths between the repeats and with the same gRNA/Cas9 site situated in the middle. Cleavage by gRNA/Cas9 would produce nonhomologous tails of 17 and 6 bp on the first reporter on either side (EGFP‐SSA‐17/6 bp), and 0.3, 0.6 kb or 1 kb to both the left and right repeats on the other reporters (EGFP‐SSA‐0.3 kb, EGFP‐SSA‐0.6 kb, and EGFP‐SSA‐1 kb). To avoid the influence of the genomic loci that the reporters are integrated in on repair efficiency, we pooled cell populations after transfection of the reporters and examined repair frequency. Consistently, SSA/RMD is significantly reduced when the distance between the repeats is increased (Fig 1B, right). These data suggest that end resection is a factor limiting SSA; SSA becomes inefficient when a DSB is at 1 kb range away from the repeats. The large distance between the repeats would thus be a major restriction for use of SSA in mammalian cells.
Interestingly, when we used sgRNA R11 to generate a DSB 11 bp away from the 3′ repeat (2,024 bp from the 5′ repeat and 11 bp from the 3′ repeat: 2024/11), we found that RMD is much higher than when using sgRNA R1011 to generate a DSB in the middle of the intervening sequence (1,024 bp from the left repeat and 1,011 bp from the right repeat: 1024/1011; Fig 1C). Since end resection of ~ 2 kb would take longer than for ~ 1 kb, it is not expected that recombination would occur more efficiently when DSBs are generated at R11 (2024/11) than at R1011 (1024/1011). This is also different from observations in yeast that the kinetics of SSA product formation are governed largely by the distance of the DSB to the further repeat (Fishman‐Lobell & Haber, 1992; Sugawara & Haber, 1992; Vaze et al, 2002). However, this is in line with the observation in mouse embryonic stem (mES) cells that RMD frequency in not changed proportionally with the distance increase of the break to the further repeat when the distance is more than 3 kb (Mendez‐Dorantes et al, 2018).
When a DSB is generated closer to one repeat than the other, in principle, ssDNA should form in that repeat first and one possibility is that ssDNA formed in the first repeat could strand‐invade the other repeat using the BIR mechanism (Fig 1D, right: BIR/RMD). After cleavage of the D‐loop, deletion products identical to those formed by SSA/RMD (Fig 1D, left) could be generated. To support this model, silencing POLD3 expression by shRNA significantly reduces the frequency of recombination between the two repeats upon cleavage at R11 (2024/11) when one end of the DSB is only 11 bp away from the right repeat, but not at R1011 (1024/1011) when both DSB ends are ~ 1 kb away from the repeats (Fig 1E). Inactivation of RAD52 by shRNA reduces recombination in both cases (Fig 1F). Since BIR depends on both POLD3 and RAD52 but SSA only requires RAD52 (Stark et al, 2004; Costantino et al, 2014), these results suggest that BIR is used when a DSB is generated in the intervening sequence close to one repeat (e.g., 11 bp away), whereas SSA is used when a DSB is located far away from both repeats. When BIR is used to mediate recombination between two repeats, resulting in a deletion, we called it BIR‐mediated RMD (BIR/RMD).
To further test the model that BIR/RMD is used when cleavage is generated close to one repeat, we tested the dependence of RMD on RAD51 by expressing RAD51 shRNA. We showed that inactivation of RAD51 results in increased RMD when cleavage is made at R1011, but causes significant reduction of RMD when a DSB is generated at R11 (Fig 1G). This is consistent with the role of RAD51 in suppressing SSA (Ivanov et al, 1996; Stark et al, 2004) and suggests that BIR/RMD is RAD51‐dependent, which is comparable to the observation in bacteria and in yeast that the major BIR pathway is RecA/RAD51‐dependent (Formosa & Alberts, 1986; Asai et al, 1993; Davis & Symington, 2004; Malkova et al, 2005). In addition to POLD3, BIR/RMD induced by R11 cleavage is also dependent on CDC45, PCNA, and RFC (Appendix Fig S2), further supporting the involvement of a replication mechanism in BIR/RMD.
The usage of BIR/RMD or SSA/RMD is governed by the distance between the DSB and the repeats
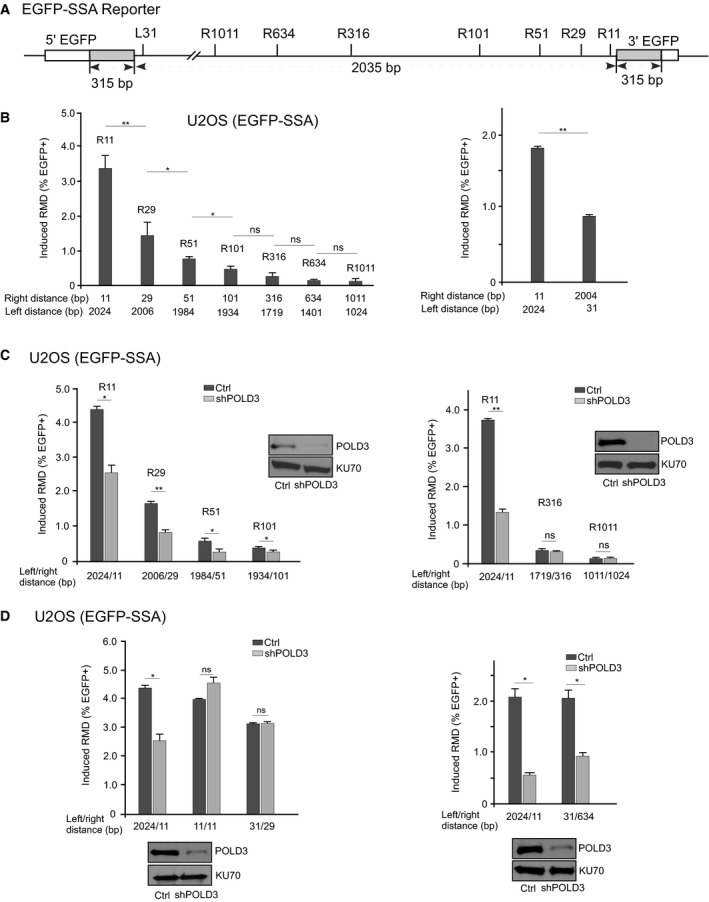
To understand the mechanism governing the usage of BIR and SSA for RMD, we created DSBs at different positions along the intervening sequence of repeats using a series of sgRNAs (Fig 2A). We demonstrated that RMD is efficiently induced when DSBs are generated at a position 29 bp away from the 3′ repeat (R29: 2006/29) and 31 bp away from the left repeat (L31: 31/2004), although the frequency is reduced by half compared to R11 (Fig 2B). RMD decreases more when DSBs are generated further away from the 3′ repeat, with an about 10‐fold drop at R316 (1719/316), and a 20‐fold drop at R634 (1401/634) and at R1011 (1024/1011) when compared to R11 (2024/11). Like at R11 (Fig 1E), depletion of POLD3 by shRNA also significantly reduces RMD when DSBs are generated at R29 and R51 (Fig 2C). However, the reduction of RMD upon depletion of POLD3 becomes minor or undetectable when DSBs are generated at R101 or R316 and R1011. These data suggest that BIR is likely involved in RMD when the DSB is close to one repeat (≤ ~100 bp). When the distance is increased to ~ 100 bp or more, BIR is not actively engaged and SSA is used instead, but with very low overall frequency.
Figure 2. Effect of the distance between the DSBs and the repeats on RMD frequency.
-
AA diagram indicating the position of DSBs generated by sgRNAs/Cas9 on the EGFP‐SSA reporter.
-
BRMD efficiency is decreased when the distance from a DSB to one repeat is increased. RMD assays were performed in U2OS (EGFP‐SSA‐3427‐17) cells using the indicated sgRNAs/Cas9. The distance between the sgRNA/Cas9 cleavage sites and the repeat sequences is shown.
-
C, DKnockdown of POLD3 impairs RMD when a DSB is close to one repeat. RMD frequency was determined in U2OS (EGFP‐SSA‐3427‐17) cells using the indicated sgRNAs/Cas9 with or without depletion of POLD3 by lentivirus‐expressed shRNA. Cells infected with a lentiviral empty vector were used as control. The distance between the sgRNA/Cas9 cleavage sites and the repeat sequences is indicated. The knockdown level of POLD3 was shown by Western blot, and KU70 was used as the loading control.
We propose that when a DSB is near one repeat, BIR is used because end resection leads to ssDNA formation first on one repeat while the other repeat is still in double‐strand form. Indeed, when two small tails, 11 bp on both sides (11/11) or 31 bp on left and 29 bp on right (31/29) are generated, RMD does not depend on POLD3 (Fig 2D, left), suggesting that SSA is used. RAD51 inhibits RMD with cleavage at both 11/11 and 31/29 sites, which is consistent with the usage of SSA (Appendix Fig S3). However, when the left tail is 31 bp but the right tail is 634 bp (31/634), RMD becomes POLD3‐dependent (Fig 2D, right). This supports the model that BIR is used when one repeat becomes ssDNA first and is used to invade the other repeat. The efficiency of RMD and the use of SSA/RMD or BIR/RMD is governed by the distance from the DSBs to the repeats.
Collectively, these studies show that BIR/RMD can efficiently launch when the DSB is close to one repeat (e.g., 2024/11), but to achieve similar efficiency of RMD by SSA, the DSB needs to be close to both repeats (e.g., 11/11 and 31/29, Fig 2D, left). Since the SSA frequency of 651/634 and 1024/1011 is very low (Fig 1B), we do not expect SSA to play a significant role in mediating RMD of the repeats at a 1–2 kb distance when one DSB is generated in the intervening sequence.
Oncogene expression and replication fork collapse stimulate BIR/RMD but not SSA/RMD
BIR is proposed to be used when DSBs are single‐ended, which often occurs when replication forks are collapsed, and thus, replication stress may potentiate BIR activity. Since oncogene expression often induces replication stress and fork collapse (Bartkova et al, 2006; Di Micco et al, 2006), we tested whether oncogene expression would modulate usage of BIR/RMD and SSA/RMD. We generated U2OS (EGFP‐SSA) cell lines carrying doxycycline‐inducible cyclin E. We showed that cyclin E overexpression leads to increased RMD when BIR/RMD is induced by DSBs generated at R29, but it has no effect on SSA after cleavage at R1011 (Fig 3A). This suggests that cyclin E overexpression promotes BIR/RMD but not SSA/RMD.
Figure 3. BIR/RMD is stimulated by oncogene overexpression and replication fork collapse.
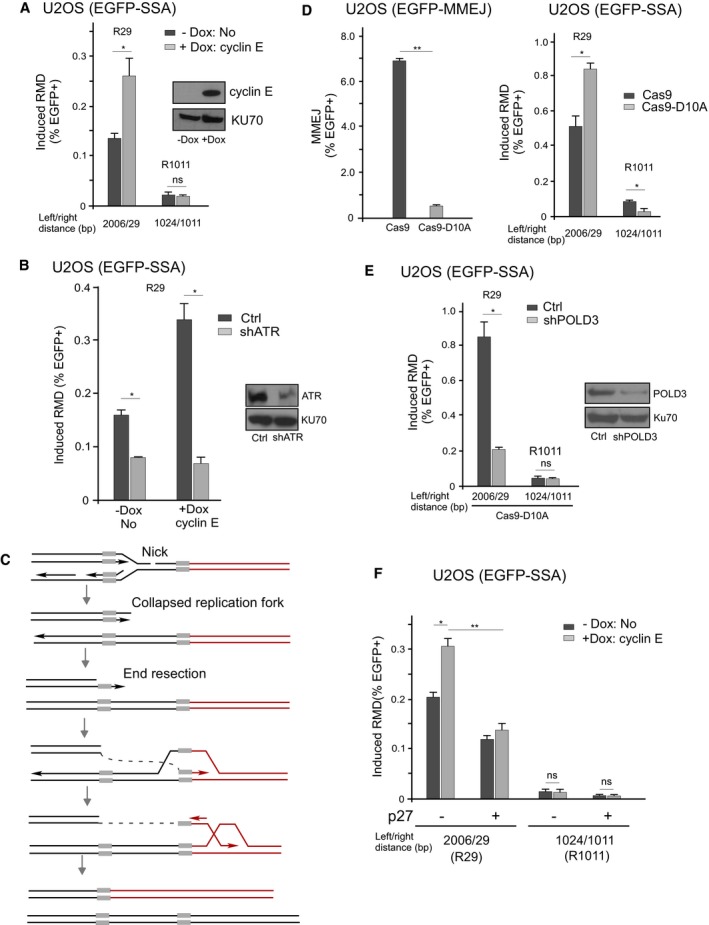
- Cyclin E overexpression results in increased RMD when the DSB is close to one repeat. The U2OS (EGFP‐SSA‐3427‐3) cell line carrying Tet‐on‐cyclin E plasmid was used, and RMD frequency was determined with or without induction of cyclin E expression by doxycycline (DOX, 0.5 μg/ml), which was added 24 h before transfection of the indicated sgRNAs/Cas9. Cyclin E expression levels were validated by Western blot, using KU70 as the loading control.
- ATR is required for BIR/RMD upon cyclin E overexpression. RMD frequency was determined in U2OS (EGFP‐SSA‐3427‐3) cells infected with lentivirus‐expressed ATR shRNA or the control vector (Ctrl) with and without induction of cyclin E expression by DOX (0.5 μg/ml) after R29 sgRNA/Cas9 cleavage. ATR knockdown was shown by Western blot with KU70 as the loading control.
- A diagram showing the BIR/RMD model when a single‐ended DSB is generated after replication fork collapse.
- RMD is stimulated when Cas9‐D10A is used to create a nick near the repeat. The repair frequencies of the U2OS (MMEJ) reporter cell line (left) and U2OS (EGFP‐SSA‐3427‐17) cell line (right) were determined after expression of the indicated sgRNAs/Cas9 and sgRNAs/Cas9‐D10A. The distance between the sgRNA cleavage sites and the left and right repeat sequences is indicated (right bottom).
- RMD induced by Cas9‐D10A is dependent on POLD3. U2OS (EGFP‐SSA‐3427‐17) cells were infected with lentiviruses expressing shRNA for POLD3 or vector control, and RMD was determined after cleavage by the indicated sgRNAs/Cas9‐D10A. The distance between sgRNA/Cas9‐D10A cleavage sites and the left and right repeat sequences is indicated at the bottom. The knockdown level of POLD3 was shown by Western blot, and KU70 was used as the loading control.
- p27 suppresses BIR/RMD and SSA/RMD. U2OS (EGFP‐SSA‐3427‐3) cells carrying the Tet‐on‐cyclin E allele were infected with and without p27 lentiviral viruses. RMD was determined before and after induction of cyclin E overexpression by DOX (0.5 μg/ml) added 24 h before transfection with the indicated sgRNAs/Cas9. Cyclin E expression levels and cell cycle profiles are shown in Appendix Fig S6.
To understand how cyclin E overexpression leads to increased BIR/RMD when DSBs have formed, we asked whether ATR is involved in this process, since oncogene expression induces replication stress and ATR activation (Bartkova et al, 2006; Di Micco et al, 2006). When ATR is depleted by shRNA, BIR/RMD induced by R29 is reduced even before induction of cyclin E expression (Fig 3B, −Dox), suggesting that ATR is needed for BIR/RMD. This is likely due to the involvement of ATR in end resection to promote BIR/RMD in a manner similar to HR (Jazayeri et al, 2006). However, BIR/RMD stimulated by cyclin E overexpression exhibits much stronger dependence on ATR than that without cyclin E expression (Fig 3B). Since under our assay conditions, DSBs are generated by Cas9, and their levels should be comparable in the presence or absence of cyclin E overexpression, we propose that besides promoting end resection, ATR plays an additional role in stimulating BIR/RMD activity upon oncogenic stress induced by cyclin E overexpression.
We then asked whether the use of BIR/RMD and SSA/RMD is modulated when single‐ended DSBs are generated specifically on replication forks compared to how they are used at double‐ended DSBs. We took advantage of the CRISPR/Cas9 system and used Cas9 nickase (Cas9‐D10A) to generate nicks on DNA (Jinek et al, 2012). When replication encounters nicks, forks would collapse, leading to single‐ended DSB formation (Fig 3C). As a control, we introduced DSBs and nicks to the previously described EGFP‐MMEJ reporter (Appendix Fig S4; Truong et al, 2013) using Cas9 and Cas9‐D10A, respectively. MMEJ frequency is much reduced when nicks are generated by Cas9‐D10A compared to by Cas9 (Fig 3D, left), and this is consistent with the idea that nicks are often efficiently repaired before conversion to DSBs for MMEJ to repair. However, when nicks are generated at R29 in the EGFP‐SSA reporter, BIR/RMD frequency is even higher than when double‐ended DSBs are created by Cas9 (Fig 3D, right and Appendix Fig S5). Cas9‐D10A/R29‐induced RMD depends on POLD3, confirming the use of BIR (Fig 3E). By contrast, RMD at R1011 is reduced when Cas9‐D10A is used compared to Cas9 (Fig 3D, right), and Cas9‐D10A/R1011‐induced RMD does not depend on POLD3 (Fig 3E). These data suggest that BIR/RMD is more efficiently used at single‐ended DSBs generated at collapsed replication forks than at double‐ended DSBs, while SSA is not stimulated upon fork collapse.
In G0/G1 cells, homology‐directed recombination is suppressed due to a lack of end resection. Indeed, when we expressed p27 to arrest cells at G0/G1 (Toyoshima & Hunter, 1994), both BIR/RMD (induced by R29) and SSA/RMD (induced by R1011) are reduced compared to those in cycling cells (Fig 3F and Appendix Fig S6). We then asked whether cyclin E overexpression could override the restriction of BIR/RMD in quiescent cells. While induction of cyclin E in cycling cells induces BIR/RMD, the same effect was not observed in cells with overexpression of p27 (Fig 3F and Appendix Fig S6), suggesting that cyclin E promotes BIR/RMD only when cells are actively proliferating.
BIR can be used to mediate RMD when repeats are separated by a long distance
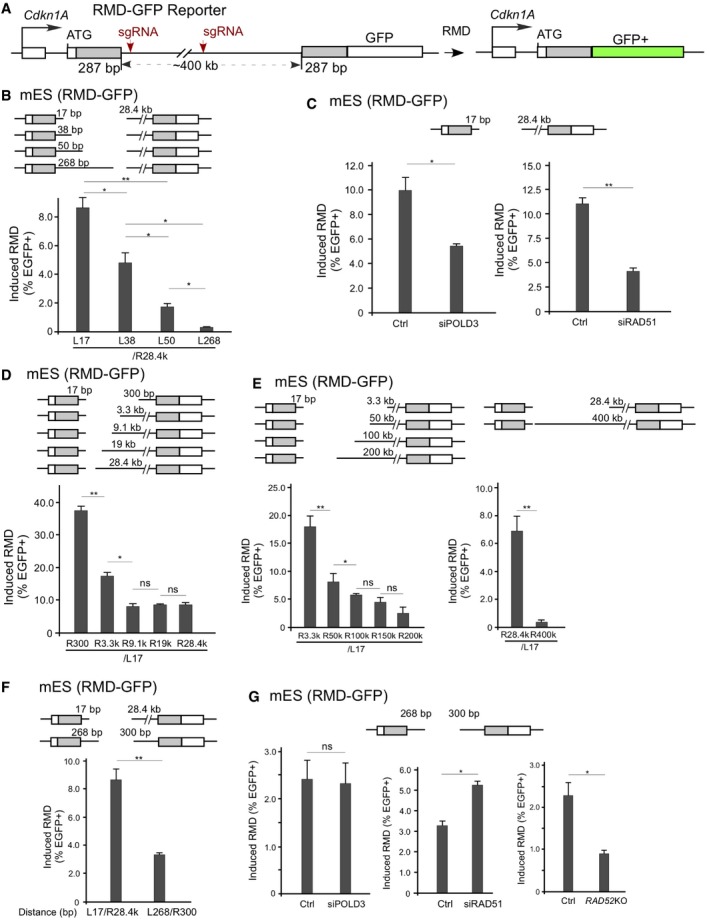
In the human genome, Alu‐associated RMD can occur over a substantial distance (Kolomietz et al, 2002; Pavlicek et al, 2004; Belancio et al, 2010). To study the mechanism involved in long‐range RMD, we used the RMD‐GFP reporter established in Dr. Stark's laboratory (Mendez‐Dorantes et al, 2018), which contains two 287 bp repeats separated by 0.4 Mb on mouse chromosome 17 (Fig 4A). RMD between these two repeats would generate a Cdkn1A‐GFP fusion gene, producing green cells. One DSB 268 bp downstream of the 5′ repeat (L268) with a second DSB 28.4 kb upstream of the 3′ repeat (R28.4k) induces low but detectable RMD in mES cells [(Mendez‐Dorantes et al, 2018) and Fig 4B]. However, when we reduced the distance of the DSB downstream of the 5′ repeat by cleaving 17, 38 bp, or 50 bp away from the 3′ repeat (L17, L38, L50), with fixed cleavage at 28.4 kb upstream of the 3′ repeat (R28.4k), RMD was strongly induced, with a more than 30‐fold increase when comparing RMD of L17/R28.4k to that of L268/R28.4k (Fig 4B). A single cleavage in the intervening sequence induces very minimal RMD (Appendix Fig S7). We further showed that RMD by L17/R28.4k requires POLD3 and RAD51 (Fig 4C and Appendix Fig S8A), suggesting the involvement of RAD51‐dependent BIR. These data suggest that when a DSB is generated close to one repeat (e.g., ~ 20 bp), RMD can be strongly induced by using BIR even when the other repeat is situated far away (e.g., 28.4 kb).
Figure 4. Long‐range RMD is mediated by BIR.
-
ASchematic drawing of the long‐range RMD‐GFP reporter in mES cells. Two 287 bp repeats are located separately in the Cdkn1A and Pim1 gene loci and are 0.4 Mbp apart. GFP+ cells would be generated after RMD.
-
BImpact of the 5′ DSB/repeat distance on RMD frequency when the 3′ DSB/repeat distance is fixed. RMD assays were performed using mES cells carrying the RMD‐GFP reporter, and RMD frequency was determined by the number of GFP+ cells normalized to transfection efficiency. The distance between the sgRNA targeting sites and the repeats is indicated.
-
CLong‐range RMD is dependent on POLD3 when one DSB is close to a repeat. mES cells carrying the RMD‐GFP reporter were transfected with non‐targeting siRNA (Ctrl) or a pool of POLD3 siRNAs (siPOLD3) 24 h before for the first time, and with transfection of sgRNAs/Cas9 the second time. The graphs show the frequency of GFP+ cells normalized to the transfection efficiency. The distance between the sgRNA targeting sites and the repeats is indicated. Depletion of POLD3 and RAD51 by transient transfection with siRNA was determined by qPCR, as shown in Appendix Fig S7A. Cells transfected with non‐targeting control siRNA were used as control.
-
D, EImpact of the 3′ DSB/repeat distance on RMD frequency when the 5′ DSB/repeat distance is fixed. RMD assays were performed using mES cells carrying the RMD‐GFP reporter, and RMD frequency was determined by the number of GFP+ cells normalized to transfection efficiency. The distance between the sgRNA targeting sites and the repeats is indicated.
-
FRMD frequency of two far‐apart repeats that are a short distance from the DSB. RMD was determined in mES cells carrying the RMD‐GFP reporter using the indicated sgRNAs/Cas9, with the frequency of GFP+ cells normalized to transfection efficiency. The distance between the sgRNA targeting sites and the repeats is indicated.
-
GThe roles of POLD3, RAD51, and RAD52 in RMD of two far‐apart repeats that are a short distance from the DSB. RMD was determined in mES cells carrying the RMD‐GFP reporter using the indicated sgRNAs/Cas9 after POLD3 (left) or RAD51 (middle) was depleted by siRNA or RAD52 was knocked out (right). The distance between the sgRNA targeting sites and the repeats is indicated.
We also increased the distance between the right‐side DSB and the 3′ repeat, with the left‐side DSB generated 17 bp downstream of the 5′ repeat. RMD is reduced 2‐fold with an increase in distance between the DSB and the 3′ repeat from 300 to 3.3 kb and from 3.3 to 9.1 kb; there is no significant decrease when the distance is increased from 9.1 to 50 kb (Fig 4D and E). When the distance is further increased from 50 to 200 kb, an about 2‐ to 3‐fold decrease was observed. However, when a single cleavage is made at L17, which generates a DSB about 400 kb away from the right repeat, very low RMD was observed (Fig 4E). These data are comparable to the previous observations from the Stark's lab (Mendez‐Dorantes et al,2018) and also suggest that BIR/RMD can act over a long distance with substantial levels of RMD (~ 2%) when one repeat is 200 kb away from the break. These results support the BIR model in which the ssDNA strand from one repeat invades the duplex DNA of the other repeat to mediate RMD, and thus, the distance between the DSB and the second repeat can be large (Fig 2D, right). We also propose that the significant decrease in RMD when the distance between the DSB and the repeat is increased to 400 kb is likely due to the requirement for domain activation by a DSB for RMD (see below).
In addition, we introduced two DSBs, each close to one repeat in the RMD‐GFP reporter. Cleavage at 268 bp downstream of the 5′ repeat and 300 bp upstream of the 3′ repeat (L268/R300) readily induces RMD, but the frequency is still lower than when BIR/RMD is induced by L17/R28.4k (Fig 4F). To analyze which pathway is involved under this condition, we depleted RAD52, POLD3, and RAD51 by transient transfection of small interfering RNA (siRNA). RMD by L268/R300 is dependent on RAD52 but independent of POLD3, and depletion of RAD51 causes an increase in RMD (Fig 4G and Appendix Fig S9A), suggesting that SSA, but not BIR, is used. Thus, when the repeats are located far apart (~ 400 kb), SSA can still be used to mediate RMD, but only when the distance from the DSBs to both repeats is short and comparable. However, BIR/SSA can mediate RMD efficiently when the DSBs are over a very long distance from one repeat (~ 100–200 kb).
ATM is required for both SSA/RMD and BIR/RMD, while H2AX exhibits different effects when repeats are close together versus far apart
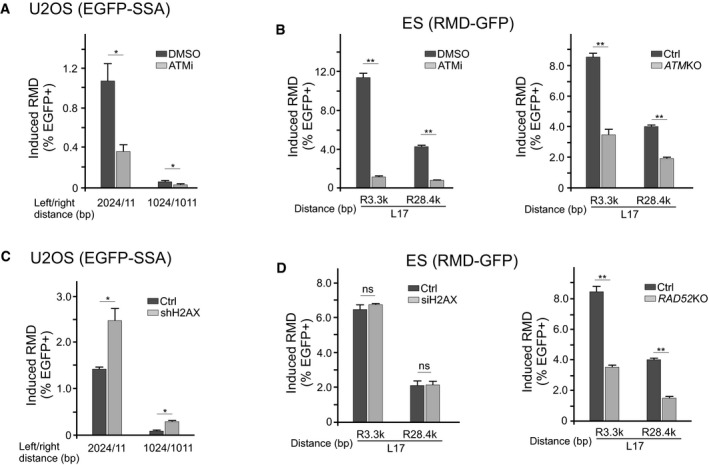
We examined the role of ATM in SSA/RMD and BIR/RMD, and showed that inhibition of ATM by the ATM inhibitor Ku55933 suppresses SSA/RMD and BIR/RMD in U2OS (EGFP‐SSA) cells when DSBs are generated at R11 or R1011, respectively. This suggests that ATM is needed for SSA/RMD and BIR/RMD (Fig 5A). By using ATM knockout (KO) cells and the ATM inhibitor, we also showed that ATM is required for BIR/RMD in ES (GFP‐RMD) cells when the repeats are situated over a long distance (~ 400 kb), with one DSB 17 bp downstream of the 5′ repeat and another DSB 3.3 kb or 28.4 kb upstream of the 3′ repeat (L17/R3.3k or L17/R28.4k, Fig 5B and Appendix Fig S9B).
Figure 5. The role of ATM and H2AX in RMD.
-
A, BSSA/RMD and BIR/SSA are both dependent on ATM. SSA/RMD and BIR/SSA frequencies in U2OS (EGFP‐SSA‐3427‐17) cells (A) and BIR/RMD in mES cells carrying the RMD‐GFP reporter (B) were determined after the addition of ATM inhibitor (20 mM ku55933) or DMSO to the cell culture medium 24 h before transfection of the indicated sgRNA/Cas9 (A and B), or in ATM KO mES cells carrying the RMD‐GFP reporter (right).
-
C, DDependence of RMD on H2AX. SSA/RMD and BIR/RMD frequencies in U2OS (EGFP‐SSA‐3427‐17) cells (C) and BIR/RMD in mES cells carrying the RMD‐GFP reporter (D) were determined using the indicated sgRNA/Cas9 after depletion of H2AX by lentivirus‐expressed shRNA (C), or by transfection with H2AX siRNA or in RAD52 KO cells (D). Depletion of H2AX by shRNA in (C) and by siRNA in (D) was determined by qPCR shown in Appendix Fig S8B and C, respectively.
Single‐strand annealing inevitably leads to deletions and thus needs to be limited. Along this line, it was shown that while H2AX is required for GC, it suppresses SSA, but the underlying mechanism is not clear (Xie et al, 2004). Using the U2OS (EGFP‐SSA) reporter cell line, we depleted H2AX by shRNA and showed that H2AX inhibits both SSA/RMD when a cleavage is made at R1011 (1024/1011) and BIR/RMD when DSBs are generated at R11 (2024/11) (Fig 5C and Appendix Fig S8B). This suggests that H2AX suppresses short‐range RMD mediated by both SSA and BIR. However, when DSBs are generated at L17/R3.3k or L17/R28.4k sites in mES (GFP‐RMD) cells with repeats located ~ 400 kb from each other, H2AX does not show a suppression effect on BIR/RMD, while RAD52 is required (Fig 5D, and Appendix Figs S8C and S9A). To exclude the possibility that H2AX regulates RMD differently in mES cells and U2OS cells, we inserted the EGFP‐SSA reporter into the ROSA locus in mES cells and showed that BIR/RMD induced by R11 (2024/11) cleavage is suppressed by H2AX (Appendix Fig S10). Thus, H2AX suppresses short‐range BIR/RMD in both mES cells and U2OS cells, but does not seem to have a role in long‐range BIR/RMD in mES cells (Fig 5D). As for long‐range BIR/RMD, a more likely model is that H2AX is required for activating the remote repeat for recombination (see below), and this requirement compensates for the suppression effect of H2AX on RMD.
A DSB generated in the vicinity of a remote, inaccessible repeat stimulates BIR/RMD
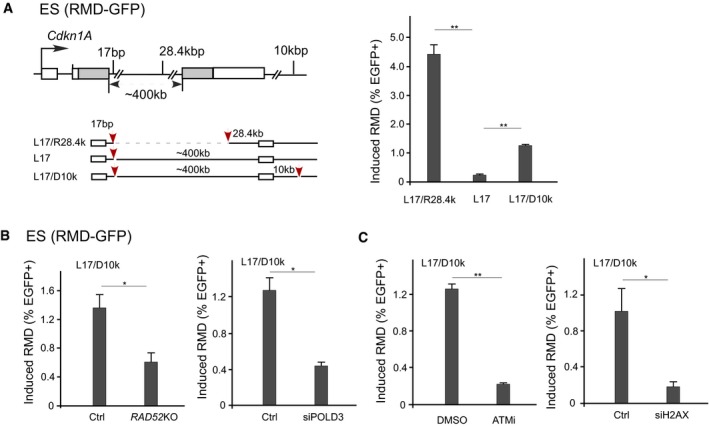
We showed that BIR/RMD can be efficiently induced when one DSB is close to the 5′ repeat (17 bp downstream) while the other DSB is as far as ~ 100–200 kb from the 3′ repeat (Fig 4E). However, when only one DSB is generated 17 bp downstream of the 5′ repeat in the RMD‐GFP reporter and the other repeat is ~ 400 kb away from the DSB (L17), RMD frequency is very low (Fig 4E, right and Fig 6A). One possibility is that when two repeats are located too far apart (~ 400 kb), it is hard for them to find each other. Alternatively, both alleles may need to be activated by the DNA damage response (DDR) in order for them to recombine. Through DDR by chromatin remodeling and/or domain organizing, a DSB may still sufficiently activate a repeat located ~ 100–200 kb away, but not ~ 400 kb away. To test the latter possibility, we generated a DSB 10 kb downstream (D10k) of the 3′ repeat along with the first DSB at L17 (L17/D10k) and found that despite only scoring the RMD of the reporter substrates that have successfully repaired the DSB at D10k (failure in repairing the DSB at D10k would cause substrate loss), the RMD frequency of the two repeats situated ~ 400 kb apart is significantly increased (Fig 6A). RMD induced by L17/D10k depends on RAD52 and POLD3 (Fig 6B), suggesting that it is mediated by BIR. We further showed that ATM is required when cleavage is generated at L17/D10k, as it is at L17/R28.4k (Fig 6C, left compare to Fig 5B). However, BIR/RMD induced by L17/D10k shows a stronger dependence on H2AX than when DSBs are generated by L17/R28.4k (Fig 6C, right, compared to 5D, left). These data support the notion that H2AX is important for RMD when one repeat is situated far away from the DSB. The difference in H2AX dependence when DSBs are made at L17/D10k versus L17/R28.4k can be attributed to the length of the chromatin between the DSB and the repeat (28.4 kb versus ~ 400 kb), and/or the DSB locations relative to the repeats (upstream or downstream) (see Discussion).
Figure 6. Efficient long‐range BIR/RMD requires DSBs that are present in the vicinity of both repeats.
-
AFrequency of long‐range RMD is increased by an additional DSB downstream of the 3′ repeat. Illustrations show the GFP‐RMD reporter, with sgRNAs/Cas9 cleavage sites indicated as red arrows (left). The frequency of BIR/SSA in mES cells carrying the RMD‐GFP reporter was determined after cleavage of the reporter by the indicated sgRNAs/Cas9 (right).
-
B, CThe effects of RAD52, POLD3, ATM, and H2AX inactivation on BIR/RMD with cleavage at L17/D10k. The frequency of BIR/RMD in mES cells carrying the RMD‐GFP reporter was determined using the sgRNAs/Cas9 at L17/D10k in RAD52 KO cells or after POLD3 depletion by siRNA (B), or after treatment with ATM inhibitor (ku55933, 15 mM) or H2AX depletion by siRNA (C).
Divergence in repeat sequences strongly inhibits BIR/RMD partially through the MSH2‐dependent pathway
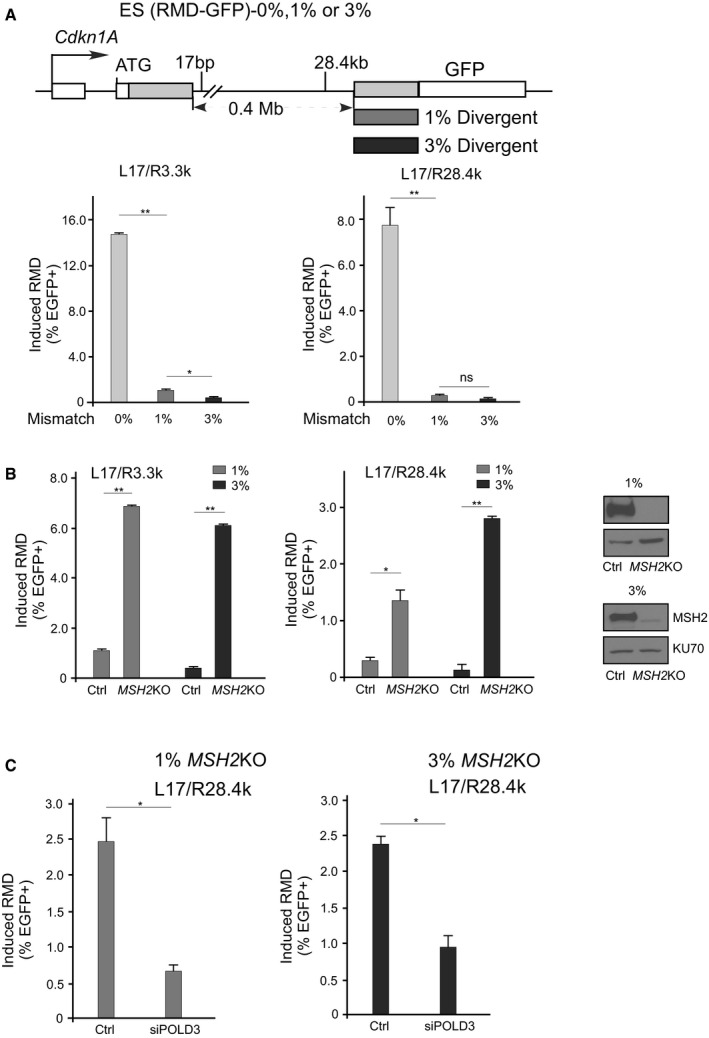
In the human genome, most repetitive sequences such as Alu elements are not identical (Batzer & Deininger, 2002; White et al, 2015). To examine the role of BIR in RMD between divergent repeat sequences, we used previously established RMD‐GFP reporters that contain 1% or 3% divergence in repeats (Mendez‐Dorantes et al, 2018). While cleavage at L17/R3.3k or L17/R28.4k readily induces RMD in the RMD‐GFP reporter containing identical repeats (0% mismatch), RMD is strongly suppressed when the repeats contain 1% or 3% mismatches (Fig 7A). Sequence divergence has been shown to suppress SSA in both yeast and mammalian cells (Sugawara et al, 2004; Mendez‐Dorantes et al, 2018), and our study suggests that the presence of mismatches also inhibits BIR/RMD.
Figure 7. Long‐range BIR/RMD is inhibited by divergence in repeat sequences.
-
ALong‐range BIR/RMD with identical repeats or divergent repeats. The frequency of BIR/RMD in mES cells carrying the RMD‐GFP reporter with identical repeats (0%) or repeats with 1 and 3% divergence was determined after cleavage at L17/R3.3k or L17/R28.4k.
-
B, CThe effect of MSH2 on BIR/RMD when repeat sequences are divergent. The frequency of BIR/RMD was determined after cleavage at L17/R3.3k or L17/R28.4k in mES cells carrying the RMD‐GFP reporter with 1 and 3% divergent repeats, and with and without MSH2 KO (B) or with MSH2 KO and POLD3 depletion by siRNAs (C). The MSH2 KO was validated by Western blot, using KU70 as the loading control as shown in (B).
We knocked out MSH2 by CRISPR/Cas9 in RMD‐GFP‐1% and RMD‐GFP‐3% reporter cell lines, and showed that MSH2 KO significantly increases RMD between divergent repeats, though not to the levels reached with identical repeats (Fig 7B and Appendix Fig S9C). This increase in RMD frequency between divergent repeats due to loss of MSH2 is consistent with previous findings using this assay system (Mendez‐Dorantes et al, 2018). We further showed that the increased RMD in MSH2 KO cells containing RMD‐GFP‐1% and RMD‐GFP‐3% reporters is dependent on POLD3 when DSBs are generated at L17/R3.3k or L17/R28.4k (Fig 7C), demonstrating that MSH2 indeed inhibits BIR/RMD between divergent repeats.
Discussion
SSA is considered as a classic pathway for repairing DSBs flanked by two repeats. However, we found that the distance between the two repeats is a major limiting factor for achieving efficient SSA in mammalian cells. BIR, on the other hand, can be efficiently used over a long distance and is stimulated upon replication fork collapse and oncogene expression. We propose that BIR is used predominantly over SSA in mediating long‐range RMD, especially under replication and oncogenic stress.
SSA is suppressed when the distance between repeats is increased in mammalian cells
In budding yeast, SSA can be efficiently used even when end resection needs to proceed over a long distance (e.g., 25 kb) to reach the repeats (Fishman‐Lobell et al, 1992; Vaze et al, 2002; Jain et al, 2009). An increase in the distance between a DSB and the repeats delays the timing of SSA product formation, but not SSA frequency, in yeast. However, in mammalian cells, SSA becomes inefficient when SSA requires end resection over a relatively short distance (~ 1 kb, Fig 1B), as assayed using the EGFP‐SSA reporter in U2OS cells. It is conceivable that inefficient SSA in mammalian cells is due to slow end resection kinetics. In yeast, the 5′‐to‐3′ resection rate is ~ 4 kb/h at HO endonuclease‐induced DSBs (Fishman‐Lobell et al, 1992; Zhu et al, 2008). However, using the SMART technique, the end resection rate in mammalian cells is estimated to be ~ 0.2 kb/h, which is 20 times slower than in yeast (Cruz‐Garcia et al, 2014). The slow kinetics of end resection may limit the extent of ssDNA formation in a timely manner and thus suppress SSA of the repeats over a large distance. Indeed, physical monitoring of ssDNA formation at DSBs has revealed that end resection most frequently occurs at a distance of < 1 kb in mammalian cells (Zhou et al, 2014).
In most cases under physiological or pathological conditions, only one DSB would be generated within the intervening sequence between the repeats. Since SSA becomes inefficient even when ~ 1 kb end resection is needed, it is not expected that SSA would play a significant role in mediating RMD of repeats with a distance over 1–2 kb.
BIR can act over a long distance to promote RMD when the DSB is close to one repeat
We propose that when a DSB is generated close to one repeat but is far away from the other, RMD is mediated by BIR but not SSA, which is supported by the dependence on POLD3 and RAD51. While the distance between repeats is a strong barrier for SSA in mediating RMD, BIR can act to generate RMD over a long distance (e.g., 100–200 kb). When end resection reaches one repeat, the ssDNA in that repeat can strand‐invade the other repeat that is still in duplex form to activate BIR (Fig 1D, right). This mechanism saves the time that is needed for long‐range end resection, especially when end resection is a slow process in mammalian cells.
One requirement for BIR/RMD, however, is that the DSB needs to be close to one repeat. We showed that an increase in the distance between a DSB and one repeat is proportional to the decrease in BIR/RMD frequency. When a DSB is ~ 50 bp away from one repeat, RMD is ~ 4‐fold lower than when it is ~ 10 bp away. However, to achieve a similar RMD efficiency, the requirement for SSA is even more stringent. For instance, to obtain a similar RMD frequency in SSA, the DSB must be close to both repeats, while in BIR/RMD the DSB only needs to be close to one repeat (Fig 2D). Thus, given that only one DSB is generated between the two repeats under most conditions, BIR/RMD is expected to be used more frequently than SSA/RMD, even when the two repeats are within a relatively short distance (such as 1–2 kb).
Strong inhibition of BIR/RMD by the distance between the DSB and one repeat is likely due to the formation of nonhomologous tails that suppress strand invasion or cause D‐loop instability. Invasion of the homologous sequences from the recipient to the donor results in an unstable paranemic joint that is converted to a more stable plectonemic joint after replication starts from the 3′ invasion strand (Riddles & Lehman, 1985). The presence of longer nonhomologous tails would make the paranemic joint more unstable, thereby reducing the BIR/RMD efficiency. When the nonhomologous tails are longer, end resection would also take more time to reach the homologous sequences in the repeats. However, considering the repeat length of 315 bp and a significant decrease in BIR/RMD frequency when the distance between the DSB and one repeat is increased from just 17 to 51 bp, we do not think end resection is a major factor in reducing BIR/RMD when the size of the short tail is increased.
It remains to be seen how flaps generated in BIR/RMD after strand invasion are removed. In yeast, Rad1/Rad10 is required for removing flaps during SSA (Fishman‐Lobell & Haber, 1992; Bardwell et al, 1994; Ivanov & Haber, 1995), and in mammalian cells, ERCC1 deficiency results in a reduction of SSA. We propose that the structure‐specific endonuclease ERCC1/XPF is also required for removing flaps formed by nonhomologous tails after strand invasion in BIR/RMD (Fig 1D, right, pink arrow). By using the GC repair substrate, we previously showed that ERCC1/XPF is required for removing nonhomologous tails 40 bp and longer, but is not involved in cleaving nonhomologous tails that are as short as 20 bp in length (Li et al, 2019). We anticipate that when the nonhomologous tails are longer than 40 bp, cleavage of flaps of different lengths does not significantly influence SSA/RMD and BIR/RMD frequency, since they all are cleaved by ERCC1/XPF through its endonuclease action. However, it remains unknown which nucleases are involved in removing flaps shorter than 20 bps and whether the cleavage efficiency of small flaps 20 bp or shorter by unknown nucleases is comparable to that of long flaps cleaved by ERCC1/XPF. The difference in cleavage efficiency of short (< 40 bp) and long (> 40 bp) flaps by different nucleases may influence BIR/RMD frequency.
Collectively, if a DSB is far away from both repeats, RMD is used at a minimal level since neither SSA nor BIR can function effectively. Such a DSB would be repaired by other pathways such as NHEJ or GC using sister chromatids. The presence of a DSB close to one repeat is needed for efficient BIR/RMD, and importantly, this mechanism can act over a large distance. The requirement for SSA is stringent, as the break needs to be in the vicinity of both repeats due to slow end resection in mammalian cells. Since most repetitive sequences are scattered across the human genome, SSA is not expected to play a major role in RMD as previously anticipated; rather, BIR is expected to play a more prominent role over SSA in mediating RMD in mammalian cells.
BIR/RMD is dependent on both RAD51 and RAD52
While SSA/RMD is suppressed by RAD51, BIR/RMD is dependent on RAD51. This suggests that BIR/RMD requires RAD51‐dependent strand invasion and is fundamentally different from SSA, which is suppressed by RAD51 (Stark et al, 2004). SSA requires the strand‐annealing activity of RAD52 and is thus dependent on RAD52 (Bhargava et al, 2016). Although BIR/RMD is initiated by RAD51‐dependent strand invasion, it is also dependent on RAD52. In mammalian cells, RAD52 is dispensable for GC (Liu & Heyer, 2011; Wang et al, 2018a), but is needed for BIR and BIR‐related mechanisms, although the underlying mechanism is still not clear (Bhowmick et al, 2016; Sotiriou et al, 2016). In addition, we showed previously that although RAD52 is not required for GC when DSB ends perfectly match the donor templates in mammalian cells, it becomes essential when the DSB ends contain nonhomologous tails (Wang et al, 2018b). We propose that when the 3′ DSB ends are blocked by nonhomologous tails, RAD52 may be needed to facilitate RAD51‐mediated strand invasion. In this respect, BIR/RMD is initiated with a small nonhomologous tail, and RAD52 may be involved in facilitating BIR/RMD in a manner similar to GC when nonhomologous tails are present at DSB ends (Fig 1D, right). Using RAD52 KO cells, we showed that BIR/RMD is substantially reduced but not eliminated (Appendix Fig S11), suggesting that a RAD52‐independent pathway for BIR/RMD also exists.
Despite the differences in mechanism between SSA/RMD and BIR/RMD, both are inhibited by KU‐dependent nonhomologous end joining (NHEJ) and microhomology‐mediated end joining (MMEJ). When we inactivated KU70 or POLQ, which are required for NHEJ and MMEJ, respectively (Lieber, 2010; Wood & Doublie, 2016), both short‐range and long‐range SSA/RMD and BIR/RMD increased (Appendix Fig S12), suggesting that SSA/RMD and BIR/RMD are in competition with NHEJ and MMEJ.
H2AX is important for activating remote, inaccessible repeats for BIR/RMD
We showed that ATM is important for both SSA/RMD and BIR/RMD. This is consistent with the role of ATM in promoting end resection (Jazayeri et al, 2006), which is required for SSA/RMD and BIR/RMD. CtIP is a key substrate of ATM in promoting end resection (Wang et al, 2013), and we have shown consistently that CtIP is important for both SSA/RMD and BIR/RMD (Appendix Fig S13). When repeats are located a large distance apart, ATM may also be needed for establishing synapsis of the repeats. In this respect, it has been shown that endonuclease‐induced DSBs cluster together in a manner dependent on ATM (Aymard et al, 2017).
We showed that in contrast to ATM, H2AX suppresses SSA/RMD and BIR/RMD when the two repeats are close to each other (e.g., 2 kb). It has been previously shown that H2AX is required for HR, but also has a role in suppressing SSA (Xie et al, 2004). The underlying mechanism, however, is not clear. The function of H2AX in suppressing SSA suggests that H2AX is not required for end resection. To this end, it was shown that H2AX prevents CtIP‐mediated end resection in G1 lymphocytes (Helmink et al, 2011), suggesting that H2AX has a role in suppressing end resection under certain conditions. The distinct activities of H2AX and ATM during end resection are likely the cause for their different roles in regulating SSA.
In both yeast and mammalian cells, γH2AX has been found to be absent in the immediate vicinity of a DSB (Shroff et al, 2004; Iacovoni et al, 2010), and the tethering process involving two ends of a chromosome break is independent of H2AX (Soutoglou et al, 2007). These findings support the notion that H2AX is not required for two close‐by repeats to find each other for recombination. However, ChIP analysis has revealed that γH2AX is spread over a large chromosomal region around a DSB site (0.5–1.7 Mb; Iacovoni et al, 2010). Hi‐C analysis has also shown that DSBs exhibiting higher levels of γH2AX demonstrate a higher ability to interact (Aymard et al, 2017), suggesting that H2AX may be important for long‐range chromatin interactions around DSBs.
In our study, we showed that when a DSB is generated close to one repeat (e.g., 17 bp) but very far away from the other (~ 400 kb), RMD operates at a very low level in the RMD‐GFP reporter (Fig 6A). However, when an additional DSB is generated 28.4 kb upstream or 10 kb downstream of the remote 3′ repeat, RMD is stimulated substantially. This suggests that it is difficult for one repeat near a DSB to find another copy hundreds of kb away. However, the remote inaccessible repeat can be activated for recombination when another DSB is generated within a certain distance (tens of kbs) of the remote repeat. Notably, chromosome conformation capture assays (capture Hi‐C) have demonstrated that DSBs can induce long‐range interactions, and the increased contact frequency is within ~ 500 kb of each side of the break (Aymard et al, 2017). In our assay with cleavage at L17, the DSB is ~ 400 kb away from the 3′ repeat, presumably at the edge of the activated domain; this is probably why RMD cannot be efficiently induced.
We propose that a γH2AX domain needs to be established for two remote repeats to synapse and interact. In our study, while SSA/RMD and BIR/RMD of close‐by repeats (EGFP‐SSA) are increased when H2AX is deficient, this suppression effect by H2AX is not observed in long‐range BIR/RMD (RMD‐GFP reporter) when DSBs are generated 17 bp downstream of the 5′ repeat and 28.4 kb upstream of the 3′ repeat (L17/R28.4k) (Fig 5D). The effects of H2AX on SSA suppression and on activation of the remote repeat may be offset in the experiment using L17/R28.4k, although we cannot exclude the possibility that H2AX simply does not have a role in long‐range RMD. In the case of L17/D10k, when a DSB is generated 10 kb downstream of the remote repeat, the dependence of RMD‐BIR on H2AX is evident (Fig 6D), revealing a stronger H2AX dependence than L17/R28.4k has. Collectively, we favor the model that H2AX is involved in the suppression of both short‐ and long‐range SSA, but is required for domain activation to promote recombination of the repeats when the repeats are far apart. The exact reason why BIR/RMD at L17/D10k relies more on H2AX than at L17/R28.4k is unclear. One possibility is that coordination of DSB repair 10 kb downstream of the 3′ repeat by end joining and RMD‐BIR between the two repeats may require more sustained γH2AX. Alternatively, chromatin structure and domain organization around the 3′ repeat may be different when the break is present at R28.4k versus D10k due to differences in chromatin length from the DSB end to the 3′ repeat (28.4 kb versus ~ 400 kb), causing varying dependency on H2AX.
BIR/RMD is stimulated upon oncogenic stress
Repeat‐mediated deletion are often found in cancers and human diseases (Carvalho & Lupski, 2016). Cyclin E overexpression‐induced processive DNA synthesis depends on POLD3, indicating activation of the BIR mechanism (Costantino et al, 2014). In this study, we observed that even after DSBs have been generated, BIR/RMD is stimulated when cyclin E is overexpressed. At this stage, the mechanism underlying the increase of BIR/RMD upon cyclin E overexpression is not completely understood, but our study suggests that replication stress and ATR activation caused by oncogene expression likely contribute to BIR/RMD stimulation. We found that ATR is also needed for BIR/RMD before cyclin E overexpression, likely due to the role of ATR in end resection (Jazayeri et al, 2006), but the dependence of BIR/RMD on ATR is much stronger after cyclin E overexpression. This supports the model that in addition to end resection, ATR also has a role in stimulating BIR/RMD activity upon oncogene expression. Identifying the key substrates of ATR in promoting BIR/RMD upon replication and oncogenic stress will be important for clarifying the underlying mechanism.
We further showed that BIR/RMD is substantially induced when replication encounters DNA nicks causing fork collapse. This is consistent with the model that BIR is a primary mechanism for repairing single‐ended DSBs at collapsed forks to mediate replication restart (Lydeard et al, 2007). Similar mechanisms may be involved in activating BIR and BIR/RMD at collapsed replication forks. Like BIR, BIR/RMD at collapsed forks can also be used to promote replication restart, resulting in the deletion of one repeat and the intervening sequence (Fig 3C). Thus, one potential source of RMD is BIR‐mediated replication restart using repetitive sequences upon replication stress.
While BIR/RMD is stimulated by oncogene expression and strongly promoted at collapsed replication forks, SSA is not activated in a similar manner. Thus, BIR/RMD plays a more significant role than SSA in inducing RMD‐associated chromosomal structure changes during oncogenesis, when both oncogenic stress and replication fork collapse are evident (Bartkova et al, 2006; Di Micco et al, 2006). The repair machineries of BIR and SSA are quite distinct, and how replication stress specifically targets BIR but not SSA is an excellent question to explore in the future.
Replicative mechanisms are often associated with the rearrangement process of repetitive sequences
Our study suggests that the BIR/RMD mechanism is likely used more often than SSA/RMD to mediate chromosomal rearrangements between repetitive sequences, especially upon replication and oncogenic stress. This provides new evidence to support the notion that a recombination‐coupled DNA replicative repair process is involved in repeat‐mediated rearrangement. Alu elements in the human genome are often divergent from one another. By analyzing Alu‐mediated copy‐number variants (CVNs) and their breakpoint junctions, Lupski's group proposed microhomology‐mediated BIR (MMBIR) model to account for divergent Alu/Alu‐mediated recombination (Gu et al, 2015; Song et al, 2018). Additionally, Jasin's group showed that translocation between divergent Alu sequences involves “in‐register” MMEJ (Elliott et al, 2005). Our study further demonstrated that a replicative BIR mechanism is also frequently used for recombination of identical DNA sequences in mammalian cells, which was thought via SSA. It has also been shown that sequence divergence between repeats suppresses RMD and MSH2 is involved in this suppression (Elliott et al, 2005; Morales et al, 2015; Mendez‐Dorantes et al, 2018). We further demonstrated that long‐range RMD is mediated by BIR/RMD, which is also limited by sequence divergence of the repeats in a manner that is at least partially dependent on MSH2. Thus, when homology sequences are not identical to each other, mismatch repair proteins act to suppress homology‐directed recombination of the repeats through BIR/RMD or SSA. Conceivably, MMBIR is used as an alternative replicative mechanism to BIR/RMD for rearrangement of divergent Alu sequences.
Collectively, our work suggests that BIR/RMD is a prominent pathway for RMD‐associated chromosomal structure changes, especially during oncogenesis. This differs from the conventional thought that SSA is the major pathway for mediating RMD. Since RMDs inevitably cause a loss of genetic information and may increase the risk of cancer, clarifying the repair pathways and genetic components responsible for RMD in humans will allow us to better understand cancer etiology and develop therapeutic interventions.
Materials and Methods
Cell cultures
U2OS and 293T cells were cultured in DMEM with 10% fetal bovine serum (FBS). Mouse embryonic stem cells were cultured on gelatin‐treated dishes in GMEM supplemented with 15% ES cell qualified FBS, 2 mM l‐glutamine, 1% non‐essential amino acid, 1 mM sodium pyruvate, 0.1 mM β‐mercaptoethanol, and 2,000 units/ml LIF. All cell lines were cultured at 37°C with 5% CO2.
Generation of RMD reporter cell lines
The EGFP‐SSA reporter substrate was generated by splitting a full‐length EGFP cassette into two fragments that share 315 bp repeat sequences. A hygromycin selection marker and an I‐SceI cleavage site along with a vector flanking sequence with a total length of 2,035 bp were inserted between 5′EGFP and 3′EGFP (Fig 1A). The reporter substrate plasmid was introduced into U2OS cells by transfection using Lipofectamine 2000. Stable integrated cells were selected by hygromycin, and single clones were picked and confirmed by Southern blot analysis for single‐copy integration. The EGFP‐SSA reporter substrate was also cloned into the mouse ROSA26 targeting vector, which was transfected into the mES cell line E14. Cell clones with single‐copy genomic integration were picked and confirmed by Southern blotting. The RMD‐GFP reporter in mES cells was a kind gift from Dr. Jeremy Stark (Mendez‐Dorantes et al, 2018).
To construct the EGFP‐SSA‐17/6 bp, EGFP‐SSA‐309/299, and EGFP‐SSA‐616/606 reporter plasmids (Fig 1B, right), the 2,035 bp fragment between the 315 bp repeats in the original EGFP‐SSA‐3427 reporter was replaced with 23 bp, 0.6 kb, and 1.2 kb fragments, respectively. These fragments were amplified by PCR using the EGFP‐SSA reporter as the template with the sgRNA‐R1011 recognition site situated in the middle. U2OS cells transfected with these reporters were pooled after drug selection and used for RMD assays to avoid the genomic position effect on repair efficiency.
RMD reporter assays
To perform RMD assays using U2OS (EGFP‐SSA) cells, 1 × 106 cells were seeded 1 day before transfection and transfected with I‐SceI or sgRNA‐Cas9 plasmids using Lipofectamine 2000. Transfected cells were selected by puromycin, the selection marker on the I‐SceI and sgRNA‐Cas9 expression vectors, and allowed to grow for 3 days before FACS analysis. The RMD‐GFP reporter assays in mES cells were performed as previously described (Mendez‐Dorantes et al, 2018). For FACS analysis, cells were collected by trypsinization and resuspended in PBS. FACS analysis was performed using a BD Accuri C6 flow cytometer. Each experiment was performed at least three times.
The sgRNAs for targeting the RMD reporter were expressed using the pSpCas9(BB)‐2A‐Puro (PX459) V2.0 vector, which was a gift from Dr. Feng Zhang [Addgene plasmid #62988 (Ran et al, 2013)]. The Cas9‐D10A mutant was generated by site mutation on Cas9 using QuickChange Site‐Directed Mutagenesis Kit (200518, Agilent). The sgRNAs sequences were listed in Appendix Table S1.
Generation of the inducible cyclin E cell line and KO cells
To generate the inducible cyclin E cell line, a human cyclin E‐coding sequence was inserted into pTRE3G‐GSX2 [a gift from Dr. Elena Cattaneo, Addgene plasmid #96964 (Faedo et al, 2017)] and transfected into U2OS (EGFP‐SSA) reporter cells, along with pCAG‐TetON‐3G [a gift from Dr. Elena Cattaneo, Addgene plasmid #96963 (Faedo et al, 2017)] and a PGK‐puromycin vector. Stable integrated clones were first picked up after puromycin selection and screened for clones that express cyclin E after addition of doxycycline (DOX). To overexpress cyclin E by lentivirus infection, cyclin E with C‐terminal HA tag was cloned into pCW‐57.1 (Addgene #41393). Cyclin E overexpression was induced by 2 μM DOX.
p27 was subcloned into the pCDH‐CMV‐MCS‐EF1‐BSD vector (made from pCDH‐CMV‐MCS‐EF1‐PURO by replacing PURO with BSD) for lentiviral infection.
To generate mES KO cell lines, sgRNA targeting the indicated genes was designed and cloned into tpSpCas9(BB)‐2A‐Puro (PX459) V2.0, which was a gift from Dr. Feng Zhang [Addgene plasmid #62988 (Ran et al, 2013)]. The sgRNA sequences are listed in Appendix Table S2 and PCR primers for screening KO cell lines are listed in Appendix Table S3. KO clones were confirmed by sequencing (Appendix Figs S9 and S11).
shRNA and siRNA interference
shRNA plasmids were constructed by inserting the shRNA target sequences into the pLKO.1‐blast vector, which was a gift from Dr. Keith Mostov [Addgene plasmid #26655 (Bryant et al, 2010)]. The shRNA sequences used were listed in Appendix Table S4. Silencing of the indicated genes in U2OS cells was performed by one round of lentiviral infection followed by blasticidin (10 μg/ml) selection for 2 days to obtain a pool of cells expressing shRNA. For independent experiments, new shRNA‐expressing cell lines were generated each time, followed by RMD analysis immediately, and the experiments were completed within a week. Silencing of the indicated genes in mES cells was performed by transient transfection of siRNA SMARTpool (obtained from Dharmacon), MOUSE‐POLD3 (M‐046305‐01000), MOUSE‐H2AX (M‐062621‐00‐0005), MOUSE‐RAD51 (M‐062730‐01‐0005), MOUSE‐POLQ (M‐050773‐00‐0005), and non‐targeting CTRL (D‐001206‐13‐05). The knockdown levels of the targeting gene were verified by qPCR using indicated primers (Appendix Table S5) or Western blot analysis. For analysis of the effect on RMD after gene depletion using both shRNAs and siRNA, experiments were performed at least three times.
Cell lysis and immunoblotting
For immunoblotting, cells were lysed with the NETN buffer containing 100 mM NaCl, 20 mM Tris–Cl (pH 8.0), 0.5 mM EDTA, 0.5% (v/v) Nonidet P‐40 (NP‐40), 0.5 mM PMSF, and 0.2 μM aprotinin. Lysis samples were boiled for 5 min after adding SDS (2×) loading buffer. Proteins were separated on 8–15% SDS–PAGE gels. Antibodies used in this paper include MSH2 (ab212188, Abcam), RAD51 (sc‐398587, Santa Cruz Biotechnology), RAD52 (sc‐365341, Santa Cruz Biotechnology), ATR (sc‐515173, Santa Cruz Biotechnology), CtIP (sc‐271339, Santa Cruz Biotechnology), KU70 (sc‐17789, Santa Cruz Biotechnology), POLD3 (ab182564, Abcam), HA (sc‐7392, Santa Cruz Biotechnology), and cyclin E (sc‐248, Santa Cruz Biotechnology).
Quantitative PCR (qPCR)
Total RNA was isolated using the RNeasy Mini Kit (Cat No. 74104, QIAGEN) according to manufacturer's instructions. cDNA was synthesized using the cDNA synthesis kit (Cat No. 1708890, Bio‐Rad). qPCR was performed using a BIO‐RAD CFX96 thermal cycler and SYBR Green Supermix (Cat No. 1725120, Bio‐Rad). qPCR primers (Appendix Table S5) used to amplify the indicated genes were designed using an online tool (https://www.idtdna.com/scitools/Applications/RealTimePCR/).
Inference of CRISPR edits (ICE)
We used ICE to compare cleavage efficiency by different sgRNAs. Plasmids coding for different sgRNAs and Cas9 were transfected into reporter cell lines. After puromycin selection, DNA fragments flanking the sgRNA/Cas9 cleavage sites were amplified and sent for sequencing. Sequencing profiles were analyzed using ICE (website: https://ice.synthego.com/). The targeting efficiency of sgRNAs measured by ICE is shown in Appendix Fig S14.
Quantification and statistical analysis
Two‐tailed non‐paired parameters were applied in t‐test (Student's t‐test) to analyze the significance of the differences between samples. In all experiments, error bars represent standard deviation (SD) of at least three independent experiments. The P value is indicated as *P < 0.05, **P < 0.01, ns, not significant.
Author contributions
XW, QH, HL, and SL designed the project. QH and HL performed most experiments. SL and HW performed some experiments and generated cell lines. LT designed and generated the EGFP‐SSA reporter plasmid. JL and SL performed some experiments. QH, HL and SL performed data analysis. XW supervised the entire project and RX contributed to some supervision. XW, QH, and SL wrote the article with input from other authors, and all authors read and revised the final article.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We would like to thank Dr. Jeremy Stark for providing the RMD‐GFP, 1%‐RMD‐GFP, and 3%‐RMD‐GFP reporter cell lines as well as sgRNA plasmids. We would like to thank Dr. Steven Reed for providing cDNA of human cyclin E. Plasmids pTRE3G‐GSX2 (#96964), pTRE3G‐GSX2 pCAG‐TetON‐3G (#96963), pSpCas9(BB)‐2A‐Puro (PX459) V2.0 (#62988), and pCW‐57.1 (Addgene # 41393) are from Addgene. We thank Zi Wang for helping with MMEJ analysis and Mingxue Ma for helping generating plasmids and reporter cell lines. This study is funded by NIH grants CA187052, CA197995, and GM080677 to X.W.
The EMBO Journal (2019) 38: e101751
References
- Anand RP, Lovett ST, Haber JE (2013) Break‐induced DNA replication. Cold Spring Harb Perspect Biol 5: a010397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Sommer S, Bailone A, Kogoma T (1993) Homologous recombination‐dependent initiation of DNA replication from DNA damage‐inducible origins in Escherichia coli . EMBO J 12: 3287–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F, Aguirrebengoa M, Guillou E, Javierre BM, Bugler B, Arnould C, Rocher V, Iacovoni JS, Biernacka A, Skrzypczak M et al (2017) Genome‐wide mapping of long‐range contacts unveils clustering of DNA double‐strand breaks at damaged active genes. Nat Struct Mol Biol 24: 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC (1994) Specific cleavage of model recombination and repair intermediates by the yeast Rad1‐Rad10 DNA endonuclease. Science 265: 2082–2085 [DOI] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC et al (2006) Oncogene‐induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633–637 [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL (2002) Alu repeats and human genomic diversity. Nat Rev Genet 3: 370–379 [DOI] [PubMed] [Google Scholar]
- Belancio VP, Roy‐Engel AM, Deininger PL (2010) All y'all need to know ‘bout retroelements in cancer. Semin Cancer Biol 20: 200–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava R, Onyango DO, Stark JM (2016) Regulation of single‐strand annealing and its role in genome maintenance. Trends Genet 32: 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick R, Minocherhomji S, Hickson ID (2016) RAD52 facilitates mitotic DNA synthesis following replication stress. Mol Cell 64: 1117–1126 [DOI] [PubMed] [Google Scholar]
- Bryant DM, Datta A, Rodriguez‐Fraticelli AE, Peranen J, Martin‐Belmonte F, Mostov KE (2010) A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol 12: 1035–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR (2016) Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR (2017) Non‐homologous DNA end joining and alternative pathways to double‐strand break repair. Nat Rev Mol Cell Biol 18: 495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD (2014) Break‐induced replication repair of damaged forks induces genomic duplications in human cells. Science 343: 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz‐Garcia A, Lopez‐Saavedra A, Huertas P (2014) BRCA1 accelerates CtIP‐mediated DNA‐end resection. Cell Rep 9: 451–459 [DOI] [PubMed] [Google Scholar]
- Davis AP, Symington LS (2004) RAD51‐dependent break‐induced replication in yeast. Mol Cell Biol 24: 2344–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger PL, Moran JV, Batzer MA, Kazazian HH Jr (2003) Mobile elements and mammalian genome evolution. Curr Opin Genet Dev 13: 651–658 [DOI] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A et al (2006) Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature 444: 638–642 [DOI] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Jasin M (2005) Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell 17: 885–894 [DOI] [PubMed] [Google Scholar]
- Faedo A, Laporta A, Segnali A, Galimberti M, Besusso D, Cesana E, Belloli S, Moresco RM, Tropiano M, Fuca E et al (2017) Differentiation of human telencephalic progenitor cells into MSNs by inducible expression of Gsx2 and Ebf1. Proc Natl Acad Sci USA 114: E1234–E1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, Powell SN (2011) Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA 108: 686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman‐Lobell J, Haber JE (1992) Removal of nonhomologous DNA ends in double‐strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science 258: 480–484 [DOI] [PubMed] [Google Scholar]
- Fishman‐Lobell J, Rudin N, Haber JE (1992) Two alternative pathways of double‐strand break repair that are kinetically separable and independently modulated. Mol Cell Biol 12: 1292–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa T, Alberts BM (1986) DNA synthesis dependent on genetic recombination: characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell 47: 793–806 [DOI] [PubMed] [Google Scholar]
- Gu S, Yuan B, Campbell IM, Beck CR, Carvalho CM, Nagamani SC, Erez A, Patel A, Bacino CA, Shaw CA et al (2015) Alu‐mediated diverse and complex pathogenic copy‐number variants within human chromosome 17 at p13.3. Hum Mol Genet 24: 4061–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, Sharma GG, McKinnon PJ, Zhang J, Bassing CH et al (2011) H2AX prevents CtIP‐mediated DNA end resection and aberrant repair in G1‐phase lymphocytes. Nature 469: 245–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, Legube G (2010) High‐resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J 29: 1446–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Haber JE (1995) RAD1 and RAD10, but not other excision repair genes, are required for double‐strand break‐induced recombination in Saccharomyces cerevisiae . Mol Cell Biol 15: 2245–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, Fishman‐Lobell J, Haber JE (1996) Genetic requirements for the single‐strand annealing pathway of double‐strand break repair in Saccharomyces cerevisiae . Genetics 142: 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Sugawara N, Lydeard J, Vaze M, Tanguy Le Gac N, Haber JE (2009) A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double‐strand break repair. Genes Dev 23: 291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Rothstein R (2013) Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5: a012740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP (2006) ATM‐ and cell cycle‐dependent regulation of ATR in response to DNA double‐strand breaks. Nat Cell Biol 8: 37–45 [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomietz E, Meyn MS, Pandita A, Squire JA (2002) The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosom Cancer 35: 97–112 [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W et al (2001) Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Li S, Lu H, Wang Z, Hu Q, Wang H, Xiang R, Chiba T, Wu X (2019) ERCC1/XPF is important for repair of DNA double‐strand breaks containing secondary structures. iScience 16: 63–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR (2010) The mechanism of double‐strand DNA break repair by the nonhomologous DNA end‐joining pathway. Annu Rev Biochem 79: 181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Heyer WD (2011) Who's who in human recombination: BRCA2 and RAD52. Proc Natl Acad Sci USA 108: 441–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE (2007) Break‐induced replication and telomerase‐independent telomere maintenance require Pol32. Nature 448: 820–823 [DOI] [PubMed] [Google Scholar]
- Malkova A, Naylor ML, Yamaguchi M, Ira G, Haber JE (2005) RAD51‐dependent break‐induced replication differs in kinetics and checkpoint responses from RAD51‐mediated gene conversion. Mol Cell Biol 25: 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Ira G (2013) Break‐induced replication: functions and molecular mechanism. Curr Opin Genet Dev 23: 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern MJ, Haber JE (2006) Break‐induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75: 111–135 [DOI] [PubMed] [Google Scholar]
- Mehta A, Haber JE (2014) Sources of DNA double‐strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 6: a016428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez‐Dorantes C, Bhargava R, Stark JM (2018) Repeat‐mediated deletions can be induced by a chromosomal break far from a repeat, but multiple pathways suppress such rearrangements. Genes Dev 32: 524–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales ME, White TB, Streva VA, DeFreece CB, Hedges DJ, Deininger PL (2015) The contribution of alu elements to mutagenic DNA double‐strand break repair. PLoS Genet 11: e1005016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Haber JE (1999) Multiple pathways of recombination induced by double‐strand breaks in Saccharomyces cerevisiae . Microbiol Mol Biol Rev 63: 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicek A, Noskov VN, Kouprina N, Barrett JC, Jurka J, Larionov V (2004) Evolution of the tumor suppressor BRCA1 locus in primates: implications for cancer predisposition. Hum Mol Genet 13: 2737–2751 [DOI] [PubMed] [Google Scholar]
- Petrij‐Bosch A, Peelen T, van Vliet M, van Eijk R, Olmer R, Drusedau M, Hogervorst FB, Hageman S, Arts PJ, Ligtenberg MJ et al (1997) BRCA1 genomic deletions are major founder mutations in Dutch breast cancer patients. Nat Genet 17: 341–345 [DOI] [PubMed] [Google Scholar]
- Puget N, Torchard D, Serova‐Sinilnikova OM, Lynch HT, Feunteun J, Lenoir GM, Mazoyer S (1997) A 1‐kb Alu‐mediated germ‐line deletion removing BRCA1 exon 17. Cancer Res 57: 828–831 [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddles PW, Lehman IR (1985) The formation of paranemic and plectonemic joints between DNA molecules by the recA and single‐stranded DNA‐binding proteins of Escherichia coli . J Biol Chem 260: 165–169 [PubMed] [Google Scholar]
- Rudin N, Haber JE (1988) Efficient repair of HO‐induced chromosomal breaks in Saccharomyces cerevisiae by recombination between flanking homologous sequences. Mol Cell Biol 8: 3918–3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff R, Arbel‐Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M (2004) Distribution and dynamics of chromatin modification induced by a defined DNA double‐strand break. Curr Biol 14: 1703–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Beck CR, Du R, Campbell IM, Coban‐Akdemir Z, Gu S, Breman AM, Stankiewicz P, Ira G, Shaw CA et al (2018) Predicting human genes susceptible to genomic instability associated with Alu/Alu‐mediated rearrangements. Genome Res 28: 1228–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou SK, Kamileri I, Lugli N, Evangelou K, Da‐Re C, Huber F, Padayachy L, Tardy S, Nicati NL, Barriot S et al (2016) Mammalian RAD52 functions in break‐induced replication repair of collapsed DNA replication forks. Mol Cell 64: 1127–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, Danuser G, Misteli T (2007) Positional stability of single double‐strand breaks in mammalian cells. Nat Cell Biol 9: 675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M (2004) Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 24: 9305–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Haber JE (1992) Characterization of double‐strand break‐induced recombination: homology requirements and single‐stranded DNA formation. Mol Cell Biol 12: 563–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE (2004) Heteroduplex rejection during single‐strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci USA 101: 9315–9320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima H, Hunter T (1994) p27, a novel inhibitor of G1 cyclin‐Cdk protein kinase activity, is related to p21. Cell 78: 67–74 [DOI] [PubMed] [Google Scholar]
- Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X (2013) Microhomology‐mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double‐strand breaks in mammalian cells. Proc Natl Acad Sci USA 110: 7720–7725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel‐Eden A, Foiani M, Haber JE (2002) Recovery from checkpoint‐mediated arrest after repair of a double‐strand break requires Srs2 helicase. Mol Cell 10: 373–385 [DOI] [PubMed] [Google Scholar]
- Wang H, Shi LZ, Wong CC, Han X, Hwang PY, Truong LN, Zhu Q, Shao Z, Chen DJ, Berns MW et al (2013) The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR‐mediated double‐strand break repair. PLoS Genet 9: e1003277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li S, Oaks J, Ren J, Li L, Wu X (2018a) The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat Commun 9: 2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li S, Zhang H, Wang Y, Hao S, Wu X (2018b) BLM prevents instability of structure‐forming DNA sequences at common fragile sites. PLoS Genet 14: e1007816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TB, Morales ME, Deininger PL (2015) Alu elements and DNA double‐strand break repair. Mob Genet Elements 5: 81–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RD, Doublie S (2016) DNA polymerase theta (POLQ), double‐strand break repair, and cancer. DNA Repair (Amst) 44: 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Puget N, Shim I, Odate S, Jarzyna I, Bassing CH, Alt FW, Scully R (2004) Control of sister chromatid recombination by histone H2AX. Mol Cell 16: 1017–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Caron P, Legube G, Paull TT (2014) Quantitation of DNA double‐strand break resection intermediates in human cells. Nucleic Acids Res 42: e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double‐strand break ends. Cell 134: 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File