

Cytomegaloviruses are betaherpesviruses that in immunocompromised individuals can lead to severe pathologies. These viruses encode various gene products that serve to evade innate immune recognition. NK cells are among the first immune cells that respond to CMV infection and use germ line-encoded NK cell receptors (NKR) to distinguish healthy from virus-infected cells. One such axis that plays a critical role in NK recognition involves the inhibitory NKR-P1B receptor, which engages the host ligand Clr-b, a molecule commonly lost on stressed cells (“missing-self”). In this study, we discovered that mouse CMV utilizes the m153 glycoprotein to circumvent host-mediated Clr-b downregulation, in order to evade NK recognition. These results highlight a novel MCMV-mediated immune evasion strategy.
KEYWORDS: natural killer cell, mouse cytomegalovirus, MCMV, Nkrp1-Clr, missing-self recognition, host-pathogen interactions, m145 family, viral immune evasion, natural killer cells
ABSTRACT
Natural killer (NK) cells are a subset of innate lymphoid cells (ILC) capable of recognizing stressed and infected cells through multiple germ line-encoded receptor-ligand interactions. Missing-self recognition involves NK cell sensing of the loss of host-encoded inhibitory ligands on target cells, including MHC class I (MHC-I) molecules and other MHC-I-independent ligands. Mouse cytomegalovirus (MCMV) infection promotes a rapid host-mediated loss of the inhibitory NKR-P1B ligand Clr-b (encoded by Clec2d) on infected cells. Here we provide evidence that an MCMV m145 family member, m153, functions to stabilize cell surface Clr-b during MCMV infection. Ectopic expression of m153 in fibroblasts augments Clr-b cell surface levels. Moreover, infections using m153-deficient MCMV mutants (Δm144-m158 and Δm153) show an accelerated and exacerbated Clr-b downregulation. Importantly, enhanced loss of Clr-b during Δm153 mutant infection reverts to wild-type levels upon exogenous m153 complementation in fibroblasts. While the effects of m153 on Clr-b levels are independent of Clec2d transcription, imaging experiments revealed that the m153 and Clr-b proteins only minimally colocalize within the same subcellular compartments, and tagged versions of the proteins were refractory to coimmunoprecipitation under mild-detergent conditions. Surprisingly, the Δm153 mutant possesses enhanced virulence in vivo, independent of both Clr-b and NKR-P1B, suggesting that m153 potentially targets additional host factors. Nevertheless, the present data highlight a unique mechanism by which MCMV modulates NK ligand expression.
IMPORTANCE Cytomegaloviruses are betaherpesviruses that in immunocompromised individuals can lead to severe pathologies. These viruses encode various gene products that serve to evade innate immune recognition. NK cells are among the first immune cells that respond to CMV infection and use germ line-encoded NK cell receptors (NKR) to distinguish healthy from virus-infected cells. One such axis that plays a critical role in NK recognition involves the inhibitory NKR-P1B receptor, which engages the host ligand Clr-b, a molecule commonly lost on stressed cells (“missing-self”). In this study, we discovered that mouse CMV utilizes the m153 glycoprotein to circumvent host-mediated Clr-b downregulation, in order to evade NK recognition. These results highlight a novel MCMV-mediated immune evasion strategy.
INTRODUCTION
Viruses are known to employ a variety of nonredundant and sometimes complementary immunoevasin strategies to circumvent host innate and adaptive immune recognition. Herpesviruses contain large double-stranded DNA (dsDNA) genomes that readily accommodate numerous immunoevasin genes. Cytomegaloviruses (CMV) are a family of betaherpesviruses with strict species specificity that have intimately coevolved with their hosts and therefore have evolved various mechanisms to subvert detection or evade immune effector mechanisms. These viruses contain ∼200 open reading frames (ORF), ∼70 of which are conserved core genes, plus numerous genes that are dispensable for replication and thought to alter the host response to the virus (1).
Mouse cytomegalovirus (MCMV) has been used as a rodent model to study human CMV (HCMV) infection and immune sequelae, as the two viruses display similar cellular tropism and pathogenesis. These viruses are well tolerated in immunocompetent hosts, in which they establish latent infections. However, they can cause severe pathologies in immunocompromised individuals, such as newborns, cancer patients undergoing chemotherapy, transplant patients, or AIDS patients (2, 3). These viruses also employ structurally divergent, yet evolutionarily convergent, mechanisms to evade immune recognition. For this reason, the study of individual MCMV immunoevasin gene products has been used to infer similar functionality of HCMV genes, although each virus does employ some unique mechanisms.
Prior to the induction of adaptive immunity, natural killer (NK) cells act as key sentinels to detect and control acute MCMV infection. They are a subset of group 1 innate lymphoid cells (ILC) that recognize pathogenic cells through receptor-ligand interactions that lead to signals transmitted via inhibitory and stimulatory NK cell receptors (NKR) (4). Illustrating the importance of NK cell-mediated control of acute MCMV infection, these NKR ligands are targeted by many MCMV-encoded immunoevasive genes (5). Some MCMV gene products act to subvert NK cell-mediated “missing-self” recognition, which involves the loss of “self” ligands (such as major histocompatibility complex class I [MHC-I]) during infection. These include surrogate ligands that directly engage inhibitory receptors, such as the interaction between m157 with Ly49C/I and between m144 and an unknown receptor (6, 7). Importantly, in some resistant mouse strains, like C57BL/6, m157 can also interact with the paralogous stimulatory receptor Ly49H, resulting in dominant immune control of MCMV infection (6, 8). MCMV also encodes m04 and MATp1, which together escort self MHC-I alleles to the cell surface to inhibit NK cells by engaging the inhibitory Ly49 receptors (9–11). However, much like the evolution between m157 and Ly49H, certain mouse strains have evolved stimulatory receptors (Ly49P/L/W2) that directly recognize the m04:MHC-I complexes (12, 13).
On the other hand, MCMV encodes numerous functionally overlapping immunoevasins that methodically target “induced-self” NK recognition via the NKG2D stimulatory receptor. Specifically, several MCMV gene products intracellularly retain distinct families of NKG2D ligands, including m138 (which targets H60, Mult-1, and Rae-1ε), m145 (targets Mult-1), m152 (targets all five Rae-1 isoforms), and m155 (targets H60) (14–20). Thus, MCMV gene products retard the expression of these ligands for the activating NK activating receptor, NKG2D. Interestingly, Rae-1 ligands themselves are actually induced by a viral protein (m18) through the release of HDAC repression (21). Recently, it has also been reported that m20.1, m166, and m154 target the DNAM-1 ligand PVR, TRAIL death receptor, and the 2B4 ligand CD48, respectively (22–24).
MHC-independent receptor-ligand interactions involving the NKR-P1 receptors also play a role in viral detection. The NKR-P1 family recognizes genetically linked C-type lectin-related (Clr) ligands and consists of five members in mice, including three stimulatory isoforms (NKR-P1A, -C, and -F) and two inhibitory isoforms (NKR-P1B and -G) (25). Mouse NKR-P1F recognizes Clr-c/d/g, whereas NKR-P1G recognizes Clr-d/f/g and NKR-P1B recognizes Clr-b (26–28). Importantly, Clr-b has been shown to represent a marker of cellular fitness that is commonly lost or downregulated during cellular pathologies. Consequently, the NKR-P1B:Clr-b axis has been shown to play a role in missing-self recognition in tumor immunosurveillance, transplantation, genotoxic and cellular stress, and viral (poxvirus and cytomegalovirus) infection (27, 29–35). Recently, we demonstrated that MCMV encodes m12, which directly engages the NKR-P1A, -B, and -C receptors in an ongoing evolutionary struggle between host and pathogen (36–38).
To extend this work, we investigated whether MCMV also targets the NKR-P1B:Clr-b recognition system independent of m12. Given that there are allelic polymorphisms in both m12 and NKR-P1 receptors, there likely exist some pairs that do not interact; therefore, we hypothesize that MCMV uses mechanisms to retain cell surface Clr-b expression during infection to evade NK recognition through NKR-P1B.
RESULTS
An MCMV m145 family member stabilizes Clr-b expression during MCMV infection.
We have previously demonstrated that MCMV infection promotes a rapid loss of the inhibitory NKR-P1B ligand Clr-b on mouse fibroblasts (29). In addition, we subsequently discovered that MCMV encodes m12, which acts to circumvent this loss in Clr-b by acting as a decoy ligand that engages NKR-P1B (36). This is synonymous with the Clr-like immunoevasin (RCTL) encoded by rat CMV (RCMV) (34). Since Clr-b acts as a marker of cellular fitness, we hypothesized that MCMV may encode gene products to counteract the host-driven downregulation that alerts NK cells. Therefore, we analyzed two large deletion mutants (Δm01-m17 for MCMV-Δ1 [Δ1 mutant virus] and Δm144-m158 for MCMV-Δ6 [Δ6 mutant virus]) to compare their phenotypes relative to that of the wild-type (WT) MCMVMW97 parental strain (Fig. 1A). Interestingly, all three viruses promoted loss of Clr-b expression on infected NIH 3T3 fibroblasts, yet a more pronounced Clr-b downregulation was consistently observed using the Δ6 mutant virus, which is missing most of the m145 gene family (Δm144-m158) (Fig. 1A and B). In contrast, the Δ1 mutant virus lacking the m02 gene family (Δm02-m16) and other genes (m01 and m17) yielded a phenotype similar to that of WT MCMV (Fig. 1A and B). The difference in magnitude of Clr-b downregulation promoted by the Δ6 virus was found to be statistically significant upon analysis of both cell surface Clr-b median fluorescence intensity (MFI) (Fig. 1C) and the percentage of cells that had fully lost Clr-b surface protein (Fig. 1D).
FIG 1.

Infection with MCMV deficient in m145 family members reveals Clr-b modulation. (A) MCMV genomic map demonstrating location of missing genes in the mutant MCMV viruses (Δ1 and Δ6). Labels correspond to the HindIII digestion map. (B to F) NIH 3T3 fibroblasts were infected with WT MCMV (MW97.01) or the Δ1 (Δm02-m17) or Δ6 (Δm144-m158) mutant at an MOI of 0.5 PFU/cell and analyzed by flow cytometry 24 h postinfection (hpi). (B) Representative histograms of Clr-b (left), MHC-I (H-2DqLq; middle), and Rae-1 (α to ε; right). Numbers on the right represent median fluorescence intensity (MFI), whereas the ones on the left represent the percentage of marker-negative cells. The shaded histogram shows stained sample, whereas the dotted histogram represents unstained sample. The vertical dotted line symbolizes mock MFI levels. Live cells were gated using propidium iodide exclusion. (C and D) Quantitation of normalized Clr-b MFI (C) and percent Clr-b– cells (D). (E and F) Quantitation of normalized MHC-I MFI (E) and Rae-1 MFI (F). MFI values were normalized to the mock MFI. Data were analyzed using 1-way ANOVA and are representative of those from 3 to 6 independent experiments. n.s., not significant.
As controls, we also investigated major histocompatibility class I (MHC-I) (H-2DqLq) and NKG2D ligand (pan-Rae-1α-ε family) cell surface expression on infected NIH 3T3 fibroblasts. Notably, the Δ1 mutant was partially deficient in MHC-I downregulation (likely due to the loss of m06), while the Δ6 mutant was surprisingly more efficient at promoting MHC-I loss (despite the loss of m152) (Fig. 1A and E), suggestive of an additional gene in this region that modulates MHC-I expression that may work in conjunction with other factors. As expected, the Δ1 virus maintained its ability to internalize Rae-1 proteins, while infection with Δ6 virus resulted in upregulation of Rae-1 ligands (likely due to loss of m152) (Fig. 1A and D). To determine the temporal stage of MCMV infection underlying these differences, we infected NIH 3T3 fibroblasts over a time course, and we observed that the Δ6 virus promoted both a more rapid and exacerbated loss of Clr-b expression, detectable as early as 12 h postinfection (Fig. 2). These results suggest that a gene in the m144-m158 region counteracts MCMV infection-mediated Clr-b loss, akin to the positive regulation of MHC-I surface expression by the m04 glycoprotein, which antagonizes the functions of m06 and m152 for certain MHC-I alleles (10).
FIG 2.

MCMV infection with MCMV-Δ6 virus reveals a more pronounced Clr-b downregulation at early time points of infection. NIH 3T3 fibroblasts were infected with WT MCMV or the Δ6 mutant at an MOI of 0.5 PFU/cell and analyzed by flow cytometry at different time points within a 24-h infection time course. (A) Representative histograms of Clr-b surface expression. The vertical dotted line symbolizes mock MFI. (B and C) Quantitation of Clr-b MFI (B) and quantitation of percent Clr-b– cells (C) from values in panel A. Data are representative of those from 3 independent experiments.
The m153 glycoprotein sustains Clr-b cell surface expression.
In order to identify candidate MCMV genes responsible for Clr-b modulation, we cloned and overexpressed individual MCMV cDNA gene products (in the pIRES2-EGFP reporter vector) and assessed their effects on cell surface Clr-b levels. To this end, we tested the m145 family members in the region from m144 to m158 (m145, m146, m150, m151, m152, m153, m154, m155, m157, and m158), in addition to m144 (which has been shown to negatively modulate NK cell function [7]), m159, and m17 (a distal m145 family member). Notably, these MCMV gene products are all related and are predicted to form MHC-I-like folds (8). Following transfection of these genes into NIH 3T3 fibroblasts, expression of surface Clr-b, MHC-I, and Rae-1 ligands was assessed by flow cytometry, using IRES-EGFP fluorescence as a marker for transfected cells (Fig. 3A and B). Interestingly, m153 was the sole gene product found to increase Clr-b cell surface expression, which it increased by roughly 50%, and although modest, this effect was reproducible and statistically significant (Fig. 3B). In contrast, the empty vector and other candidate gene products promoted a slight Clr-b downregulation at high levels of green fluorescent protein (GFP), an effect that may be due to innate recognition of high levels of cytosolic plasmid DNA or induction of cellular stress (Fig. 3A and B). Importantly, none of the m02 family members modulated Clr-b expression (Fig. 4A and B). Consistent with previous studies, we observed that m152 altered MHC-I and Rae-1 expression (Fig. 5A to D). Therefore, the m153 gene product is likely responsible for sustaining Clr-b levels following infection by MCMV.
FIG 3.

Expression of m153 in mouse cells increases cell surface Clr-b. (A) NIH 3T3 cells were transfected with cDNA encoding the m145 family and analyzed for Clr-b cell surface expression 48 h posttransfection. Numbers on the left and right represent MFI values of GFP– and GFP+ gated populations, respectively. (B) Quantitation of normalized cell surface Clr-b expression in panel A (GFP+/GFP–). Data were analyzed using 1-way ANOVA and are representative of those from 3 independent experiments. (C to E) NIH 3T3 cells were infected with MCMV viruses (WT, Δ6, or Δm153) at different MOI and assessed for cell surface Clr-b expression by flow cytometry 24 hpi. (C) Histograms showing Clr-b expression at the different MOI. (D) Quantitation of normalized Clr-b MFI. (E) Quantitation of % Clr-b– cells. Data were analyzed using 2-way ANOVA and are representative of those from 3 independent experiments. (F) Transcript levels of m153 measured during time course of MCMV infection. All data are representative of those from 3 or 4 independent experiments.
FIG 4.
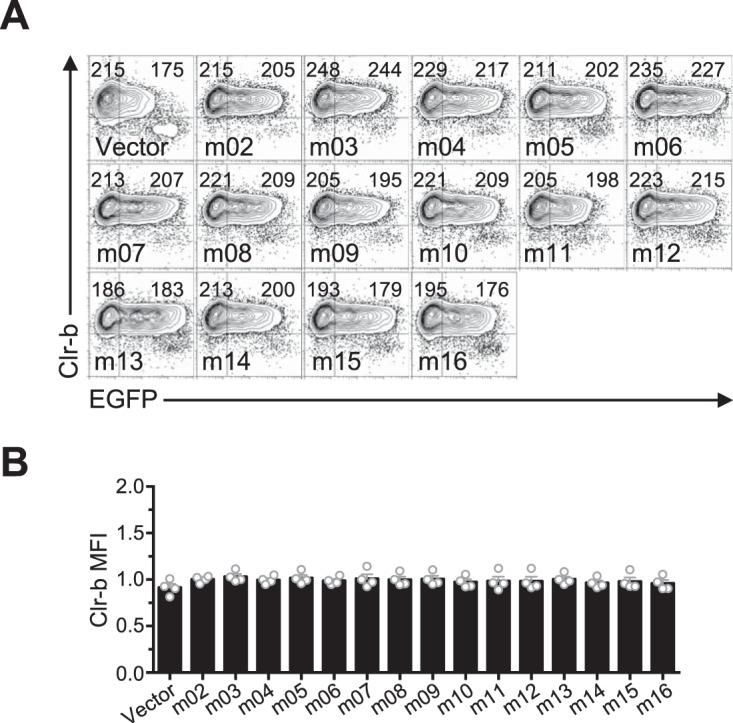
Ectopic expression of the m02 family of immunoevasins does not affect Clr-b expression. (A) NIH 3T3 cells were transfected with m02 ORF and analyzed for Clr-b cell surface expression 48 h posttransfection. Numbers on the left and right represent MFI values of GFP– and GFP+ populations, respectively. (B) Quantitation of Clr-b surface expression in panel A. Data were analyzed using 1-way ANOVA and are representative of those from 4 independent experiments.
FIG 5.

Ectopic expression of m02 and m145 family members confirms effects of immunoevasins on MHC-I and Rae-1 expression. (A) Representative flow plots of MHC-I expression upon transfection of cDNA encoding MCMV genes. (B) Quantitation of results in panel A. (C) Representative flow plots of Rae-1 expression upon transfection of cDNA for MCMV genes. (D) Quantitation of results in panel C. All data are representative of those from 4 independent experiments and were analyzed using 1-way ANOVA.
To confirm this, we extended analysis using an m153-deficient MCMV mutant (Δm153). Upon infection of mouse NIH 3T3 fibroblasts at different multiplicities of infection (MOI) and comparison to WT MCMV, the Δm153 and Δ6 mutant viruses were found to similarly downregulate Clr-b expression (Fig. 3C). Furthermore, the extent of Clr-b loss correlated well with viral MOI, revealing greater Clr-b downregulation (MFI) and increased percentages of cells completely lacking Clr-b expression (Clr-b– cells) at higher MOI (Fig. 3D and E). Quantitative real-time reverse transcription-PCR (qRT-PCR) analysis of m153 mRNA levels revealed that m153 expression peaked during the early phase of infection, around 3 to 6 h postinfection (hpi) (Fig. 3F). Consistently, exacerbated loss of surface Clr-b expression was detected in infected cells as early as 12 hpi with the Δ6 virus (Fig. 2) MHC-I and Rae-1 levels were not different when cells were infected with Δm153 or WT virus (Fig. 6). These data demonstrate that m153 selectively sustains cell surface Clr-b levels during MCMV infection, countering the opposite effect of viral infection.
FIG 6.

Infection with MCMV Δm153 virus does not alter MHC-I or Rae-1 expression. NIH 3T3 fibroblasts were infected with WT MCMV or the Δm153 mutant at an MOI of 0.5 PFU/cell and analyzed by flow cytometry 24 h later. (A) Histograms of cell surface expression of Clr-b (left), MHC-I (middle), and Rae-1 (right). The vertical dotted line symbolizes mock MFI, whereas numbers on the left represent the percent marker– cells in gate, and numbers on the right represent MFI values. (B to D) Quantitation of cell surface levels of Clr-b (B), MHC-I (C), and Rae-1 expression (D). Data were analyzed using 1-way ANOVA and are representative of those from 3 to 6 independent experiments.
Exogenous m153 complementation upon MCMV-Δm153 infection rescues Clr-b levels.
To further determine the effects of m153 expression on Clr-b levels, we generated stable NIH 3T3 transductants possessing tetracycline-inducible (Tet-On) m153 expression. To monitor cell surface m153 levels, we cloned the m153 cDNA with an N-terminal hemagglutinin (HA) tag (m153HA) into a modified pTRIPZ Tet-On lentiviral vector and used this construct to stably transduce NIH 3T3 fibroblasts. Interestingly, in comparison to control cells transduced using the empty vector (NIH 3T3.vector), transductants expressing m153 (NIH 3T3.m153HA) constitutively expressed higher basal levels of cell surface Clr-b, even in the absence of doxycycline (Dox) treatment (∼2.3-fold increase [Fig. 7A and B]). This effect was likely due to low-level m153HA expression by the minimal Tet-On promoter or long terminal repeat (LTR)-driven expression since we could detect m153HA transcripts and cell surface staining (by both HA tag and an anti-m153 monoclonal antibody [MAb]) in the absence of Dox treatment (data not shown). However, in agreement with the lack of an observed diagonal correlation between Clr-b and enhanced GFP (EGFP) levels in m153 transient-transfection experiments (Fig. 3A and B), we observed no further increase in Clr-b expression in stable NIH 3T3.m153HA transductants upon treatment with titrated Dox, while HA tag expression increased dramatically at the cell surface of NIH 3T3.m153HA transductants in the presence of Dox (data not shown). Taken together, these findings suggest that low levels of m153 protein are sufficient to increase Clr-b cell surface expression.
FIG 7.

Complementation of m153 abrogates Clr-b loss observed in m153-deficient virus. NIH 3T3 cells were transduced with pTRIPZ lentivirus expressing m153 or empty vector and infected with MCMV. (A) Comparison of the Clr-b expression on resting transduced cells. (B) Histograms of Clr-b levels on NIH 3T3.vector and NIH 3T3.m153HA fibroblasts upon infection with MCMV (WT, Δ6, or Δm153). (C and D) Analysis of NIH 3T3.vector cells by quantitation of Clr-b MFI (C) and quantitation of percent Clr-b– cells at different MOI (D). (E and F) Analysis of NIH 3T3.m153HA cells by quantitation of Clr-b MFI (E) and quantitation of percent Clr-b– cells at different MOI (F). Data were analyzed using 2-way ANOVA and are representative of those from 3 independent experiments.
To determine if low-level m153 expression was sufficient to exogenously complement and stabilize Clr-b levels following infection with Δm153 and Δ6 mutants, we infected NIH 3T3.m153HA and control NIH 3T3.vector transductants and compared Clr-b levels to that of cells infected with WT MCMV. As expected, infections at different MOI revealed that NIH 3T3.vector transductants behaved similarly to parental NIH 3T3 cells (Fig. 3C and 7B), in that Δm153 and Δ6 mutants had a more pronounced Clr-b loss relative to that with WT MCMV infection, as determined by both Clr-b MFI levels (Fig. 7C) and the percentage of Clr-b– cells (Fig. 7D). In contrast, NIH 3T3.m153HA transductants uniformly expressed similar Clr-b levels regardless of infection using the WT MCMVMW97 or MCMV-Δm153, or MCMV-Δ6 mutants, across all MOI tested (Fig. 7B, E, and F). Similar results were also obtained in the presence of Dox treatment (data not shown). These data confirm that the presence of m153 stabilizes Clr-b expression and antagonizes MCMV infection-mediated Clr-b downregulation.
m153 may stabilize Clr-b surface levels by an indirect mechanism.
Collectively, the above-described findings suggest that m153 sustains Clr-b surface protein expression in a nonlinear manner. To gain insight into the mechanism, we first assessed whether transcriptional regulation was involved. We measured Clr-b (Clec2d) transcript levels during infection with WT or Δm153 virus yet observed similar losses of Clec2d mRNA following infection (Fig. 8A). We also observed similar steady-state Clec2d mRNA levels in stable NIH 3T3.m153HA and control NIH 3T3.vector transductants, in both the absence and presence of Dox induction (Fig. 8B). These findings demonstrate that m153 does not modulate Clr-b at the transcript level.
FIG 8.

Clr-b is stabilized by m153 through an indirect mechanism. (A and B) Clec2d transcript levels in NIH 3T3 cells infected with WT or Δm153 virus (A) and in NIH 3T3 cells transduced with m153HA or empty control pTRIPZ vector (B). (C and D) Immunoprecipitations using anti-FLAG or anti-HA MAb from lysates of NIH 3T3.Clr-bFLAG cells transfected with m153HA or empty control vector. These lysates were immunoblotted for anti-FLAG (C) or anti-HA (D). (E) NIH 3T3 cells were transfected with constructs of m153 and Clr-b fused to either EGFP or mCherry and analyzed for cellular localization using fluorescence microscopy 48 h posttransfection. All data are representative of those from at least 3 independent experiments.
Because both MHC-I molecules and NKG2D ligands are targeted by multiple nonredundant MCMV proteins that involve direct interactions, and because both Clr-b and m153 can be expressed at the cell surface, we tested whether m153 may stabilize Clr-b expression by direct interaction. Since previous attempts to immunoprecipitate Clr-b protein using 4A6 MAb (rat IgM) have been unsuccessful, we generated stable NIH 3T3 transductants that expressed Clr-b with a C-terminal FLAG tag (NIH 3T3.Clr-bFLAG) using the inducible pTRIPZ lentiviral vector. Using these cells, Clr-bFLAG protein could be visualized upon FLAG tag immunoprecipitation (IP) followed by anti-FLAG Western blotting (WB) upon Dox induction (Fig. 8C, left lanes). Therefore, we transiently transfected these cells with the m153HA construct, or empty vector, to determine if IP for Clr-bFLAG could coimmunoprecipitate m153HA. However, FLAG tag IP followed by anti-HA WB failed to detect any m153HA protein, even though HA tag IP followed by anti-HA WB of the same cells was able to detect m153HA (Fig. 8D). HA tag IP followed by anti-FLAG WB failed to detect any Clr-bFLAG protein (Fig. 8C, right lanes). These results suggest that either the two proteins do not interact directly or the IP conditions may have disrupted noncovalent (direct or indirect) interactions between Clr-b and m153. However, similar experiments using a milder detergent, digitonin, also failed to detect an interaction (data not shown).
We also attempted to visualize whether Clr-b and m153 may colocalize to the same subcellular compartments upon overexpression. We generated vectors encoding EGFP or mCherry fusion proteins of either Clr-bFLAG or m153HA and then analyzed transient NIH 3T3 transfectants using fluorescence microscopy. Upon visualization, m153 constructs (m153HA-EGFP/mCherry) were primarily detected in intracellular punctate structures, whereas Clr-b constructs (Clr-bFLAG-EGFP/mCherry) were detected more diffusely throughout the cell and at the cell surface. Notably, this was reproducibly independent of the fluorophore combinations used (Fig. 8E, top left and middle images). To validate colocalization, we cotransfected NIH 3T3 cells with constructs encoding each protein fused to two independent fluorophores; importantly, these showed overlapping expression, as expected (Fig. 8E, right images). In contrast, cotransfection of NIH 3T3 cells with constructs encoding the two proteins fused to alternate fluorophores revealed limited colocalization of m153HA and Clr-bFLAG (Fig. 8E, bottom images). Although ambiguous, these results suggest that the majority of the m153 and Clr-b proteins largely fail to colocalize to the same subcellular compartments upon transfection; consequently, they may not directly interact with one another in a heteromeric complex, or m153 may target alternative Clr family members or other proteins.
Nonetheless, to further investigate interaction between m153 and Clr-b at the posttranslational level, we conducted MCMV infections of our stable NIH 3T3.Clr-bFLAG transductants. Basal endogenous Clr-b expression in the absence of Dox was similar to parental NIH 3T3 cells, while an ∼15-fold increase in Clr-b levels was detected using 4A6 MAb upon Dox induction (Fig. 9A). Upon infection using WT or Δm153 MCMV, uninduced NIH 3T3.Clr-bFLAG cells downregulated Clr-b levels similar to parental NIH 3T3 cells, with the Δm153 mutant promoting a more pronounced Clr-b loss (Fig. 9A). Moreover, in the presence of Dox induction, the ability of m153 to sustain Clr-b expression was clearly evident, as only an ∼4-fold reduction in Clr-b MFI levels was observed using WT MCMV, while an ∼70-fold reduction was observed using the Δm153 mutant (Fig. 9A and B, right histograms and graph, respectively). This was further evident when comparing the percentage of Clr-b– cells, where the Δm153 mutant approached ≥40% Clr-b– cells, a magnitude not seen using WT MCMV (Fig. 9C, right graph).
FIG 9.

Prominent Clr-b loss in cells overexpressing Clr-b when infected with MCMV lacking m153. NIH 3T3.Clr-b fibroblasts were cultured in the absence or presence of Dox and infected with either WT or Δm153 MCMV virus and analyzed using flow cytometry 24 hpi. (A) Clr-b cell surface expression measured by anti-Clr-b (4A6 MAb). The vertical dotted line symbolizes mock MFI, whereas numbers on the left represent the percent Clr-b– cells in the gate and numbers on the right represent MFI values. (B) Quantitation of Clr-b MFI in panel A, normalized to Clr-b levels on resting uninduced cells. (C) Measurements of percent Clr-b– cells with different viruses in panel A. (D) Clr-b cell surface expression measured by anti-FLAG (M2 MAb). (E) Quantitation of Clr-b MFI in panel D. (F) Measurements of percent Clr-b– cells with different viruses in panel D. Data were analyzed using 1-way ANOVA and are representative of those from 3 to 6 independent experiments.
Using the same cells, we also analyzed ectopic Clr-bFLAG expression in the absence of endogenous Clr-b using anti-FLAG MAb. While minimal basal FLAG tag staining was observed on uninduced NIH 3T3.Clr-bFLAG cells, Dox induction promoted high levels of the exogenous Clr-bFLAG protein (Fig. 9D). Following infection, Clr-b downregulation was clearly exacerbated using the Δm153 mutant in comparison to that with WT MCMV (Fig. 9D to F). These results demonstrate the efficacy of MCMV infection in promoting a loss of cell surface Clr-b protein and highlight the clear impact of m153 in stabilizing Clr-b levels during MCMV infection.
In vitro and in vivo analysis of WT and m153-deficient MCMV.
In order to test the functional role of m153, we first assessed NKR-P1B–Clr-b interactions in the context of m153 expression via BWZ reporter cell assays using stimulator cells varying in m153 expression. Briefly, BWZ.P1B reporter cells induce NFAT (nuclear factor of activated T cells)-driven β-galactosidase (lacZ) activity upon ligation of a CD3ζ/NKR-P1B chimeric fusion receptor by cognate Clr-b ligand, relative to parental BWZ.36 cells. In order to determine if we could detect differences in receptor interactions upon MCMV infection with WT and Δm153 viruses we infected NIH 3T3.Clr-bFLAG in the presence of Dox and used these cells as stimulator cells (Fig. 10A). Using this approach, the functional effect of m153 in sustaining Clr-b levels was revealed, whereby a significant increase in BWZ.P1B reporter cell activity was observed using WT-infected versus Δm153-infected stimulator cells.
FIG 10.

In vivo and in vitro analyses reveal additional modes of immunomodulation for m153. (A) NIH 3T3.Clr-bFLAG cells were infected with WT or Δm153 virus and cocultured with BWZ.NKR-P1B reporter cells to measure receptor-ligand interactions using the BWZ reporter assay. (B to C) NIH 3T3 cells were infected with WT and Δm153 virus and then used as targets in a 51Cr release assay using NKR-P1B+ (B) and NKR-P1B– (C) splenic NK-LAKs (NKp46+CD3–). Data were analyzed using 2-way ANOVA. (D and E) In vivo MCMV infection using WT or Δm153 viruses on WT/B6, Clrb–/–, and Nkrp1b–/– mice measuring viral titers in spleen (D) and liver (E) 3 days postinfection. Data are cumulative of those from 2 independent experiments including 3 or 4 mice/group. (F) Tetramers of m153 and HLA-B*57 were used to stain the DC2.4 (DC) and MNK-3 (ILC3) cell lines. Numbers in italics on the top right corner represent MFI values; numbers above gates represent positive gated cell percentages; the dotted line represents negative-control MFI values. Data are representative of those from 2 independent experiments.
We tested the MCMV variants in NK cell-mediated cytotoxicity assays. To this end, we sorted NKR-P1B+ and NKR-P1B– splenic NK-LAK (NKp46+) effectors for use in 51Cr release cytotoxicity assays against MCMV-infected NIH 3T3 target cells. In agreement with a previous report, NKR-P1B+ NK-LAK cells displayed slightly higher levels of cytotoxicity in general than did NKR-P1B– NK-LAK cells (39). Interestingly, cytotoxicity of Δm153-infected targets was slightly augmented in comparison to that for cells infected with WT virus (Fig. 10B and C). Surprisingly, this result was consistent regardless of NKR-P1B receptor expression on the NK-LAK effectors, and statistically significant, although the magnitude was more pronounced using NKR-P1B+ NK-LAK cells. Together, these results demonstrate that expression of m153 protects infected cells from NK-mediated cytotoxicity in vitro.
To determine the role of m153 in vivo, and in the context of NKR-P1B–Clr-b interactions, we used cohorts of WT B6 mice, ligand-deficient Clr-b–/– mice, and receptor-deficient Nkrp1b–/– mice and infected all three cohorts using either WT MCMV or Δm153 mutant. Consistent with previous results (36, 40), infection using WT MCMV demonstrates that Nkrp1b–/– mice possess significantly lower splenic viral titers than do WT/B6 mice, while Clr-b–/– mice show a trend (albeit not significant) toward higher viral titers (Fig. 10D). Interestingly, infection using the Δm153 mutant resulted in similar overall phenotypes among the three host cohorts, with significantly lower viral titers in Nkrp1b–/– mice, and an insignificant trend toward higher titers in Clr-b–/– mice, versus those in WT B6 mice. Unexpectedly, however, splenic viral titers in all three host strains were elevated using Δm153 mutant versus WT MCMV (Fig. 10D). No differences were observed between WT and Δm153 viruses in livers of infected mice (Fig. 10E). These results demonstrate that while m153 stabilizes Clr-b levels during MCMV infection, it appears to be deleterious for overall viral fitness in the B6 host strain. Thus, the elevated viral titers seen using the Δm153 mutant suggest that m153 may engage an unknown stimulatory receptor; while this could involve alternative NKR-P1–Clr family interactions, we cannot rule out the involvement of other receptor-ligand systems.
To examine direct interactions between m153 and cellular receptors or ligands in trans, we used m153 tetramers to stain various cell lines in vitro. Interestingly, m153 tetramers reproducibly stained cells of the DC2.4 dendritic cell (DC) line at high levels, as well as a subset of the mouse ILC3-like MNK-3 cell line at lower levels (Fig. 10F). These findings suggest that additional interactions between m153 and an unknown receptor or ligand on innate DCs and ILC may influence the immune response to MCMV in vivo. The generation of a cDNA library from DC2.4 cells combined with the use of m153 tetramers should facilitate the cloning of this putative m153 receptor.
Nonetheless, the reduced viral titers observed in Nkrp1b–/– mice in this study and elsewhere (40) are consistent with the effects of the Clr-b-independent MCMV m12 decoy ligand recognized by the inhibitory NKR-P1B receptor (36). In contrast, the trend toward higher viral titers in Clr-b–/– mice suggests a more complex, perhaps m153-independent, role for Clr-b in restraining viral fitness, perhaps due to NK cell education effects (30). Collectively, these data suggest that MCMV employs multiple strategies to modulate NKR-P1B–Clr-b interactions, in addition to multiple roles for m153 in regulating viral fitness in vivo, making it difficult to decipher the direct influence of m153 in isolation.
DISCUSSION
Betaherpesviruses dedicate a significant portion of their genome to viral immunoevasive genes that subvert host immune recognition. Here we report that the viral MHC-I homolog m153 plays a role in limiting the downregulation of the host inhibitory NKR-P1B ligand Clr-b during MCMV infection. Host cells infected with m153-deficient (Δm153) MCMV display an exacerbated “missing-self” loss of Clr-b surface expression, with faster kinetics and to a greater extent than WT MCMV. In addition, cells overexpressing m153 have increased basal levels of Clr-b compared to those of control cells. Despite efforts in this study to identify a direct mechanism, the effect of m153 on Clr-b expression appears to be indirect, as the two proteins fail to interact upon coimmunoprecipitation (co-IP), and they colocalize only minimally upon transfection. However, the effect of m153 on Clr-b levels is independent of transcription and likely posttranslational in nature.
Earlier work described m153 as a glycoprotein whose cell surface expression is independent of β2-microgloblin and TAP expression (41). The resolution of the m153 crystal structure revealed several features. First, m153 exhibits a closed binding cleft, unlikely to accommodate peptides or other small molecular ligands (41). Second, it exists as m153 homodimers, a finding that was corroborated by intracellular complementation experiments, and consistent with our observations (Fig. 8). Nevertheless, functions for this protein have not previously been described.
Previous work has shown that Clr-b is lost in response to a number of cellular pathologies. These include transformation, genotoxic and cellular stress, and viral infection (27, 31). The last finding has been documented in a variety of infection models, including poxviruses (vaccinia and ectromelia viruses) (35), rat CMV (RCMV) English (34), MCMV (29), and RNA viruses (influenza and encephalomyocarditis virus [unpublished observations]). Thus, Clr-b appears to be a “healthy-self” marker on normal cells, and Clr-b downregulation appears to be a host response to viral infection that promotes “missing-self” recognition via loss of NKR-P1B inhibition. In support of this hypothesis, both RCMV and MCMV encode viral immunoevasins that act as surrogate ligands to functionally engage NKR-P1B and circumvent this missing-self NK cell recognition axis (34, 36). In turn, cell surface Clr-b expression appears to be regulated by sensors that detect cellular fitness versus pathology, and thus the ligand behaves as an innate relay mechanism to alert NK cells, where loss of Clr-b acts as a “pattern of pathogenesis” (42). This missing-self role is also consistent with reports of short-lived Clec2d transcripts, as well as Clr-b protein, in the absence of constitutive or basal nascent transcription in healthy cells (29, 43).
Given that m153 expression stabilizes cell surface Clr-b expression, this is suggestive that m153 functions to prevent host-mediated Clr-b loss. Despite our attempts at resolving this matter, the mechanism behind Clr-b stabilization by m153 remains ambiguous. We hypothesized that m153 may function similarly to the m04 gene product (gp34 glycoprotein), perhaps binding directly to Clr-b and escorting it to the cell surface, in order to maintain inhibitory signals via NKR-P1B. However, direct protein-protein interactions and substantial direct cellular colocalization could not be demonstrated between m153 and Clr-b. The fact that the Δm153 virus is capable of enhancing Clr-b downregulation, even on cells that express ectopic Clr-bFLAG, demonstrates that this mechanism is likely posttranslational in nature. Thus, m153 might interfere with the host ubiquitin proteasomal degradation, autophagic internalization machinery, or cytoskeletal interruptions that lead to loss of self ligands from the cell surface upon infection. However, similar stabilization effects on MHC-I or Rae-1 expression were not observed upon transfection of m153, or upon infection with m153-deficient MCMV.
Despite the differential Clr-b expression on fibroblasts upon infection with m153-sufficient or -deficient viruses, in vivo infection of mice reveals a more complex role for m153. Our in vitro evidence suggests that an m153-deficient virus, lacking the ability to sustain Clr-b expression during the acute phase of infection, might result in lower viral titers, as this virus may be eliminated through more efficient NKR-P1B-mediated missing-self recognition. However, we observed a contrasting phenotype, whereby removing m153 yielded a more virulent virus. This is independent of Clr-b and NKR-P1B, as similar trends were observed in Clec2d–/– and Klrb1b–/– mice; thus, these data highlight the potential for additional functions of the m153 protein, one of which may include direct engagement of a host activating receptor. Our data using m153 tetramers support this possibility, and in fact, it was previously reported that m153 binds to an unknown ligand or receptor expressed on DCs and NK cells (44). In light of this, to truly determine the role of m153-driven Clr-b modulation during MCMV infection, it is necessary to first identify the m153-interacting partner(s). These findings would also help navigate the interplay between m153 and Clr-b in the context of the m12 immunoevasin.
In conclusion, these findings reveal a novel putative immune evasion mechanism by which MCMV targets the host innate immune response. The human genome encodes a single conserved inhibitory NKR-P1 family member, NKR-P1A (CD161; KLRB1), suggesting that HCMV or other viruses may also employ analogous mechanisms to evade NK cell recognition by modulating expression of the NKR-P1A ligand LLT-1 (CLEC2D).
MATERIALS AND METHODS
Animals.
C57BL/6 (B6) mice were obtained from Jackson Laboratories. B6.Clec2d–/– (Clr-b–/–) mice were provided by M. T. Gillespie (Monash University, Australia) (30, 45). B6.Klrb1b–/– (Nkrp1b–/–) mice were generated as described previously (32). All animals were maintained in accordance with approved animal care protocols within the University of Toronto network (including Sunnybrook Research Institute) or the University of Ottawa.
Cells and viruses.
Mouse embryonic fibroblasts (NIH 3T3) were obtained from the ATCC. Human embryonic kidney cells (HEK293T) were obtained from D. H. Raulet (University of California, Berkeley, CA). C57BL/6 mouse embryonic fibroblasts (MEF) were provided by T. W. Mak (University of Toronto, Canada). Cells were cultured in complete Dulbecco modified Eagle medium (DMEM) supplemented with 2 mM glutamine, 100 U/ml of penicillin, 100 μg/ml of streptomycin, 50 μg/ml of gentamicin, 110 μg/ml of sodium pyruvate, 50 μM 2-mercaptoethanol, 10 mM HEPES, and 10% fetal bovine serum (FBS).
The MCMV MW97.01 (WT) strain was generated using bacterial artificial chromosome (BAC) technology of a cloned MCMV Smith genome (46). The MCMV-Δ1 and -Δ6 viruses were generated as described previously using BAC cloning. The m153-deficient MCMV virus (Δm153) was constructed by removing the m153 gene using ET cloning from the full-length MCMV BAC pSM3fr (47). All viruses were passaged and titers were determined using MEF as described previously, without centrifuging the supernatant (48). Unless otherwise noted, all in vitro MCMV infections were done by exposing cells to 0.5 PFU/cell followed by centrifugal enhancement at 800 × g for 30 min (corresponding to effective MOI of 10). For in vivo infections, mice were infected with 2 × 106 PFU/mouse by intraperitoneal injection.
Generation of Dox-inducible cell lines.
The pTRIPZ-Empty vector was generated by inserting the multiple-cloning site from pEGFP-N1 (AgeI to XhoI; Clontech) and placing it into the parental pTRIPZ lentiviral vector (Thermo Fisher Scientific), which was digested with AgeI and XhoI. Similarly, the pTRIPZ-m153HA vector was constructed by PCR amplification of an N-terminally HA-tagged m153 (m153HA) cDNA using 5′ AgeI and 3′ XhoI primers and subcloning into the digested pTRIPZ vector. pTRIPZ-Clr-bFLAG was synthesized by a similar approach by amplifying a C-terminally FLAG-tagged Clr-b (Clr-bFLAG) cDNA. All PCR primers are listed in Table 1. These vectors were transfected into HEK293T cells with packaging vectors to generate lentivirus, which were then used to transduce NIH 3T3 fibroblasts. Transduced cells were selected using media containing 2.5 μg/ml of puromycin (Sigma-Aldrich).
TABLE 1.
Primers used in this study
| Gene | Primer | Sequence (5′–3′) |
|---|---|---|
| Cloning of m02 family and m145 familya | ||
| N-terminally HA tagging m153 | ||
| m153HA | m153 NT-HA Fwd1 | TTGCTTACCCATACGATGTTCCAGATTACGCTGGATCAGGATCAGAGGTCGTGCGGCCCGAAGT |
| m153 NT-HA Xho F2 | gcctcgagATGTCTGCACTTCTGATCCTAGCTCTTGTTGGAGCTGCAGTTGCTTACCCATACGATGTT | |
| m153 NotI R | ggcggccgcTCACACCACATTCTCCTCCGTA | |
| Construction of type I m153 reporters | ||
| m153 EC | m153 XhoI ATG Fwd | gctcgagATGATTCCCCTTCTCCTTCTGCCG |
| m153 NF-PSP Fwd | gcctcgagATGTCTGCACTTCTGATCCTAGCT | |
| m153 EC Rev | tagcggccgcGGTCAGTCTCGAATCGTTGATCGTCCTCTGG | |
| Construction of Tet-inducible pTRIPZ vectors | ||
| m153 | m153 NT-HA AgeI-ATG F | ataaccggtcgccaccATGTCTGCACTTCTGATCCTAGC |
| m153 XhoI-TGA R | tatctcgagTCACACCACATTCTCCTCCGTATCCG | |
| Clr-b | Clr-b AgeI-ATG F | ataaccggtcgccaccATGTGTGTCACAAAGGCTTCC |
| Clr-b C-FLAG XhoI-TGA R | tatctcgagCTACTTGTCGTCGTCGTCCTTGTAGTCTGATCCTGATCCGGAAGGAAAAAAAGGAGTTTGG | |
| Construction of EGFP/mCherry fusion constructs | ||
| m153 | m153 fusion XhoI F | ggactcagatctcgagcgccaccATGTCTGCACTTCTGATCCTAGCTCTTGTTGGAGC |
| m153 fusion BamHI R | ggcgaccggtggatccCCTGATCCTGATCCCACCACATTCTCCTCCGTATCCGAGCACTCG | |
| Clr-b | Clr-b fusion XhoI F | atctcgagGATCAGGATCAATGTGTGTCACAAAGGCTTCC |
| Clr-b fusion BamHI R | gtggatccTCACTTGTCGTCGTCGTCCTTGTAGTCTGATCCTGATCCGGAAGGAAAAAAAGGAGTTTGGCAGTGG | |
| qRT-PCR primers | ||
| m153 | m153 qPCR Fwd | GTGTCAGATGACGACCCAGG |
| m153 qPCR Rev | TCTGACTTCTGTTGACCGGC |
The complete m02 and m145 family were cloned as previously described (36).
Flow cytometry and antibodies.
Cells were stained in flow buffer (0.5% bovine serum albumin [BSA] and 0.03% NaN3 in Hanks’ balanced salt solution [HBSS]) on ice with primary monoclonal antibodies (MAb) for 25 to 30 min, or secondary streptavidin (SA) conjugates for 15 to 20 min, washed, and then analyzed using a FACSCalibur flow cytometer (BD Biosciences). Live cells were gated using propidium iodide exclusion. Data were analyzed using FlowJo 9 software (FlowJo, LLC). Anti-Clr-b MAb (4A6; rat IgM), anti-NKR-P1B (2D12), and anti-m153 MAb (clone m153.16) have been previously described (26, 27, 41). Anti-NK1.1 (PK136), anti-NKp46 (29A1.4), and anti-CD3ε (145-2C11) were purchased from BioLegend. Biotin-conjugated H-2DqLq MAb (KH117; mouse IgG2a) was purchased from BD Pharmingen, purified pan-Rae1 MAb (186107; rat IgG2a) was purchased from R&D Systems, and SA-allophycocyanin (APC) was purchased from Thermo Fisher Scientific. Anti-FLAG (M2 MAb) was purchased from Sigma-Aldrich, and anti-HA (C29F4 MAb) was purchased from Cell Signaling Technologies.
RNA isolation, cloning, and qRT-PCR.
Total RNA isolation was performed using an RNA isolation kit (Norgen Biotek) and then quantitated and assessed for integrity on a 1% agarose gel using ethidium bromide. First-strand cDNA synthesis was accomplished using the Superscript III kit (Thermo Fisher Scientific). Cloning of MCMV genes was performed using cDNA from MCMV-infected NIH 3T3 cells, a Q5 high-fidelity PCR kit (New England BioLabs [NEB]), and gene-specific primers (Table 1), followed by cloning into the pIRES2-EGFP vector (Clontech). Quantitative real-time reverse transcription-PCR (qRT-PCR) was performed on a CFX-96 system (Bio-Rad) using 20 to 50 ng of cDNA, PerfeCTa mix (Quanta), and gene-specific primers designed using Primer-BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast). Data were analyzed using CFX Manager software (Bio-Rad).
Immunoprecipitation, Western blotting, and immunofluorescence.
Immunoprecipitations (IP) were performed by transfecting NIH 3T3.Clr-bFLAG cells with the pIRES2-EGFP vector containing either m153HA or an empty control. Forty-eight hours later, cells were lysed in lysis buffer (0.5% NP-40, 50 mM Tris-HCl, 150 mM NaCl, 10% glycerol) and IP was performed using specific antibodies bound to protein A/G agarose resin (ThermoFisher Scientific). Following IP, lysates were incubated with 2× Laemmli buffer, boiled, run on SDS-PAGE gels, and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore) for immunoblotting using anti-FLAG–HRP or anti-HA–HRP MAb conjugates.
Immunofluorescence experiments were performed by transfection of NIH 3T3 fibroblasts (grown on poly-l-lysine-treated coverslips) with pEGFP-N1/C1 or pmCherry-N1/C1 vectors containing Clr-b or m153 fused in frame with either EGFP or mCherry. After 48 h, cells were fixed, stained with 4′,6-diamidino-2-phenylindole (DAPI), mounted onto microscope slides, and imaged for fluorescence using an Axiovert 200M wide-field fluorescence microscope (Zeiss).
BWZ reporter cell assays.
BWZ.CD3ζ-NKR-P1B reporter cells were generated as previously described (28). BWZ assays were performed by coculturing stimulator cells in 3-fold dilutions with reporter cells (5 × 104/well) in 96-well flat-bottom plates overnight at 37°C. Positive-control cells were incubated with 10 ng/ml of phorbol myristate acetate (PMA) plus 0.5 μM ionomycin. Cells were subsequently washed using phosphate-buffered saline (PBS) and resuspended in 100 μl of 1× CPRG buffer (90 mg/liter of chlorophenol-red-β-d-galactopyranoside [Sigma-Aldrich], 9 mM MgCl2, and 0.1% NP-40 in PBS), incubated at room temperature, and analyzed using a Varioskan microplate reader (Thermo Scientific) using absorbance readings (optical density [OD] at 595 to 655 nm).
Chromium release assays.
Cytotoxicity assays were performed as previously described (30). B6 splenic lymphokine-activated killer (LAK) effector cells were cultured in 10% complete RPMI 1640 supplemented with 2,500 U/ml of human recombinant interleukin 2 (rIL-2; Proleukin; Novartis). On day 4, these cells were sorted for CD3ε– NKp46+ cells, further fractionated for NKR-P1B+/– expression, and used as effectors in 51Cr release assays on day 7. Target cells were incubated with 50 μCi of Na251CrO4 (PerkinElmer) in FBS at 37°C for 1 h, washed, plated in V-bottom wells in combination with serially diluted effectors, and incubated for 4 h at 37°C. Supernatants (100 μl) were transferred to LumaPlate-96 scintillation plates (PerkinElmer), dried, and counted using a Top Count NXT microplate scintillation counter (Packard Instrument Company). Percent specific lysis values were calculated relative to maximum release (2% Triton X-100 in media) and spontaneous release (media) values.
Statistical analysis.
All data were analyzed using Prism 5 (GraphPad), employing either a paired Student two-tailed t test or one- or two-way analysis of variance (ANOVA) applying Bonferroni’s post hoc tests (see figure legends for details). Graphs show mean values ± standard errors of the means (SEM). Statistical significance is shown in figures as follows: *, P < 0.05; **, P < 0.01; and ***, P < 0.001. Data are representative of those from at least 3 independent experiments.
ACKNOWLEDGMENTS
We thank L. L. Lanier for critical readings of the manuscript.
O.A.A. was supported by a Postgraduate Scholarship-Doctoral Award from the National Sciences and Engineering Research Council of Canada. Oscar A. Aguilar holds a Postdoctoral Enrichment Program Award from the Burroughs Wellcome Fund (BWF) and is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute. I.S.S. was supported by a Science Without Borders scholarship from the Brazilian National Council for Scientific and Technological Development (CNPq). This work was supported by operating grants from the Canadian Institute of Health Research (number 86630 to A.P.M. and J.R.C., number 106491 to J.R.C., and number 136808 to B.T.), by an Investigator in the Pathogenesis of Infectious Disease from the Burroughs Wellcome Fund (number 1007761 to J.R.C.), a European Regional and Development Fund (K.01.1.1.01.0006) awarded to the Scientific Centre of Excellence for Virus Immunology and Vaccines and cofinanced by the European Regional Development Fund (S.J. and A.K.), and by the intramural research program of the NIAID, NIH (ZIA AI001028-11 to D.H.M.).
REFERENCES
- 1.Rawlinson WD, Farrell HE, Barrell BG. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol 70:8833–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffiths P, Baraniak I, Reeves M. 2015. The pathogenesis of human cytomegalovirus. J Pathol 235:288–297. doi: 10.1002/path.4437. [DOI] [PubMed] [Google Scholar]
- 3.Krmpotic A, Bubic I, Polic B, Lucin P, Jonjic S. 2003. Pathogenesis of murine cytomegalovirus infection. Microbes Infect 5:1263–1277. doi: 10.1016/j.micinf.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Lanier LL. 2005. NK cell recognition. Annu Rev Immunol 23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 5.Lanier LL. 2008. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol 8:259–268. doi: 10.1038/nri2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. 2002. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 7.Farrell HE, Vally H, Lynch DM, Fleming P, Shellam GR, Scalzo AA, Davis-Poynter NJ. 1997. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature 386:510–514. doi: 10.1038/386510a0. [DOI] [PubMed] [Google Scholar]
- 8.Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM. 2002. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A 99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeleznjak J, Lisnic VJ, Popovic B, Lisnic B, Babic M, Halenius A, L’Hernault A, Rovis TL, Hengel H, Erhard F, Redwood AJ, Vidal SM, Dolken L, Krmpotic A, Jonjic S. 29 May 2019. The complex of MCMV proteins and MHC class I evades NK cell control and drives the evolution of virus-specific activating Ly49 receptors. J Exp Med doi: 10.1084/jem.20182213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleijnen MF, Huppa JB, Lucin P, Mukherjee S, Farrell H, Campbell AE, Koszinowski UH, Hill AB, Ploegh HL. 1997. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J 16:685–694. doi: 10.1093/emboj/16.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babic M, Pyzik M, Zafirova B, Mitrovic M, Butorac V, Lanier LL, Krmpotic A, Vidal SM, Jonjic S. 2010. Cytomegalovirus immunoevasin reveals the physiological role of “missing self” recognition in natural killer cell dependent virus control in vivo. J Exp Med 207:2663–2673. doi: 10.1084/jem.20100921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pyzik M, Charbonneau B, Gendron-Pontbriand E-M, Babić M, Krmpotić A, Jonjić S, Vidal SM. 2011. Distinct MHC class I-dependent NK cell-activating receptors control cytomegalovirus infection in different mouse strains. J Exp Med 208:1105–1117. doi: 10.1084/jem.20101831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kielczewska A, Pyzik M, Sun T, Krmpotic A, Lodoen MB, Munks MW, Babic M, Hill AB, Koszinowski UH, Jonjic S, Lanier LL, Vidal SM. 2009. Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J Exp Med 206:515–523. doi: 10.1084/jem.20080954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lenac T, Budt M, Arapovic J, Hasan M, Zimmermann A, Simic H, Krmpotic A, Messerle M, Ruzsics Z, Koszinowski UH, Hengel H, Jonjic S. 2006. The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J Exp Med 203:1843–1850. doi: 10.1084/jem.20060514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krmpotic A, Busch DH, Bubic I, Gebhardt F, Hengel H, Hasan M, Scalzo AA, Koszinowski UH, Jonjic S. 2002. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat Immunol 3:529–535. doi: 10.1038/ni799. [DOI] [PubMed] [Google Scholar]
- 16.Arapović J, Lenac Rovis T, Reddy AB, Krmpotić A, Jonjić S. 2009. Promiscuity of MCMV immunoevasin of NKG2D: m138/fcr-1 down-modulates RAE-1epsilon in addition to MULT-1 and H60. Mol Immunol 47:114–122. doi: 10.1016/j.molimm.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 17.Krmpotic A, Hasan M, Loewendorf A, Saulig T, Halenius A, Lenac T, Polic B, Bubic I, Kriegeskorte A, Pernjak-Pugel E, Messerle M, Hengel H, Busch DH, Koszinowski UH, Jonjic S. 2005. NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J Exp Med 201:211–220. doi: 10.1084/jem.20041617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL. 2003. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med 197:1245–1253. doi: 10.1084/jem.20021973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasan M, Krmpotic A, Ruzsics Z, Bubic I, Lenac T, Halenius A, Loewendorf A, Messerle M, Hengel H, Jonjic S, Koszinowski UH. 2005. Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J Virol 79:2920–2930. doi: 10.1128/JVI.79.5.2920-2930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arapovic J, Lenac T, Antulov R, Polic B, Ruzsics Z, Carayannopoulos LN, Koszinowski UH, Krmpotic A, Jonjic S. 2009. Differential susceptibility of RAE-1 isoforms to mouse cytomegalovirus. J Virol 83:8198–8207. doi: 10.1128/JVI.02549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greene TT, Tokuyama M, Knudsen GM, Kunz M, Lin J, Greninger AL, DeFilippis VR, DeRisi JL, Raulet DH, Coscoy L. 2016. A herpesviral induction of RAE-1 NKG2D ligand expression occurs through release of HDAC mediated repression. Elife 5:e14749. doi: 10.7554/eLife.14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lenac Rovis T, Kucan Brlic P, Kaynan N, Juranic Lisnic V, Brizic I, Jordan S, Tomic A, Kvestak D, Babic M, Tsukerman P, Colonna M, Koszinowski U, Messerle M, Mandelboim O, Krmpotic A, Jonjic S. 2016. Inflammatory monocytes and NK cells play a crucial role in DNAM-1-dependent control of cytomegalovirus infection. J Exp Med 213:1835–1850. doi: 10.1084/jem.20151899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verma S, Loewendorf A, Wang Q, McDonald B, Redwood A, Benedict CA. 2014. Inhibition of the TRAIL death receptor by CMV reveals its importance in NK cell-mediated antiviral defense. PLoS Pathog 10:e1004268. doi: 10.1371/journal.ppat.1004268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarama A, Perez-Carmona N, Farre D, Tomic A, Borst EM, Messerle M, Jonjic S, Engel P, Angulo A. 2014. Cytomegalovirus m154 hinders CD48 cell-surface expression and promotes viral escape from host natural killer cell control. PLoS Pathog 10:e1004000. doi: 10.1371/journal.ppat.1004000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirkham CL, Carlyle JR. 2014. Complexity and diversity of the NKR-P1:Clr (Klrb1:Clec2) recognition systems. Front Immunol 5:214. doi: 10.3389/fimmu.2014.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iizuka K, Naidenko OV, Plougastel BF, Fremont DH, Yokoyama WM. 2003. Genetically linked C-type lectin-related ligands for the NKRP1 family of natural killer cell receptors. Nat Immunol 4:801–807. doi: 10.1038/ni954. [DOI] [PubMed] [Google Scholar]
- 27.Carlyle JR, Jamieson AM, Gasser S, Clingan CS, Arase H, Raulet DH. 2004. Missing self-recognition of Ocil/Clr-b by inhibitory NKR-P1 natural killer cell receptors. Proc Natl Acad Sci U S A 101:3527–3532. doi: 10.1073/pnas.0308304101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen P, Belanger S, Aguilar OA, Zhang Q, St-Laurent A, Rahim MM, Makrigiannis AP, Carlyle JR. 2011. Analysis of the mouse 129-strain Nkrp1-Clr gene cluster reveals conservation of genomic organization and functional receptor-ligand interactions despite significant allelic polymorphism. Immunogenetics 63:627–640. doi: 10.1007/s00251-011-0542-8. [DOI] [PubMed] [Google Scholar]
- 29.Aguilar OA, Mesci A, Ma J, Chen P, Kirkham CL, Hundrieser J, Voigt S, Allan DS, Carlyle JR. 2015. Modulation of Clr ligand expression and NKR-P1 receptor function during murine cytomegalovirus infection. J Innate Immun 7:584–600. doi: 10.1159/000382032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen P, Aguilar OA, Rahim MM, Allan DS, Fine JH, Kirkham CL, Ma J, Tanaka M, Tu MM, Wight A, Kartsogiannis V, Gillespie MT, Makrigiannis AP, Carlyle JR. 2015. Genetic investigation of MHC-independent missing-self recognition by mouse NK cells using an in vivo bone marrow transplantation model. J Immunol 194:2909–2918. doi: 10.4049/jimmunol.1401523. [DOI] [PubMed] [Google Scholar]
- 31.Fine JH, Chen P, Mesci A, Allan DS, Gasser S, Raulet DH, Carlyle JR. 2010. Chemotherapy-induced genotoxic stress promotes sensitivity to natural killer cell cytotoxicity by enabling missing-self recognition. Cancer Res 70:7102–7113. doi: 10.1158/0008-5472.CAN-10-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rahim MM, Chen P, Mottashed AN, Mahmoud AB, Thomas MJ, Zhu Q, Brooks CG, Kartsogiannis V, Gillespie MT, Carlyle JR, Makrigiannis AP. 2015. The mouse NKR-P1B:Clr-b recognition system is a negative regulator of innate immune responses. Blood 125:2217–2227. doi: 10.1182/blood-2014-02-556142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka M, Fine JH, Kirkham CL, Aguilar OA, Belcheva A, Martin A, Ketela T, Moffat J, Allan DSJ, Carlyle JR. 2018. The inhibitory NKR-P1B:Clr-b recognition axis facilitates detection of oncogenic transformation and cancer immunosurveillance. Cancer Res 78:3589–3603. doi: 10.1158/0008-5472.CAN-17-1688. [DOI] [PubMed] [Google Scholar]
- 34.Voigt S, Mesci A, Ettinger J, Fine JH, Chen P, Chou W, Carlyle JR. 2007. Cytomegalovirus evasion of innate immunity by subversion of the NKR-P1B:Clr-b missing-self axis. Immunity 26:617–627. doi: 10.1016/j.immuni.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 35.Williams KJ, Wilson E, Davidson CL, Aguilar OA, Fu L, Carlyle JR, Burshtyn DN. 2012. Poxvirus infection-associated downregulation of C-type lectin-related-b prevents NK cell inhibition by NK receptor protein-1B. J Immunol 188:4980–4991. doi: 10.4049/jimmunol.1103425. [DOI] [PubMed] [Google Scholar]
- 36.Aguilar OA, Berry R, Rahim MM, Reichel JJ, Popovic B, Tanaka M, Fu Z, Balaji GR, Lau TN, Tu MM, Kirkham CL, Mahmoud AB, Mesci A, Krmpotic A, Allan DS, Makrigiannis AP, Jonjic S, Rossjohn J, Carlyle JR. 2017. A viral immunoevasin controls innate immunity by targeting the prototypical natural killer cell receptor family. Cell 169:58–71.e14. doi: 10.1016/j.cell.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Balaji GR, Aguilar OA, Tanaka M, Shingu-Vazquez MA, Fu Z, Gully BS, Lanier LL, Carlyle JR, Rossjohn J, Berry R. 2018. Recognition of host Clr-b by the inhibitory NKR-P1B receptor provides a basis for missing-self recognition. Nat Commun 9:4623. doi: 10.1038/s41467-018-06989-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weizman OE, Song E, Adams NM, Hildreth AD, Riggan L, Krishna C, Aguilar OA, Leslie CS, Carlyle JR, Sun JC, O’Sullivan TE. 2019. Mouse cytomegalovirus-experienced ILC1s acquire a memory response dependent on the viral glycoprotein m12. Nat Immunol 20:1004. doi: 10.1038/s41590-019-0430-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aust JG, Gays F, Mickiewicz KM, Buchanan E, Brooks CG. 2009. The expression and function of the NKRP1 receptor family in C57BL/6 mice. J Immunol 183:106–116. doi: 10.4049/jimmunol.0804281. [DOI] [PubMed] [Google Scholar]
- 40.Rahim MM, Wight A, Mahmoud AB, Aguilar OA, Lee SH, Vidal SM, Carlyle JR, Makrigiannis AP. 2016. Expansion and protection by a virus-specific NK cell subset lacking expression of the inhibitory NKR-P1B receptor during murine cytomegalovirus infection. J Immunol 197:2325–2337. doi: 10.4049/jimmunol.1600776. [DOI] [PubMed] [Google Scholar]
- 41.Mans J, Natarajan K, Balbo A, Schuck P, Eikel D, Hess S, Robinson H, Simic H, Jonjic S, Tiemessen CT, Margulies DH. 2007. Cellular expression and crystal structure of the murine cytomegalovirus major histocompatibility complex class I-like glycoprotein, m153. J Biol Chem 282:35247–35258. doi: 10.1074/jbc.M706782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vance RE, Isberg RR, Portnoy DA. 2009. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6:10–21. doi: 10.1016/j.chom.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martick M, Horan LH, Noller HF, Scott WG. 2008. A discontinuous hammerhead ribozyme embedded in a mammalian messenger RNA. Nature 454:899–902. doi: 10.1038/nature07117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mans J. 2008. Characterization of mouse cytomegalovirus MHC-1 homologs. PhD dissertation. University of Witwatersrand, Johannesburg, South Africa. [Google Scholar]
- 45.Kartsogiannis V, Sims NA, Quinn JM, Ly C, Cipetic M, Poulton IJ, Walker EC, Saleh H, McGregor NE, Wallace ME, Smyth MJ, Martin TJ, Zhou H, Ng KW, Gillespie MT. 2008. Osteoclast inhibitory lectin, an immune cell product that is required for normal bone physiology in vivo. J Biol Chem 283:30850–30860. doi: 10.1074/jbc.M801761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner M, Jonjic S, Koszinowski UH, Messerle M. 1999. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol 73:7056–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wagner M, Koszinowski UH. 2004. Mutagenesis of viral BACs with linear PCR fragments (ET recombination). Methods Mol Biol 256:257–268. doi: 10.1385/1-59259-753-X:257. [DOI] [PubMed] [Google Scholar]
- 48.Brune W, Hengel H, Koszinowski UH. 2001. A mouse model for cytomegalovirus infection. Curr Protoc Immunol Chapter 19:Unit 19.17. [DOI] [PubMed] [Google Scholar]