

Type I interferons (IFNs) possess a range of biological activity and protect against a number of viruses, including alphaviruses. Despite signaling through a shared receptor, there are established biochemical and functional differences among the IFN subtypes. The significance of our research is in demonstrating that IFN-α and IFN-β both have protective roles during acute chikungunya virus (CHIKV) infection but do so by distinct mechanisms. IFN-α limits CHIKV replication and dissemination, whereas IFN-β protects from CHIKV pathogenesis by limiting inflammation mediated by neutrophils. Our findings support the premise that the IFN subtypes have distinct biological activities in the antiviral response.
KEYWORDS: chikungunya virus, host-pathogen interactions, innate immunity, interferons
ABSTRACT
Type I interferons (IFNs) are key mediators of the innate immune response. Although members of this family of cytokines signal through a single shared receptor, biochemical and functional variation exists in response to different IFN subtypes. While previous work has demonstrated that type I IFNs are essential to control infection by chikungunya virus (CHIKV), a globally emerging alphavirus, the contributions of individual IFN subtypes remain undefined. To address this question, we evaluated CHIKV pathogenesis in mice lacking IFN-β (IFN-β knockout [IFN-β-KO] mice or mice treated with an IFN-β-blocking antibody) or IFN-α (IFN regulatory factor 7 knockout [IRF7-KO] mice or mice treated with a pan-IFN-α-blocking antibody). Mice lacking either IFN-α or IFN-β developed severe clinical disease following infection with CHIKV, with a marked increase in foot swelling compared to wild-type mice. Virological analysis revealed that mice lacking IFN-α sustained elevated infection in the infected ankle and in distant tissues. In contrast, IFN-β-KO mice displayed minimal differences in viral burdens within the ankle or at distal sites and instead had an altered cellular immune response. Mice lacking IFN-β had increased neutrophil infiltration into musculoskeletal tissues, and depletion of neutrophils in IFN-β-KO but not IRF7-KO mice mitigated musculoskeletal disease caused by CHIKV. Our findings suggest disparate roles for the IFN subtypes during CHIKV infection, with IFN-α limiting early viral replication and dissemination and IFN-β modulating neutrophil-mediated inflammation.
IMPORTANCE Type I interferons (IFNs) possess a range of biological activity and protect against a number of viruses, including alphaviruses. Despite signaling through a shared receptor, there are established biochemical and functional differences among the IFN subtypes. The significance of our research is in demonstrating that IFN-α and IFN-β both have protective roles during acute chikungunya virus (CHIKV) infection but do so by distinct mechanisms. IFN-α limits CHIKV replication and dissemination, whereas IFN-β protects from CHIKV pathogenesis by limiting inflammation mediated by neutrophils. Our findings support the premise that the IFN subtypes have distinct biological activities in the antiviral response.
INTRODUCTION
Type I interferons (IFNs) are a family of multifunctional cytokines that consists of 14 IFN-α subtypes and single forms of IFN-β, IFN-ε, IFN-κ, and IFN-ω in mice. Their broad, pleiotropic properties include upregulating cell-intrinsic antiviral defense mechanisms, modulating proinflammatory cytokine production, and augmenting innate and adaptive cellular immune responses (1). IFNs are induced rapidly in response to infection with viruses and other pathogens. Host pattern recognition receptors (PRRs) that recognize nucleic acids are important sensors of viral infection. For many cells, the PRR signaling response leads to the production of the IFN-β and IFN-α4 subtypes via activation of IFN regulatory factor 3 (IRF3) (2–6). These IFN subtypes signal in an autocrine and paracrine manner to modulate the expression of IFN-stimulated genes (ISGs), which include antiviral effectors and immunoregulatory molecules. IRF7 is among the transcriptional regulators induced by IFN-β and IFN-α4 and participates in a positive-feedback loop that induces the other IFN-α subtypes, thus amplifying and diversifying the response (7–9). The central role of IRF7 in inducing the IFN-α subtypes is demonstrated in IRF7-deficient mice, which fail to produce significant levels of IFN-α in response to multiple viral infections (10–16).
IFNs exert their effects by binding to the shared heterodimeric IFN-α/β receptor (IFNAR), which is composed of the IFNAR1 and IFNAR2 subunits. Engagement of IFNAR activates Janus kinase (JAK) and signal transducer and activator of transcription (STAT) signaling programs to induce the expression of hundreds of ISGs (17). Despite signaling through a single, shared receptor, the IFN subtypes have distinct properties, presumably due to different binding affinities and receptor dissociation rates for the individual IFN subtypes. These parameters ultimately affect receptor internalization, intracellular regulators, and feedback loops. Biochemical and functional studies have revealed that some IFN properties, such as antiproliferative activity, depend on cellular context and affinity for the receptors. Others, such as antiviral activity, improve only marginally with increased affinity and appear programmed for maximal output by most subtypes irrespective of affinity or cellular context (18, 19). The ability of the IFN receptor to have graded responses to multiple ligands likely explains the pleiotropic activities ascribed to different IFN subtypes.
Some of the first attempts to delineate properties of individual IFN subtypes in vivo came with the generation of mice that specifically lack IFN-β (20). IFN-β knockout (IFN-β-KO) mice are more susceptible to vaccinia virus, influenza A virus, and West Nile virus (WNV) (21–24). For these viruses, IFN-β appears to restrict viral replication in a number of tissues and cell types. The lack of globally IFN-α-deficient mice had hindered the direct functional comparison of IFN-α versus IFN-β in vivo, although the recent development of blocking monoclonal antibodies (mAbs) specific for IFN-α or IFN-β has made such studies possible (13). The use of these blocking antibodies in a study with persistent lymphocytic choriomeningitis virus (LCMV) infection demonstrated that IFN-α but not IFN-β was the dominant subtype controlling early LCMV replication and spread. In comparison, IFN-β unexpectedly was detrimental to the host response and responsible for promoting LCMV persistence. Blockade of IFN-β improved antiviral T cell responses and allowed for clearance of persistent LCMV (25–27). These studies with LCMV demonstrated distinguishable biological activities of IFN subtypes, with profound implications for viral pathogenesis and immunity.
Chikungunya virus (CHIKV) is a mosquito-transmitted alphavirus that causes explosive outbreaks of acute fever, rash, polyarthritis, arthralgia, and myositis (28, 29). Studies with a mouse model of CHIKV arthritis have demonstrated that these symptoms reflect an interplay between extensive viral replication and damage mediated by immune cells, such as monocytes, macrophages, and activated CD4+ T cells (30–34). However, not all immune responses are detrimental. Neutralizing antibodies and IFNs are important in controlling CHIKV infection, and mice lacking IFNAR1 expression rapidly succumb to disseminated infection (11, 35). Despite their essential role in limiting CHIKV infection, little is known about the contributions of individual IFN subtypes to protection. To explore this question, we used genetic deletion mutants and mAb blockade to determine the functions of IFN-α and IFN-β during acute CHIKV infection. While both IFN-α and IFN-β protected the host from clinical disease induced during acute CHIKV infection, distinct roles for the IFN subtypes were observed, with IFN-α limiting early viral replication and spread and IFN-β controlling neutrophil-mediated inflammation. These results define a mechanism by which IFN-α can have biological activity that is distinguished from that of IFN-β.
RESULTS
IFN-α and IFN-β protect against CHIKV-induced clinical disease.
Although type I IFN signaling protects against CHIKV pathogenesis, the function of individual IFN subtypes remains poorly characterized. To begin to address this question, we evaluated the pathogenesis of CHIKV in wild-type, IFN-β-KO, and IRF7-KO C57BL/6 mice. As IRF7-KO mice lose amplification of IFN-α during CHIKV infection, with a minimal impact on IFN-β production (11), these mice were utilized, since pan-IFN-α knockout mice are not available. Animals were inoculated with 103 PFU of CHIKV (LR2006 OPY1 strain) and monitored daily for foot swelling. Previous studies have described a biphasic swelling pattern in the foot of CHIKV-infected wild-type mice, with a first peak observed at 2 to 3 days postinfection (dpi) and a second, larger peak at 6 to 7 dpi. Mice lacking IFN-β developed more severe foot swelling, with kinetics similar to those seen in wild-type mice (Fig. 1A). In contrast, mice lacking IRF7 developed a large initial peak of swelling at 3 to 4 dpi and a delayed second peak at 11 dpi. Both IFN-β-KO and IRF7-KO mice also displayed prolonged foot swelling through 28 dpi compared to wild-type mice, which typically recover by about 12 dpi (Fig. 1A). These data suggest that IFN-α and IFN-β have protective roles in limiting musculoskeletal disease during CHIKV infection.
FIG 1.
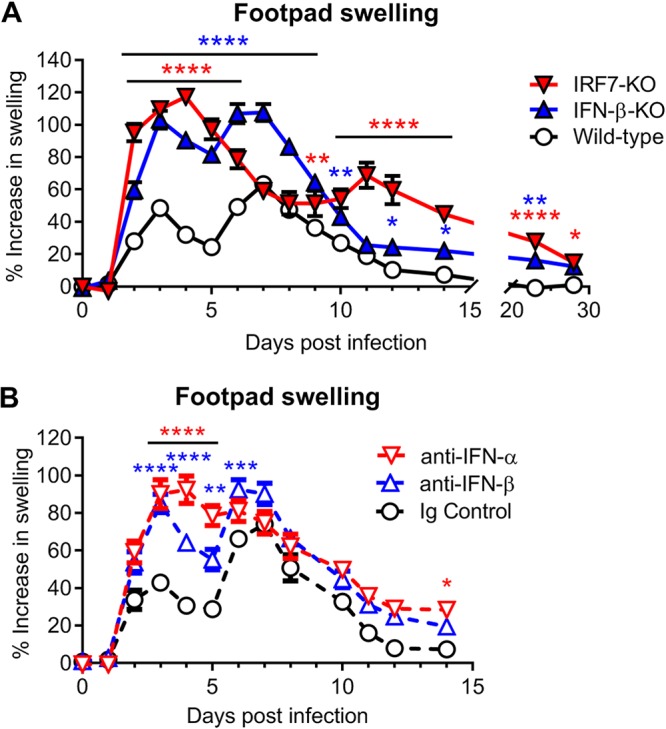
IFN-α and IFN-β protect against CHIKV-induced clinical disease. Shown are data for swelling of the ipsilateral feet of mice inoculated with 103 PFU of CHIKV. Foot swelling was measured daily for wild-type, IFN-β-KO, or IRF7-KO mice (A) or for wild-type mice treated with anti-mouse IFN-α (TIF-3C5), anti-mouse IFN-β (HDβ-4A7), or isotype control antibody 1 day before and 1 day after infection (B), as described in Materials and Methods. Data are reported as percent increases in foot area (vertical × horizontal [square millimeters]) over baseline. Data are pooled from at least two experiments with 18 to 22 mice per group (A) or 10 mice per group (B) and expressed as means ± standard errors of the means (SEM) (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 [by two-way ANOVA with Dunnett’s posttest]).
Although IRF7 transcriptionally activates IFN-α production, it also regulates antiviral gene expression independently of IFN production (36–38). To evaluate if the phenotype in IRF7-deficient mice was due to a loss of IFN-α, we utilized a pan-IFN-α-blocking mAb (TIF-3C5). In parallel, we also used an IFN-β-specific blocking mAb (HDβ-4A7) to assess the impact of individual IFN subtypes on CHIKV pathogenesis (13, 27). Wild-type mice were treated with the blocking antibodies 1 day before and 1 day after CHIKV infection and monitored for swelling in the ipsilateral foot. Wild-type mice treated with anti-IFN-β mAb blockade had similarly increased foot swelling as seen in IFN-β-KO mice (Fig. 1B). Moreover, administration of the pan-IFN-α-blocking antibody phenocopied the foot swelling of IRF7-KO mice until approximately 9 to 10 dpi, with a severe increase in foot swelling compared to isotype control mAb-treated mice observed at 3 to 5 dpi. Pan-IFN-α mAb treatment, however, failed to reproduce the second swelling peak at 11 dpi observed in the IRF7-KO mice (Fig. 1B). This discrepancy may arise from an incomplete blockade of IFN-α due to the administration of only two doses of blocking antibody or from IFN-α-independent effects of IRF7. Regardless, these data confirm that the early foot swelling phenotypes in IFN-β-KO and IRF7-KO mice are due to the loss of IFN-β and IFN-α, respectively, and suggest that the differences in swelling are conferred by distinct activities of IFN-α and IFN-β.
IFN-α but not IFN-β limits CHIKV replication and dissemination.
To further investigate the differential functions of IFN-α and IFN-β during CHIKV infection, we determined the viral burden in the ipsilateral foot and distal tissues at several times postinfection. Mice lacking IFN-β had viral loads in the ipsilateral foot that were similar to those in wild-type mice at each time point analyzed, and clearance of replicating virus occurred in both strains by 10 dpi (Fig. 2A). In contrast, mice deficient for IRF7 sustained increased viral loads compared to wild-type mice, with an ∼8-fold increase in viral titers at 2 and 3 dpi and an ∼137-fold increase in viral titers at 7 dpi (Fig. 2A).
FIG 2.
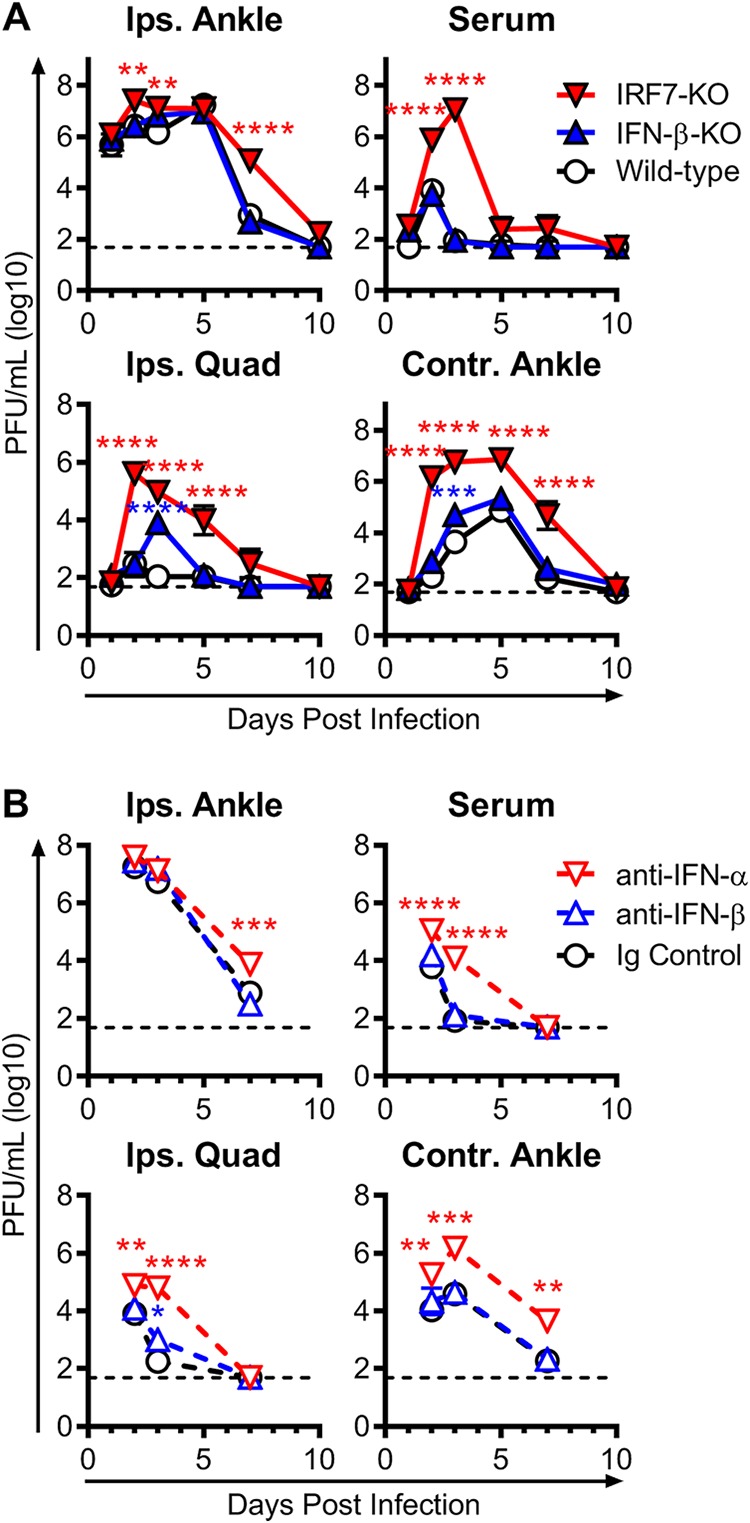
IFN-α but not IFN-β limits CHIKV replication and dissemination. Mice were inoculated with 103 PFU of CHIKV, and the levels of infectious virus were measured by a plaque assay in the ipsilateral ankle (Ips. Ankle), serum, ipsilateral quadriceps muscle (Ips. Quad), and contralateral ankle (Contr. Ankle) at the indicated time points from wild-type, IFN-β-KO, or IRF7-KO CHIKV-infected mice (A) or from wild-type mice treated with anti-mouse IFN-α (TIF-3C5), anti-mouse IFN-β (HDβ-4A7), or isotype control antibody 1 day before and 1 day after infection (B), as described in Materials and Methods. The dashed lines represent the limit of detection. Data are pooled from 2 to 3 experiments with 6 to 8 mice per group and are expressed as medians ± SEM (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 [by two-way ANOVA with Dunnett’s posttest]).
We evaluated the impact of the loss of IFN-β or IFN-α on dissemination of CHIKV to distant tissues. The loss of IFN-β had little effect on viral dissemination. Viral burdens in the serum were similar between wild-type and IFN-β-KO mice at all time points examined. Similarly, we observed no differences in the dissemination to and clearance from distant sites, except for an increase in viral titers in the quadriceps muscle (∼72-fold) and contralateral ankle (∼6-fold) of IFN-β-KO mice only at 3 dpi (Fig. 2A). Analogously, blockade of IFN-β with mAb treatment in wild-type mice had a minimal impact on viral burden in the ipsilateral foot or distal sites compared to mice treated with an isotype control mAb (Fig. 2B).
In contrast, IFN-α had a substantive role in regulating viral replication and dissemination. IRF7-KO mice had high and prolonged viremia compared to wild-type and IFN-β-KO mice, with ∼92-fold and ∼128,000-fold increases at 2 and 3 dpi, respectively (Fig. 2A). Markedly elevated viral titers were also seen in the contralateral ankle (∼7,190-fold) and the quadriceps (∼1,350-fold) of IRF7-KO mice at 2 dpi, at a time when replication is only just detectable in wild-type and IFN-β-KO mice. These increased viral loads were still present in the quadriceps and contralateral ankles of the IRF7-KO mice at 5 and 7 dpi, respectively, when both IFN-β-KO and wild-type mice had cleared infectious virus. This increase in viral replication and dissemination was due to the loss of IFN-α since treatment of wild-type mice with the pan-IFN-α-blocking mAb also resulted in an elevated viral load in the inoculated ankle at 7 dpi, severe viremia, and substantially increased dissemination to the muscle and distal joints (Fig. 2B).
Previous studies using bone marrow chimeras demonstrated that IFN signaling is required in nonhematopoietic cells to survive CHIKV infection (39). To determine if IFN signaling on hematopoietic cells had any role in CHIKV-induced clinical disease, replication, or dissemination, we utilized Vav-Cre transgenic mice crossed onto the IFNAR1flox/flox background, which removes IFN signaling from cells of hematopoietic origin (Fig. 3A and B) (40, 41). Consistent with the bone marrow chimera studies, infection of Vav-Cre+ mice with 103 PFU of CHIKV in the foot resulted in no lethality and did not yield any significant differences in foot swelling, compared to Cre− littermate controls (Fig. 3C). We also observed no significant differences in viral replication in the ipsilateral ankle or distant tissues, although there was a slight delay in clearance of CHIKV from the serum in the Vav-Cre+ mice (Fig. 3D). These data confirm that type I IFNs principally signal on nonhematopoietic cells to exert their protective effects on CHIKV arthritis. Altogether, these data suggest distinct antiviral mechanisms of action, with IFN-α controlling early replication and spread and IFN-β being largely dispensable for these effects.
FIG 3.

IFN-α and IFN-β protect against CHIKV-induced clinical disease by signaling on nonhematopoietic cells. Vav-Cre+ mice crossed to IFNAR1flox/flox mice or Cre− littermate controls were inoculated with 103 PFU of CHIKV. (A and B) Splenic single-cell suspensions were stained and analyzed by flow cytometry to confirm IFNAR1 deletion. (A) Representative histograms for IFNAR1 from Vav-Cre− or Vav-Cre+ mice compared to a fluorescence-minus-one (FMO) control. (B) Quantification of the mean fluorescence intensity (MFI) of IFNAR1 on CD45+ splenic cells normalized to the FMO control MFI. Flow cytometry results are representative of data from 2 independent experiments with 8 total mice per group, and data are presented as means ± SEM (***, P < 0.0001 [by an unpaired t test]). (C) Foot swelling in Vav-Cre+ and Vav-Cre− mice was measured daily with digital calipers. Results are pooled from two experiments with 6 to 9 mice per group, and swelling data are presented as means ± SEM (not significant [P > 0.05] by two-way ANOVA with Sidak’s posttest). (D) Infectious virus in the joints, muscle, and serum was determined by a plaque assay at the indicated time points. Data are presented as medians ± SEM (*, P < 0.05 [by two-way ANOVA with Sidak’s posttest]).
IFN-β-KO but not IRF7-KO mice have increased immune cell infiltration in the joints during acute CHIKV infection.
In a wild-type mouse, acute CHIKV infection causes edema and swelling within the skin and subcutaneous tissues that are driven by viral replication, cell death, and local proinflammatory cytokine production, and immune cells infiltrating into the joint spaces, muscle, and surrounding soft tissues (30–32, 42). We determined the impact of the loss of IFN-α or IFN-β on inflammatory infiltrates in the joint-associated tissues. At 7 dpi, histology revealed that 100% of the IFN-β-KO mice showed severe inflammation in the infected foot, whereas more variability existed in the wild-type and IRF7-KO mice (Fig. 4A to D and Q). All infected mice showed edema in the hypodermis of the skin, and this was especially evident in the IRF7-KO mice (Fig. 4E to H). Additionally, all infected mice showed evidence of cellular infiltrates in muscle tissues (Fig. 4I to L). Cellular infiltration into the joint spaces was evident in wild-type, IFN-β-KO, and IRF7-KO mice (Fig. 4M to P), but only the IFN-β-KO mice showed a significant increase in cellular infiltration into the midfoot joint spaces (Fig. 4R). These data suggest that IFN-β modulates immune cell recruitment to a greater degree than IFN-α during acute CHIKV infection.
FIG 4.

IFN-β-KO but not IRF7-KO mice have increased immune cell infiltration in the joints during acute CHIKV infection. Wild-type, IFN-β-KO, or IRF7-KO mice were mock treated or inoculated with 103 PFU of CHIKV, and ipsilateral ankles/feet were processed at 3 or 7 dpi for histological analysis by hematoxylin and eosin staining. (A to P) Representative images of sections of the midfoot (tiled), skin, muscle, and synovium at 7 dpi. (E to H) The skin and associated tissue sections show the epidermis (e), dermis (d), and hypodermis (h). (M to P) The synovium sections show synovium (s) and bone (b), with arrows indicating immune infiltrates into the synovial cavity. Bars = 200 μm (A to D), 50 μm (E to H and M to P), and 100 μm (I to L). (Q) Feet and ankles were scored for histological damage as described in Materials and Methods. (R) Quantification of inflammatory cells per high-power field (HPF) in the midfoot joint spaces as described in Materials and Methods. For panels Q and R, data are pooled from at least two experiments with 4 to 6 mice per group. Data were analyzed using one-way ANOVA (Q) or two-way ANOVA (R) with Tukey’s posttest. All data are presented as means ± SEM. (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
IFN-β-deficient mice have increased neutrophil recruitment in the foot during CHIKV infection.
Macrophages, monocytes, and neutrophils are among the early cells recruited to the infected foot that mediate clinical disease (30, 34, 43, 44). Because mice lacking IFN-β developed increased foot swelling by 2 dpi and increased cellular infiltrates in the joint spaces by as early as 3 dpi (Fig. 1 and 4), we hypothesized that IFN-β might protect against host-mediated inflammation during CHIKV infection. To evaluate this idea, cellular infiltrates in the foot of infected wild-type or IFN-β-KO mice were isolated and analyzed at 2 or 3 dpi by flow cytometry to determine the total number of live leukocytes (CD45+), neutrophils (Ly6G+ CD11b+), monocytes (Ly6C+ CD11b+ Ly6G−), and macrophages (F4/80+ MERTK+) (Fig. 5A) (45). Although we detected no significant difference in the total numbers of CD45+ cells, monocytes, or macrophages between wild-type and IFN-β-KO mice at 2 or 3 dpi, we observed increases in the fraction and number of neutrophils (4-fold) in mice lacking IFN-β compared to wild-type mice (Fig. 5B and C). By 3 dpi, there was no longer a difference in neutrophil numbers, although there was still a small increase in the percentage of neutrophils infiltrating the IFN-β-KO foot.
FIG 5.

IFN-β-deficient mice have increased neutrophil recruitment in the foot during CHIKV infection. Wild-type or IFN-β-KO mice were inoculated with 103 PFU of CHIKV, and cells were isolated from the ipsilateral foot at 2 or 3 dpi and analyzed by flow cytometry as described in Materials and Methods. (A) Gating strategy showing subpopulations of live CD45+ cells in wild-type (WT) and IFN-β-KO mice. (B and C) Total number of isolated CD45+ leukocytes and total number (B) or percentage (C) of CD11b+ Ly6G+ neutrophils, CD11b+ Ly6C+ Ly6G− monocytes, and F4/80+ MERTK+ macrophages. Results are pooled from two experiments with 6 to 7 mice per group and represent the means ± SEM (*, P < 0.05; **, P < 0.01; ****, P < 0.0001 [by two-way ANOVA with Sidak’s posttest]).
To determine the potential basis for the increased neutrophil numbers in the IFN-β-KO mice, we next assessed local cytokine and chemokine levels in the ipsilateral foot at 0.5, 1, 2, and 3 dpi. Unexpectedly, we did not observe elevated levels of common neutrophil-attracting chemokines, such as CXCL1 (KC) or CXCL2 (MIP-2), in IFN-β-KO mice compared to wild-type mice, nor was there an increase in proinflammatory mediators, such as interleukin-1α/β (IL-1α/β) or tumor necrosis factor alpha (TNF-α) (Fig. 6), which can modulate cellular recruitment by upregulating endothelial adhesion molecules. Instead, in mice lacking IFN-β, there were significant decreases in IL-1β, TNF-α, CXCL9 (MIG), CXCL10 (IP-10), CCL2 (MCP-1), CCL3 (MIP-1α), CCL5 (RANTES), and eotaxin at 2 or 3 dpi (Fig. 6). Taken together with the Vav-Cre findings (Fig. 3), these data suggest that IFN-β acts on nonhematopoietic cells to influence neutrophil recruitment and/or accumulation in the foot during acute CHIKV infection by an unclear mechanism.
FIG 6.
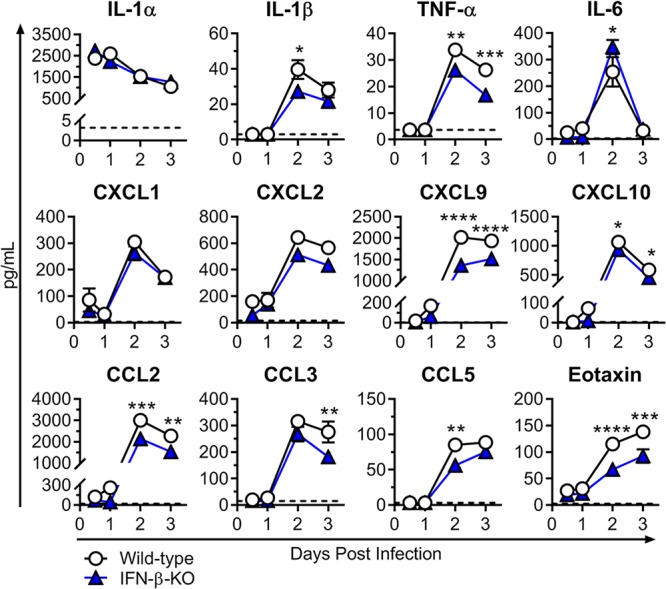
Cytokine and chemokine levels in the ipsilateral feet of wild-type and IFN-β-KO mice. Mice were inoculated with 103 PFU of CHIKV via the subcutaneous route. Cytokine and chemokine levels in the ipsilateral foot were determined using Luminex technology (Millipore) at the indicated time points as described in Materials and Methods. Results are pooled from two experiments with 6 to 7 mice per group and represent the means ± SEM (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 [by two-way ANOVA with Sidak’s posttest]). Dashed lines indicate the limit of detection.
Depletion of neutrophils alleviates the increased foot swelling in CHIKV-infected IFN-β-KO mice but not IRF7-KO mice.
We next assessed whether greater numbers of infiltrating neutrophils were responsible for the enhanced clinical disease severity in the IFN-β-KO mice. We depleted neutrophils by intraperitoneal administration of anti-Ly6G mAb (clone 1A8) 1 day before infection and every other day through 7 dpi in wild-type, IFN-β-KO, and IRF7-KO mice. Neutrophil depletion was confirmed by flow cytometry of cells isolated from peripheral blood and the ipsilateral foot at 2 dpi (Fig. 7A and B). Whereas neutrophil depletion had no effect on foot swelling in wild-type mice, it alleviated the increased foot swelling observed in the IFN-β-KO mice (Fig. 7C and D). The beneficial effects of neutrophil depletion in the IFN-β-KO mice began at 2 dpi, when differences in foot swelling are first observed, and this protective effect persisted throughout the course of acute infection (Fig. 7D). In contrast, neutrophil depletion had no effect on the increased foot swelling observed in the IRF7-KO mice (Fig. 7E). Thus, neutrophils are required for the exacerbated clinical disease observed in IFN-β-deficient mice during CHIKV infection, and this further highlights the distinct mechanisms that drive pathogenesis in IFN-β-KO versus IRF7-KO mice.
FIG 7.
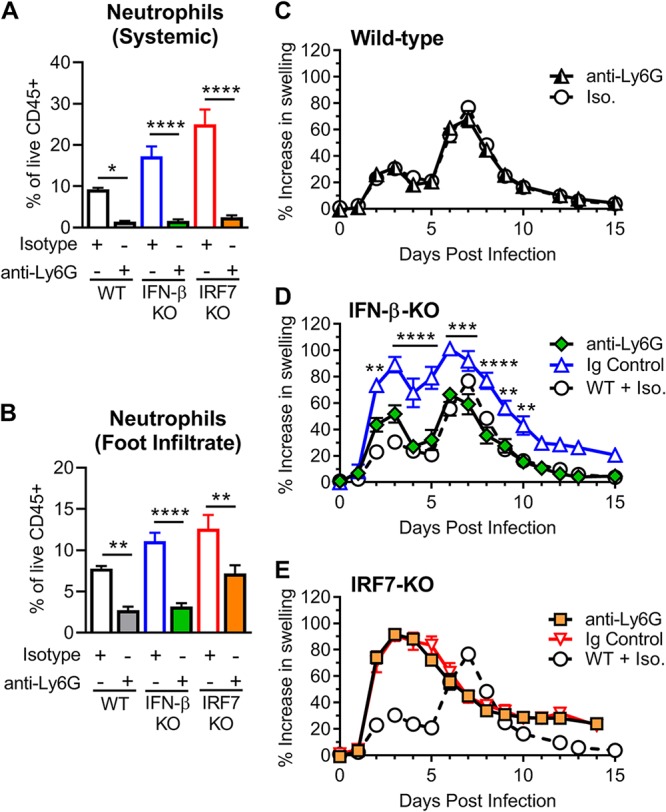
Depletion of neutrophils alleviates the increased foot swelling in CHIKV-infected IFN-β-KO mice but not in IRF7-KO mice. Wild-type, IFN-β-KO, or IRF7-KO mice were inoculated with 103 PFU of CHIKV and administered anti-Ly6G (1A8) or isotype control (Iso.) antibody beginning 1 day before infection and then every other day through 7 dpi. (A and B) Cells were isolated from blood (A) or foot tissue (B) at 2 dpi and analyzed by flow cytometry to confirm neutrophil depletion as described in Materials and Methods. Results are pooled from two experiments with 4 to 6 mice per group and reported as the means ± SEM (*, P < 0.05; **, P < 0.01; ****, P < 0.0001 [by one-way ANOVA with Tukey’s posttest]). (C to E) Swelling of the ipsilateral feet/ankles of wild-type (WT) (C), IFN-β-KO (D), or IRF7-KO (E) mice. Swelling data are pooled from two experiments with 5 to 8 mice per group and reported as the means ± SEM (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001 [by two-way ANOVA with Dunnett’s posttest]).
DISCUSSION
Much of what is known about type I IFN effects on viral replication and pathogenesis in vivo comes from the use of animals lacking IFN signaling through IFNAR1 deletion, which has not allowed delineation of the roles of individual IFN subtypes. Advances in generating mice that specifically lack IFN-β and mAbs that block specific IFN subtypes have enabled investigation of the functional differences between IFN-α and IFN-β. In this study, we show that IFN-α and IFN-β have distinct protective roles during acute CHIKV infection. IFN-α subtypes limited CHIKV replication and spread, whereas IFN-β functioned primarily to limit inflammation by modulating neutrophil accumulation at the site of infection.
We demonstrate an important role for IFN-α in controlling CHIKV replication and dissemination. IRF7-KO mice developed worse clinical disease and sustained higher viral loads in the inoculated foot. The most striking phenotype observed in the IRF7-KO mice was the markedly increased viremia and dissemination to distal sites. This severe disseminated CHIKV infection may be the result of decreased IFN-α activity in the serum of IRF7-KO mice (11, 12). Indeed, multiple studies have demonstrated that IRF7-KO mice produce little to no systemic IFN-α during infection with LCMV, WNV, dengue virus (DENV), herpes simplex virus 1 (HSV-1), and encephalomyocarditis virus (EMCV), and IRF7-KO mice display increased susceptibility to these infections (10–16). We confirmed that the major protective function of IRF7 during CHIKV infection was the production and amplification of IFN-α, since pan-IFN-α mAb blockade mimicked the clinical and virological phenotypes observed in IRF7-KO mice during acute CHIKV infection. Other studies have also demonstrated an antiviral role of IFN-α in vivo. During persistent LCMV (clone 13) infection, the blockade of IFNAR led to increased early dissemination, and this was attributed to a loss of IFN-α, as the loss of IFN-β did not have this effect. Moreover, the specific blockade of IFN-α with a pan-IFN-α antibody led to increased viral loads late in infection, which was not observed with blockade or genetic deletion of IFN-β (27). Our findings are consistent with evidence pointing to the importance of IFN-α in restricting viral replication and spread.
The cell types involved in IFN-α antiviral responses depend on the model. For example, IFN signaling on CD11c+ and LysM+ myeloid cells is critical for controlling WNV and DENV infections (46, 47), and IFN signaling in astrocytes regulates blood-brain barrier permeability in the hindbrain region during WNV infection (48). Loss of IFNAR1 expression during persistent LCMV infection led to early increased viral replication in splenic dendritic cells and macrophages, and this was attributed to a loss of IFN-α, since the loss of IFN-β had no impact on viral loads at these early time points (27). In addition, IFNAR1 deletion during infection with Sindbis virus (SINV), an alphavirus related to CHIKV, resulted in increased susceptibility to infection and altered tropism of myeloid cells in the draining lymph node and spleen (49). We show that deletion of IFNAR expression on all immune cells through the utilization of Vav-Cre+ IFNAR1flox/flox mice did not impact CHIKV-induced foot swelling and had a minimal impact on CHIKV replication and spread, indicating that IFN-α and IFN-β exert their protective effects through their action on nonhematopoietic cells. These findings agree with bone marrow chimera studies that demonstrated that IFN signaling is required on nonhematopoietic cells to survive CHIKV infection (39). These nonhematopoietic cells likely include muscle fibers, myoblasts, muscle satellite cells, dermal and synovial fibroblasts, and osteoblasts, all of which are targets of CHIKV infection (11, 50, 51).
While most published studies on IFN-α support its important role in limiting viral replication and spread, the functions attributed to IFN-β vary with the virus studied. In some models, IFN-β limits viral replication. For example, loss of IFN-β during infection with WNV or vaccinia virus led to higher viral loads in some but not all tissues (21, 24). Loss of IFN-β resulted in a significant increase in WNV replication in dendritic cells, macrophages, some neuron populations, and, to a lesser extent, fibroblasts. In our study, the loss of IFN-β exacerbated acute CHIKV clinical disease, with a minimal impact on the viral load at the site of inoculation or in distant tissues, suggesting that IFN-β functions dominantly as an immunomodulatory molecule. IFN-β has documented immunomodulatory activities, and the therapeutic benefits of this property are demonstrated in its efficacy against multiple sclerosis. In this context, IFN-β treatment improves the frequency and suppressive function of regulatory T cells and also downregulates antigen presentation and T cell stimulation in myeloid cells (52, 53). However, there is growing appreciation that IFNs can also have detrimental consequences, especially during chronic viral infections and some bacterial infections (1). During chronic LCMV infection, IFN-β was dispensable for controlling early viral replication but had a detrimental role for the host in promoting LCMV persistence (27). Selective blockade of IFN-β led to accelerated virus clearance through improved antiviral T cell responses (27, 54). Even though CD4+ T cells contribute to acute CHIKV pathogenesis (31), we found that the loss of IFN-β led to an early (2 dpi) worsening of clinical disease that correlated with increased neutrophil accumulation at the site of infection. Moreover, neutrophils are required for the phenotype in IFN-β-KO mice, as their depletion reversed the disease exacerbation.
Dysregulated neutrophil recruitment has been associated with acute CHIKV disease. Mice deficient for chemokine receptor 2 (CCR2) or Toll-like receptor 3 (TLR3) developed more severe CHIKV arthritis, and this was correlated with early and severe neutrophil infiltration (44, 55). The loss of CCR2 did not impact viral loads, and the increased neutrophils seen in the feet of infected mice were accompanied by a decrease in the recruitment of monocytes and increases in CXCL1, CXCL2, and IL-1β mRNA levels in the infected foot (44). In the case of TLR3 deficiency, viral loads were increased at distant tissue sites but were less affected in the infected foot, and increased neutrophils were associated with decreased CCR2 expression and increased IL-1β expression in the foot tissue (55). The mechanism by which IFN-β regulates neutrophil recruitment to the joints during CHIKV infection remains unclear. Although neutrophil numbers were increased, we did not observe a significant decrease in monocyte recruitment or alterations in other cells types or in total immune cells. Our antibody staining panel was not exhaustive, and there could be other cell types altered in the immune infiltrate of IFN-β-KO mice, such as mast cells or basophils. Neutrophil recruitment to inflamed tissues requires coordination between multiple proinflammatory cytokines and chemokines, especially the CXCR2 ligands CXCL1 (KC) and CXCL2 (MIP-2) (56). In a cancer model, the loss of IFN-β resulted in increases in the expression of CXCL1 and CXCL2, altering neutrophil infiltration into the tumor environment (57), yet our analysis revealed no such increase in levels of these chemokines or proinflammatory cytokines, including TNF-α, IL-1, or IL-6, in the foot tissue of IFN-β-KO mice. Instead, we observed decreased levels of many proinflammatory cytokines and chemokines, which may reflect that the ligands for CXCR3—CXCL9, CXCL10, and CXCL11—are all ISGs induced downstream of type I IFN and type II IFN (IFN-γ) signaling (17, 58–60). While we did not observe an increase in cytokine or chemokine levels, there could be other mediators of inflammation, such as leukotrienes or complement, contributing to neutrophil accumulation in the IFN-β-KO mice (56, 61, 62).
Neutrophils can eliminate microbial agents through multiple mechanisms, including phagocytosis, the production of neutrophil extracellular traps (NETs), and degranulation of antimicrobial peptides and enzymes, which kill microbial agents but can also damage the host (63). Type I IFNs can prime neutrophils to produce NETs and may therefore alter their function (64). In addition, recruited neutrophils die through various mechanisms after infiltrating the site of inflammation, and the loss of IFN-β has been previously described to promote the longevity of tumor-associated neutrophils (57). We cannot rule out the possibility that the loss of IFN-β qualitatively alters neutrophil activation, function, or survival akin to that seen with IFN-λ (65–67). However, it is important to point out that the removal of IFN signaling from immune cells did not alter CHIKV-induced foot swelling, suggesting that if neutrophil function or longevity is altered, it would be from indirect effects of IFN-β signaling on nonimmune cells in musculoskeletal tissues. Studies exploring the responses of muscle cells and synovial and dermal fibroblasts to type I IFNs may be needed to understand how this microenvironment regulates the recruitment and the function of immune cells, including neutrophils, in the inflamed joint.
Our studies add to the accumulating evidence that IFN subtypes have distinct activities in vivo, and the mechanisms underlying the differential functions can vary depending on the biological context. We propose a model in which IFN-α is the dominant subtype eliciting antiviral responses, thus controlling CHIKV replication and spread even in the absence of IFN-β. In contrast, IFN-β modulates host immunity to protect against inflammation mediated by neutrophils during acute CHIKV infection. Both IFN-α and IFN-β exert their effects by signaling on nonhematopoietic cells, which may include dermal and synovial fibroblasts and muscle cells. While the antiviral effects of IFN-α on these cell types are direct and limit viral replication, IFN-β signals on nonhematopoietic cells to influence neutrophil recruitment and/or activation, making its activity an indirect effect. Our studies highlight the need for continued work to delineate the biological activities of specific IFN subtypes. These efforts may clarify why the multigene nature of type I IFN is conserved across multiple species and may lead to more effective IFN therapies with fewer off-target effects.
MATERIALS AND METHODS
Ethics statement.
Experiments were approved and performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (68). The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (assurance number A-3381-01).
Mice.
All animal experiments were performed in accordance with Washington University Institutional Animal Care and Use Committee guidelines, and all CHIKV infection studies were performed in an animal biosafety level 3 laboratory. Experiments were performed with 4-week-old male mice, unless otherwise specified. C57BL/6J wild-type, IFN-β-KO (Ifnb−/−) (20), and IRF7-KO (Irf7−/−) (14) mice were bred and maintained on the C57BL/6J background in our mouse colony before being transferred to the animal biosafety level 3 laboratory for infection experiments. In some experiments, 4-week-old male and female Vav-iCre+/− [B6.Cg-Tg(Vav1-cre)A2Kio/J] × Ifnar1flox/flox mice (Vav-Cre+) or Vav-iCre− × Ifnar1flox/flox littermate controls (Vav-Cre−) were used, which were also bred and maintained in our colony and genotyped prior to use in experiments (40, 41).
Virus experiments.
A recombinant strain of CHIKV (LR2006 OPY1) was provided by S. Higgs (Kansas State University) and generated from in vitro-transcribed cDNA, as previously described (69). At 4 weeks of age, mice were inoculated in the left rear footpad with 103 PFU of the LR2006 OPY1 strain of CHIKV in a volume of 10 or 20 μl. Infected mice were monitored daily for foot swelling with digital calipers for 14 to 28 days. At the termination of experiments, mice were sedated with a ketamine-xylazine cocktail, euthanized, and perfused via intracardiac injection with phosphate-buffered saline (PBS). Tissues (ipsilateral left ankle, contralateral right ankle, and ipsilateral quadriceps muscle) were harvested into 0.65 ml PBS and then stored at −80°C until processing for viral burden. “Ankle” refers to the distal foot with the skin and digits removed, and “foot” refers to the distal foot with cutaneous and subcutaneous tissues everted but still attached to the distal digits. For serum analysis, blood was collected at the time of sacrifice. After clotting, blood was centrifuged for 10 min at 10,000 × g and stored at −80°C. Samples harvested for cytokine and chemokine analysis were collected into 0.5 ml PBS with 0.1% bovine serum albumin (BSA) added and stored at −80°C until processing.
Tissue viral burden.
Organs were harvested into 0.65 ml PBS and homogenized with 1.0-mm-diameter zirconia-silica beads (BioSpec Products) with 2 pulses of 3,000 rpm for 30 s with a MagNA Lyser (Roche) prior to plaque assays on BHK21 cells. Two-hundred-microliter serial dilutions of organ homogenates or serum in Dulbecco’s modified Eagle’s medium (DMEM) with 1% fetal bovine serum (FBS) was added to BHK21 cells (5 × 105 cells for 6-well plates), and the cells were incubated for 1 h at 37°C, with rocking every 20 min. A minimal essential medium (MEM) agar overlay was then added to the cells, and the cells were incubated for approximately 60 h at 37°C or until plaques were visible by visual examination. Plates were then fixed with 1% formaldehyde (at least 30 min at room temperature), and agar plugs were removed. Plaques were visualized using a 1% crystal violet solution and counted.
Histopathological analysis.
The ipsilateral feet of infected mice were treated with Nair to remove fur, and the mice were then sacrificed with a ketamine-xylazine cocktail and perfused by intracardiac injection of PBS at the indicated time point. The whole foot tissue (with the skin intact) was dissected above the ankle and fixed in 4% paraformaldehyde for 48 h, followed by decalcification in 10 ml of 14% acid-free EDTA (pH 7.2) for 10 to 14 days, with the buffer changed every couple of days. Decalcified tissues were dehydrated with increasing ethanol gradients (30%, 50%, and then 70%) for 30 min each and subsequently embedded in paraffin with 5-μm sections prepared. Tissue sections were stained with hematoxylin and eosin (H&E). Embedding, sectioning, and staining were performed by the Washington University Musculoskeletal Histology and Morphometry Core.
For quantification of cells in the joint space, slides were imaged with a Zeiss Axio Imager Z2 microscope equipped with a color charge-coupled-device (CCD) camera (Washington University Center for Cellular Imaging). The investigator was not blind to the identity of the samples at the time of imaging, but all midfoot synovial caps for each section were imaged and then blinded for quantification by a second investigator. The data are presented as the number of cells in the synovial space per high-power field (HPF). For overall inflammation scoring, the slides were scored by a pathologist blind to the group and time point. The pathologist noted the absence or presence of joint space inflammation, myositis, and tenosynovitis and then gave an overall inflammation score based on the following criteria: 0 for no inflammation, 1 for mild inflammation, 2 for moderate inflammation, and 3 for severe inflammation.
Immune cell flow cytometry analysis.
Mice were sacrificed 2 or 3 days after inoculation and perfused with PBS. The foot was disarticulated at the ankle without fracturing the bone. Cutaneous and subcutaneous tissues were everted but still attached to the distal foot and digits during digestion. Tissues were incubated for 2 h at 37°C in 5 ml digestion buffer, with manual shaking every 20 to 30 min. Digestion buffer consisted of RPMI (Sigma), type IV collagenase (2.5 mg/ml; Sigma), DNase I (10 mg/ml; Sigma), 15 mM HEPES buffer (Corning), and 10% FBS (BioWest). Digested tissues were passed through a 70-μm cell strainer and washed once with 40 ml PBS containing 4% FBS. The number of viable cells was quantified by trypan blue staining. For other experiments, whole blood was collected into BD Microtainer tubes with K2E (K2EDTA) (Becton, Dickinson) and then treated with two rounds of red blood cell lysis buffer (Sigma).
The single-cell suspension was transferred to a 96-well V-bottom plate, incubated with anti-mouse CD16/CD32 (clone 93; BioLegend) for 10 min at 4°C, and then surface stained in PBS containing 4% FBS for 1 h at 4°C. The following antibodies, all from BioLegend unless otherwise specified, were used: anti-CD45 FITC (fluorescein isothiocyanate) (1:400) (clone 30-F11), anti-CD11b brilliant violet 605 (1:200) (M1/70), anti-CD3e brilliant violet 510 (1:200) (145-2C11), anti-CD19 brilliant violet 510 (1:400) (1D3; BD Biosciences), anti-F4/80 APC (allophycocyanin)-Cy7 (1:200) (BM8), anti-Ly6C Alexa Fluor 700 (1:200) (HK1.4), anti-MERTK PE (phycoerythrin) (1:200) (2B10C42), anti-Ly6G PE-Cy7 (1:200) (1A8), anti-IFNAR1 APC (1:200) (MAR1-5A3; Leinco Technologies), and eFluor 506 fixable viability dye (1:500) (eBioscience).
After staining, cells were washed and fixed at 4°C for 10 min in 4% paraformaldehyde (Electron Microscopy Sciences). The fixed cells were washed and resuspended in PBS containing 4% FBS. Cells were processed on an LSR Fortessa flow cytometer (Becton, Dickinson) managed by the Flow Cytometry and Fluorescence Activated Cell Sorting Core at Washington University and analyzed using BD FACSDiva and FlowJo V10 software.
In vivo IFN blockade.
Wild-type mice were administered 1 mg of anti-mouse pan-IFN-α (TIF-3C5), 1 mg of anti-mouse IFN-β (HDβ-4A7), or 1 mg of the isotype control (PIP) (all from Leinco Technologies) 1 day before and 1 day after infection by the intraperitoneal route (13, 27).
In vivo neutrophil depletion.
Wild-type, IFN-β-KO, or IRF7-KO mice were treated with 0.25 mg of anti-mouse Ly6G (1A8) or the isotype control (rat IgG2a clone 1-1) (Leinco) 1 day before infection and every other day through 7 dpi by the intraperitoneal route. Neutrophil depletion was verified by flow cytometry of peripheral blood cells (Fig. 7A) and foot infiltrates (Fig. 7B) at 2 dpi. Because anti-Ly6G antibody was utilized to deplete neutrophils, an alternative gating scheme was used for these experiments. In the blood, we first identified CD3+ T cells and CD19+ B cells from live CD45+ cells, and the remaining CD3− CD19− cells were analyzed for Ly6C and CD11b expression, which revealed three distinct populations of cells. The cells with intermediate Ly6C expression were Ly6G+ neutrophils. In the foot, MERTK+ F4/80+ macrophages were gated from live CD45+ cells, and the remaining cells were analyzed for Ly6C and CD11b expression, revealing two populations of cells. Similar to what was observed in the blood, the Ly6C-intermediate group contained Ly6G+ neutrophils. These analyses demonstrate significant neutrophil depletion from both the blood and the foot tissue.
Cytokine and chemokine analysis.
Foot tissues (with the cutaneous and subcutaneous tissues everted but still attached to the distal foot and digits) were harvested from euthanized infected mice at 0.5, 1, 2, and 3 dpi and collected in 500 μl PBS with 0.1% BSA added. At the time of the assay, the samples were homogenized with 1.0-mm-diameter zirconia-silica beads (BioSpec Products) with two pulses of 3,000 rpm for 30 s with a MagNA Lyser (Roche). Cytokine and chemokine levels in the homogenates were measured using Luminex technology with a mouse 25-plex assay (Millipore), according to the manufacturer’s instructions, with modifications recommended for “sticky” samples in the Millipore “tips and tricks” brochure (version 1.0 2017-01180). In brief, these modifications included pelleting tissue debris with a 5-min centrifugation step (600 × g) before adding samples to the 96-well assay plate and running the plate in 1× wash buffer instead of sheath fluid.
Quantification and statistical analysis.
All statistical analyses were performed with GraphPad Prism 8 software. For foot swelling over time (3 experimental groups; multiple time points) two-way analysis of variance (ANOVA) with Dunnett’s posttest was used. For viral burden analysis (3 experimental groups; multiple time points), two-way ANOVA with Dunnett’s posttest was used. For cytokine/chemokine and immune infiltrate analyses (2 experimental groups; multiple time points), two-way ANOVA with Sidak’s posttest was used. For histological scoring and quantification of cells per HPF (3 experimental groups; 1 time point), one-way ANOVA with Tukey’s posttest was used. A P value of <0.05 indicated statistically significant differences.
ACKNOWLEDGMENTS
We thank the Washington University Musculoskeletal Research Center for performing paraffin embedding and sectioning and the H&E staining used for these studies. We also acknowledge that microscopy imaging was performed in part through the use of the Washington University Center for Cellular Imaging (WUCCI). We also thank Robert D. Schreiber (Washington University School of Medicine) for his helpful discussion and assistance with the IFN-α- and IFN-β-blocking antibodies.
L.E.C. was supported by a National Institutes of Health (NIH) postdoctoral research training grant (T32 CA009547) (https://www.nih.gov/). A.R.Y. was supported by a National Institute of General Medical Sciences Cellular, Biochemical, and Molecular (CMB) Sciences predoctoral research training grant (T32 GM007067) (https://www.nigms.nih.gov/). D.J.V. was supported by the NIH (R01 AR070030 and R21 AR073507) (https://www.nih.gov/) and Shriners Hospitals for Children—St. Louis (85117) (https://www.shrinershospitalsforchildren.org/st-louis). M.S.D. was supported by the NIH (R01 AI143673, R01 AI127513, and R01 AI123348) (https://www.nih.gov/). D.J.L. was supported by the NIH (R01 AI127513 and R21 AI135490) (https://www.nih.gov/). The Washington University Musculoskeletal Research Center received funding from the NIH (P30 AR057235) (https://www.nih.gov/). The Washington University Center for Cellular Imaging was supported by the Washington University School of Medicine, The Children’s Discovery Institute of Washington University, and St. Louis Children’s Hospital (CDI-CORE-2015-505) (http://www.childrensdiscovery.org/grants) and by the Foundation for Barnes-Jewish Hospital (3770) (https://www.foundationbarnesjewish.org/Grants). Funding for this project was provided in part by the Washington University Rheumatic Diseases Research Resource-Based Center funded by the NIH (P30 AR073752) (https://www.nih.gov/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We declare the following conflict of interest: M.S.D. is a consultant for Inbios and Atreca and is on the scientific advisory board of Moderna.
REFERENCES
- 1.McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. 2015. Type I interferons in infectious disease. Nat Rev Immunol 15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol 18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. 1998. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-β gene. FEBS Lett 425:112–116. doi: 10.1016/s0014-5793(98)00210-5. [DOI] [PubMed] [Google Scholar]
- 4.Schafer SL, Lin R, Moore PA, Hiscott J, Pitha PM. 1998. Regulation of type I interferon gene expression by interferon regulatory factor-3. J Biol Chem 273:2714–2720. doi: 10.1074/jbc.273.5.2714. [DOI] [PubMed] [Google Scholar]
- 5.Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. 1998. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol Cell 1:507–518. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- 6.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J 17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marié I, Durbin JE, Levy DE. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J 17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honda K, Takaoka A, Taniguchi T. 2006. Type I inteferon [sic] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. 1998. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett 441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- 10.Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M, Diamond MS. 2008. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J Virol 82:8465–8475. doi: 10.1128/JVI.00918-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, Le TT, Anraku I, Kumagai Y, Loo Y-M, Gale M, Akira S, Khromykh AA, Suhrbier A. 2012. Interferon response factors 3 and 7 protect against chikungunya virus hemorrhagic fever and shock. J Virol 86:9888–9898. doi: 10.1128/JVI.00956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schilte C, Buckwalter MR, Laird ME, Diamond MS, Schwartz O, Albert ML. 2012. Cutting edge: independent roles for IRF-3 and IRF-7 in hematopoietic and nonhematopoietic cells during host response to chikungunya infection. J Immunol 188:2967–2971. doi: 10.4049/jimmunol.1103185. [DOI] [PubMed] [Google Scholar]
- 13.Sheehan KCF, Lazear HM, Diamond MS, Schreiber RD. 2015. Selective blockade of interferon-α and -β reveals their non-redundant functions in a mouse model of West Nile virus infection. PLoS One 10:e0128636. doi: 10.1371/journal.pone.0128636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 15.Chen H-W, King K, Tu J, Sanchez M, Luster AD, Shresta S. 2013. The roles of IRF-3 and IRF-7 in innate antiviral immunity against dengue virus. J Immunol 191:4194–4201. doi: 10.4049/jimmunol.1300799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lang PA, Cervantes-Barragan L, Verschoor A, Navarini AA, Recher M, Pellegrini M, Flatz L, Bergthaler A, Honda K, Ludewig B, Ohashi PS, Lang KS. 2009. Hematopoietic cell-derived interferon controls viral replication and virus-induced disease. Blood 113:1045–1052. doi: 10.1182/blood-2007-10-117861. [DOI] [PubMed] [Google Scholar]
- 17.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 18.Schreiber G, Piehler J. 2015. The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol 36:139–149. doi: 10.1016/j.it.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, Nolan GP, Piehler J, Schreiber G, Garcia KC. 2011. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 146:621–632. doi: 10.1016/j.cell.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, Tanaka N, Taniguchi T. 2000. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science 288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- 21.Deonarain R, Alcamí A, Alexiou M, Dallman MJ, Gewert DR, Porter ACG. 2000. Impaired antiviral response and alpha/beta interferon induction in mice lacking beta interferon. J Virol 74:3404–3409. doi: 10.1128/jvi.74.7.3404-3409.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deonarain R, Cerullo D, Fuse K, Liu PP, Fish EN. 2004. Protective role for interferon-β in coxsackievirus B3 infection. Circulation 110:3540–3543. doi: 10.1161/01.CIR.0000136824.73458.20. [DOI] [PubMed] [Google Scholar]
- 23.Davidson S, Maini MK, Wack A. 2015. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J Interferon Cytokine Res 35:252–264. doi: 10.1089/jir.2014.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lazear HM, Pinto AK, Vogt MR, Gale M, Diamond MS. 2011. Beta interferon controls West Nile virus infection and pathogenesis in mice. J Virol 85:7186–7194. doi: 10.1128/JVI.00396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. 2013. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KCF, Welch M, Schreiber RD, de la Torre JC, Oldstone MBA. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, Sheehan KCF, Schreiber RD, Oldstone MBA. 2015. Blockade of interferon beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 17:653–661. doi: 10.1016/j.chom.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staples JE, Breiman RF, Powers AM. 2009. Chikungunya fever: an epidemiological review of a re‐emerging infectious disease. Clin Infect Dis 49:942–948. doi: 10.1086/605496. [DOI] [PubMed] [Google Scholar]
- 29.Morrison TE. 2014. Reemergence of chikungunya virus. J Virol 88:11644–11647. doi: 10.1128/JVI.01432-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, Schroder WA, Higgs S, Suhrbier A. 2010. Chikungunya virus arthritis in adult wild-type mice. J Virol 84:8021–8032. doi: 10.1128/JVI.02603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teo T-H, Lum F-M, Claser C, Lulla V, Lulla A, Merits A, Renia L, Ng LPF. 2013. A pathogenic role for CD4+ T cells during chikungunya virus infection in mice. J Immunol 190:259–269. doi: 10.4049/jimmunol.1202177. [DOI] [PubMed] [Google Scholar]
- 32.Fox JM, Diamond MS. 2016. Immune-mediated protection and pathogenesis of chikungunya virus. J Immunol 197:4210–4218. doi: 10.4049/jimmunol.1601426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawman DW, Stoermer KA, Montgomery SA, Pal P, Oko L, Diamond MS, Morrison TE. 2013. Chronic joint disease caused by persistent chikungunya virus infection is controlled by the adaptive immune response. J Virol 87:13878–13888. doi: 10.1128/JVI.02666-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM, Elmore SA, Heise MT. 2011. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am J Pathol 178:32–40. doi: 10.1016/j.ajpath.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suhrbier A, Jaffar-Bandjee MC, Gasque P. 2012. Arthritogenic alphaviruses—an overview. Nat Rev Rheumatol 8:420–429. doi: 10.1038/nrrheum.2012.64. [DOI] [PubMed] [Google Scholar]
- 36.Bego MG, Mercier J, Cohen EA. 2012. Virus-activated interferon regulatory factor 7 upregulates expression of the interferon-regulated BST2 gene independently of interferon signaling. J Virol 86:3513–3527. doi: 10.1128/JVI.06971-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones PH, Maric M, Madison MN, Maury W, Roller RJ, Okeoma CM. 2013. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology 438:37–49. doi: 10.1016/j.virol.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahauad-Fernandez WD, Jones PH, Okeoma CM. 2014. Critical role for bone marrow stromal antigen 2 in acute chikungunya virus infection. J Gen Virol 95:2450–2461. doi: 10.1099/vir.0.068643-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F, Kraxner A, Tschopp J, Higgs S, Michault A, Arenzana-Seisdedos F, Colonna M, Peduto L, Schwartz O, Lecuit M, Albert ML. 2010. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med 207:429–442. doi: 10.1084/jem.20090851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. 2003. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- 41.Kamphuis E, Junt T, Waibler Z, Forster R, Kalinke U. 2006. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 108:3253–3261. doi: 10.1182/blood-2006-06-027599. [DOI] [PubMed] [Google Scholar]
- 42.Poo YS, Rudd PA, Gardner J, Wilson JAC, Larcher T, Colle MA, Le TT, Nakaya HI, Warrilow D, Allcock R, Bielefeldt-Ohmann H, Schroder WA, Khromykh AA, Lopez JA, Suhrbier A. 2014. Multiple immune factors are involved in controlling acute and chronic chikungunya virus infection. PLoS Negl Trop Dis 8:e3354. doi: 10.1371/journal.pntd.0003354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haist KC, Burrack KS, Davenport BJ, Morrison TE. 2017. Inflammatory monocytes mediate control of acute alphavirus infection in mice. PLoS Pathog 13:e1006748. doi: 10.1371/journal.ppat.1006748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poo YS, Nakaya H, Gardner J, Larcher T, Schroder WA, Le TT, Major LD, Suhrbier A. 2014. CCR2 deficiency promotes exacerbated chronic erosive neutrophil-dominated chikungunya virus arthritis. J Virol 88:6862–6872. doi: 10.1128/JVI.03364-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miner JJ, Cook LE, Hong JP, Smith AM, Richner JM, Shimak RM, Young AR, Monte K, Poddar S, Crowe JE Jr, Lenschow DJ, Diamond MS. 2017. Therapy with CTLA4-Ig and an antiviral monoclonal antibody controls chikungunya virus arthritis. Sci Transl Med 9:eaah3438. doi: 10.1126/scitranslmed.aah3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinto AK, Ramos HJ, Wu X, Aggarwal S, Shrestha B, Gorman M, Kim KY, Suthar MS, Atkinson JP, Gale M Jr, Diamond MS. 2014. Deficient IFN signaling by myeloid cells leads to MAVS-dependent virus-induced sepsis. PLoS Pathog 10:e1004086. doi: 10.1371/journal.ppat.1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zust R, Toh Y-X, Valdes I, Cerny D, Heinrich J, Hermida L, Marcos E, Guillen G, Kalinke U, Shi P-Y, Fink K. 2014. Type I interferon signals in macrophages and dendritic cells control dengue virus infection: implications for a new mouse model to test dengue vaccines. J Virol 88:7276–7285. doi: 10.1128/JVI.03827-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daniels BP, Jujjavarapu H, Durrant DM, Williams JL, Green RR, White JP, Lazear HM, Gale M, Diamond MS, Klein RS. 2017. Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J Clin Invest 127:843–856. doi: 10.1172/JCI88720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. 2000. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol 74:3366–3378. doi: 10.1128/jvi.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, Rudnicka D, Sol-Foulon N, Le Roux K, Prevost M-C, Fsihi H, Frenkiel M-P, Blanchet F, Afonso PV, Ceccaldi P-E, Ozden S, Gessain A, Schuffenecker I, Verhasselt B, Zamborlini A, Saïb A, Rey FA, Arenzana-Seisdedos F, Desprès P, Michault A, Albert ML, Schwartz O. 2007. Characterization of reemerging chikungunya virus. PLoS Pathog 3:e89. doi: 10.1371/journal.ppat.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noret M, Herrero L, Rulli N, Rolph M, Smith PN, Li RW, Roques P, Gras G, Mahalingam S. 2012. Interleukin 6, RANKL, and osteoprotegerin expression by chikungunya virus-infected human osteoblasts. J Infect Dis 206:455–457. doi: 10.1093/infdis/jis368. [DOI] [PubMed] [Google Scholar]
- 52.Kasper LH, Reder AT. 2014. Immunomodulatory activity of interferon-beta. Ann Clin Transl Neurol 1:622–631. doi: 10.1002/acn3.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.González-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ng CT, Oldstone MBA. 2012. Infected CD8α− dendritic cells are the predominant source of IL-10 during establishment of persistent viral infection. Proc Natl Acad Sci U S A 109:14116–14121. doi: 10.1073/pnas.1211910109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Her Z, Teng T-S, Tan JJ, Teo T-H, Kam Y-W, Lum F-M, Lee WW, Gabriel C, Melchiotti R, Andiappan AK, Lulla V, Lulla A, Win MK, Chow A, Biswas SK, Leo Y-S, Lecuit M, Merits A, Renia L, Ng LF. 2015. Loss of TLR3 aggravates CHIKV replication and pathology due to an altered virus-specific neutralizing antibody response. EMBO Mol Med 7:24–41. doi: 10.15252/emmm.201404459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sadik CD, Kim ND, Luster AD. 2011. Neutrophils cascading their way to inflammation. Trends Immunol 32:452–460. doi: 10.1016/j.it.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jablonska J, Wu C-F, Andzinski L, Leschner S, Weiss S. 2014. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int J Cancer 134:1346–1358. doi: 10.1002/ijc.28551. [DOI] [PubMed] [Google Scholar]
- 58.Metzemaekers M, Vanheule V, Janssens R, Struyf S, Proost P. 2017. Overview of the mechanisms that may contribute to the non-redundant activities of interferon-inducible CXC chemokine receptor 3 ligands. Front Immunol 8:1970. doi: 10.3389/fimmu.2017.01970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jia T, Leiner I, Dorothee G, Brandl K, Pamer EG. 2009. MyD88 and type I interferon receptor-mediated chemokine induction and monocyte recruitment during Listeria monocytogenes infection. J Immunol 183:1271–1278. doi: 10.4049/jimmunol.0900460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Génin P, Algarté M, Roof P, Lin R, Hiscott J. 2000. Regulation of RANTES chemokine gene expression requires cooperativity between NF-kappa B and IFN-regulatory factor transcription factors. J Immunol 164:5352–5361. doi: 10.4049/jimmunol.164.10.5352. [DOI] [PubMed] [Google Scholar]
- 61.Sadik CD, Luster AD. 2012. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol 91:207–215. doi: 10.1189/jlb.0811402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miyabe Y, Miyabe C, Murooka TT, Kim EY, Newton GA, Kim ND, Haribabu B, Luscinskas FW, Mempel TR, Luster AD. 2017. Complement C5a receptor is the key initiator of neutrophil adhesion igniting immune complex-induced arthritis. Sci Immunol 2:eaaj2195. doi: 10.1126/sciimmunol.aaj2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. 2014. The balancing act of neutrophils. Cell Host Microbe 15:526–536. doi: 10.1016/j.chom.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 64.Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, Dummer R, Simon H-U, Yousefi S. 2004. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem 279:44123–44132. doi: 10.1074/jbc.M405883200. [DOI] [PubMed] [Google Scholar]
- 65.Broggi A, Tan Y, Granucci F, Zanoni I. 2017. IFN-λ suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat Immunol 18:1084–1093. doi: 10.1038/ni.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Galani IE, Triantafyllia V, Eleminiadou E-E, Koltsida O, Stavropoulos A, Manioudaki M, Thanos D, Doyle SE, Kotenko SV, Thanopoulou K, Andreakos E. 2017. Interferon-λ mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity 46:875–890.e6. doi: 10.1016/j.immuni.2017.04.025. [DOI] [PubMed] [Google Scholar]
- 67.Blazek K, Eames HL, Weiss M, Byrne AJ, Perocheau D, Pease JE, Doyle S, McCann F, Williams RO, Udalova IA. 2015. IFN-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J Exp Med 212:845–853. doi: 10.1084/jem.20140995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 69.Vanlandingham DL, Tsetsarkin K, Hong C, Klingler K, McElroy KL, Lehane MJ, Higgs S. 2005. Development and characterization of a double subgenomic chikungunya virus infectious clone to express heterologous genes in Aedes aegypti mosquitoes. Insect Biochem Mol Biol 35:1162–1170. doi: 10.1016/j.ibmb.2005.05.008. [DOI] [PubMed] [Google Scholar]