

Gammaherpesviruses establish lifelong infection in a majority of adults worldwide and are associated with a number of malignancies, including B cell lymphomas. These viruses infect naive B cells and manipulate B cell differentiation to achieve a lifelong infection of memory B cells. The germinal center stage of B cell differentiation is important as both an amplifier of the viral latent reservoir and the target of malignant transformation. In this study, we demonstrate that expression of tyrosine phosphatase SHP1, a negative regulator that normally limits the activation and proliferation of hematopoietic cells, enhances the gammaherpesvirus-driven germinal center response and the establishment of chronic infection. The results of this study uncover an intriguing beneficial interaction between gammaherpesviruses that are presumed to profit from B cell activation and a cellular phosphatase that is traditionally perceived to be a negative regulator of the same processes.
KEYWORDS: SHP1, gammaherpesvirus, germinal center B cell
ABSTRACT
Gammaherpesviruses are ubiquitous pathogens that establish lifelong infections in the majority of adults worldwide. Chronic gammaherpesvirus infection has been implicated in both lymphomagenesis and, somewhat controversially, autoimmune disease development. Pathogenesis is largely associated with the unique ability of gammaherpesviruses to usurp B cell differentiation, specifically, the germinal center response, to establish long-term latency in memory B cells. The host tyrosine phosphatase SHP1 is known as a brake on immune cell activation and is downregulated in several gammaherpesvirus-driven malignancies. However, here we demonstrate that B cell- but not T cell-intrinsic SHP1 expression supports the gammaherpesvirus-driven germinal center response and the establishment of viral latency. Furthermore, B cell-intrinsic SHP1 deficiency cooperated with gammaherpesvirus infection to increase the levels of double-stranded DNA-reactive antibodies at the peak of viral latency. Thus, in spite of decreased SHP1 levels in gammaherpesvirus-driven B cell lymphomas, B cell-intrinsic SHP1 expression plays a proviral role during the establishment of chronic infection, suggesting that the gammaherpesvirus-SHP1 interaction is more nuanced and is modified by the stage of infection and pathogenesis.
IMPORTANCE Gammaherpesviruses establish lifelong infection in a majority of adults worldwide and are associated with a number of malignancies, including B cell lymphomas. These viruses infect naive B cells and manipulate B cell differentiation to achieve a lifelong infection of memory B cells. The germinal center stage of B cell differentiation is important as both an amplifier of the viral latent reservoir and the target of malignant transformation. In this study, we demonstrate that expression of tyrosine phosphatase SHP1, a negative regulator that normally limits the activation and proliferation of hematopoietic cells, enhances the gammaherpesvirus-driven germinal center response and the establishment of chronic infection. The results of this study uncover an intriguing beneficial interaction between gammaherpesviruses that are presumed to profit from B cell activation and a cellular phosphatase that is traditionally perceived to be a negative regulator of the same processes.
INTRODUCTION
Gammaherpesviruses are ubiquitous pathogens that establish lifelong infections in the majority of adults worldwide. Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) are two known human gammaherpesviruses that are associated with several cancers, including B cell lymphomas (1, 2). Although more controversial, gammaherpesvirus infection also likely contributes to select autoimmune diseases (3–5). Long-term gammaherpesvirus latency is maintained in memory B cells, which presents a logistical challenge early in infection, as memory B cells represent a very small proportion of the overall B cell population. Thus, unlike other viruses that only transiently infect B cells, if they infect them at all, gammaherpesvirus chronic infection and viral pathogenesis are intimately tied to the unique ability of gammaherpesviruses to manipulate the differentiation of infected B cells in order to establish long-term latency. Specifically, EBV and murine gammaherpesvirus 68 (MHV68) infect highly abundant naive B cells, with the subsequent progression of infected and bystander B cells to the germinal center stage of differentiation (6–10). While in the germinal center, latent virus achieves an exponential expansion of its reservoir via the rapid proliferation of infected germinal center B cells. Importantly, germinal center B cells are thought to be the target of viral transformation, as a majority of EBV-positive B cell lymphomas are of germinal center or post-germinal center origin (11, 12). Following further differentiation, gammaherpesvirus-infected germinal center B cells transition to memory B cells that support lifelong latent infection or plasma cells, where the latent-to-lytic switch occurs (13–15).
Importantly, the gammaherpesvirus-induced germinal center responses are qualitatively distinct from the physiological B cell differentiation induced during other virus infections. While MHV68-specific class-switched antibody titers rise slowly and do not peak until at least a month after infection, both MHV68 and EBV induce a robust increase in antibodies directed against self-antigens and antigens of other species early in chronic infection (16, 17). The level to which autoantibodies are induced is significantly higher during gammaherpesvirus infection than during infections with other viruses (18). This induction of irrelevant, virus-nonspecific antibodies during gammaherpesvirus infection is taken advantage of in the clinic, where the detection of high titers of antibodies against horse red blood cells serves as a diagnostic of a recent EBV infection (19). Induction of irrelevant B cell differentiation is likely of benefit for gammaherpesviruses, as viral latency and reactivation predominantly occur in B cells that do not encode a gammaherpesvirus-specific B cell receptor (BCR) (20, 21). While the host and viral mechanisms underlying gammaherpesvirus-induced irrelevant B cell differentiation are poorly understood, these may promote lymphomagenesis or the development of autoimmune disease in a susceptible host.
Not surprisingly, given the intimate relationship between gammaherpesviruses and B cell differentiation, the activation of naive B cells is important for the establishment of latent infection. In an elegant study from the Speck group, infected cell-specific attenuation of NF-κB signaling led to decreased B cell activation and attenuated gammaherpesvirus latency (22, 23). Thus, we hypothesized that host factors that limit B cell activation may also limit the gammaherpesvirus-driven germinal center response and, by extension, viral pathogenesis.
SHP1 (encoded by PTPN6) is a tyrosine phosphatase that is primarily expressed in hematopoietic cells and that negatively regulates immune cell activation (24, 25). In B cells, SHP1 is a cytoplasmic protein that localizes to the B cell receptor (BCR) to dephosphorylate several substrates, including Igα-Igβ (CD79a/CD79b) subunits, Syk, and BLNK, ultimately attenuating BCR-proximal signaling (reviewed in reference 26). Intriguingly, B cell-specific SHP1 deficiency leads to the decreased maintenance of germinal center responses following immunization (25). Because gammaherpesvirus-driven B cell differentiation is qualitatively distinct from the physiological response, the role of SHP1 during the establishment of gammaherpesvirus infection remained unclear. Consistent with the potential role of SHP1 as an antagonist of chronic gammaherpesvirus infection and pathogenesis, SHP1 expression is reduced in multiple EBV- or KSHV-driven B cell lymphomas (27–30). Mice with global SHP1 deficiency present with myeloid hyperproliferation and hypersensitive immune activation (31, 32). The widespread immune dysfunction and mortality by 3 to 4 weeks of age make it difficult to evaluate the role of SHP1 during gammaherpesvirus infection using the global deficiency mouse model. To circumvent this limitation, this study utilized mouse models of conditional SHP1 deficiency (33, 34).
In contrast to the predicted outcome, the establishment of chronic gammaherpesvirus infection was attenuated in mice with SHP1-deficient B cells, along with an attenuated germinal center response, a stage of B cell differentiation critical for the optimal establishment of latent infection. Interestingly, a T cell-specific deficiency of SHP1 did not alter the gammaherpesvirus-driven germinal center response or the establishment of chronic infection, indicating that it is the B cell-intrinsic SHP1 expression that functions in a proviral manner. Despite an attenuated germinal center response, the levels of self-reactive antibodies were elevated at the baseline and further exacerbated at the peak of gammaherpesvirus latency in mice with B cell-specific SHP1 deficiency. However, the levels of self-reactive antibodies were no longer increased above those at the baseline in long-term-infected mice with SHP1-deficient B cells, suggesting that the positive effect of gammaherpesvirus infection on the development of autoimmunity may be temporally restricted by B cell-intrinsic SHP1 deficiency.
RESULTS
Loss of SHP1 expression in B cells leads to attenuated establishment of MHV68 chronic infection.
Due to coevolution with their host, gammaherpesviruses are extremely species specific, which limits in vivo studies of human gammaherpesviruses. Thus, the current study utilizes MHV68, a natural rodent pathogen that is genetically and biologically similar to EBV and KSHV (35–37). After a brief acute lytic replication in a naive host, MHV68 establishes latency in several organs, including the spleen (38, 39). Viral latency in the spleen peaks at 14 to 18 days postinfection, with most of the latent virus being present in the germinal center B cells (40, 41). To define the role of SHP1 in gammaherpesvirus infection while overcoming the deleterious effects of global SHP1 deficiency, a published mouse model of B cell-specific SHP1 deficiency was used (33). To determine the effect of B cell-specific SHP1 deficiency on the establishment of MHV68 latency, SHP1flox/flox (SHP1fl/fl) mice heterozygous for CD19 promoter-driven Cre recombinase or homozygous for wild-type (wt) CD19 allele (referred to as CD19 Cre-positive and CD19 Cre-negative mice, respectively, throughout this article) were infected with MHV68, and parameters of viral latency were determined at 16 days postinfection.
In spite of the known role of SHP1 as a negative regulator of B cell activation, with the latter supporting the establishment of chronic gammaherpesvirus infection, CD19 Cre-positive mice displayed a significantly lower frequency of MHV68 DNA-positive splenocytes than CD19 Cre-negative mice (11-fold; Fig. 1A), along with a decrease in the absolute number of MHV68 DNA-positive splenocytes (∼7-fold; Fig. 1B). Similarly, the frequency of ex vivo reactivation from CD19 Cre-positive splenocytes was decreased compared to that in the control group (Fig. 1C). Thus, B cell-specific SHP1 deficiency resulted in the overall attenuation of MHV68 latency and reactivation.
FIG 1.

Loss of SHP1 expression in B cells leads to attenuated establishment of MHV68 chronic infection. CD19 Cre-negative or CD19 Cre-positive mice were intranasally infected with 500 PFU of MHV68, and splenocytes were harvested at 16 days postinfection. As described in Materials and Methods, limiting dilution assays were used to measure the frequency (A) and, subsequently, the absolute number (B) of MHV68 genome-positive splenocytes and the frequency of viral reactivation (C). Splenocytes from 3 to 5 mice were pooled within an individual group in each experiment, and data from at least 3 independent experiments were pooled. Error bars here and throughout the figures represent the standard error of the measurement. The dashed lines in panels A and C are drawn at 63% to define the frequency of a positive event. CPE, cytopathic effect.
B cell-intrinsic SHP1 expression supports the MHV68-driven germinal center response.
Gammaherpesviruses exploit B cell differentiation via latent infection of naive B cells, with the subsequent entry of both infected and uninfected naive B cells into the germinal center response. The rapid proliferation of germinal center B cells passively expands the latent viral reservoir (8), such that the germinal center B cells host a majority of latent MHV68 at 16 days postinfection. Having observed a decreased frequency of MHV68 DNA-positive splenocytes, we next examined the germinal center response. As previously published (33), B cell-specific SHP1 deficiency results in an increase in the splenic B-1 B cell population that expresses intermediate levels of B220, in contrast to classical splenic B-2 B cells, which are B220high. When the gating strategy was adjusted to include intermediate B220 expression (Fig. 2A), the frequency of splenic B cells was similar in all examined groups, regardless of infection or SHP1 status (Fig. 2B). Further, the MHV68-induced increase in the absolute number of splenic B cells was independent of SHP1 expression (Fig. 2C). In contrast to the overall splenic B cell population, the frequency and absolute number of germinal center B cells were significantly decreased in CD19 Cre-positive mice compared to CD19 Cre-negative mice (Fig. 2D to F), consistent with the decrease in the MHV68 latent reservoir (Fig. 1A).
FIG 2.

B cell-intrinsic SHP1 expression supports MHV68-driven germinal center response. Mice of the indicated genotypes were infected as described in the legend to Fig. 1, and splenocytes were harvested at 16 days postinfection. (A to C) The surface expression of B220 (a representative result is shown in panel A, including a gating strategy for B220+ cells used throughout this study) was used to identify the percentage of B cells (B) and the total number of B cells (C) per spleen. Germinal center B cells were identified by positive surface staining of GL7 and CD95 of pregated, B220+ splenocytes (a representative result is shown in panel D) and expressed as the frequency (E) and absolute number (F) of B220+ cells. The same analyses were used to determine the frequency (G) and absolute number (H) of germinal center B cells at the indicated times postinfection. (B to F) Each symbol represents the results for an individual spleen, and data from 2 to 4 independent experiments were pooled. (G, H) Each symbol represents the mean from 2 to 4 independent experiments with 3 to 5 mice per experiment. The levels of significance are indicated: *, P = 0.05; **, P = 0.01; ***, P = 0.001; ****, P = 0.0001.
Because SHP1 is a negative regulator of B cell activation, it was possible that the kinetics of the germinal center response were accelerated in CD19 Cre-positive mice, with the response peaking before day 16. To define the kinetics of the germinal center response, the frequency and absolute number of germinal center B cells were examined at several time points throughout the first 16 days of infection and at 42 days, when long-term MHV68 infection is established. B cell-specific SHP1 deficiency did not alter germinal center B cell kinetics following MHV68 infection (Fig. 2G and H), supporting the conclusion that B cell-specific SHP1 deficiency attenuates the MHV68-driven expansion of germinal center B cells.
Because CD19 is an important component of the B cell receptor complex and the hemizygosity of wild-type CD19 in the CD19 Cre-positive mice, the effects of a potential decrease in CD19 expression on the induction of the germinal center response were explored. Importantly, the MHV68-driven induction of germinal center B cells was similar in SHP1fl/wt mice that were either CD19 Cre positive or negative (see Fig. S1A and B in the supplemental material), indicating that CD19 hemizygosity does not affect the MHV68-driven germinal center response. Thus, the germinal center B cell phenotypes observed in SHP1fl/fl CD19 Cre-positive mice can be attributed to the loss of SHP1 expression in B cells.
T cell-specific deficiency of SHP1 does not alter the germinal center response.
During the germinal center response, the expansion of germinal center B cells is mediated via interaction with T follicular helper (TFH) cells (42), and both cell types signal in a positive feedback to promote germinal center expansion (43). Similar to the physiological response, the gammaherpesvirus-driven expansion of germinal center B cells is critically dependent on TFH cell help (44, 45). Having observed decreased germinal center B cells in MHV68-infected CD19 Cre-positive mice, TFH cells were examined next. Surprisingly, the frequency and the absolute number of TFH cells were significantly elevated in CD19 Cre-positive mice, such that the baseline levels matched those observed in MHV68-infected control mice (Fig. 3A to C). The total number, but not the frequency, of TFH cells was further increased following MHV68 infection of CD19 Cre-positive mice. Similar to the germinal center B cell phenotype, the increase in TFH cells observed in CD19 Cre-positive mice was not a result of CD19 hemizygosity (Fig. S1C and D). Thus, B cell-specific SHP1 deficiency resulted in a dysbalance between germinal center B cells and TFH cells, a dysbalance that was exacerbated by MHV68 infection.
FIG 3.

A T cell-specific deficiency of SHP1 does not alter the germinal center response. Mice of the indicated genotypes were infected as described in the legend to Fig. 1. At 16 days postinfection, splenocytes were harvested and subjected to flow cytometry. TFH cells were identified by CXCR5- and PD-1-positive surface staining (a representative result is shown in panel A) and expressed as the frequency (B, D) and absolute number (C, E) of CD3+ CD4+ cells. As described in the legend to Fig. 2, germinal center B cells were defined as the B220+ GL7+ CD95+ population and are expressed as the frequency (F) and absolute number (G) of B220+ cells. Each symbol represents the results for an individual spleen, and data from 3 independent experiments were pooled. The levels of significance are indicated: *, P = 0.05; **, P = 0.01; ****, P = 0.0001.
Given the importance of germinal center B cells and TFH cells for the establishment of MHV68 latency and the surprising, opposite direction of the phenotypes in these two immune populations observed in CD19 Cre-positive mice, the effect of T cell-specific SHP1 deficiency was assessed. Shp1fl/fl mice heterozygous for distal Lck (dLck) promoter-driven Cre recombinase or homozygous for the wild-type dLck allele (34) (referred to as dLck Cre-positive and dLck Cre-negative mice, respectively) were infected with MHV68 and assessed at 16 days postinfection.
Surprisingly, the baseline levels of TFH cells were similar in dLck Cre-positive and Cre-negative mice and T cell-specific SHP1 deficiency had no effect on the MHV68-driven induction of either TFH cells (Fig. 3D and E) or germinal center B cells (Fig. 3F and G). Consistent with the lack of a germinal center phenotype, the frequencies of MHV68 DNA-positive splenocytes were similar in dLck Cre-positive and -negative mice (data not shown). Thus, B cell-intrinsic but not T cell-intrinsic SHP1 expression facilitated the gammaherpesvirus-driven germinal center response and the establishment of latent infection.
B cell-intrinsic SHP1 expression limits both the proliferation and apoptosis of germinal center B cells during MHV68 infection.
The counterbalance of cellular proliferation and apoptosis determines the magnitude of the germinal center response. Having observed an increase in the TFH cell population with a concurrent decrease in germinal center B cells in the CD19 Cre-positive mice, the proliferation and apoptosis of the two populations were measured at 16 days postinfection. While more CD19 Cre-positive germinal center B cells than Cre-negative B cells were Ki67 positive (Fig. 4A), there was also an increase in the frequency of germinal center B cells with active caspase 3 in CD19 Cre-positive mice (Fig. 4B). In contrast, the proportion of either Ki67-positive or active caspase 3-positive TFH cells was independent of the mouse CD19 Cre genotype (Fig. 4C and D). Thus, the increased proliferation of germinal center B cells in the absence of SHP1 was coupled to increased apoptosis in this cell population, affecting the magnitude of the germinal center B cell population.
FIG 4.
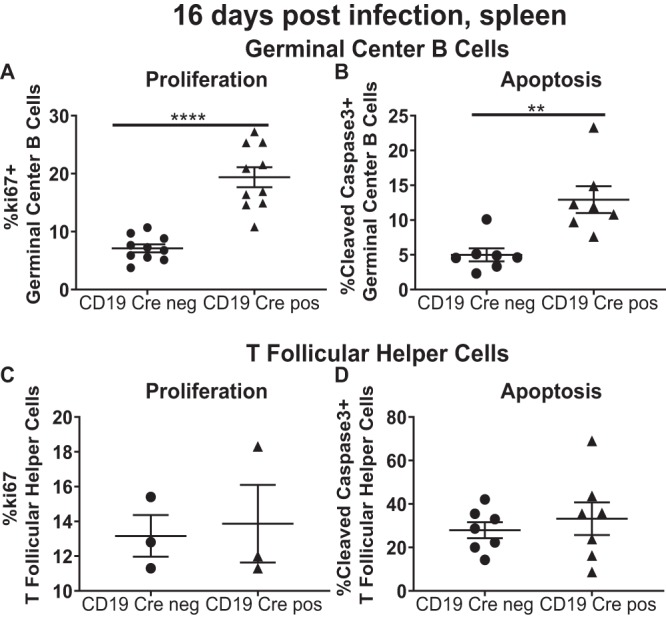
B cell-intrinsic SHP1 expression limits both the proliferation and apoptosis of germinal center B cells during MHV68 infection. Mice of the indicated genotypes were infected as described in the legend to Fig. 1. Splenocytes were analyzed at 16 days postinfection. Intracellular staining with Ki67 was used to identify proliferating germinal center B cells (A) and T follicular helper cells (C), identified as described in the legends to Fig. 2 and 3. Similarly, the levels of active caspase 3 were measured in germinal center B cells (B) and T follicular helper cells (D). Each symbol represents the results for an individual spleen. The levels of significance are indicated: **, P = 0.01; ****, P = 0.0001.
Peak MHV68 latency exacerbates the production of self-reactive antibodies.
Germinal center B cells undergo class switching and differentiate into antibody-secreting plasma cells. Along with slowly increasing titers of gammaherpesvirus-specific antibodies, MHV68, like EBV, drives a robust, irrelevant B cell differentiation that results in the peak titers of class-switched antibodies being directed against self-antigens at 16 days postinfection (17). Having observed the decreased induction of germinal center B cells in CD19 Cre-positive mice, further B cell differentiation along with humoral responses was examined next.
CD19 Cre-positive mice had an elevated frequency and an elevated number of class-switched plasma cells at the baseline (Fig. 5A to C), consistent with the propensity to develop autoimmune disease upon aging (33). As expected, MHV68 infection of control, CD19 Cre-negative mice stimulated an increase in class-switched plasma cells. In contrast, the abundance of class-switched plasma cells in MHV68-infected CD19 Cre-positive mice was similar to the already high abundance observed at the baseline.
FIG 5.

Peak MHV68 latency exacerbates the production of self-reactive antibodies. Mice of the indicated genotypes were infected as described in the legend to Fig. 1, and splenocytes were analyzed at 16 days postinfection. (A to C) Class-switched plasma cells were identified by CD138+ surface staining and IgG-positive intracellular staining of a B220-positive population (a representative plot is shown in panel A) and expressed as the frequency (B) and the absolute number (C). (D, E) Sera from mock- and MHV68-infected (16 days) mice of the indicated genotypes were subjected to ELISA to determine total IgG (D) and anti-dsDNA IgG (E) levels. (F) The data from panel E are presented as the fold change relative to the mock-infected controls. (G, H). Sera were subjected to ANA analyses using anti-mouse IgG fluorophore-conjugated antibody (G), and the fluorescence was quantified (H). Data from 2 to 3 independent experiments were pooled. The levels of significance are indicated: *, P = 0.05; **, P = 0.01. dpi, days postinfection; int/hi, intermediate/high.
Interestingly, in spite of the higher abundance of class-switched plasma cells both at the baseline and following infection of CD19 Cre-positive mice, the total serum IgG levels were similar between the two CD19 Cre genotypes, including at the baseline and at 16 days after MHV68 infection (Fig. 5D). Because gammaherpesvirus infection induces a robust, albeit transient, increase in self-directed antibody titers, the levels of class-switched antibodies directed against double-stranded DNA (dsDNA) were measured next. As expected, anti-dsDNA antibodies were induced by MHV68 infection in control, CD19 Cre-negative mice (Fig. 5E and F). The baseline levels of anti-dsDNA antibodies were elevated in CD19 Cre-positive mice (Fig. 5E), despite the relatively young age of these mice (8 to 10 weeks) and consistent with a previously described tendency of these mice to develop autoimmune disease (33). Interestingly, MHV68 infection produced a further increase in the titers of dsDNA-directed class-switched antibodies in CD19 Cre-positive mice. Because double-stranded DNA represents one of many self-antigens recognized by gammaherpesvirus-induced antibodies, a broader analysis of self-directed responses was performed using an antinuclear antibody (ANA) assay that detects class-switched antibodies reacting with monolayers of permeabilized epithelial cells. As expected, MHV68 infection of control mice increased the levels of self-reactive antibodies (Fig. 5G and H). Similar to what was observed for anti-dsDNA antibodies, MHV68 infection of CD19 Cre-positive mice further increased the elevated baseline levels of broadly self-reactive class-switched antibodies, suggesting that gammaherpesvirus infection may exacerbate the autoimmune predisposition of this mouse strain.
Long-term MHV68 infection does not further elevate the production of self-reactive antibodies in CD19 Cre-positive mice.
The MHV68-driven germinal center response and the generation of self-reactive antibodies peak at 16 days postinfection and are subsequently attenuated via poorly understood mechanisms by the time that long-term infection is reached (42 days postinfection). As expected, by 42 days after MHV68 infection, CD19 Cre-negative mice demonstrated a significant contraction of both the germinal center B cell and TFH cell populations (Fig. 6A to D). CD19 Cre-positive MHV68-infected mice continued to demonstrate a decreased abundance of germinal center B cells (Fig. 6A and B), although this trend did not reach statistical significance at this time point. Intriguingly, the high baseline TFH cell population observed in naive CD19 Cre-positive mice and at 16 days postinfection was completely reversed by 42 days of MHV68 infection (Fig. 6C and D).
FIG 6.
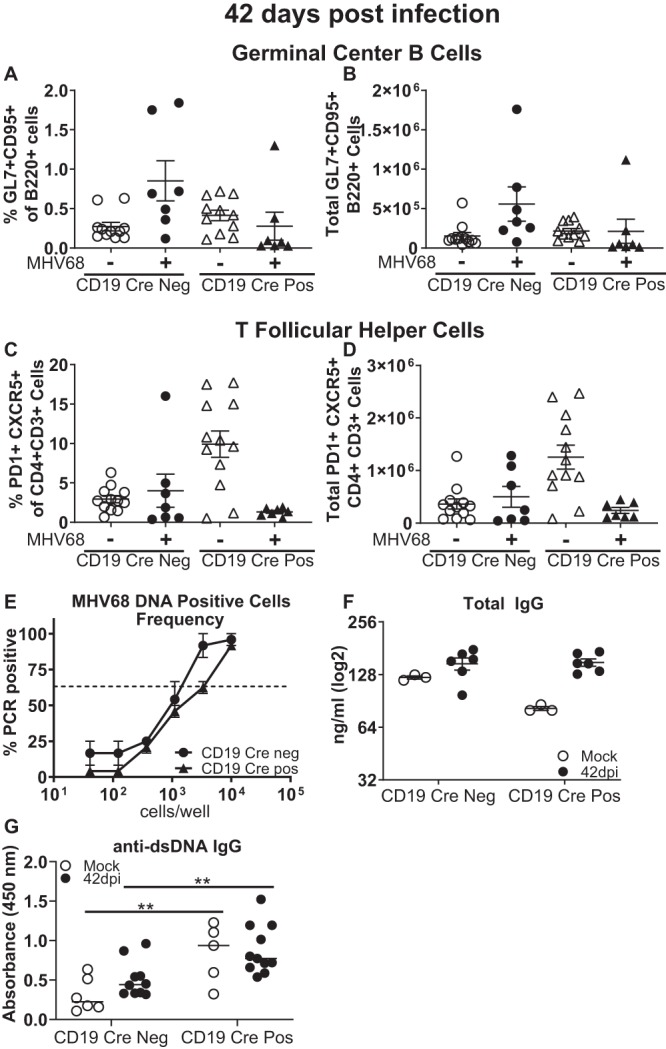
Long-term MHV68 infection does not further elevate the production of self-reactive antibodies in CD19 Cre-positive mice. Mice of the indicated genotypes were infected as described in the legend to Fig. 1 and examined at 42 days postinfection. Age-matched naive mice were used as controls. (A, B) Splenic germinal center B cells were identified as described in the legends to Fig. 2 and 3 and are expressed as a frequency (A) and the absolute number (B) of B220+ cells. (C, D) T follicular helper cells were identified as described in the legends to Fig. 2 and 3 and expressed as a frequency (C) and the absolute number (D) of CD3+ CD4+ cells. Each symbol represents the results for an individual spleen, and data from 2 to 4 independent experiments were pooled. (E) The frequencies of MHV68 DNA-positive splenocytes were measured using a limiting dilution assay as described in the legend to Fig. 1. Data for three to five mice within each group in each experiment were pooled, and data from 2 to 3 independent experiments were pooled. (F, G) Total IgG (F) and anti-dsDNA IgG (G) were measured by ELISA. Data from 2 to 4 independent experiments were pooled, and each symbol represents an individual mouse. The levels of significance are indicated: **, P = 0.01.
While MHV68 relies on the germinal center response to build up the peak latent reservoir at 16 days postinfection, the maintenance of long-term, lower levels of viral latency becomes more dependent on the infection of developing B cells in the bone marrow (46). Consistent with this switch, the frequency of MHV68 DNA-positive splenocytes was decreased compared to that observed at 16 days postinfection and was similar in CD19 Cre-positive and -negative mice at 42 days postinfection (Fig. 6E).
Finally, given the exacerbated generation of dsDNA-reactive antibodies at 16 days postinfection observed in CD19 Cre-positive mice, we wanted to test the hypothesis that long-term gammaherpesvirus infection cooperates with B cell-intrinsic SHP1 deficiency to enhance the generation of self-reactive antibodies, eventually accelerating autoimmune disease in this susceptible host. However, the levels of total and anti-dsDNA IgG antibodies were similar to the respective baseline levels in both CD19 Cre-negative and -positive mice (Fig. 6F and G). Thus, the generation of self-reactive antibodies exacerbated by B cell-intrinsic SHP1 deficiency was no longer observed during long-term MHV68 infection.
DISCUSSION
Gammaherpesviruses have a unique relationship with B cells: not only do they infect B cells, but they also usurp B cell differentiation to establish lifelong infection and, in a susceptible host, promote B cell malignancies and autoimmune disease. Identification of the cellular and molecular mechanisms that modify chronic gammaherpesvirus infection and pathogenesis remains challenging due to the exquisite species specificity of human gammaherpesviruses. In this study, we used mice infected with MHV68, a robust animal model of gammaherpesvirus infection and pathogenesis, to demonstrate that B cell- but not T cell-intrinsic SHP1 expression facilitates the gammaherpesvirus-driven germinal center response and the establishment of peak viral latency. SHP1 is a tyrosine phosphatase that attenuates B cell activation and proliferation. Correspondingly, SHP1 expression is significantly decreased in EBV- or KSHV-positive B cell lymphomas, including Burkitt’s lymphoma, diffuse large B cell lymphoma, monomorphic posttransplant lymphoproliferative disease, and primary effusion lymphoma (27–30). Intriguingly, the SHP1 protein is readily detectable in B cells transformed by EBV in vitro (29). These studies and our results suggest that the interaction between gammaherpesviruses and host SHP1 is much more nuanced than anticipated and that targeting SHP1 may have opposite effects, depending on the stage of virus infection and pathogenesis.
While the association between gammaherpesvirus infection and lymphomagenesis is well established, the connection between infection and autoimmunity is more controversial due to the heterogeneous nature of the latter disease and the high seroprevalence of EBV. However, the emerging consensus is that gammaherpesvirus infection may cooperate with the genetic or environmental susceptibility of the host to promote autoimmunity. We observed that MHV68 infection further enhanced the generation of class-switched anti-dsDNA and pan-self-reactive antibodies in autoimmunity-prone CD19 Cre-positive mice at the peak of viral latency (Fig. 5E to H). Unexpectedly, this cooperation between MHV68 infection and B cell-specific SHP1 deficiency was no longer observed by 42 days postinfection (Fig. 6G). A potential explanation for the temporally limited effect of MHV68 infection is offered by a report where SHP1 depletion in class-switched B cells led to a significant decrease in the abundance of long-lived, bone marrow-resident plasma cells (47). Additionally, the increased vulnerability of SHP1-depleted germinal center B cells to cell death may also explain why long-term MHV68 infection did not continue to exacerbate the already elevated production of dsDNA-targeted antibodies in CD19 Cre-positive mice. Because significant aging (8 months) is required to generate autoimmune disease in CD19 Cre-positive mice (33), examining additional time points beyond the first 6 weeks of MHV68 infection could offer an interesting insight into the potential of lifelong gammaherpesvirus infection to affect autoimmune disease in this susceptible host.
Our studies indicate that in spite of the decreased SHP1 expression observed in several gammaherpesvirus-driven B cell malignancies, in the context of natural infection SHP1 supports the establishment of peak viral latency by promoting the gammaherpesvirus-driven germinal B cell response. While there seems to be a lack of consensus regarding overall SHP1 protein levels in naive, germinal center, and memory B cells (25, 29), it is clear that, in contrast to other B cell differentiation stages, SHP1 is constitutively associated with BCR in germinal center B cells and promotes maintenance of the germinal center response following immunization with a T cell-dependent antigen (25). Interestingly, EBV reactivation from a Burkitt’s lymphoma cell line increases SHP1 expression (48). This observation, combined with normal SHP1 expression in EBV-transformed B cells, suggests that, in addition to the indirect proviral effects of SHP1 on the germinal center response, SHP1 may also have infected cell-intrinsic proviral effects, effects that become less relevant during lymphomagenesis in vivo.
The proviral effect of SHP1 that we observed during gammaherpesvirus infection was dependent on B cell-intrinsic SHP1 expression. The loss of SHP1 expression in T cells had no effect on any of the examined parameters, in spite of the critical importance of TFH cells for the MHV68-driven germinal center response and peak viral latent reservoir (44, 45). The SHP1-deficient T cells in the dLck Cre mouse model utilized in this study are resistant to suppression mediated by classical regulatory CD4 T cells (34). Unfortunately, very little, if anything, is known about the role of classical regulatory CD4 T cells during chronic gammaherpesvirus infection.
Intriguingly, the TFH cell population was increased in mice with B cell-specific SHP1 deficiency, in spite of the fact that the T cells in these mice have an intact PTPN6 gene, suggesting that the SHP1-deficient B cells might constitutively produce a survival factor(s) that selectively acts on the TFH cell population. Correspondingly, this factor must be present and/or functional only at the baseline and through the peak of MHV68 infection, as the levels of TFH cells in long-term MHV68-infected mice were low and independent of the CD19 Cre genotype. In contrast, SHP1-deficient germinal center B cells displayed higher levels of active caspase 3, indicating that the survival/growth factor in the germinal center milieu was not sufficient to maintain the viability of these SHP1-deficient B cells.
How might B cell-intrinsic SHP1 expression support the viability of germinal center B cells? One potential explanation is tied to the known role of SHP1 in the attenuation of BCR signaling. While BCR cross-linking is one of the steps required for naive B cell entry into the germinal center reaction, further differentiation into a germinal center B cell is associated with the constitutive association of SHP1 with the BCR and the attenuation of BCR signaling (25). In the absence of SHP1, the activation threshold for B cells is lower, resulting in low-affinity B cells entering the germinal center. These lower-affinity germinal center B cells are at a higher risk for being selected against during the germinal center response, which may account for the observed elevated cell death of SHP1-deficient germinal center B cells.
MATERIALS AND METHODS
Ethics statement.
All experimental manipulations of mice were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin (MCW; approval AUA971).
Animal studies.
All mouse strains used in this study were maintained at the MCW animal facility. Shp1fl/fl and dLck Cre-positive Shp1fl/fl mice were provided by Ulrike Lorenz. To regenerate the published model of B cell-specific SHP1 deficiency (33) that was no longer available from the original investigator, Shp1fl/fl mice were bred to CD19 Cre-positive (C.129P2-Cd19tm1(cre)Cgn/J) mice from The Jackson Laboratory (Bar Harbor, ME). At 6 to 10 weeks of age, the mice were intranasally inoculated with 500 PFU of MHV68 (WUMS) in 15 μl serum-free Dulbecco modified Eagle medium or mock infected. Males and females were interchangeably used in replicate experiments. Data were analyzed with respect to gender, and no significant differences were found. Mice were euthanized by CO2 inhalation from a compressed gas source in a nonovercrowded chamber as mandated by the American Veterinary Medical Association Panel on Euthanasia. Mice were bled at the indicated time points via the submandibular route, and serum was isolated using BD Microtainer blood collection tubes (Becton, Dickinson and Company, Franklin Lakes, NJ).
Limiting dilution assays.
The frequency of MHV68 DNA-positive cells and the frequency of ex vivo reactivation of MHV68 were determined as previously described (49). Briefly, to determine the frequency of cells harboring viral DNA, splenocytes were pooled from all mice within each experimental group (3 to 5 mice/group), and 6 serial 3-fold dilutions were subjected to a nested PCR (12 replicates/dilution) using primers against the viral genome. To determine the frequency of cells reactivating virus ex vivo, splenocytes were pooled from all mice within each experimental group (3 to 5 mice/group). and 8 to 12 serial 2-fold dilutions of splenocyte suspensions from each group were plated onto monolayers of mouse embryonic fibroblasts (MEFs) at 24 replicates per dilution. In order to control for preformed virus, 2-fold serial dilutions of mechanically disrupted splenocytes were plated as described above. Cytopathic clearing of MEFs was scored at 21 days postplating. The use of primary MEFs to amplify virus lowers the sensitivity of lytic MHV68 detection to a single PFU.
Flow cytometry.
Single-cell suspensions of splenocytes were prepared in fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline [PBS], 2% fetal bovine serum, 0.05% sodium azide) at 1 × 107 nucleated cells/ml. A total of 1.5 × 106 cells were prestained with Fc block (24G2 clone) and then incubated with an optimal dilution of antibody on ice. When necessary, following surface staining, the cells were fixed and permeabilized with BD Cytofix/Cytoperm solution (Fisher Scientific, Hampton, NH) for the detection of intracellular antigens. The following antibodies and reagents were used in this study and purchased from BioLegend (San Diego, CA), unless indicated otherwise: B220-phycoerythrin (PE)-Cy7, CD95-PE, GL7-fluorescein isothiocyanate (FITC), CD3-allophycocyanin (APC) A700, CD4-Pacific Blue, CXCR5 (a triple amplification with CXCR5-rat anti-mouse immunoglobulin, biotin-goat anti-rat immunoglobulin, streptavidin-APC was used), PD-1–PE, CD138-APC, IgG-PE, Ki67-PE, and cleaved caspase 3-FITC (CaspGLOW; Invitrogen). Data acquisition was performed on an LSR II flow cytometer (BD Biosciences, Sane Jose, CA), and the data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Enzyme-linked immunosorbent assay (ELISA).
Sera were collected from uninfected or infected mice of both genotypes at 16 or 42 days postinfection. Serum levels of total IgG was detected by coating Nunc MaxiSorp immunoplates (Fisher Scientific, Pittsburgh, PA) with goat anti-mouse IgG (Fcγ fragment specific; Jackson ImmunoResearch, West Grove, PA) at 10 μg/ml in PBS. The plates were washed with PBS-Tween (0.05%), blocked for 1 h with PBS-Tween (0.05%)-bovine serum albumin (BSA; 3%), incubated with 3-fold dilutions of serum in PBS-Tween (0.05%)-BSA (0.5%) for 1 h, and washed with PBS-Tween (0.05%). Bound antibody was detected with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Fcγ fragment specific) using 3,3′,5,5′-tetramethylbenzidine substrate (Life Technologies, Gaithersburg, MD). HRP enzymatic activity was stopped by the addition of 1 N HCl, and the absorbance was read at 450 nm on a model 1420 Victor3V multilabel plate reader (PerkinElmer, Waltham, MA). Anti-double-stranded DNA antibodies were detected by precoating Nunc MaxiSorp immunoplates (Fisher Scientific, Pittsburgh, PA) with 5 μg/ml methyl-BSA in H2O and were then washed with PBS and coated with 12.5 μg/ml DNA in PBS at 4°C. The wells were washed 2 times with PBS and then blocked with blocking buffer (1× PBS, 3% BSA, 3 mM EDTA, 0.1% gelatin) for 1 h. The wells were then incubated with 3-fold dilutions of serum in blocking buffer for 1 h, washed 2 times with PBS, and then washed 3 times with blocking buffer. Goat anti-mouse IgG (H+L)-HRP diluted in blocking buffer was added for 1 h at room temperature. Samples were washed 3 times with blocking buffer and then 2 times with PBS, followed by 3,3′,5,5′-tetramethylbenzidine substrate addition (Life Technologies, Gaithersburg, MD). HRP enzymatic activity was stopped by the addition of 1 N HCl, and the absorbance was read as described above.
ANA panels.
An antinuclear antibody (ANA) test kit was purchased from Antibodies Incorporated (Davis, CA), and serum was quantitatively analyzed following the manufacturer’s protocol. Diluted serum (1:40 in PBS) was incubated over slides coated with fixed HEp-2 cells. The slides were rinsed with PBS, followed by staining with anti-mouse IgG-Alexa Fluor 488 (H+L; Thermo Scientific). Fluorescent images were captured using NIS Elements software. The corrected fluorescence was quantified, using ImageJ software, from a randomly chosen field of ∼20 cells in each sample.
Statistical analyses.
Statistical analyses were performed using Student's t test (Prism software; GraphPad Software, Inc.). Significance was determined when P was ≤0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank Aaron Dirk for his technical support in this study. We thank the members of John Corbett's laboratory for the lively discussions of this study.
This study was supported by grant 18PRE33960455 (to P.T.L.) and grants CA183593 and CA203923 and a Rosenberg award (to V.L.T.).
K.E.J. and V.L.T. wrote the manuscript. K.E.J., P.T.L., V.L.T., B.N.D., and W.C. contributed to the design of the experiments. K.E.J., P.T.L., C.N.J., and P.J.V. performed and analyzed the results of the experiments. U.M.L. contributed mouse models and provided advice on the design of the studies and data interpretation.
We declare no competing interests.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Jha HC, Banerjee S, Robertson ES. 2016. The role of gammaherpesviruses in cancer pathogenesis. Pathogens 5:E18. doi: 10.3390/pathogens5010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cesarman E. 2014. Gammaherpesviruses and lymphoproliferative disorders. Annu Rev Pathol 9:349–372. doi: 10.1146/annurev-pathol-012513-104656. [DOI] [PubMed] [Google Scholar]
- 3.Namjou B, Kilpatrick J, Harley JB. 2007. Genetics of clinical expression in SLE. Autoimmunity 40:602–612. doi: 10.1080/08916930701510962. [DOI] [PubMed] [Google Scholar]
- 4.Draborg AH, Duus K, Houen G. 2012. Epstein-Barr virus and systemic lupus erythematosus. Clin Dev Immunol 2012:370516. doi: 10.1155/2012/370516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lossius A, Johansen J, Torkildsen Ø, Vartdal F, Holmøy T. 2012. Epstein-Barr virus in systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis—association and causation. Viruses 4:3701–3730. doi: 10.3390/v4123701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coleman CB, Nealy MS, Tibbetts SA. 2010. Immature and transitional B cells are latency reservoirs for a gammaherpesvirus. J Virol 84:13045–13052. doi: 10.1128/JVI.01455-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. 2000. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol 165:1074–1081. doi: 10.4049/jimmunol.165.2.1074. [DOI] [PubMed] [Google Scholar]
- 8.Stevenson PG, Doherty PC. 1999. Non-antigen-specific B-cell activation following murine gammaherpesvirus infection is CD4 independent in vitro but CD4 dependent in vivo. J Virol 73:1075–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorley-Lawson DA. 2001. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol 1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 10.Collins CM, Boss JM, Speck SH. 2009. Identification of infected B-cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J Virol 83:6484–6493. doi: 10.1128/JVI.00297-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuppers R, Klein U, Hansmann ML, Rajewsky K. 1999. Cellular origin of human B-cell lymphomas. N Engl J Med 341:1520–1529. doi: 10.1056/NEJM199911113412007. [DOI] [PubMed] [Google Scholar]
- 12.Thorley-Lawson DA, Gross A. 2004. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med 350:1328–1337. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- 13.Flano E, Kim IJ, Woodland DL, Blackman MA. 2002. gamma-Herpesvirus Latency Is preferentially maintained in splenic germinal center and memory B cells. J Exp Med 196:1363–1372. doi: 10.1084/jem.20020890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matar CG, Rangaswamy US, Wakeman BS, Iwakoshi N, Speck SH. 2014. Murine gammaherpesvirus 68 reactivation from B cells requires IRF4 but not XBP-1. J Virol 88:11600–11610. doi: 10.1128/JVI.01876-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. 2009. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog 5:e1000677. doi: 10.1371/journal.ppat.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sangster MY, Topham DJ, D'Costa S, Cardin RD, Marion TN, Myers LK, Doherty PC. 2000. Analysis of the virus-specific and nonspecific B cell response to a persistent B-lymphotropic gammaherpesvirus. J Immunol 164:1820–1828. doi: 10.4049/jimmunol.164.4.1820. [DOI] [PubMed] [Google Scholar]
- 17.Gauld SB, De Santis JL, Kulinski JM, McGraw JA, Leonardo SM, Ruder EA, Maier W, Tarakanova VL. 2013. Modulation of B-cell tolerance by murine gammaherpesvirus 68 infection: requirement for Orf73 viral gene expression and follicular helper T cells. Immunology 139:197–204. doi: 10.1111/imm.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darrah EJ, Jondle CN, Johnson KE, Xin G, Lange PT, Cui W, Olteanu H, Tarakanova VL. 2019. Conserved gammaherpesvirus protein kinase selectively promotes irrelevant B cell responses. J Virol 93:e01760-18. doi: 10.1128/JVI.01760-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleisher GR, Collins M, Fager S. 1983. Limitations of available tests for diagnosis of infectious mononucleosis. J Clin Microbiol 17:619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Decalf J, Godinho-Silva C, Fontinha D, Marques S, Simas JP. 2014. Establishment of murine gammaherpesvirus latency in B cells is not a stochastic event. PLoS Pathog 10:e1004269. doi: 10.1371/journal.ppat.1004269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tracy SI, Kakalacheva K, Lunemann JD, Luzuriaga K, Middeldorp J, Thorley-Lawson DA. 2012. Persistence of Epstein-Barr virus in self-reactive memory B cells. J Virol 86:12330–12340. doi: 10.1128/JVI.01699-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krug LT, Moser JM, Dickerson SM, Speck SH. 2007. Inhibition of NF-κB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog 3:e11–e118. doi: 10.1371/journal.ppat.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krug LT, Collins CM, Gargano LM, Speck SH. 2009. NF-kappaB p50 plays distinct roles in the establishment and control of murine gammaherpesvirus 68 latency. J Virol 83:4732–4748. doi: 10.1128/JVI.00111-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenz U. 2009. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev 228:342–359. doi: 10.1111/j.1600-065X.2008.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khalil AM, Cambier JC, Shlomchik MJ. 2012. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science 336:1178–1181. doi: 10.1126/science.1213368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamir I, Dal Porto JM, Cambier JC. 2000. Cytoplasmic protein tyrosine phosphatases SHP-1 and SHP-2: regulators of B cell signal transduction. Curr Opin Immunol 12:307–315. doi: 10.1016/s0952-7915(00)00092-3. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Zhu C, Wei F, Zhang L, Mo X, Feng Y, Xu J, Yuan Z, Robertson E, Cai Q. 2015. Constitutive activation of interleukin-13/STAT6 contributes to Kaposi’s sarcoma-associated herpesvirus-related primary effusion lymphoma cell proliferation and survival. J Virol 89:10416–10426. doi: 10.1128/JVI.01525-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ok CY, Li L, Xu-Monette ZY, Visco C, Tzankov A, Manyam GC, Montes-Moreno S, Dybkaer K, Dybaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Chen J, Richards KL, Hsi ED, Choi WWL, van Krieken JH, Huh J, Ai W, Ponzoni M, Ferreri AJM, Farnen JP, Møller MB, Bueso-Ramos CE, Miranda RN, Winter JN, Piris MA, Medeiros LJ, Young KH. 2014. Prevalence and clinical implications of Epstein-Barr virus infection in de novo diffuse large B-cell lymphoma in Western countries. Clin Cancer Res 20:2338–2349. doi: 10.1158/1078-0432.CCR-13-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delibrias CC, Floettmann JE, Rowe M, Fearon DT. 1997. Downregulated expression of SHP-1 in Burkitt lymphomas and germinal center B lymphocytes. J Exp Med 186:1575–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morscio J, Dierickx D, Tousseyn T. 2013. Molecular pathogenesis of B-cell posttransplant lymphoproliferative disorder: what do we know so far? Clin Dev Immunol 2013:150835. doi: 10.1155/2013/150835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsui HW, Siminovitch KA, de Souza L, Tsui FW. 1993. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet 4:124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 32.Shultz LD, Sidman CL. 1987. Genetically determined murine models of immunodeficiency. Annu Rev Immunol 5:367–403. doi: 10.1146/annurev.iy.05.040187.002055. [DOI] [PubMed] [Google Scholar]
- 33.Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, Thomas ML, Neel BG, Rajewsky K. 2007. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity 27:35–48. doi: 10.1016/j.immuni.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 34.Mercadante ER, Lorenz UM. 2017. T cells deficient in the tyrosine phosphatase SHP-1 resist suppression by regulatory T cells. J Immunol 199:129–137. doi: 10.4049/jimmunol.1602171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Efstathiou S, Ho Y, Hall S, Styles C, Scott SD, Gompels U. 1990. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J Gen Virol 71:1365–1372. doi: 10.1099/0022-1317-71-6-1365. [DOI] [PubMed] [Google Scholar]
- 36.Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71:5894–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarakanova VL, Suarez F, Tibbetts SA, Jacoby MA, Weck KE, Hess JL, Speck SH, Virgin HW. 2005. Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB β2 microglobulin-deficient mice. J Virol 79:14668–14679. doi: 10.1128/JVI.79.23.14668-14679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weck KE, Kim SS, Virgin HW, Speck SH. 1999. B cells regulate murine gammaherpesvirus 68 latency. J Virol 73:4651–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weck KE, Kim SS, Virgin HW, Speck SH. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol 73:3273–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willer DO, Speck SH. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J Virol 77:8310–8321. doi: 10.1128/jvi.77.15.8310-8321.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collins CM, Speck SH. 2012. Tracking murine gammaherpesvirus 68 infection of germinal center B cells in vivo. PLoS One 7:e33230. doi: 10.1371/journal.pone.0033230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. 1998. Visualization of specific B and T lymphocyte interactions in the lymph node. Science 281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 43.Yusuf I, Stern J, McCaughtry TM, Gallagher S, Sun H, Gao C, Tedder T, Carlesso G, Carter L, Herbst R, Wang Y. 2014. Germinal center B cell depletion diminishes CD4+ follicular T helper cells in autoimmune mice. PLoS One 9:e102791. doi: 10.1371/journal.pone.0102791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins CM, Speck SH. 2015. Interleukin 21 signaling in B cells is required for efficient establishment of murine gammaherpesvirus latency. PLoS Pathog 11:e1004831. doi: 10.1371/journal.ppat.1004831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collins CM, Speck SH. 2014. Expansion of murine gammaherpesvirus latently infected B cells requires T follicular help. PLoS Pathog 10:e1004106. doi: 10.1371/journal.ppat.1004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coleman CB, McGraw JE, Feldman ER, Roth AN, Keyes LR, Grau KR, Cochran SL, Waldschmidt TJ, Liang C, Forrest JC, Tibbetts SA. 2014. A gammaherpesvirus Bcl-2 ortholog blocks B cell receptor-mediated apoptosis and promotes the survival of developing B cells in vivo. PLoS Pathog 10:e1003916. doi: 10.1371/journal.ppat.1003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li YF, Xu S, Ou X, Lam KP. 2014. Shp1 signalling is required to establish the long-lived bone marrow plasma cell pool. Nat Commun 5:4273. doi: 10.1038/ncomms5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan J, Cahir-McFarland E, Zhao B, Kieff E. 2006. Virus and cell RNAs expressed during Epstein-Barr virus replication. J Virol 80:2548–2565. doi: 10.1128/JVI.80.5.2548-2565.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HW. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol 70:6775–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.