

Abstract
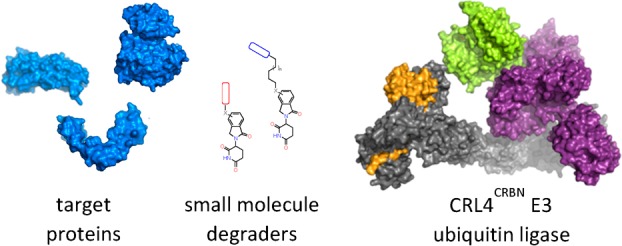
Many cellular processes and pathways are mediated by the regulation of protein–protein complexes. For example, E3 ubiquitin ligases recruit substrate proteins and transfer a ubiquitin tag to target those proteins for destruction by the proteasome. It has now been shown that this cellular process for protein destruction can be redirected by small molecules in both laboratory and clinical settings. This presents a new paradigm in drug discovery, enabling the rapid removal of target proteins linked to disease. In this Innovations review, we will describe the work done on cereblon as a case study on the different strategies available for targeted protein degradation.
Keywords: Cereblon, targeted protein degradation, CELMoD, molecular glue, E3 ligase
Protein–protein interactions are critical in many cellular processes and targeting them with small molecule therapeutics remains one of the most elusive of drug discovery endeavors, though there is precedent for small molecules enhancing protein–protein interactions to significant biological effect.1 Brefeldin A, a fungal metabolite stabilizing the interaction between ARF1 and Sec7, inhibits protein secretion.1,2 The structure of the ARF1-Brefeldin A-Sec7 complex revealed that Brefeldin A is bound at the interface between ARF1 and Sec7, explaining the molecular basis for stabilization.3,4 Other early examples, rapamycin and FK506,5,6 in contrast to Brefeldin A, bind tightly to one protein, FKBP12. The complex of FKBP12–rapamycyin then inhibits mTOR, while the FKBP12–FK506 complex inhibits calcineurin.7,8 Rapamycin and FK506 drive a substantial proportion of the direct interactions in the ternary complexes,9 though neither rapamycin nor FK506 bind to the second binding partner, mTOR, or calcineurin, in the absence of FKBP12. Small molecules that can act to bring two proteins together resulting in modulation of a downstream signaling cascade, or even to trigger the degradation of the protein harnessing the body’s natural proteosomal machinery, have the potential to expand the druggable proteome beyond the more classical protein families possessing small molecule binding sites. Protein degradation in particular is emerging as a powerful therapeutic modality, with interest from both academic and biopharma institutions in this relatively nascent field skyrocketing over the past 10 years. Although recent advances in understanding of mechanistic aspects of protein degradation necessary for realizing the full potential of this modality have garnered a great deal of attention in the drug discovery arena, a rich history, including many approved drugs has been quietly evolving for decades. Selective Estrogen Receptor Degraders (SERDs) are some of the earliest known small molecule protein degraders. Approved in 2002, fulvestrant was the first pure antiestrogen mechanistically linked to the decrease of intracellular ERα levels.10 Upon binding the ER, fulvestrant causes a destabilization of the ER and consequent proteasomal degradation. This early example of protein degradation has served as an important advance in the field of breast cancer treatment, and illustrated the potential benefits of protein degradation over antagonism/inhibition in overcoming common resistance mechanisms.11 A resurgence in this field has led to a number of next generation clinical stage molecules, with more favorable pharmacokinetic profiles and oral routes of administration.12
The Ubiquitin Proteasome System
Targeted protein degradation utilizing the ubiquitin proteasome system (UPS) has emerged as a therapeutic modality with broad potential utility, whereby small molecules are able to hijack the cell’s natural protein degradation machinery to regulate proteolysis in eukaryotic cells by tagging proteins to be degraded with ubiquitin.13,14 Ubiquitin is post-translationally added to lysine residues on the surface of the target protein by a ubiquitin ligase enzyme, and can be subsequently conjugated to lysine residues on ubiquitin itself, leading to the formation of ubiquitin chains. Specific ubiquitin chain linkages then direct the conjugated protein to the 26S proteasome for ATP-dependent proteolytic destruction (Figure 1).15 Ubiquitination occurs by way of a three-enzyme cascade, where ubiquitin is first activated by a ubiquitin activating enzyme, or E1, and then transferred to one of many ubiquitin conjugating enzymes, or E2s. Finally, the ubiquitin ligase, or E3 brings together a ubiquitin-conjugated E2 and the target protein (termed the substrate), and facilitates the transfer of ubiquitin from the E2 to the substrate.16 The spectrum of activity of a particular ligase is determined by the substrate-binding subunits or domains, which recognize specific features including amino acid sequence and post-translational modifications. E2s, E3s, and substrates can all be differentially regulated to allow the cell to control specific target protein levels in response to varying cellular conditions.17 There are multiple approaches to hijacking the UPS to degrade specific proteins of interest for both therapeutic and research objectives. The goal of the small molecule in this context is to confer the appropriate proximity and residence time of the substrate to the ubiquitin ligase complex to facilitate tagging, and there are distinct classes of ligands capable of redirecting E3 ubiquitin ligases toward new proteins, each with different applications and pharmacological properties.
Figure 1.

CRL4–cereblon as an example of an E3 ubiquitin ligase that can be targeted with small molecules. The CRL4–cereblon ubiquitin ligase is a protein complex containing cereblon (CRBN), DDB1, Cul4, and Rbx1. Cereblon modulators (CM) like lenalidomide (len) bind to cereblon and modify its surface to create a new interface for target substrate (S) binding. Target substrates bind CRBN + CM, allowing a substrate lysine side chain to attack the E2–ubiquitin bond and leading to ubiquitin transfer from E2 to substrate.
Small molecule modulators of E3 ligases that scaffold protein–protein interactions exist in nature, a mechanism termed “molecular glue”.18 For example, the plant hormone auxin (Figure 2) binds to the SCF–TIR1 complex and triggers the degradation of the Aux/IAA proteins,19 transcription factors sharing a common “degron” that bind with an increased affinity to auxin-bound SCF–TIR1. Structural studies revealed that auxin binds in a surface cavity on TIR1 and provides extensive interactions to the degron peptide,20 providing a striking proof-of-concept for the regulation of E3 ligases by very small, efficient ligands. A similar mechanism is found with the plant hormone jasmonate, which binds to the ligase SCF–COI1 to drive recruitment of JAZ proteins.21 Along with plant hormones, further examples of small molecules that induce targeted protein degradation include indisulam,22 the aforementioned SERDs, specific androgen receptor degraders (SARDs),23 a recently reported BCL6 degrader,24 and cereblon modulators, which will be discussed extensively herein.
Figure 2.

The plant hormone auxin promotes the interaction between TIR1 and IAA7. TIR1 (green) is a ubiquitin ligase substrate adapter to which TIR1 (blue) is recruited. Auxin (orange) binds to TIR1 at the interface with IAA7 and increases the affinity for the interaction of E3 ligase and substrate to promote substrate ubiquitination and degradation.
Among the E3 ligase family of proteins, cereblon (CRBN) has emerged as a particularly tractable target for low molecular weight therapeutics. Small molecule cereblon modulators scaffold direct protein–protein interactions between the CRL4–CRBN E3 ubiquitin ligase and substrate, exemplifying the molecular glue mechanism. The first known cereblon modulator is the infamous drug thalidomide, first commercialized by Chemie Grunenthal in the 1950s for over-the-counter use in various indications including morning sickness, causing thousands of cases of severe birth defects.25 Thalidomide was withdrawn from the market in the early 1960s, but later found clinical use in treating leprosy and mutliple myeloma. The thalidomide analogues lenalidomide and pomalidomide (Figure 3) were subsequently developed and approved for hematological malignancies including multiple myeloma. These molecules were discovered and developed based on phenotypic observations in cellular assays and whole animals well before the exact molecular mechanisms underlying their therapeutic action was identified to be through cereblon.26 Cereblon was found to associate with the protein DNA damage binding protein-1 (DDB1), an adaptor protein component of the DDB1–CUL4–Rbx1 ubiquitin ligase (CRL4), establishing cereblon as a one of ∼70 CRL4 substrate adapters, or DCAFs (DDB1 and Cul4 associated factors).18,27−29
Figure 3.

Chemical structures of thalidomide, lenalidomide, and pomalidomide.
Unlocking the Cereblon Mechanism of Action
Further mechanistic understanding emerged when multiple groups demonstrated that lenalidomide and pomalidomide and other cereblon modulating drugs were not inhibiting, but instead activating the cereblon-CRL4 ubiquitin ligase toward specific proteins targets, termed neosubstrates.30−33 The first neosubstrates to be identified were Ikaros (IKZF1) and Aiolos (IKZF3), two C2H2 Zn-finger transcription factors that regulate hematopoetic cell differentiation.34 The protein kinase casein kinase 1α (CK1α) was also shown to also be a neosubstrate recruited to cereblon by lenalidomide, but not pomalidomide, indicating the potential for ligand-differentiated neosubstrate recruitment.35
The ability to selectively and differentially degrade substrates with different chemical matter is critical to the potential for cereblon E3 Ligase Modulators (CELMoDs) in drug discovery. For example, CC-885 exhibits highly potent antiproliferative activity against a broad panel of tumor cell lines (Figure 4).36 Immunoprecipitation of cereblon in the presence of CC-885 identified G to S Phase Transition Protein 1 (GSPT1) as the target protein, and GSPT1 degradation by CC-885 was the driver for the potent antiproliferative activity. Of the panel of cell lines tested, acute myeloid leukemia (AML) lines showed extreme sensitivity to GSPT1 degradation, a finding that was reproducible in patient-derived ex vivo AML cells. In a demonstration of the potential versatility of CELMoDs, GSPT1 recruitment is not mediated by lenalidomide or pomalidomide. However, the molecular mechanisms of ligand-dependent substrate recruitment remained unclear, and it was unknown whether Ikaros/Aiolos, CK1α, and GSPT1 degradation would function through a common mechanism, given their apparent lack of any overlapping sequence, function, or unifying theme.
Figure 4.

Chemical structures of CC-885, iberdomide (CC-220), and avadomide (CC-122).
Structure–Function Studies on Neosubstrate Ternary Complexes
The cocrystal structure of lenalidomide bound to cereblon–DDB1 confirmed that the glutarimide ring of lenalidomide was a common moiety critical for functional activity, and multiple X-ray structures revealed that this glutarimide ring docks in a hydrophobic tritryptophan pocket on the cereblon surface, leaving the isoindolinone exposed on the protein surface.37,38 It was hypothesized that drug binding altered the surface of cereblon such that a planar hydrophobic group was positioned among available hydrogen bond donors and acceptors, creating a hotspot for the nucleation of protein–protein interactions. In this way, the surface of cereblon can be modified by ligands to form a neomorphic interface—an interface that imparts new features and activities. Two structural studies of substrates bound to cereblon and ligand confirmed the molecular mechanism of substrate recruitment for these compounds: CK1α was crystallized bound to cereblon through lenalidomide, and GSPT1 was crystallized bound to cereblon and CC-885 (Figure 5).39,36 In both cases, the major substrate–ubiquitin ligase interface involves a surface turn on the substrate that contributes three hydrogen bonds to cereblon from backbone carbonyl oxygens and contains a key glycine residue that packs directly against compound and takes on backbone torsion angles only accessible to glycine (Figure 5).
Figure 5.

CRBN interacts with a similar feature on the substrate proteins GSPT1, CK1α, and Ikaros. CRBN (green) bound to GSPT1 (a, blue, PDB ID: 5HXB), CK1α (b, blue, PDB ID: 5FQD), or Ikaros (c, blue, PDB ID: 6H0F) interacts with a hairpin on the substrate protein. Ligands are represented as sticks (maroon) with CC-885 present in panel a, lenalidomide present in panel b, and pomalidomide present in panel c. The same pattern of three direct hydrogen bonds from residues N351, H357 and W400 is present between CRBN and GSPT1, CK1α, or Ikaros.
Both accounts were able to predict that Ikaros and Aiolos contained a similar molecular feature that mediated their recruitment to cereblon, leading to the startling finding that even though CK1α, GSPT1, and Ikaros/Aiolos are structurally and functionally unrelated and share no obvious sequence homology, they are recruited through a homologous interacting structural feature, termed a “degron”, defined by a specific loop containing a glycine residue at a key position. As predicted by the model, the key glycine residue in each case could not be mutated (even to an alanine) with preservation of function.36 Both crystal structures show that these substrates directly contact both cereblon and ligand, with the ligand scaffolding protein–protein interactions in a mechanism reminiscent of auxin and jasmonate. The role of the protein–protein interactions in scaffolding the recruitment of GSPT1 was confirmed by alanine-scanning mutagenesis of residues on the cereblon surface, showing that regions outside of the CELMoD binding pocket are critical for protein–protein complex formation and degradation of substrate. Importantly, neither GSPT1 nor CK1α was observed to bind cereblon in the absence of a CELMoD using biochemical and biophysical techniques, and there is no detectable binding between the CELMoD and the substrate protein. The structures of cereblon in complex with the zinc finger domains from the substrates Ikaros and ZNF692 showed that the degron in zinc fingers is recruited to cereblon in the analogous manner to GSPT1 and CK1α,40 further confirming the degron hypothesis. It is clear from the structural and biophysical studies that molecular glue offers therapeutic accessibility to a broad family of proteins previously thought to be undruggable, and by scaffolding protein–protein interactions, degradation can be achieved without the need for canonical small molecule binding sites on the target protein.
Teratogenicity: Elucidating Pleiotropy of Thalidomide
Once a common mechanism of neosubstrate recruitment was elucidated, it became possible to search for new substrates through the degron feature, and the degradation of an unidentified neosubstrate was hypothesized to be responsible for the teratogenic effects of thalidomide and its analogues. A candidate-based approach of testing developmental regulators with potential glycine-containing structural degrons identified the embryonic zinc finger transcription factor SALL4 as a new thalidomide-dependent neosubstrate.41 Thalidomide was found to induce the ubiquitination of SALL4 by cereblon-CRL4 in vitro as well as proteasomal degradation of SALL4 in human iPSCs, both dependent upon a glycine-containing degron zinc finger within the protein. A separate unbiased approach used mass spectrometry on human embryonic stem cells treated with thalidomide and thalidomide analogs, also leading to the identification of SALL4 as a cereblon neosubstrate.42 Loss-of-function mutations in SALL4 cause human syndromes characterized by forearm malformations as well as varying degrees of ear, eye, kidney, and heart defects.43 The effects of SALL4 mutation are sufficiently overlapping with those of thalidomide embryopathy that patients misdiagnosed with thalidomide embryopathy have later been found to harbor heritable mutations in SALL4.44,45
SALL4 degradation has also been assessed in animal models, where a confounding factor in thalidomide teratogenicity studies has been a strong species-specificity. As early as the 1960s there were reports of differential teratogenic effects in rabbits, mice, and rats, with rabbits being sensitive to teratogenicity, while mice and rats are resistant.46 Amino acid differences between species in SALL4 and cereblon led to SALL4 degradation in rabbits, while sequence variations in both mouse cereblon and mouse SALL4 prevent SALL4 from being targeted in mice or mice engineered to express human cereblon, which are both insensitive to teratogenicity. SALL4 degradation was confirmed in rabbit embryos treated with thalidomide during the sensitive window of development, correlating with the observed limb defects and gross developmental abnormalities.41 While these studies do not conclude that SALL4 is the only neosubstrate whose degradation is capable of causing thalidomide embyropathy, the strong link to human genetics indicates that SALL4 degradation is likely a major contributor. Importantly, SALL4 degradation does not always correlate with degradation of therapeutic targets. Clinical molecules iberomide (CC-220) and avadomide (CC-122) showed enhanced degradation of Ikaros and Aiolos, but significantly weaker activity toward SALL4, demonstrating an important SAR break and proof of principle for increasing desired efficiency on a target while reducing SALL4 degradation.
From Phenotypic to Target-based Approaches
Although thalidomide and lenalidomide were discovered and developed without knowledge of the direct molecular targets, understanding of substrates such as Ikaros and Aiolos, GSPT1, and SALL4 have opened new opportunities for target-based approaches. The identification of mechanism-of-action and subsequent biophysical characterization of cereblon in ternary complexes, both as molecular glue and in more recent heterobifunctional degraders, are transformative to the application of hypothesis-driven rational drug design for cereblon-accessible targets. Knowledge of molecular mechanism enables the discovery of novel substrates with potentially new clinical utility and enables the differentiation of clinical molecules with shared or overlapping target degradation mechanisms. For example, iberdomide (CC-220) is an Aiolos and Ikaros degrader in clinical development for lupus and relapsed refractory multiple myeloma47 and is roughly 30-times more potent at engaging cereblon compared to lenalidomide.48 Correspondingly, iberdomide has a similar increase in potency for the degradation of Ikaros and Aiolos. A crystal structure of cereblon in complex with iberdomide showed that increased interactions with the surface of cereblon underlie the observed potency enhancement (Figure 6).
Figure 6.
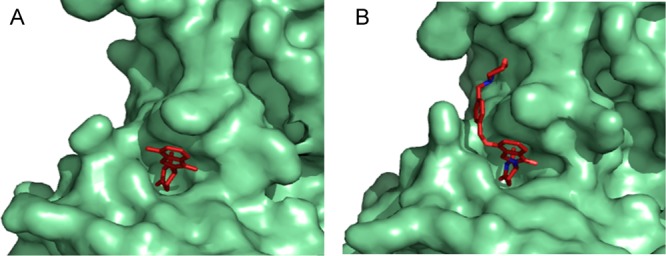
Iberdomide makes more extensive interactions with the cereblon surface than lenalidomide. Crystal structures of cereblon (green) bound to either lenalidomide (a, red, PDB ID: 4TZ4) or iberdomide (b, red, PDB ID: 5V3O) demonstrate the increased surface interactions of iberdomide compared to lenalidomide, contributing to the increased potency of iberdomide.
The Emergence of Heterobifunctional Degraders
Building upon historical learnings in protein degradation, a conceptually simple yet operationally complex modality, has emerged, which may be capable of bringing a wider swath of the human proteome into the realm of druggable. Heterobifunctional degraders, or Proteolysis TArgeting Chimeras (PROTACs), provide an approach for targeting proteins that lack a functional degron.49 In contrast to molecular glue, heterobifunctional degraders comprise multiple structurally and functionally differentiated components: one end of the molecule binds to an E3–ligase, the second to a binding interface of a target protein, and these are connected by a linker of sufficient length and architecture to span the binding pockets and promote ternary complex formation. Following initial proof-of-principle experiments using peptidic E3–ligase binding moieties,50 the field advanced by optimization of the peptidic ligands into more drug-like chemical matter.51,52 This work converged with the cereblon mechanism of action learnings as it was demonstrated that thalidomide analogs could function as ligase binding moieties in heterobifunctional degraders. The seminal report of an effective heterobifunctional degrader leveraging a thalidomide analog as the E3–ligase binder demonstrated the degradation of BRD4 using dBET1 (Figure 7).53 This approach has proven to be broadly applicable, with success subsequently demonstrated in the degradation of BTK, CDK6, p38, etc.54−77
Figure 7.

Conceptual framework of the components of a heterobifunctional degrader and chemical structure of dBET1.
Heterobifunctional degraders in the broadest definition comprise a target binding moiety (TBM), a ubiquitin E3 ligase binder, and an adjoining linker. Heterobifunctional degraders are designed to maximize specific interactions with the proteins of interest through conformational optimization, while balancing pharmacokinetic and physicochemical properties. Each component brings with it a set of unique and tunable properties, the culmination of which results in a concerted functionality that is unlikely to be purely additive or easily predicted based upon first principles. (1) TBM. There are numerous factors to consider in the identification and optimization of a TBM. Transient association of the TBM can induce irreversible degradation; therefore, an expanded range of properties have utility in heterobifunctional degraders compared to functional inhibitors. In addition, there is opportunity to override intrinsic functional activity and selectivity of autonomous binders. The affinity of the TBM to the target of interest need not be of the same potency range typically required of an inhibitor, and below a certain threshold, considerations of ligand efficiency and selectivity need to be balanced in order to impart suitable pharmaceutics properties to the overall heterobifunctional molecule. (2) Ubiquitin E3–ligase binding moiety. Ligands have been identified for recognition of Mouse Double Minute 2 homologue (MDM2), Von Hippel–Lindau (VHL), inhibitor-of-apoptosis proteins (IAP), and cereblon, and employed in the degradation of a variety of protein classes including proteases, nuclear hormone receptors, epigenetic regulators, and kinases. Utility of a ligand for a given ubiquitin E3 ligase depends upon its ability to promote ternary complex formation, ubiquitination, and degradation, as well as its physiochemical properties, the latter of which is particularly important for systemic administration and oral bioavailability. Furthermore, considerations regarding subcellular colocalization of the E3 ligase with the target protein of interest can be an important factor driving effectiveness of protein depletion. (3) Linker. Linker design is guided by the same principles as TBMs and E3 ligase binders. However, linker architecture presents an expanded opportunity for diversification and optimization toward improved drug-like properties. Strategies for linker optimization include increasing rigidity/minimization of entropic cost, increasing atom economy, modulating physicochemical properties by thoughtful introduction of heteroatoms and lipophilicity, and fine-tuning of the trajectory from target protein to ubiquitin ligase binding sites. Depending on the depth of the binding pocket within the protein of interest, the linker may also be engaged in binding contacts in a channel on the way to the pocket, and as such, can also impact affinity and selectivity.
Molecular glue and heterobifunctional degradation are on a conceptual continuum; in practice, there are notable distinctions in the way these strategies are deployed in drug discovery. Target proteins for molecular glue degraders are neo- or hypermorphic in their ability to interact with hotspot surfaces of ubiquitin ligases. Target protein identification requires a nuanced understanding of protein–protein and protein–small molecule structural interactions. Historically, molecular glue degraders were discovered serendipitously and phenotypically, followed by deconvolution of the participants and mechanism of degradation. More recently, rational design of molecular glues have relied on identification of target proteins with a conserved glycine-containing structural degron, and their optimization can be achieved through structure-guided medicinal chemistry. An understanding of off-target neosubstrate activity is paramount for managing safety and tolerability.
There are numerous complexities in the application of rational design principles for heterobifunctional degraders compared to molecular glue. In molecular glue, there is the presumption of a positive cooperative interaction with the neosubstrate protein leading to ternary complex formation resulting in ubiquitination/degradation. With heterobifunctional degraders, a substrate protein in ternary complex with a heterobifunctional ligand and an E3–ligase can be either positively or negatively cooperatively stabilized by interaction with the E3–ligase. And, while X-ray crystallography has resulted in detailed molecular views of the ternary complexes,78,79 protein–protein interactions can be differentially modulated by different target and ligase binding moieties, respectively, possessing different vectors and linker geometries. Due to the wider relative degree-of-freedom between the protein surfaces by virtue of the lack of a discrete degron, new methods have developed to rationally model the complexes. Some focus on designing complexes to active ligands, effectively aligning the protein structures to active ligand conformations, whereas others focus on obtaining ternary models of the most highly cooperative pairing between E3–ligase and neosubstrate, using molecular dynamics and/or protein–protein docking.
Target selection for molecular glue or heterobifunctional degradation is guided by the biologic phenotype and safety profile associated with transient removal of the protein of interest. There are, however, a number of target attributes for which ligand directed degradation (LDD) is uniquely advantageous compared to small molecule or biologic therapeutics. Additionally, there are special considerations for assessing tractability of LDD for a given protein of interest. The most notable and differentiating aspect of LDD compared to small molecules and biologics is that LDD results in removal of entire proteins. As a result, LDD can elicit amplified biological responses and provide a means of targeting proteins that are catalytically inactive and/or function exclusively, or in part, through scaffolding. In cases of novel biology, LDD can be readily compared with human genetic data and genetic knockdown (KD), knockout (KO), or inducible KO in preclinical in vitro or in vivo systems. Additionally, LDD expands the binding sites that can be leveraged to modulate functional effect beyond catalytic sites and protein–protein interfaces. Because LDD is catalytic and irreversible, degraders can provide chemical KD that persists beyond the circulating or tissue resident half-life of the degrader. Accordingly, de novo synthesis rates of target and off-target proteins should be considered. Selectivity with respect to both inhibition and degradation of off-target proteins and the potential for proximity induced off-target degradation are also important considerations. Lastly, spaciotemporal colocalization of the target protein and E3 in disease relevant tissues, cells, and intracellular compartments is critical.
Pharmacological Profiling of LDDs
The suite of assays required to prosecute LDD as a pharmacological approach differ from and are more labor intensive than a canonical small molecule effort. Typically, an on-target screening cascade to enable a drug discovery program is supported by a biochemical binding assay to assess on-target intrinsic potency as well as a cellular functional assay to account for permeability and potential potency shifts relevant to the target of interest and its proximal biological interactions. There are numerous additional considerations to further inform on-target biologically relevant potency, off-target selectivity, and pharmacokinetic–pharmacodynamic–efficacy relationships. A binding assay to the target of interest is a useful starting point and should be paired with a relevant functional assay, degradation of a protein of interest in the case of a heterobifunctional degrader, or neosubstrate(s) of interest in the case of molecular glue. For cereblon, a host of neosubstrates have been identified and characterized allowing selectivity profiling for a phenotypic degradation fingerprint to be established and optimized. The intermediate steps between binding and degradation: ternary complex formation, ubiquitination, and proteasomal trafficking, can each be assessed to inform design and optimize degradation.80 There are a number of methods available to characterize these processes and the resultant degradation with varying levels of investment required to operationalize and generate data. Critical metrics include the potency, or concentration needed to induce degradation of a particular level (e.g., DC50 representing a 50% reduction of the target protein present), as well as the corresponding depth of degradation achieved (e.g., Ymin) (Figure 8). As a relevant example, thalidomide, lenalidomide, and pomalidomide interact with cereblon to induce degradation of neosubstrates, including Aiolos. These three small molecules differentiate on DC50 and Ymin, and this differentiated pharmacology is clinically validated with greater efficacy achieved with lenalidomide versus thalidomide and still further affect pomalidomide in lenalidomide refractory patients. Unlike a pure sigmoidal potency continuum for a typical inhibitor, there is often a threshold effect for tipping cells into apoptosis with degradation mechanisms. As noted in the literature for heterobifunctional degraders, a “hook effect” or “prozone region” can be observed when the system is oversaturated in vitro and ternary complex formation is reduced in preference to separate binding events to the target of interest and the ligase of interest.81 Degradation is often assessed via Western blot in a cell type of interest, though technologies continue to advance enabling higher throughput, quantitative data generation in overexpressing, and endogenous tagged systems (Table 1). Global proteomics can also be leveraged to probe off-target selectivity for reduction in protein levels.
Figure 8.
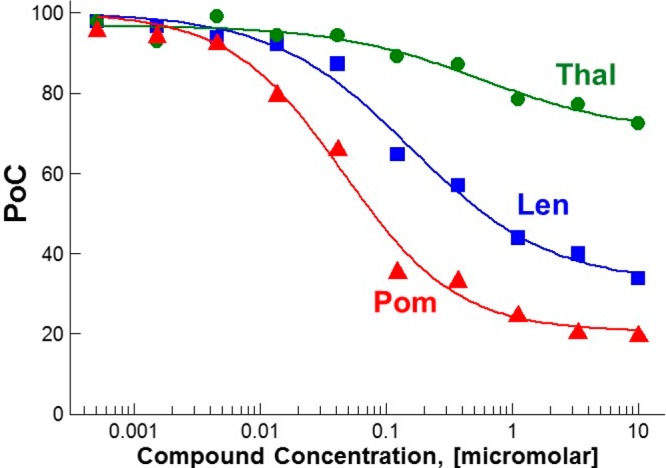
In vitro Aiolos degradation via molecular glue with thalidomide (Thal), lenalidomide (Len), and pomalidomide (Pom) in DF-15 cells at 4 h.
Table 1. Representative Assays and Techniques for Assessment LDD in Cells.
| assay | cell lysates | live cells | target engagement | ternary complex formation | ubiquitination | proteosome trafficking | degradation |
|---|---|---|---|---|---|---|---|
| Western blot | √ | √ | |||||
| enzyme-linked immunosorbent assay (ELISA) | √ | √ | |||||
| multiplexed immunoassays | √ | √ | |||||
| proteomics | √ | √ | √ | ||||
| enzyme fragment complementation | √ | √ | √ | ||||
| protein fusion tags | √ | √ | √ | ||||
| bioluminescence | √ | √ | √ | √ | √ | √ | √ |
Once degradation and the preceding intermediary steps are characterized in vitro, LDD can be assessed in vivo to introduce the additional interdependencies of plasma levels and tissue distribution. Heterobifunctional degraders present challenges for achieving oral bioavailability, though since they can function in a substoichiometric catalytic manifold, careful consideration to target coverage and downstream functional implications is critical. Depending upon the target or neosubstrate of interest along with its corresponding resynthesis rate,82 endogenous local levels of the E3 ligase, and kinetics of ternary complex formation, ubiquitination, and proteasomal trafficking, a single molecular glue or LDD can participate in multiple degradation events. Further, since the downstream proteasome-mediated degradation is independent of small molecule binding, functional effects may persist beyond systemic exposure of the degrader (Figure 9). This characteristic may invoke safety implications if off-target proteins are degraded, even transiently at Cmax.
Figure 9.
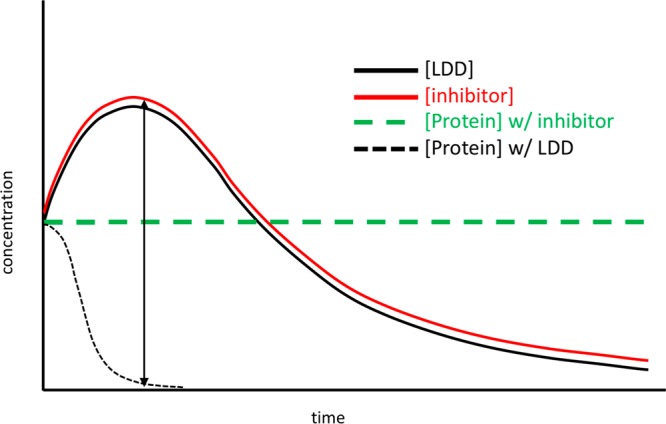
Representative indirect PK–PD–efficacy–safety relationship in vivo of degraders compared with conventional inhibitors.
The development of LDDs since first conceptualized and described in 1999 has evolved from rudimentary chemical biology tools to molecules with real therapeutic potential. An early heterobifunctional degrader of androgen receptor with a peptidic binding moiety had consequently poor physicochemical properties, requiring microinjection into the cell to observe degradation.83 Advancing toward therapeutic relevance, a CRBN-based heterobifunctional BRD4 degrader (dBET1) showed in vivo efficacy when dosed at 50 mg/kg QD for 14 days, delaying leukemia progression in mice,53 though using intraperitoneal (IP) injection. To date, most accounts assessing PD and/or efficacy of heterobifunctional degraders in vivo have been administered via IP or subcutaneous (SC) injection. While oral bioavailability data for heterobifunctional degraders have not yet appeared in the literature, it has been reported that androgen receptor degrader ARV-110 was dosed orally in a mouse xenograft model and entered clinical trials in 2019.
For continued progression of LDDs toward clinical utility, favorable pharmacokinetics will be critical. In an effort to categorize molecular properties that could guide development of oral bioavailability, a recent review evaluated a representative set of published heterobifunctional degraders that included a table of calculated properties for each entry as well as averaged scores for a set of CRBN-based degraders.84 Notably among the 38 molecules cited, of the 13 degraders that have been evaluated in vivo, only one was reported with oral administration, without mention of fraction absorbed. The challenge of designing heterobifunctional degraders with oral bioavailability was noted early in the evolution of this field,85 although realization of this goal should be achievable through highly ligand-efficient degraders with reduced molecular weight and lipophilicity.
Conclusion
While both the heterobifunctional and molecular glue classes of degraders share the advantages described above, their unique molecular characteristics engender significant differences in their scope of utility and in their physicochemical and pharmacological properties. Molecular glues have significantly lower molecular weights than heterobifunctional molecules, a substantial advantage in the development of drug-like therapeutics. This increased molecular efficiency is possible because binding affinity is also contributed from the protein surfaces within the complex, placing considerable constraint on the molecular glue approach: the protein surfaces must be able to complement one another and the ligand. However, this means that the ligand need not have binding affinity for each of the individual complex members. Consequently proteins can be targeted to the complex, which entirely lacks small molecule binding affinity, as in the case of the Ikaros family of zinc fingers, which would generally be considered “undruggable” to conventional modalities.
A key question for the future of cereblon modulators is the breadth of protein targets that will be accessible given sufficiently elaborated chemical libraries. Recent studies have identified more neosubstrate targets of even the clinically approved cereblon modulators thalidomide, lenalidomide, and pomalidomide,40−42 encompassing both zinc finger and nonzinc finger domain proteins. While heterobifunctional linker-based molecules offer expanded possibilities, these may require optimization for substrate and ligase compatibility. The ligand-directed protein–protein interaction approach, as typified by the CELMoDs, offers the possibility of recruiting and degrading proteins for which there are no ligands, but requires specific complementarity between the proteins and ligand. The identification of proteins with a potentially compatible degron paired with expanded CELMoD libraries offers wide potential to address previously undruggable targets. Given the ∼600 ubiquitin ligases in the genome86 and the potential for both tissue- and organelle-specific ubiquitin ligases, there remains great potential for diversity in targeting options. These approaches in the emerging modality of targeted protein degradation should be expected to considerably expand the “druggable proteome” to provide countless new therapeutic opportunities.
The authors declare the following competing financial interest(s): All authors are employees of Celgene Corporation.
References
- Thiel P.; Kaiser M.; Ottmann C. Small-molecule stabilization of protein-protein interactions: an underestimated concept in drug discovery?. Angew. Chem., Int. Ed. 2012, 51 (9), 2012–2018. 10.1002/anie.201107616. [DOI] [PubMed] [Google Scholar]
- Chardin P.; McCormick F. Brefeldin A: the advantage of being uncompetitive. Cell 1999, 97, 153–155. 10.1016/S0092-8674(00)80724-2. [DOI] [PubMed] [Google Scholar]
- Mossessova E.; Corpina R. A.; Goldberg J. Crystal structure of ARF1*Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol. Cell 2003, 12, 1403–1411. 10.1016/S1097-2765(03)00475-1. [DOI] [PubMed] [Google Scholar]
- Renault L.; Guibert B.; Cherfils J. Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature 2003, 426, 525–530. 10.1038/nature02197. [DOI] [PubMed] [Google Scholar]
- Liu J.; Farmer J. D. Jr.; Lane W. S.; Friedman J.; Weissman I.; Schreiber S. L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. 10.1016/0092-8674(91)90124-H. [DOI] [PubMed] [Google Scholar]
- Brown E. J.; Albers M. W.; Shin T. B.; Ichikawa K.; Keith C. T.; Lane W. S.; Schreiber S. L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Griffith J. P.; Kim J. L.; Kim E. E.; Sintchak M. D.; Thomson J. A.; Fitzgibbon M. J.; Fleming M. A.; Caron P. R.; Hsiao K.; Navia M. A. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 1995, 82, 507–522. 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- Kissinger C. R.; Parge H. E.; Knighton D. R.; Lewis C. T.; Pelletier L. A.; Tempczyk A.; Kalish V. J.; Tucker K. D.; Showalter R. E.; Moomaw E. W.; Gastinel L. N.; Habuka N.; Chen X.; Maldonado F.; Barker J. E.; Bacquet R.; Villafranca J. E. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature 1995, 378, 641–644. 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- Choi J.; Chen J.; Schreiber S. L.; Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273, 239–242. 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- Dauvois S.; Danielian P. S.; White R.; Parker M. G. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 4037–4041. 10.1073/pnas.89.9.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell D. P.; Wardell S. E.; Norris J. D. Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. J. Med. Chem. 2015, 58, 4883–4887. 10.1021/acs.jmedchem.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombos A. Selective oestrogen receptor degraders in breast cancer: a review and perspectives. Curr. Opin. Oncol. 2019, 31, 424–429. 10.1097/CCO.0000000000000567. [DOI] [PubMed] [Google Scholar]
- Zheng N.; Shabek N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. 10.1146/annurev-biochem-060815-014922. [DOI] [PubMed] [Google Scholar]
- Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau R.; Rape M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- Berndsen C. E.; Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. 10.1038/nsmb.2780. [DOI] [PubMed] [Google Scholar]
- Duda D. M.; Scott D. C.; Calabrese M. F.; Zimmerman E. S.; Zheng N.; Schulman B. A. Structural regulation of cullin-RING ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 2011, 21, 257–264. 10.1016/j.sbi.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers S.; Li T.; Yi X.; MacCoss M. J.; Moon R. T.; Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- Gray W. M.; Kepinski S.; Rouse D.; Leyser O.; Estelle M. Auxin regulates SCF(TIR1)-dependent degradation of AUX/IAA proteins. Nature 2001, 414, 271–276. 10.1038/35104500. [DOI] [PubMed] [Google Scholar]
- Tan X.; Calderon-Villalobos L. I.; Sharon M.; Zheng C.; Robinson C. V.; Estelle M.; Zheng N. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 2007, 446, 640–645. 10.1038/nature05731. [DOI] [PubMed] [Google Scholar]
- Sheard L. B.; Tan X.; Mao H.; Withers J.; Ben-Nissan G.; Hinds T. R.; Kobayashi Y.; Hsu F. F.; Sharon M.; Browse J.; He S. Y.; Rizo J.; Howe G. A.; Zheng N. Jasmonate perception by inositol-phosphate-potentiated COI1-JAZ co-receptor. Nature 2010, 468, 400–405. 10.1038/nature09430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Indisulam: an anticancer sulfonamide in clinical development. Expert Opin. Invest. Drugs 2003, 12, 283–287. 10.1517/13543784.12.2.283. [DOI] [PubMed] [Google Scholar]
- Loddick S. A.; Ross S. J.; Thomason A. G.; Robinson D. M.; Walker G. E.; Dunkley T. P.; Brave S. R.; Broadbent N.; Stratton N. C.; Trueman D.; Mouchet E.; Shaheen F. S.; Jacobs V. N.; Cumberbatch M.; Wilson J.; Jones R. D.; Bradbury R. H.; Rabow A.; Gaughan L.; Womack C.; Barry S. T.; Robson C. N.; Critchlow S. E.; Wedge S. R.; Brooks A. N. AZD3514: a small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 1715–1727. 10.1158/1535-7163.MCT-12-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerres N.; Steurer S.; Schlager S.; Bader G.; Berger H.; Caligiuri M.; Dank C.; Engen J. R.; Ettmayer P.; Fischerauer B.; Flotzinger G.; Gerlach D.; Gerstberger T.; Gmaschitz T.; Greb P.; Han B.; Heyes E.; Iacob R. E.; Kessler D.; Kolle H.; Lamarre L.; Lancia D. R.; Lucas S.; Mayer M.; Mayr K.; Mischerikow N.; Muck K.; Peinsipp C.; Petermann O.; Reiser U.; Rudolph D.; Rumpel K.; Salomon C.; Scharn D.; Schnitzer R.; Schrenk A.; Schweifer N.; Thompson D.; Traxler E.; Varecka R.; Voss T.; Weiss-Puxbaum A.; Winkler S.; Zheng X.; Zoephel A.; Kraut N.; McConnell D.; Pearson M.; Koegl M. Chemically Induced Degradation of the Oncogenic Transcription Factor BCL6. Cell Rep. 2017, 20, 2860–2875. 10.1016/j.celrep.2017.08.081. [DOI] [PubMed] [Google Scholar]
- Vargesson N. Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res., Part C 2015, 105, 140–156. 10.1002/bdrc.21096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T.; Sakamoto S.; Handa H. Finally solved! Mystery of teratogenesis caused by thalidomide. FG bead determined the target molecule cereblon. Kagaku 2010, 65, 47–51. [Google Scholar]
- He Y. J.; McCall C. M.; Hu J.; Zeng Y.; Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006, 20, 2949–2954. 10.1101/gad.1483206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa L. A.; Wu M.; Ye T.; Kobayashi R.; Sun H.; Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 2006, 8, 1277–1283. 10.1038/ncb1490. [DOI] [PubMed] [Google Scholar]
- Jin J.; Arias E. E.; Chen J.; Harper J. W.; Walter J. C. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 2006, 23, 709–721. 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Gandhi A. K.; Kang J.; Havens C. G.; Conklin T.; Ning Y.; Wu L.; Ito T.; Ando H.; Waldman M. F.; Thakurta A.; Klippel A.; Handa H.; Daniel T. O.; Schafer P. H.; Chopra R. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br. J. Haematol. 2014, 164, 811–821. 10.1111/bjh.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; Middleton R. E.; Sun H.; Naniong M.; Ott C. J.; Mitsiades C. S.; Wong K. K.; Bradner J. E.; Kaelin W. G. Jr The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A.; Mendy D.; Ito T.; Miller K.; Gandhi A. K.; Kang J.; Karasawa S.; Carmel G.; Jackson P.; Abbasian M.; Mahmoudi A.; Cathers B.; Rychak E.; Gaidarova S.; Chen R.; Schafer P. H.; Handa H.; Daniel T. O.; Evans J. F.; Chopra R. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. 10.1038/leu.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. X.; Braggio E.; Shi C. X.; Bruins L. A.; Schmidt J. E.; Van Wier S.; Chang X. B.; Bjorklund C. C.; Fonseca R.; Bergsagel P. L.; Orlowski R. Z.; Stewart A. K. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. 10.1182/blood-2011-05-356063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink E. C.; McConkey M.; Adams D. N.; Haldar S. D.; Kennedy J. A.; Guirguis A. A.; Udeshi N. D.; Mani D. R.; Chen M.; Liddicoat B.; Svinkina T.; Nguyen A. T.; Carr S. A.; Ebert B. L. Crbn (I391V) is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice. Blood 2018, 132, 1535–1544. 10.1182/blood-2018-05-852798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke J.; Fink E. C.; Hollenbach P. W.; MacBeth K. J.; Hurst S. N.; Udeshi N. D.; Chamberlain P. P.; Mani D. R.; Man H. W.; Gandhi A. K.; Svinkina T.; Schneider R. K.; McConkey M.; Jaras M.; Griffiths E.; Wetzler M.; Bullinger L.; Cathers B. E.; Carr S. A.; Chopra R.; Ebert B. L. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015, 523, 183–188. 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela M. E.; Lu G.; Ito T.; Pagarigan B.; Lu C. C.; Miller K.; Fang W.; Wang N. Y.; Nguyen D.; Houston J.; Carmel G.; Tran T.; Riley M.; Nosaka L.; Lander G. C.; Gaidarova S.; Xu S.; Ruchelman A. L.; Handa H.; Carmichael J.; Daniel T. O.; Cathers B. E.; Lopez-Girona A.; Chamberlain P. P. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 2016, 535, 252–257. 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- Chamberlain P. P.; Lopez-Girona A.; Miller K.; Carmel G.; Pagarigan B.; Chie-Leon B.; Rychak E.; Corral L. G.; Ren Y. J.; Wang M.; Riley M.; Delker S. L.; Ito T.; Ando H.; Mori T.; Hirano Y.; Handa H.; Hakoshima T.; Daniel T. O.; Cathers B. E. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 2014, 21, 803–809. 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- Fischer E. S.; Bohm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E.; Harper J. W.; Jenkins J. L.; Thoma N. H. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold G.; Fischer E. S.; Thoma N. H. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature 2016, 532, 127–130. 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- Sievers Q. L.; Petzold G.; Bunker R. D.; Renneville A.; Slabicki M.; Liddicoat B. J.; Abdulrahman W.; Mikkelsen T.; Ebert B. L.; Thoma N. H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362, eaat0572. 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela M. E.; Couto S.; Zheng X.; Lu G.; Hui J.; Stamp K.; Drew C.; Ren Y.; Wang M.; Carpenter A.; Lee C. W.; Clayton T.; Fang W.; Lu C. C.; Riley M.; Abdubek P.; Blease K.; Hartke J.; Kumar G.; Vessey R.; Rolfe M.; Hamann L. G.; Chamberlain P. P. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol. 2018, 14, 981–987. 10.1038/s41589-018-0129-x. [DOI] [PubMed] [Google Scholar]
- Donovan K. A.; An J.; Nowak R. P.; Yuan J. C.; Fink E. C.; Berry B. C.; Ebert B. L.; Fischer E. S. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife 2018, 7, 38430. 10.7554/eLife.38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhase J.; Chitayat D.; Kotzot D.; Ceylaner S.; Froster U. G.; Fuchs S.; Montgomery T.; Rosler B. SALL4 mutations in Okihiro syndrome (Duane-radial ray syndrome), acro-renal-ocular syndrome, and related disorders. Hum. Mutat. 2005, 26, 176–183. 10.1002/humu.20215. [DOI] [PubMed] [Google Scholar]
- Kohlhase J.; Holmes L. B. Mutations in SALL4 in malformed father and daughter postulated previously due to reflect mutagenesis by thalidomide. Birth Defects Res., Part A 2004, 70, 550–551. 10.1002/bdra.20050. [DOI] [PubMed] [Google Scholar]
- Kohlhase J.; Schubert L.; Liebers M.; Rauch A.; Becker K.; Mohammed S. N.; Newbury-Ecob R.; Reardon W. Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J. Med. Genet. 2003, 40, 473–478. 10.1136/jmg.40.7.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratta I. D.; Sigg E. B.; Maiorana K. Teratogenic effects of Thalidomide in rabbits, rats, hamsters, and mice. Toxicol. Appl. Pharmacol. 1965, 7, 268–286. 10.1016/0041-008X(65)90095-5. [DOI] [PubMed] [Google Scholar]
- Schafer P. H.; Ye Y.; Wu L.; Kosek J.; Ringheim G.; Yang Z.; Liu L.; Thomas M.; Palmisano M.; Chopra R. Cereblon modulator iberdomide induces degradation of the transcription factors Ikaros and Aiolos: immunomodulation in healthy volunteers and relevance to systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1516–1523. 10.1136/annrheumdis-2017-212916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela M. E.; Zhang W.; Man H. W.; Muller G.; Khambatta G.; Baculi F.; Hickman M.; LeBrun L.; Pagarigan B.; Carmel G.; Lu C. C.; Lu G.; Riley M.; Satoh Y.; Schafer P.; Daniel T. O.; Carmichael J.; Cathers B. E.; Chamberlain P. P. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2018, 61, 535–542. 10.1021/acs.jmedchem.6b01921. [DOI] [PubMed] [Google Scholar]
- Vu P. K.; Sakamoto K. M. Ubiquitin-mediated proteolysis and human disease. Mol. Genet. Metab. 2000, 71, 261–266. 10.1006/mgme.2000.3058. [DOI] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung J.; Kim K. B.; Lindsten K.; Dantuma N. P.; Crews C. M. Lack of proteasome active site allostery as revealed by subunit-specific inhibitors. Mol. Cell 2001, 7, 411–420. 10.1016/S1097-2765(01)00188-5. [DOI] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Drug Development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Puppala D.; Choi E.-Y.; Swanson H.; Kim K.-B. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: a useful chemical genetic tool. ChemBioChem 2007, 8, 2058–2062. 10.1002/cbic.200700438. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Gonzalez A.; Cyrus K.; Salcius M.; Kim K.; Crews C. M.; Deshaies R. J.; Sakamoto K. M. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008, 27, 7201–7211. 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Winkler J. D.; Crew A. P.; Coleman K.; Hines J.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55, 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Lu J.; Qian Y.; Altieri M.; Gordon D.; Rossi A. M. K.; Wang J.; Chen X.; Dong H.; Siu K.; Winkler J. D.; Crew A. P.; Crews C. M.; Coleman K. G. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 7124–7129. 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhimschi A. D.; Armstrong H. A.; Toure M.; Jaime-Figueroa S.; Chen T. L.; Lehman A. M.; Woyach J. A.; Johnson A. J.; Byrd J. C.; Crews C. M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57, 3564–3575. 10.1021/acs.biochem.8b00391. [DOI] [PubMed] [Google Scholar]
- Burslem G. M.; Smith B. E.; Lai A. C.; Jaime-Figueroa S.; McQuaid D. C.; Bondeson D. P.; Toure M.; Dong H.; Qian Y.; Wang J.; Crew A. P.; Hines J.; Crews C. M. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77. 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem G. M.; Song J.; Chen X.; Hines J.; Crews C. M. Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc. 2018, 140, 16428–16432. 10.1021/jacs.8b10320. [DOI] [PubMed] [Google Scholar]
- Cromm P. M.; Samarasinghe K. T. G.; Hines J.; Crews C. M. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. 10.1021/jacs.8b08008. [DOI] [PubMed] [Google Scholar]
- McCoull W.; Cheung T.; Anderson E.; Barton P.; Burgess J.; Byth K.; Cao Q.; Castaldi M. P.; Chen H.; Chiarparin E.; Carbajo R. J.; Code E.; Cowan S.; Davey P. R.; Ferguson A. D.; Fillery S.; Fuller N. O.; Gao N.; Hargreaves D.; Howard M. R.; Hu J.; Kawatkar A.; Kemmitt P. D.; Leo E.; Molina D. M.; O’Connell N.; Petteruti P.; Rasmusson T.; Raubo P.; Rawlins P. B.; Ricchiuto P.; Robb G. R.; Schenone M.; Waring M. J.; Zinda M.; Fawell S.; Wilson D. M. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem. Biol. 2018, 13, 3131–3141. 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
- Schiedel M.; Herp D.; Hammelmann S.; Swyter S.; Lehotzky A.; Robaa D.; Olah J.; Ovadi J.; Sippl W.; Jung M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- Yang K.; Xie H.; Wu H.; Leisten E. D.; Song Y.; Wu Y.-T.; Tang W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. 10.1016/j.bmcl.2018.05.057. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Han X.-R.; Yang X.; Jiang B.; Liu J.; Xiong Y.; Jin J. Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur. J. Med. Chem. 2018, 151, 304–314. 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargbo R. B. Treatment of Cancer and Alzheimer’s Disease by PROTAC Degradation of EGFR. ACS Med. Chem. Lett. 2019, 10, 1098–1099. 10.1021/acsmedchemlett.9b00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargbo R. B. Treatment of Alzheimer’s by PROTAC-Tau Protein Degradation. ACS Med. Chem. Lett. 2019, 10, 699–700. 10.1021/acsmedchemlett.9b00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Yang J.; Aguilar A.; McEachern D.; Przybranowski S.; Liu L.; Yang C.-Y.; Wang M.; Han X.; Wang S. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem. 2019, 62, 448–466. 10.1021/acs.jmedchem.8b00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes J.; McGonagle G. A.; Eden J.; Kiritharan G.; Touzet M.; Lewell X.; Emery J.; Eidam H.; Harling J. D.; Anderson N. A. Targeting IRAK4 for Degradation with PROTACs. ACS Med. Chem. Lett. 2019, 10, 1081–1085. 10.1021/acsmedchemlett.9b00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatzimas J. W.; Gorobets E.; Maity R.; Muniyat M. I.; MacCallum J. L.; Neri P.; Bahlis N. J.; Derksen D. J. From Inhibition to Degradation: Targeting the Antiapoptotic Protein Myeloid Cell Leukemia 1 (MCL1). J. Med. Chem. 2019, 62, 5522–5540. 10.1021/acs.jmedchem.9b00455. [DOI] [PubMed] [Google Scholar]
- Rana S.; Bendjennat M.; Kour S.; King H. M.; Kizhake S.; Zahid M.; Natarajan A. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg. Med. Chem. Lett. 2019, 29, 1375–1379. 10.1016/j.bmcl.2019.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovell H.; Testa A.; Zhou H.; Shpiro N.; Crafter C.; Ciulli A.; Alessi D. R. Design and characterization of SGK3-PROTAC1, an isoform specific SGK3 kinase PROTAC degrader. ACS Chem. Biol. 2019, 14, 2024. 10.1021/acschembio.9b00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; He N.; Guo Z.; Niu C.; Song T.; Guo Y.; Cao K.; Wang A.; Zhu J.; Zhang X.; Zhang Z. Proteolysis Targeting Chimeras for the Selective Degradation of Mcl-1/Bcl-2 Derived from Nonselective Target Binding Ligands. J. Med. Chem. 2019, 62, 8152. 10.1021/acs.jmedchem.9b00919. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Lan T.; Su S.; Rao Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. 2019, 55, 369–372. 10.1039/C8CC07813K. [DOI] [PubMed] [Google Scholar]
- Smith B. E.; Wang S. L.; Jaime-Figueroa S.; Harbin A.; Wang J.; Hamman B. D.; Crews C. M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K. H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak R. P.; DeAngelo S. L.; Buckley D.; He Z.; Donovan K. A.; An J.; Safaee N.; Jedrychowski M. P.; Ponthier C. M.; Ishoey M.; Zhang T.; Mancias J. D.; Gray N. S.; Bradner J. E.; Fischer E. S. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018, 14, 706–714. 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S. L.; Phillips A. J. Targeted protein degradation and the enzymology of degraders. Curr. Opin. Chem. Biol. 2018, 44, 47–55. 10.1016/j.cbpa.2018.05.004. [DOI] [PubMed] [Google Scholar]
- Douglass E. F. Jr.; Miller C. J.; Sparer G.; Shapiro H.; Spiegel D. A. A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc. 2013, 135, 6092–9099. 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B.; Gossen M.; Dittmar G.; Selbach M. Global analysis of cellular protein translation by pulsed SILAC. Proteomics 2009, 9, 205–209. 10.1002/pmic.200800275. [DOI] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Verma R.; Ransick A.; Stein B.; Crews C. M.; Deshaies R. J. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteomics 2003, 2, 1350–1358. 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- Edmondson S. D.; Yang B.; Fallan C. Proteolysis targeting chimeras (PROTACs) in ’beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- Gilbert A.; Noe M.. Protein Degraders: Moving towards therapeutically viable agents. In Medicinal Chemistry Reviews; Desai M. C., Ed.; VCH: New York, 2016; Vol. 51, 347–371. [Google Scholar]
- Morreale F. E.; Walden H. Types of Ubiquitin Ligases. Cell 2016, 165 (1), 248–248. 10.1016/j.cell.2016.03.003. [DOI] [PubMed] [Google Scholar]