

Abstract
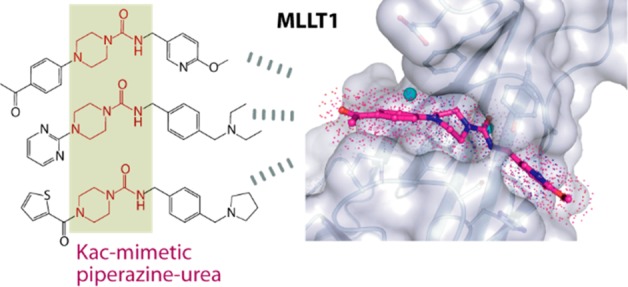
YEATS-domain-containing MLLT1 is an acetyl/acyl-lysine reader domain, which is structurally distinct from well-studied bromodomains and has been strongly associated in development of cancer. Here, we characterized piperazine-urea derivatives as an acetyl/acyl-lysine mimetic moiety for MLLT1. Crystal structures revealed distinct interaction mechanisms of this chemotype compared to the recently described benzimidazole-amide based inhibitors, exploiting different binding pockets within the protein. Thus, the piperazine-urea scaffold offers an alternative strategy for targeting the YEATS domain family.
Keywords: YEATS domains, MLLT1, piperazine-urea, inhibitor development, cancer
Epigenetic signaling plays crucial functions in chromatin-dependent transcription and often requires tight regulation involving post-translational modifications essentially at specific lysines of several key proteins, including histone proteins. Recognition of acetylated lysine, one of the most common epigenetic marks, is one of the hallmark events in epigenetic signaling. In addition to the well-established readers such as bromodomains and some double PHD finger domains,1,2 YEATS domain, which has been described in four human proteins including MLLT1 (ENL), YEATS2, MLLT3 (AF9), and glioma amplified sequence 41 (GAS41 or YEATS4), has been classified as a protein class capable of recognizing histone acetylation as well as bulkier lysine posttranslational modifications such as propionylation, butyrylation, crotonylation, and succinylation.3−9 YEATS-domain-containing proteins have potentially distinct preferences toward different lysine modifications on histone proteins. For instance, MLLT1/3 prefer acetylated Lys9, Lys18, and Lys27 of histone H3,4,10 while YEATS2 has been demonstrated to have a stronger affinity for crotonylation marks.11,12 In addition to moderate affinities for acetyl-lysine, some unusual reader activities of GAS41 have been reported including the recognition of histone succinylation in a pH-dependent manner,13 as well as its ability to form a dimer through the C-terminal coiled-coil domain for cooperatively bivalent binding to diacetylated histone.14
Several reports have associated YEATS-domain-containing proteins as a key driver for the development of diseases, in particular cancer.3 For example, chromosomal rearrangements involving the MLLT1 as well as MLLT3 YEATs domains promote development of leukemia.10,15,16 The YEATS2 gene is highly amplified in non-small cell lung cancer (NSCLC), and its role regulating transcription is essential for tumorigenesis.17 In addition, overexpression of growth-promoting GAS41 has been associated with malignancy of many cancer types, including gastric18 and hepatic carcinomas19 as well as NSCLC.20 These consistent lines of evidence have suggested, therefore, YEATS domains as a potential target for chemotherapeutic treatment.
YEATS domains exhibit highly distinct beta-sheet topology compared to other acetylation readers such as helical bromodomains. The YEATS acetyl-lysine binding site comprises a more open, shallow, surface-exposed binding channel, which is open on both sites allowing accommodation of larger lysine modifications.4,21 These distinct structural features suggest that YEATS domain inhibitors require different structural features compared to acetyl-lysine mimetic groups explored for the development of bromodomain inhibitors.22,23 An early attempt exploiting a peptide-based strategy with 2-furancarbonyl lysine led to a competitive nanomolar binder,24 demonstrating potential inhibitions of the YEATS protein module. Our recent screening efforts alternatively revealed a number of small molecule binders for MLLT1/3.21,25 One chemotype identified was benzimidazole-amide, and extensive modification focused on this scaffold led to the development of the potent and selective MLLT1/3 inhibitor (SGC-iMLLT) with IC50 of ∼0.26 μM and KD of 113 nM.26 Unfortunately, moderate metabolic stability of this SGC-iMLLT chemical probe has limited its further use in chemotherapeutics. Nevertheless, the discovery of this chemical probe has demonstrated, therefore, a potential of targeting YEATS domains using small molecule inhibitor.
Alternative MLLT1/3 binders with different chemical moieties may offer a benefit in cellular activities of MLLT1 inhibitors. We therefore aimed to search for other scaffolds distinct from benzimidazole-amide, which is the only characterized binder to date. Analyses of the results from our previous screening campaign revealed a number of piperazine-1-carboxamide hits,25 suggesting that this piperazine-urea scaffold could serve an alternative acetyl-lysine mimetic moiety for MLLT1/3. Interestingly, this chemical class has been extensively used previously in development of inhibitors with good pharmacokinetic properties for diverse targets, such as fatty acid amide hydrolase (FAAH),27 melanocortin subtype-4 receptor (MC4R),28 and the receptor for the chemokine CCL2 (CCR2 or known also as MCP-1).29 For the potential application as an alternative scaffold for YEATS domain inhibitors, we investigated interaction mechanisms of the piperazine-urea chemotype in MLLT1.
Based on our previous work,25,26 we selected three piperazine-1-carboxamide hits (1, 2, and 3) and for comparison three benzimidazole-amide derivatives of similar sizes (4, 5, and 6), and first determined the crystal structures of their complexes with MLLT1 (Figure 1). We observed surprisingly different binding modes and interactions between these two chemotypes to the protein. The most remarkable change was the orientation of their central primary amide cores. This functional group of the piperazine-urea derivatives flipped horizontally in comparison to that of the benzimidazole-amide compounds and instead assumed a similar binding orientation to that of acetyl-lysine.21 Nevertheless, similar β-sheet type hydrogen bonding patterns between the amide carbonyl atom to the backbone amine of Tyr78 as well as contacts with the conserved water molecule and Ser58 were highly maintained. Comparable binding modes between the secondary amide central core of the piperazine-1-carboxamide compounds and acetyl-lysine confirmed our previous hypothesis on the use of the amide group as an acetyl-lysine mimetic moiety for YEATS domains.21
Figure 1.

Piperazine-urea and benzimidazole-amide derivatives and their binding modes in MLLT1. (A) The binding mode of acetyllysine in MLLT1 (pdb id: 6hpz) revealing potential front and rear pockets that could be targeted by small molecules. Chemical structures and the binding modes in MLLT1-complexed crystal structures of three piperazine-1-carboxamide 1, 2, and 3 (B–D; pdb ids: 6t1i, 6t1j, and 6t1l) and three benzimidazole-amide 4, 5, and 6 (E–G; pdb ids: 6t1m, 6t1n, and 6t1o). Dashed lines indicate hydrogen bonds, while bar, dashed lines are for potential aromatic π-stacking. Water molecules are shown in spheres. Electron density maps contoured at 1σ for the bound ligands are shown in the insets below the chemical structures in each panel. See Supplementary Figure S1 for omitted electron density maps.
Earlier crystal structures suggested an important role of aromatic moieties on both ends of the amide central core for achieving aromatic stacking with Phe28, Phe59, and His56,21,26 as exemplified also here in the interactions of 4, 5, and 6 (Figure 1E–G). However, in contrast to benzimidazoles, all piperazine-urea 1, 2, and 3 did not follow this scheme and contained instead an sp3 piperazine on the C-linked region of the amide, whereas a benzyl substituent was featured on the N-linked portion of the amide. This resulted in slightly different accommodation of their decorations within the protein (Figure 1B–D). At the front pocket, despite being elongated by one carbon spacing, the phenyl moiety of the benzyl ring was still located in proximity to His56 for aromatic stacking with this residue. Apart from this, there was no direct contact with the protein observed for the decorations at the other rear end. Although sharing the same binding pocket as the phenyl part of benzimidazole, the lack of aromaticity of the piperazine moiety restricted a π-stacking contact with Phe59. In addition, the orientation of the amide core in a canonical acetyl-lysine binding mode forced the aromatic decorations on the piperazine ring at the 4-position to assume a different trajectory than that observed for the benzimidazole counterparts, and to occupy otherwise space adjacent to loop4, which was not exploited previously by other MLLT1 binders. However, this binding mode diminished a π-stacking contact with Phe28 and feasibly resulted in more solvent-exposed for these extended groups.
Structural comparison between the piperazine-1-carboxamide- and benzimidazole-amide-MLLT1 complexes revealed that in addition to different binding modes of the compounds there were a number of conformational rearrangements within the protein (Figure 2A,B). Consistent with our previous observation for fragment binding,21 the flipped amide with different binding positions likely affected slight adjustment of Ser58 side chain, which was required for maintaining their contacts. More dramatic structural alteration was evident for loop1, which was feasibly a consequence of different ligand binding at the rear pocket. In comparison, the “closed” conformation of loop1 in the piperazine-1-caboxamide-MLLT1 complexes remained similar to that in the apo form,21 albeit with a slight shift of Phe28 side chain likely due to steric effect from the piperazine moiety (Figure 2A). This suggested that the space adjacent to loop4 might typically exist in the ligand-free state, and binding of the decorated piperazine moieties within this cavity did not significantly perturb structural integrity of the protein. In contrast, the presence of the benzimidazole ring within a cavity located in the rear pocket toward β5, which was filled by water in the MLLT1-1 complex, led to significant structural alterations. This was evident not only by ∼34° twist of Phe28 side chain for an optimal aromatic stacking, but an outward movement of loop1 by ∼3–4 Å, adopting an open conformation (Figure 2B). These structural alterations remodeled the shape of the rear pocket, which nevertheless could accommodate both ligands (Figure 2C). The binding modes of the piperazine-urea and benzimidazole-amide derivatives revealed therefore not only cavities that can be targeted for the design of more potent ligands interacting with the rear pocket, but they demonstrated also significant plasticity of the MLLT1 pocket.
Figure 2.
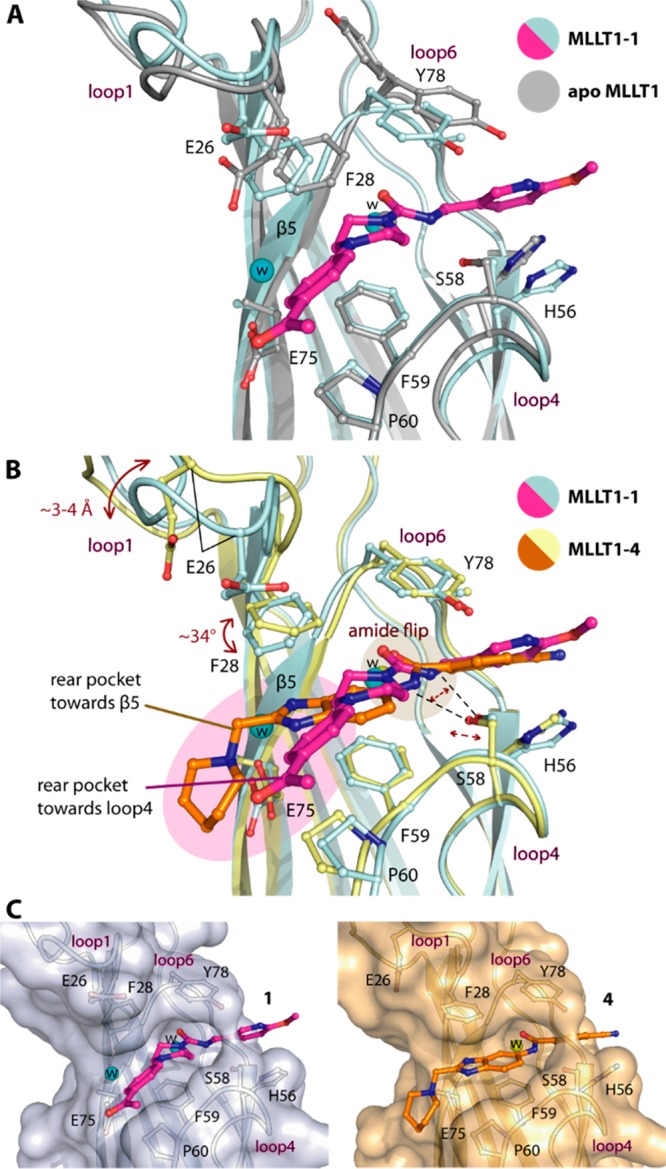
Structural comparison of interaction mechanisms of piperazine-1-carboxamide 1 and benzimidazole-amide 4 in MLLT1. (A) Superimposition of the apo (pdb id 6hq0) and the 1-complexed structures demonstrates no significant conformational changes for MLLT1. (B) Comparison of 1- and 4-complexed structures reveals alterations of loop1, Phe28, and Ser58 potentially due to the orientation of the amide central core and different binding modes of the ligands that occupy different space at the rear pocket. (C) Surface representation shows that conformational changes of MLLT1 adjust the shapes of the binding pocket complimentarily for different compounds.
We next assessed binding affinities of all six derivatives for MLLT1 and performed isothermal titration calorimetry (ITC) to analyze binding constants and thermodynamics. All compounds exhibited binding potencies in a micromolar range (Figure 3A,B). Among the piperazine-urea derivatives, 1 was the most potent binder with KD of ∼5.5 μM, which was ∼3-fold lower than those of 2 and 3 and was comparable to those of the benzimidazole counterparts that showed also low micromolar affinities. Interestingly, we observed also that the trend of unfavorable entropy was characterized for the binding of all piperazine-urea compounds, which was in contrast to a gain in entropic contribution measured for benzimidazole-amide inhibitors. This change in thermodynamic signatures was in agreement with their diverse interaction mechanisms. However, the thermodynamics parameters observed from moderate binding in ITC remained highly estimated, and, therefore, further experiment might be required to confirm such differences.
Figure 3.
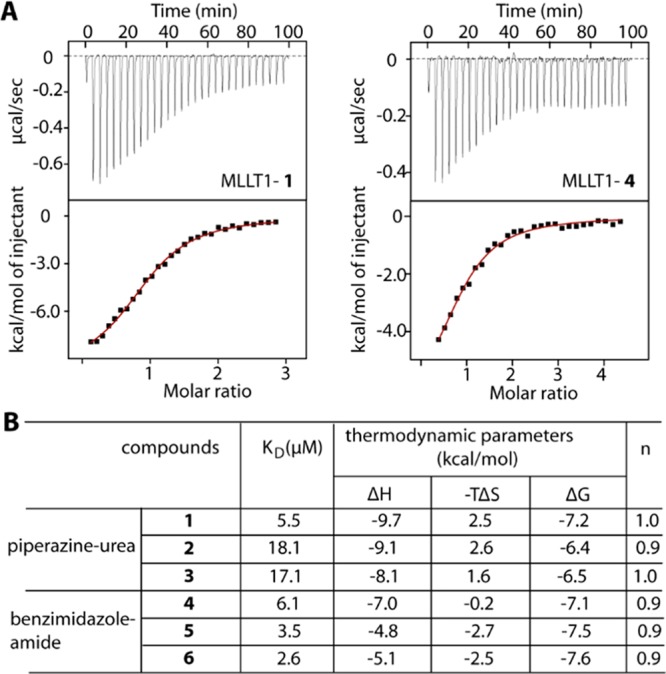
Binding of the derivatives in MLLT1 measured by ITC. (A) Examples of ITC data for 1 and 4. Shown are the isotherms of raw titration heat (top) and the normalized binding heat with the single-site fitting (red line; bottom). (B) Summary of binding constants and thermodynamic parameters.
We next performed orthogonal AlphaScreen assays to confirm the binding of all compounds in MLLT1. In agreement, the measured IC50 values for all compounds correlated well with their KD values obtained from ITC (Figure 4A,B). In essence, 1 was the most potent binder among the piperazine-urea derivatives, and its low micromolar potency was similar to that observed for the benzimidazole-amide counterparts. In addition, we observed weak binding of these compounds to MLLT3, and much lower affinities or no detectable activity for YEATS2 and YEATS4 (Supplementary Table S2). In general, a trend of lower affinities of piperazine-urea compounds than the benzimidazole-amide derivatives could be due to the lack of potential aromatic stacking with Phe28 and Phe59. Nevertheless, the different binding mode of the piperazine-urea compounds indicated a possibility of targeting the previously unseen rear pocket toward loop4. Hybrid compounds between these two scaffolds exploiting a larger area of the rear pocket as well as maintaining aromatic stacking with Phe28 and Phe59 might offer a strategy for potent inhibitors for targeting MLLT1.
Figure 4.
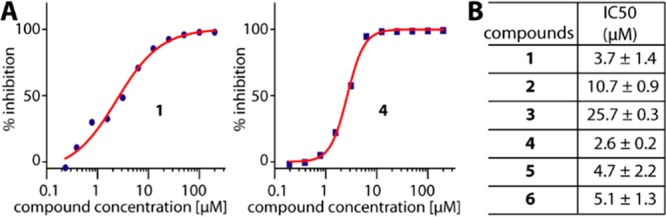
AlphaScreen assays for inhibition of MLLT1. (A) Examples of MLLT1 inhibition with 1 and 4. (B) Summary of IC50s.
YEATS-domain-containing proteins plays a key role in development of many diseases, presenting a potential target for chemotherapeutic treatment. Although sharing similar functions in recognition of acetyl-lysine, targeting this reader family requires distinct classes of small molecule binders to those for bromodomains. Our previous effort has identified benzimidazole-amide as an acetyl-lysine mimetic scaffold for MLLT1/3, which led to development of the first chemical probe for MLLT1/3, albeit with limited pharmacokinetic properties. Here we characterized another chemotype in piperazine-urea of which the derivatives demonstrated also affinities in low micromolar range for MLLT1. Comparative structural analyses suggested that this alternative scaffold likely exhibited different interaction mechanisms and exploited diverse binding cavities. The piperazine-urea scaffold not only presents an alternative chemical starting point, but expands the existing knowledge on landscape of potential druggable pockets of the protein, which would benefit future programs on the development of inhibitors for this attractive epigenetic targets.
Experimental Procedures
Piperazine-urea compound 1–3 were purchased from Enamine,25 while benzimidazole-amide derivatives 4–6 were obtained from our previous study.26
Protein purification and crystallization: MLLT1 YEATS domain (aa 1–148) was purified using the procedure described previously.21 Apo crystals were produced using sitting drop vapor diffusion method at 20 °C and the condition containing 25% PEG 3350, 0.2 M NaCl, and either 0.1 M bis-tris, pH 6.5, or 0.1 M HEPES, pH 7.5. Ligands were soaked into crystals. Diffraction data were collected at SLS X06SA, processed with XDS30 and scaled with aimless.31 Molecular replacement was performed with Phaser32 and the MLLT1 apo structure (pdb id 6hq0). All structures were rebuilt in COOT,33 refined using REFMAC,34 and validated with molprobity.35 Data collection and refinement statistics are summarized in Supplementary Table S1.
Isothermal calorimetry (ITC) experiments were performed using NanoITC instrument (TA Instruments) at 25 °C with protein at 300 μM in 25 mM Tris pH 7.5, 500 mM NaCl, 0.5 TCEP, 5% glycerol titrating into 20–30 μM compounds. Data analyses were carried out using the software and protocols provided by TA Instruments.
AlphaScreen assays for all compounds with MLLT1 were performed as previously described.25
Acknowledgments
The SGC is a registered charity that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative [ULTRA-DD 115766], Wellcome Trust, Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Centre of Excellence Macromolecular complexes (CEF). S.K. and A.C. are supported by the Sonderforschungsbereich SFB1177 Autophagy. M.M. is grateful to the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for a studentship, supported by AstraZeneca, Diamond, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB, and Vertex. The authors thank staff at Swiss Light Source for their assistance during data collection, which has been supported also by the funding from the European Union’s Horizon 2020 research and innovation program under grant agreement number 730872, project CALIPSOplus.
Glossary
ABBREVIATIONS
- YEATS domain
Yaf9, ENL, AF9, Taf14, Sas5 (YEATS) domain
- ENL
eleven-nineteen-leukemia protein
- MLLT1
myeloid/lymphoid or mixed-lineage leukemia translocated to chromosome 1 protein
- AF9
ALL1-fused gene from chromosome 9 protein
- MLLT3
myeloid/lymphoid or mixed-lineage leukemia translocated to chromosome 3 protein.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00460.
Crystallographic statistics and additional supplemental data (PDF)
Accession Codes
Coordinates and structure factors have been deposited with accession codes 6t1i, 6t1j, 6t1l, 6t1m, 6t1n, and 6t1o.
Author Present Address
∥ C4 Therapeutics Inc., 490 Arsenal Way, Suite 200, Watertown, MA 02472.
Author Contributions
X.N., D.H., T.C., and J.M. conducted the experiment. M.M. and P.B. contributed inhibitors. A.C., S.K., O.F., and P.B. supervised research. A.C. and S.K. wrote the paper. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Dhalluin C.; Carlson J. E.; Zeng L.; He C.; Aggarwal A. K.; Zhou M. M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399 (6735), 491–496. 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Muller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149 (1), 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; Li Y.; Xiong X.; Chen Z.; Li H. YEATS Domain-A Histone Acylation Reader in Health and Disease. J. Mol. Biol. 2017, 429 (13), 1994–2002. 10.1016/j.jmb.2017.03.010. [DOI] [PubMed] [Google Scholar]
- Li Y.; Wen H.; Xi Y.; Tanaka K.; Wang H.; Peng D.; Ren Y.; Jin Q.; Dent S. Y.; Li W.; Li H.; Shi X. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 2014, 159 (3), 558–571. 10.1016/j.cell.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Sabari B. R.; Panchenko T.; Wen H.; Zhao D.; Guan H.; Wan L.; Huang H.; Tang Z.; Zhao Y.; Roeder R. G.; Shi X.; Allis C. D.; Li H. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol. Cell 2016, 62 (2), 181–193. 10.1016/j.molcel.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Zeng L.; Zhao C.; Ju Y.; Konuma T.; Zhou M. M. Structural Insights into Histone Crotonyl-Lysine Recognition by the AF9 YEATS Domain. Structure 2016, 24 (9), 1606–12. 10.1016/j.str.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze J. M.; Wang A. Y.; Kobor M. S. YEATS domain proteins: a diverse family with many links to chromatin modification and transcription. Biochem. Cell Biol. 2009, 87 (1), 65–75. 10.1139/O08-111. [DOI] [PubMed] [Google Scholar]
- Dutta A.; Abmayr S. M.; Workman J. L. Diverse Activities of Histone Acylations Connect Metabolism to Chromatin Function. Mol. Cell 2016, 63 (4), 547–552. 10.1016/j.molcel.2016.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein B. J.; Vann K. R.; Andrews F. H.; Wang W. W.; Zhang J.; Zhang Y.; Beloglazkina A. A.; Mi W.; Li Y.; Li H.; Shi X.; Kutateladze A. G.; Strahl B. D.; Liu W. R.; Kutateladze T. G. Structural insights into the pi-pi-pi stacking mechanism and DNA-binding activity of the YEATS domain. Nat. Commun. 2018, 9 (1), 4574. 10.1038/s41467-018-07072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L.; Wen H.; Li Y.; Lyu J.; Xi Y.; Hoshii T.; Joseph J. K.; Wang X.; Loh Y. E.; Erb M. A.; Souza A. L.; Bradner J. E.; Shen L.; Li W.; Li H.; Allis C. D.; Armstrong S. A.; Shi X. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 2017, 543 (7644), 265–269. 10.1038/nature21687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; Guan H.; Zhao S.; Mi W.; Wen H.; Li Y.; Zhao Y.; Allis C. D.; Shi X.; Li H. YEATS2 is a selective histone crotonylation reader. Cell Res. 2016, 26 (5), 629–632. 10.1038/cr.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Zhao D.; Chen Z.; Li H. YEATS domain: Linking histone crotonylation to gene regulation. Transcription 2017, 8 (1), 9–14. 10.1080/21541264.2016.1239602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Jin J.; Chung M. W. H.; Feng L.; Sun H.; Hao Q. Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (10), 2365–2370. 10.1073/pnas.1717664115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. J.; Li H.; Linhares B. M.; Kim E.; Ndoj J.; Miao H.; Grembecka J.; Cierpicki T. GAS41 Recognizes Diacetylated Histone H3 through a Bivalent Binding Mode. ACS Chem. Biol. 2018, 13 (9), 2739–2746. 10.1021/acschembio.8b00674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb M. A.; Scott T. G.; Li B. E.; Xie H.; Paulk J.; Seo H. S.; Souza A.; Roberts J. M.; Dastjerdi S.; Buckley D. L.; Sanjana N. E.; Shalem O.; Nabet B.; Zeid R.; Offei-Addo N. K.; Dhe-Paganon S.; Zhang F.; Orkin S. H.; Winter G. E.; Bradner J. E. Transcription control by the ENL YEATS domain in acute leukaemia. Nature 2017, 543 (7644), 270–274. 10.1038/nature21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulou V.; Peters A.; Schwaller J. Aggressive leukemia driven by MLL-AF9. Mol. Cell Oncol 2017, 5 (3), e1241854 10.1080/23723556.2016.1241854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi W.; Guan H.; Lyu J.; Zhao D.; Xi Y.; Jiang S.; Andrews F. H.; Wang X.; Gagea M.; Wen H.; Tora L.; Dent S. Y. R.; Kutateladze T. G.; Li W.; Li H.; Shi X. YEATS2 links histone acetylation to tumorigenesis of non-small cell lung cancer. Nat. Commun. 2017, 8 (1), 1088. 10.1038/s41467-017-01173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiuchi J.; Komatsu S.; Imamura T.; Nishibeppu K.; Shoda K.; Arita T.; Kosuga T.; Konishi H.; Shiozaki A.; Kubota T.; Okamoto K.; Fujiwara H.; Ichikawa D.; Tsuda H.; Otsuji E. Overexpression of YEATS4 contributes to malignant outcomes in gastric carcinoma. Am. J. Cancer Res. 2018, 8 (12), 2436–2452. [PMC free article] [PubMed] [Google Scholar]
- You S.; Wang F.; Hu Q.; Li P.; Zhang C.; Yu Y.; Zhang Y.; Li Q.; Bao Q.; Liu P.; Li J. Abnormal expression of YEATS4 associates with poor prognosis and promotes cell proliferation of hepatic carcinoma cell by regulation the TCEA1/DDX3 axis. Am. J. Cancer Res. 2018, 8 (10), 2076–2087. [PMC free article] [PubMed] [Google Scholar]
- Hsu C. C.; Shi J.; Yuan C.; Zhao D.; Jiang S.; Lyu J.; Wang X.; Li H.; Wen H.; Li W.; Shi X. Recognition of histone acetylation by the GAS41 YEATS domain promotes H2A.Z. deposition in non-small cell lung cancer. Genes Dev. 2018, 32 (1), 58–69. 10.1101/gad.303784.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidenreich D.; Moustakim M.; Schmidt J.; Merk D.; Brennan P. E.; Fedorov O.; Chaikuad A.; Knapp S. Structure-Based Approach toward Identification of Inhibitory Fragments for Eleven-Nineteen-Leukemia Protein (ENL). J. Med. Chem. 2018, 61 (23), 10929–10934. 10.1021/acs.jmedchem.8b01457. [DOI] [PubMed] [Google Scholar]
- Wu Q.; Heidenreich D.; Zhou S.; Ackloo S.; Kramer A.; Nakka K.; Lima-Fernandes E.; Deblois G.; Duan S.; Vellanki R. N.; Li F.; Vedadi M.; Dilworth J.; Lupien M.; Brennan P. E.; Arrowsmith C. H.; Muller S.; Fedorov O.; Filippakopoulos P.; Knapp S. A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat. Commun. 2019, 10 (1), 1915. 10.1038/s41467-019-09672-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13 (5), 337–356. 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Li X.; Li X. M.; Jiang Y.; Liu Z.; Cui Y.; Fung K. Y.; van der Beelen S. H. E.; Tian G.; Wan L.; Shi X.; Allis C. D.; Li H.; Li Y.; Li X. D. Structure-guided development of YEATS domain inhibitors by targeting pi-pi-pi stacking. Nat. Chem. Biol. 2018, 14 (12), 1140–1149. 10.1038/s41589-018-0144-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christott T.; Bennett J.; Coxon C.; Monteiro O.; Giroud C.; Beke V.; Felce S. L.; Gamble V.; Gileadi C.; Poda G.; Al-Awar R.; Farnie G.; Fedorov O. Discovery of a Selective Inhibitor for the YEATS Domains of ENL/AF9. SLAS Discov 2019, 24 (2), 133–141. 10.1177/2472555218809904. [DOI] [PubMed] [Google Scholar]
- Moustakim M.; Christott T.; Monteiro O. P.; Bennett J.; Giroud C.; Ward J.; Rogers C. M.; Smith P.; Panagakou I.; Diaz-Saez L.; Felce S. L.; Gamble V.; Gileadi C.; Halidi N.; Heidenreich D.; Chaikuad A.; Knapp S.; Huber K. V. M.; Farnie G.; Heer J.; Manevski N.; Poda G.; Al-Awar R.; Dixon D. J.; Brennan P. E.; Fedorov O. Discovery of an MLLT1/3 YEATS Domain Chemical Probe. Angew. Chem., Int. Ed. 2018, 57 (50), 16302–16307. 10.1002/anie.201810617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith J. M.; Apodaca R.; Xiao W.; Seierstad M.; Pattabiraman K.; Wu J.; Webb M.; Karbarz M. J.; Brown S.; Wilson S.; Scott B.; Tham C. S.; Luo L.; Palmer J.; Wennerholm M.; Chaplan S.; Breitenbucher J. G. Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2008, 18 (17), 4838–43. 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]
- Hong Q.; Bakshi R. K.; Palucki B. L.; Park M. K.; Ye Z.; He S.; Pollard P. G.; Sebhat I. K.; Liu J.; Guo L.; Cashen D. E.; Martin W. J.; Weinberg D. H.; MacNeil T.; Tang R.; Tamvakopoulos C.; Peng Q.; Miller R. R.; Stearns R. A.; Chen H. Y.; Chen A. S.; Strack A. M.; Fong T. M.; MacIntyre D. E.; Wyvratt M. J.; Nargund R. P. Discovery of a piperazine urea based compound as a potent, selective, orally bioavailable melanocortin subtype-4 receptor partial agonist. Bioorg. Med. Chem. Lett. 2011, 21 (8), 2330–4. 10.1016/j.bmcl.2011.02.090. [DOI] [PubMed] [Google Scholar]
- Cumming J. G.; Bower J. F.; Waterson D.; Faull A.; Poyser P. J.; Turner P.; McDermott B.; Campbell A. D.; Hudson J.; James M.; Winter J.; Wood C. The design and synthesis of novel, potent and orally bioavailable N-aryl piperazine-1-carboxamide CCR2 antagonists with very high hERG selectivity. Bioorg. Med. Chem. Lett. 2012, 22 (12), 3895–9. 10.1016/j.bmcl.2012.04.118. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (Pt 2), 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69 (Pt 7), 1204–14. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. Acknowledging Errors: Advanced Molecular Replacement with Phaser. Methods Mol. Biol. 2017, 1607, 421–453. 10.1007/978-1-4939-7000-1_18. [DOI] [PubMed] [Google Scholar]
- Emsley P. Tools for ligand validation in Coot. Acta Crystallogr. D Struct Biol. 2017, 73 (Pt 3), 203–210. 10.1107/S2059798317003382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. A.; Steiner R. A.; Lebedev A. A.; Potterton L.; McNicholas S.; Long F.; Murshudov G. N. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60 (Pt 12 Pt 1), 2184–2195. 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- Williams C. J.; Headd J. J.; Moriarty N. W.; Prisant M. G.; Videau L. L.; Deis L. N.; Verma V.; Keedy D. A.; Hintze B. J.; Chen V. B.; Jain S.; Lewis S. M.; Arendall W. B. 3rd; Snoeyink J.; Adams P. D.; Lovell S. C.; Richardson J. S.; Richardson D. C. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27 (1), 293–315. 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.