

Abstract

The ability of G protein-coupled receptor (GPCR) kinases (GRKs) to regulate desensitization of GPCRs has made GRK2 and GRK5 attractive targets for treating heart failure and other diseases such as cancer. Although advances have been made toward developing inhibitors that are selective for GRK2, there have been far fewer reports of GRK5 selective compounds. Herein, we describe the development of GRK5 subfamily selective inhibitors, 5 and 16d that covalently interact with a nonconserved cysteine (Cys474) unique to this subfamily. Compounds 5 and 16d feature a highly amenable pyrrolopyrimidine scaffold that affords high nanomolar to low micromolar activity that can be easily modified with Michael acceptors with various reactivities and geometries. Our work thereby establishes a new pathway toward further development of subfamily selective GRK inhibitors and establishes Cys474 as a new and useful covalent handle in GRK5 drug discovery.
Keywords: covalent inhibitor, kinase inhibitor, GPCR kinase, heart failure
Many cellular events are modulated in response to extracellular signals by G protein-coupled receptors (GPCRs).1 GPCR kinases (GRKs) selectively recognize and phosphorylate activated GPCRs, leading to their desensitization and internalization, which is critical for a normal return to cellular homeostasis.2 Based on their phylogeny, the seven mammalian GRKs are divided into the GRK1 (GRK1 and 7), GRK2 (GRK2 and 3), and GRK4 (GRK4, 5, and 6) subfamilies.3 GRK1 and 7 are expressed primarily in the retina and GRK4 in the testes, whereas GRK2, 3, 5, and 6 are more ubiquitously expressed.4
Among the GRKs, the β-adrenergic receptor (βAR) kinases (GRK2 and GRK3) are the most widely studied, due to their role in various disease states such as cardiovascular disease, cancer, and inflammation.5,6 Although GRK5 has been studied for its role in multiple disease states including cancer, neurodegeneration, type 2 diabetes, and cardiovascular disease, few examples of GRK5 selective inhibitors are found in the literature.7,8 Given its cross-functionality, a selective and potent probe is needed to further investigate the role of GRK5 in the disease states it is implicated in.
Cardiac function is controlled in part by βARs. Under physiological conditions, βARs at the cardiomyocyte cell surface are activated in response to increased circulating levels of the fight-or-flight hormones, epinephrine and norepinephrine, leading to an increase in cardiac output.9 GRK2 and GRK5, the predominant GRKs expressed in the heart, then regulate signal termination through phosphorylation leading to subsequent internalization of the βARs.10 In the failing heart, epinephrine and norepinephrine levels remain high in an attempt to compensate for decreased cardiac output.11 Although initially beneficial for increasing heart contractility, prolonged exposure to catecholamines exacerbates the problem as evidenced by increased GRK2 and GRK5 levels, a decreased number of βARs at the cell surface, and initiation of a pathological hypertrophic stress response.12 βAR antagonists (β-blockers) are used to treat heart failure, but there is growing evidence that inhibition of GRK2, GRK5, or both could improve the currently available heart failure therapies.13−20
There is growing evidence that GRK2 and GRK5 have distinct pathological roles within the failing heart.21−25 Increased GRK2 levels are thought to mediate the decrease in cell-surface βARs and the prolonged sympathetic nervous system activation, leading to decreased contractility.22 GRK5 is unique among GRKs in that it undergoes Ca2+·calmodulin-dependent nuclear localization that allows GRK5 to translocate into the nucleus where it phosphorylates histone deacetylase 5, turning on the transcription of hypertrophic genes.24 Indeed, cardiac-specific GRK2 knockout mice have improved contractility and increased cell-surface βARs postmyocardial infarction,16 and GRK5 knockout mice are protected from cardiac hypertrophy following controlled cardiac stress. The extent of the functional differences in GRK2 and GRK5 within cardiomyocytes remains to be elucidated, but selective inhibition of each of these kinases would offer the opportunity to further understand their distinct roles in the progression of heart failure. In addition, the selective inhibition of GRK2 or GRK5 presents the possibility of treating different aspects of heart failure without affecting the entire cardiac regulatory system.
Despite high structural similarity in the active site among GRKs, we have had success in developing potent and selective small molecule inhibitors for the GRK2 subfamily that improve contractility in isolated adult mouse cardiomyocytes, in part because GRK2, and presumably GRK3, adopts a distinct inactive pose from other GRKs and other protein kinases.26−29 Comparison of GRK2 and GRK5 crystal structures revealed a more spacious ATP-binding pocket in GRK2/3 that was able to accommodate bulkier chemical substituents, which allowed us to build out GRK5 binding.26 We have also developed pan GRK-selective compounds (CCG-215022 and CCG-258748) with nanomolar potency for both GRK2 and GRK5,26,27,29 but thus far we have not been successful in developing GRK5 selective (or GRK4 subfamily selective) inhibitors using the canonical reversible binding model. Others have reported GRK4 subfamily selective compounds, but they are also potent inhibitors of other kinase families8 and have not been independently confirmed.
Examination of the crystal structure of GRK6 in what is believed to be an active conformation (PDB 3NYN(30)) revealed that the thiol group of Cys474, located within the flexible active site tether (AST) loop of the kinase domain, is positioned adjacent to the ATP-binding site, at least when the kinase adopts a more closed conformation (Figure 1A). Because this cysteine is unique to GRK4 subfamily members, it could be exploited as a handle for covalent inhibition to gain selectivity for GRK5 over GRK2. In recent years, the popularity of covalent warheads has risen because they offer the possibility of more potency and selectivity than reversible inhibitors.31 In particular, specifically targeting nonconserved cysteines in the ATP-binding pocket of kinases has demonstrated utility.31−33 The most successful irreversible modifiers have come from designing a reversibly binding compound with low or sub-μM affinity to also contain a covalent warhead that is within reach and in the proper orientation to interact with the free thiol of a nearby cysteine. The most widely used reaction to achieve irreversible covalent attachment onto a cysteine is a Michael addition using electrophilic warheads such as acrylamides, vinyl sulfones, and alkynes.33 Recent advances have also been made with the use of an N,N-dimethyl-butenoic amide, which improves solubility and contains an internal base that can deprotonate and activate the thiol group.33−35
Figure 1.
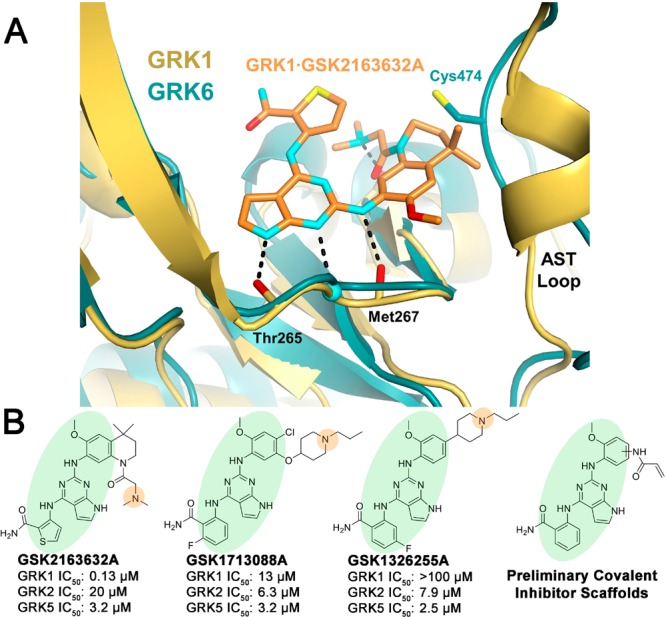
GSK2163632A and related compounds suggest a route to selective inhibition of GRK5 via covalent modification of Cys474. (A) GSK2163632A (green) bound to GRK1 (yellow, PDB entry 4PNI) superimposed with the active conformation of GRK6 (blue, PDB entry 3NYN). (B) Previously identified pyrrolopyrimidine based GRK inhibitors and covalent inhibitor design rationale. Green ovals highlight the base of the scaffold that is generally conserved, and orange circles represent a basic nitrogen that was removed except in the case of analogs where it is replicated potentially by a N,N-dimethyl-butenoic amide.
In a previous screen, GSK2163632A was identified as a modestly potent GRK5 inhibitor (IC50 = 3.2 μM) with high potency for GRK1 (IC50 = 130 nM) and lower potency for PKA (IC50 > 500 μM). Two related compounds, GSK1713088A and GSK1326255A (Figure 1B), were shown to have similar potency for GRK5 (IC50 = 3.2 and 2.5 μM, respectively), but also modest selectivity over both GRK1 and GRK2.36 All three GSK compounds share a common pyrrolopyrimidine core, which binds within the ATP pocket with the nitrogens from the core forming hydrogen bonds with the hinge of the kinase domain in a donor–acceptor–donor motif, as observed for GSK2163632A in complex with GRK1 (PDB entry 4PNI, Figure 1A). We therefore hypothesized that we could append covalent warheads to the methoxyphenyl of GSK1713088A or GSK1326255A that could react with Cys474 (Figure 1B). To avoid a highly substituted ring, we replaced the tertiary amine appendages with our covalent warheads in hopes that the potency gained by the covalent bond would overcome the loss of a hydrogen bond accepted by the tertiary amine. The N,N-dimethyl-butenoic amide warhead would, however, place a basic tertiary amine in a similar position.
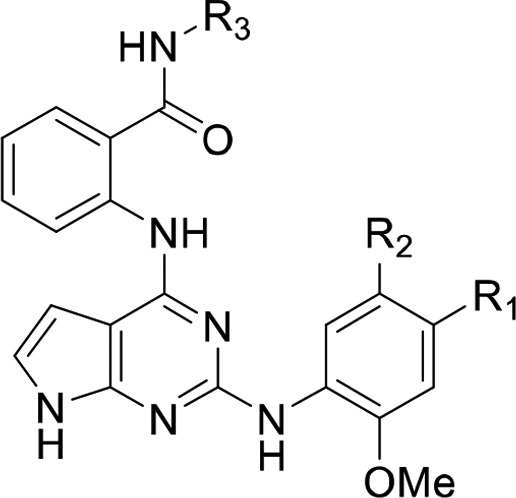
We first rationally designed six different variants of the GSK inhibitor series with short amide linkers. We overlaid a GRK5 crystal structure (PDB entry 4WNK) with that of the active conformation of GRK6 (PDB entry 3NYN) (Figure 1A). Building a covalent warhead meta to the aniline (ring C) likely retains the hydrogen bond of the amide carbonyl that GSK2163632A forms with the backbone nitrogen of GRK1-Asp271 (GRK5-Asp270), but this may position the Michael acceptor too distant from Cys474. Alternatively, building the warhead para to the aniline would likely hinder the hydrogen bond with Asp271, but may put the Michael acceptor within 1.5 Å of the Cys474 thiol group. However, it is not possible to accurately model these interactions because the degree of kinase domain closure and the conformations of the flexible AST and P loops cannot be predicted a priori. Therefore, both para and meta substituents were synthesized, including unreactive saturated ethyl amide analogs as negative controls (Table 1).
Table 1. IC50 Values for Pyrrolopyrimidine Compounds (μM ± SD)a.

Experimental values derived from this report were run three times in duplicate with the exception of assays involving 5 against GRK5, GRK6, and GRK5-C474S with 4 h incubations, which were performed two times in duplicate. Inhibitor incubation times are given in parentheses.
From Homan et al.38 Note that these compounds were not assayed after 4 h incubations and are listed here for comparison purposes only.
Selectivity for GRK5 over GRK2 based on IC50 ratio. The IC50 value for the longest incubation with GRK5 was used for the calculation.
Measured at 100 min.
Synthesis of para analogs 4, 5, and 6 (Table 1) and meta substituted analogs 7, 8, and 9 are described in Schemes S1 and S2, respectively. Tosyl protection of commercially available 4, 6-dichloropyrrolopyrimidine S1 followed by base-catalyzed aromatic substitution with 2-aminobenzamide gave S3.37 Acid promoted lactam cyclization activated the 2-chloro for substitution with either aniline S8 or S12 to give lactams S4 and S9. Hydrolysis using ammonium hydroxide afforded amides S5 and S10. Tosyl deprotection followed by amide coupling provided analogs 4–9.
The para and meta ethylamide controls, 4 and 7, respectively, showed substantially different biochemical results indicating a possible structure–activity relationship (SAR) cliff. The para substituted ethyl amide 7 showed no activity up to 100 μM against all GRKs tested, whereas 4 showed low micromolar inhibition for GRK5 with an IC50 of 5.9 μM. There was no consistent time-dependent change in inhibition of GRK5 by 4 or 7 as a function of time, as expected. The para substituted acrylamide, 5, exhibited an IC50 of 6.2 μM for GRK5 after 1 h of preincubation on ice, with no potency against GRK1 or GRK2. After 4 h, 5 showed a convincing increase in potency as a function of time consistent with covalent inhibition, with an IC50 of 0.2 μm (Table 1, Figure 2). The >16-fold difference in GRK5 potency for 5 relative to its noncovalent control 4 at 1 h also suggests a covalent mechanism of action. When 5 was also tested using light activated rod outer segments (ROS) as a substrate instead of tubulin, the inhibitory potency was comparable across all incubation times (Figure S5). This result is consistent with our prior GRK inhibitor studies.27,38 The compounds also exhibited >450 selectivity over GRK2. The meta acrylamide analog 8 had similar potency for GRK5 at all incubation times with respect to its noncovalent control 7 suggesting that it is not acting covalently. It also showed no inhibition of GRK1 but modest GRK2 inhibition (IC50 = 39 μM).
Figure 2.
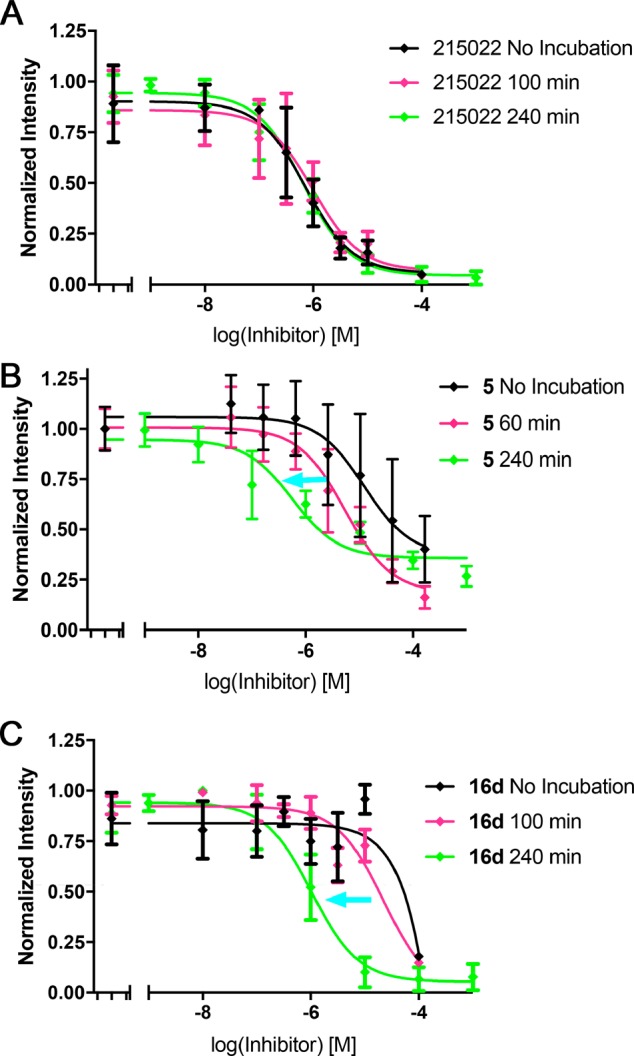
Time-dependent inhibition of GRK5 by 5 and 16d but not CCG-215022. Compounds were preincubated for times of 0 min (black), 100 min (magenta), and 240 min (green). (A) CCG-215022, which has no covalent modifier, does not show a significant change in IC50 (380 nM) over time. Compounds (B) 5 and (C) 16d exhibit the expected leftward shift in IC50 for covalent inhibitors as preincubation times increase, as indicated by the blue arrows. Each curve is the average of either three (215022 and 16d) or four (5) experiments. Error bars represent standard deviation.
Both 6 and 9, featuring the N,N-dimethyl-butenoic amide in para and meta positions, respectively, had increased potency for all three GRKs relative to the acrylamide inhibitors 5 and 8, indicating that addition of the basic nitrogen augments potency against all three kinases. The para substituted analog, 6, was predicted to be closer to the AST loop and thus more likely to form a covalent bond with Cys474. In our time-dependent inhibition of GRK5, it shows some improvement from 0 min to 1 h (GRK5 IC50 from 0.57 to 0.35 μM, respectively) but exhibits no selectivity over GRK1 or GRK2 (IC50 = 0.76 and 0.68 μM, respectively). The meta substituted analog, 9, did not show time-dependent GRK5 inhibition (IC50 range = 0.22–0.27 μM), although it showed ∼10-fold selectivity for GRK5 over GRK1 and GRK2 (IC50 = 2.1 and 2.7 μM, respectively). This difference in GRK selectivity between the meta and para substituted analogs 6 and 9 indicates that the position of the amide linked appendage from the methoxyphenyl ring is one route for gaining GRK5 selectivity. Despite their GRK5 potency, 6 and 9 were not further pursued due to their lack of selectivity against GRK1 and 2.
We next tested whether homologation of the covalent warhead linkers to the methoxyphenyl ring would allow them to better engage Cys474 by adding in an additional rotatable bond. For each series of para and meta substituted analogs, we expanded our search to test five unique covalent warheads of varying softness (acrylamides through vinyl sulfones; Table 1). In Scheme S3, the benzamide on Ring A was methylated to avoid an acid catalyzed intramolecular ring closure.37 Synthesis of 16a–j starts with SEM protection of 4,6-dicholopyrrolopyrimidine (10) to give starting material 11. Compound 11 underwent a base catalyzed aromatic substitution to give 12. Using a traditional Buchwald–Hartwig cross coupling with 2-methoxy-4-cyanoaniline, compound 13 was achieved. From 13, the nitrile was reduced using Raney Nickel in methanolic ammonia to give precursor 14. Using standard amide coupling conditions, compounds 15a–e were achieved in moderate yields (50–80%). Final compounds 16a–e were then produced by acid catalyzed SEM deprotection. The meta series of compounds 16f–j were accomplished through a modified route (Scheme S4). Coupling partner 17 was achieved through reduction of the amide (18) to the 3-amino-aniline, 19. Compound 19 was then protected with Cbz-chloride to then yield 17 in 75% yield. Then, using coupling partners 12 and 17, 13a′ was achieved via a standard Buchwald coupling. Precursor 14a′ was achieved by a reductive Cbz deprotection. From 14a′, compounds 15f–j and 16f–j were furnished using the same coupling and deprotection conditions used in Scheme S4.
We then determined IC50 values of the homologated compounds after a 4 h incubation for GRK2, GRK5, and GRK6 (Table 1). GRK6 was included as a positive control for GRK4 subfamily selectivity. As in the nonhomologated series, the ethylamide compounds (16a and 16f) showed an SAR cliff, with the para ethylamide unable to inhibit any of the GRKs tested. The reactive para-substituted series (16b–e) were more selective for GRK5/6 over GRK2 than their meta analogs with the exception of 16e. GRK5 tolerated both the small substituents of the acrylamide 16b (IC50 = 78 μM) and vinyl sulfone, 16e (IC50 = 19 μM), but also the large N,N-dimethyl-butenoic amide of 16c (IC50 = 17 μM). The para-alkyne (16d) was the most potent and selective GRK5 inhibitor from this series (IC50 = 1.1 μM, 90-fold selectivity over GRK2 after 4 h incubation). Compound 16d also demonstrated comparable inhibitory potency for GRK5/6 when using light-activated ROS as a substrate instead of tubulin (Figure S5).
Interestingly, potency for GRK2 was lost for all compounds with the para-substitution. It is possible that the slightly longer AST-loop in the GRK2 subfamily allows it to reach further in the active site, as seen for residues 476–479 in the GRK2-Gβγ·GSK180736A cocrystal structure (PDB entry 4PNK), which would collide with substituents in the para-position.
Overall, the meta substituted compounds (16g–j) tended to have higher potency against GRK5 but lower or even reversed selectivity versus GRK2 (the exception being the vinyl sulfone 16j). Small polar groups, specifically those with hydrogen bond acceptor capability, were well tolerated in GRK5. For example, both 16g and 16j inhibited GRK5, but the polar vinyl sulfone of 16j was more potent (IC50 = 7.1 μM) than the more lipophilic acrylamide of 16g (IC50 = 22 μM). The meta-alkyne (16i) had no potency for GRK5, suggesting that this more rigid covalent modifier may collide with the P-loop or AST-loop. The 16h compound had low micromolar potency for GRK5.
Although these compounds selectively inhibit GRK5, they have only modest potency. Our two most potent analogs, 5 and 16d, have IC50 values of 0.22 and 1.1 μM, respectively, after 4 h incubation using tubulin as a substrate (Table 1). We attribute this to one of two reasons. Either the core scaffold has a suboptimal engagement with the hinge region, or the entropic cost of locking down the flexible AST loop via a covalent bond is high enough to limit the binding affinity.
Of the homologated series, only 16g, 16h, and 16f showed activity against GRK2. Our modeling suggested that shorter warheads may be more easily accommodated within the shallow GRK2 ribose pocket. All the other meta-substituted materials have larger, less flexible warheads. Our most GRK2 selective analog, 16f, has only modest potency compared to previously developed GRK2 inhibitors, and we therefore are not pursuing it as a GRK2 lead compound.26,29
As expected, inhibition of GRK6 was similar to that of GRK5 in most cases, the exception being 16d, which was unable to inhibit GRK6. It is unclear whether this represents true intrasubfamily selectivity or a vagary of the experimental conditions for this combination. The most potent compound 16d was then additionally tested at 0 and 100 min incubations to assess if there was a time-dependent decrease in IC50, the hallmark of covalent inhibition. Indeed, 16d displayed a marked increase in apparent potency as a function of time (Figure 2). Further confirmation of covalent inhibition of GRK5 was achieved through intact protein mass spectrometry (MS). Compounds 5 and 16a–e were tested, but only 5, 16b, and 16d showed significant amounts of covalent linkage after a 3 h incubation (Figures 3 and S1), consistent with the results of our radiometric assays. Compounds 16f–j were also tested but showed no covalent inhibition (Figure S2).
Figure 3.
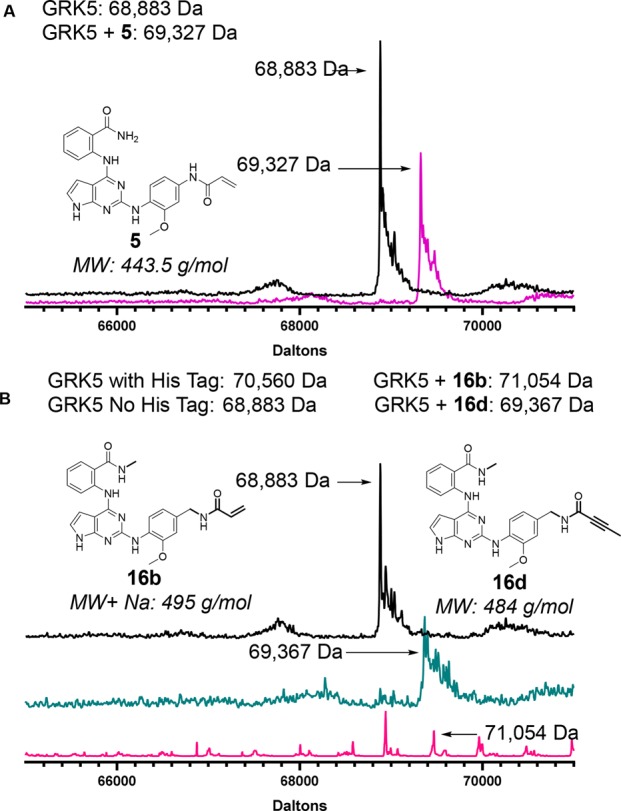
Evidence from intact mass spectrometry for covalent modification of GRK5 by 5, 16b, and 16d: (A) GRK5 only (black trace), GRK5·5 (purple). (B) MS traces for GRK5 (black trace), GRK5·16d (ocean blue), and GRK5·16b (pink). The latter trace indicates that 16b only partially labeled GRK5.
For 5, 16b, and 16d, we also tested whether inhibition is affected when Cys474 is mutated to serine (GRK5-C474S). A decrease in GRK5-C474S potency relative to wild-type GRK5 would thus be consistent with a covalent inhibition mechanism. Compound 5 lost all potency against the mutant protein, whereas 16b and 16d retained comparable activity. The reason is unclear, but a structure of GRK5-C474S bound to 16d would help resolve that discrepancy by illuminating how this particular compound interacts with the Ser474 point mutation.
We next tested whether 16d engaged GRK5 in a covalent bond specifically at Cys474 using intact protein MS and showed that GRK5-C474S mutant does not react (Figure 4). Using tandem mass spectrometry (MS/MS) we further observed that 16d labels Cys474 but also a cysteine located in a solvent-exposed position in its regulator of G protein signaling homology domain, remote from the active site (Figures S3–S4). This additional labeling event is indicative of a concentration-dependent covalent engagement, and we do not believe that its presence is indicative of a biologically relevant interaction.
Figure 4.

Evidence for GRK5-Cys474 covalent engagement of 16d. (A) MS trace for GRK5, (B) MS trace for GRK5 incubated with 16d indicating covalent modification, (C) MS trace for GRK5-C474S, and (D) MS trace for GRK5-C474 incubated with 16d indicates that 16d cannot covalently modify GRK5-C474S. Each peak shown is representative of n = 3 experiments.
In summary, we report what we believe to be the first examples of covalent inhibitors of GRK5, including some that are GRK5 subfamily selective (5 and 16d) with high nanomolar to low micromolar potency. We have leveraged the pyrrolopyrimidine scaffold to install para-substituted linkers that can interact covalently with the AST from GRK5 but not from GRK2. Additionally, we have shown that Cys474 is selectively targeted by our covalent warheads, validating our design strategy. Moving forward, we aim to further improve the potency of these compounds against GRK5 and GRK6, and to pursue crystal structures of the GRK5/6·5 and 16d complexes to confirm their binding poses and the effect of covalent modification on the overall conformation of GRK5. Before examining the effects of our compounds in vivo on cardiomyocyte contractility and, ultimately, to parse the role of GRK5 in heart failure, these compounds will also need to be tested for selectivity against other protein kinases.
Acknowledgments
We thank Dr. Venky Bashar of the University of Michigan Proteomic Resource Facility for assistance with tandem mass spectrometry efforts. We also thank Dr. Pil Lee for her technical assistance in building a GRK5 homology model.
Glossary
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- βAR
β-adrenergic receptor
- AST
active site tether
- P-loop
phosphate-binding loop
- MS
mass spectrometry
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00365.
Experimental procedures and supplementary figures (PDF)
Author Present Address
∥ (H.V.W.) 130 Scripps Way, Jupiter, FL 33458, USA
Author Contributions
The manuscript was written primarily by R.A.R., M.C.C., and J.G.G.T. All authors have given approval to the final version of the manuscript. H.V.W. synthesized compounds 4–9 and determined their IC50 values for GRKs 1, 2, and 5. R.A.R. synthesized compounds 16a–j and performed MS experiments. M.C.C. determined IC50 values for GRK2, 5, 6, and GRK5-C474S. M.C.C. purified GRK2. R.A.B. purified GRK5, 6, and GRK5-C474S and contributed some of the IC50 dose–response experiments. Q.C. and L.A. performed ROS phosphorylation assays.
This work was supported by NIH grants HL071818 and HL122416 and the Walther Cancer Foundation (to J.J.G.T.), American Heart Association grants 15PRE22730028 (to H.V.W.) and 19POST3445019 (to Q.C.), and a pilot grant from the University of Michigan Center for Discovery of New Medicines (to A.D.W.). M.C.C. acknowledges training grant support from the University of Michigan Chemistry–Biology Interface (CBI) training program (NIH grant 5T32GM008597).
The authors declare no competing financial interest.
Supplementary Material
References
- Katritch V.; Cherezov V.; Stevens R. C. Structure-Function of the G Protein–Coupled Receptor Superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53 (1), 531–556. 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich E. V.; Tesmer J. J. G.; Mushegian A.; Gurevich V. V. G. Protein-Coupled Receptor Kinases: More than Just Kinases and Not Only for GPCRs. Pharmacol. Ther. 2012, 133 (1), 40–69. 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher J. A.; Freedman N. J.; Lefkowitz R. J. G. Protein-Coupled Receptor Kinases. Annu. Rev. Biochem. 1998, 67 (1), 653–692. 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- Ferguson S. S. Evolving Concepts in G Protein-Coupled Receptor Endocytosis: The Role in Receptor Desensitization and Signaling. Pharmacol. Rev. 2001, 53 (1), 1–24. [PubMed] [Google Scholar]
- Nogués L.; Palacios-García J.; Reglero C.; Rivas V.; Neves M.; Ribas C.; Penela P.; Mayor F. G. Protein-Coupled Receptor Kinases (GRKs) in Tumorigenesis and Cancer Progression: GPCR Regulators and Signaling Hubs. Semin. Cancer Biol. 2018, 48, 78–90. 10.1016/j.semcancer.2017.04.013. [DOI] [PubMed] [Google Scholar]
- Steury M. D.; McCabe L. R.; Parameswaran N. G. Protein-Coupled Receptor Kinases in the Inflammatory Response and Signaling. Adv. Immunol. 2017, 136, 227–277. 10.1016/bs.ai.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickx J. O.; van Gastel J.; Leysen H.; Santos-Otte P.; Premont R. T.; Martin B.; Maudsley S. GRK5 – A Functional Bridge Between Cardiovascular and Neurodegenerative Disorders. Front. Pharmacol. 2018, 9, 04184. 10.3389/fphar.2018.01484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S. Y.; Lee B. H.; Jung H.; Yun C. S.; Ha J. D.; Kim H. R.; Chae C. H.; Lee J. H.; Seo H. W.; Oh K.-S. Design and Synthesis of Novel 3-(Benzo[d]Oxazol-2-Yl)-5-(1-(Piperidin-4-Yl)-1H-Pyrazol-4-Yl)Pyridin-2-Amine Derivatives as Selective G-Protein-Coupled Receptor Kinase-2 and −5 Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23 (24), 6711–6716. 10.1016/j.bmcl.2013.10.036. [DOI] [PubMed] [Google Scholar]
- Huang Z. M.; Gold J. I.; Koch W. J. G. Protein-Coupled Receptor Kinases in Normal and Failing Myocardium. Front. Biosci., Landmark Ed. 2011, 16 (1), 3057–3060. 10.2741/3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M. J.; Engelhardt S.; Eschenhagen T. What Is the Role of β-Adrenergic Signaling in Heart Failure?. Circ. Res. 2003, 93, 896–906. 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- Triposkiadis F.; Karayannis G.; Giamouzis G.; Skoularigis J.; Louridas G.; Butler J. The Sympathetic Nervous System in Heart Failure. J. Am. Coll. Cardiol. 2009, 54 (19), 1747–1762. 10.1016/j.jacc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Port J. D.; Bristow M. R. Altered Beta-Adrenergic Receptor Gene Regulation and Signaling in Chronic Heart Failure. J. Mol. Cell. Cardiol. 2001, 33 (5), 887–905. 10.1006/jmcc.2001.1358. [DOI] [PubMed] [Google Scholar]
- Rockman H. A.; Chien K. R.; Choi D.-J.; Iaccarino G.; Hunter J. J.; Ross J.; Lefkowitz R. J.; Koch W. J. Expression of a -Adrenergic Receptor Kinase 1 Inhibitor Prevents the Development of Myocardial Failure in Gene-Targeted Mice. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (12), 7000–7005. 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhart A. D.; Ozaki T.; Tevaearai H.; Rockman H. A.; Koch W. J. Vascular-Targeted Overexpression of G Protein-Coupled Receptor Kinase-2 in Transgenic Mice Attenuates Beta-Adrenergic Receptor Signaling and Increases Resting Blood Pressure. Mol. Pharmacol. 2002, 61 (4), 749–758. 10.1124/mol.61.4.749. [DOI] [PubMed] [Google Scholar]
- Kohout T. A. Regulation of G Protein-Coupled Receptor Kinases and Arrestins During Receptor Desensitization. Mol. Pharmacol. 2003, 63 (1), 9–18. 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- Raake P. W.; Vinge L. E.; Gao E.; Boucher M.; Rengo G.; Chen X.; DeGeorge B. R.; Matkovich S.; Houser S. R.; Most P.; et al. Protein–Coupled Receptor Kinase 2 Ablation in Cardiac Myocytes Before or After Myocardial Infarction Prevents Heart Failure. Circ. Res. 2008, 103 (4), 413–422. 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal D. M.; Homan K. T.; Chen J.; Wu E. K.; Hinkle P. M.; Huang Z. M.; Chuprun J. K.; Song J.; Gao E.; Cheung J. Y.; et al. Paroxetine Is a Direct Inhibitor of G Protein-Coupled Receptor Kinase 2 and Increases Myocardial Contractility. ACS Chem. Biol. 2012, 7 (11), 1830–1839. 10.1021/cb3003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A.; Rengo G.; J. Koch W. GRK2 Inhibition in Heart Failure: Something Old, Something New. Curr. Pharm. Des. 2012, 18 (2), 186–191. 10.2174/138161212799040510. [DOI] [PubMed] [Google Scholar]
- Raake P. W. J.; Schlegel P.; Ksienzyk J.; Reinkober J.; Barthelmes J.; Schinkel S.; Pleger S.; Mier W.; Haberkorn U.; Koch W. J.; et al. AAV6.ΒARKct Cardiac Gene Therapy Ameliorates Cardiac Function and Normalizes the Catecholaminergic Axis in a Clinically Relevant Large Animal Heart Failure Model. Eur. Heart J. 2013, 34, 1437–1447. 10.1093/eurheartj/ehr447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo A.; Liccardo D.; Koch W. J. Targeting Cardiac β-Adrenergic Signaling via GRK2 Inhibition for Heart Failure Therapy. Front. Physiol. 2013, 4, 1–7. 10.3389/fphys.2013.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penela P.; Murga C.; Ribas C.; Tutor A.; Peregrin S.; Mayorjr F. Mechanisms of Regulation of G Protein-Coupled Receptor Kinases (GRKs) and Cardiovascular Disease. Cardiovasc. Res. 2006, 69 (1), 46–56. 10.1016/j.cardiores.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A.; Rengo G.; Funakoshi H.; Eckhart A. D.; Koch W. J. Adrenal GRK2 Upregulation Mediates Sympathetic Overdrive in Heart Failure. Nat. Med. 2007, 13 (3), 315–323. 10.1038/nm1553. [DOI] [PubMed] [Google Scholar]
- Kemp C. D.; Conte J. V. The Pathophysiology of Heart Failure. Cardiovasc. Pathol. 2012, 21 (5), 365–371. 10.1016/j.carpath.2011.11.007. [DOI] [PubMed] [Google Scholar]
- Traynham C. J.; Hullmann J.; Koch W. J. Canonical and Non-Canonical Actions of GRK5 in the Heart. J. Mol. Cell. Cardiol. 2016, 92, 196–202. 10.1016/j.yjmcc.2016.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hullmann J.; Traynham C. J.; Coleman R. C.; Koch W. J. The Expanding GRK Interactome: Implications in Cardiovascular Disease and Potential for Therapeutic Development. Pharmacol. Res. 2016, 110, 52–64. 10.1016/j.phrs.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldschmidt H. V.; Homan K. T.; Cruz-Rodríguez O.; Cato M. C.; Waninger-Saroni J.; Larimore K. M.; Cannavo A.; Song J.; Cheung J. Y.; Kirchhoff P. D.; et al. Structure-Based Design, Synthesis, and Biological Evaluation of Highly Selective and Potent G Protein-Coupled Receptor Kinase 2 Inhibitors. J. Med. Chem. 2016, 59 (8), 3793–3807. 10.1021/acs.jmedchem.5b02000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldschmidt H. V.; Homan K. T.; Cato M. C.; Cruz-Rodríguez O.; Cannavo A.; Wilson M. W.; Song J.; Cheung J. Y.; Koch W. J.; Tesmer J. J. G.; et al. Structure-Based Design of Highly Selective and Potent G Protein-Coupled Receptor Kinase 2 Inhibitors Based on Paroxetine. J. Med. Chem. 2017, 60 (7), 3052–3069. 10.1021/acs.jmedchem.7b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.-Q.; Cato M. C.; Labudde E.; Beyett T. S.; Tesmer J. J. G.; Grant B. J. Navigating the Conformational Landscape of G Protein–Coupled Receptor Kinases during Allosteric Activation. J. Biol. Chem. 2017, 292 (39), 16032–16043. 10.1074/jbc.M117.807461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouley R.; Waldschmidt H. V.; Cato M. C.; Cannavo A.; Song J.; Cheung J. Y.; Yao X.-Q.; Koch W. J.; Larsen S. D.; Tesmer J. J. G. Structural Determinants Influencing the Potency and Selectivity of Indazole-Paroxetine Hybrid G Protein–Coupled Receptor Kinase 2 Inhibitors. Mol. Pharmacol. 2017, 92 (6), 707–717. 10.1124/mol.117.110130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguth C. A.; Singh P.; Huang C.; Tesmer J. J. G. Molecular Basis for Activation of G Protein-Coupled Receptor Kinases. EMBO J. 2010, 29 (19), 3249–3259. 10.1038/emboj.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discovery 2011, 10 (4), 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Leproult E.; Barluenga S.; Moras D.; Wurtz J.-M.; Winssinger N. Cysteine Mapping in Conformationally Distinct Kinase Nucleotide Binding Sites: Application to the Design of Selective Covalent Inhibitors. J. Med. Chem. 2011, 54 (5), 1347–1355. 10.1021/jm101396q. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20 (2), 146–159. 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Inesta-Vaquera F.; Niepel M.; Zhang J.; Ficarro S. B.; Machleidt T.; Xie T.; Marto J. A.; Kim N.; Sim T.; et al. Discovery of Potent and Selective Covalent Inhibitors of JNK. Chem. Biol. 2012, 19 (1), 140–154. 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating G. M. Afatinib: A Review of Its Use in the Treatment of Advanced Non-Small Cell Lung Cancer. Drugs 2014, 74 (2), 207–221. 10.1007/s40265-013-0170-8. [DOI] [PubMed] [Google Scholar]
- Homan K. T.; Larimore K. M.; Elkins J. M.; Szklarz M.; Knapp S.; Tesmer J. J. G. Identification and Structure–Function Analysis of Subfamily Selective G Protein-Coupled Receptor Kinase Inhibitors. ACS Chem. Biol. 2015, 10 (1), 310–319. 10.1021/cb5006323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens K.; Shotwell J. B.; Redman A.; Wilson J. W.; Lei H.; Gerding R.; Patnaik S. Pyrrolopyrimidine Compounds. WO 2010045451 A1, April 22, 2010.
- Homan K. T.; Wu E.; Wilson M. W.; Singh P.; Larsen S. D.; Tesmer J. J. G. Structural and Functional Analysis of G Protein–Coupled Receptor Kinase Inhibition by Paroxetine and a Rationally Designed Analog. Mol. Pharmacol. 2014, 85 (2), 237–248. 10.1124/mol.113.089631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.