

Abstract

In MLL-rearranged cancer cells, disruptor of telomeric silencing 1-like protein (DOT1L) is aberrantly recruited to ectopic loci leading to local hypermethylation of H3K79 and consequently misexpression of leukemogenic genes. A structure-guided optimization of a HTS hit led to the discovery of DOT1L inhibitors with subnanomolar potency, allowing testing of the therapeutic principle of DOT1L inhibition in a preclinical mouse tumor xenograft model. Compounds displaying good exposure in mouse and nanomolar inhibition of target gene expression in cells were obtained and tested in vivo.
Keywords: Dot1L, lysine histone methyltransferase, inhibitor, HTS hit, structure-based design
Chromosomal rearrangements of the MLL (MLL1, KMT2A) gene can lead to a variety of fusion proteins that cause an aggressive form of acute leukemia. In these fusion proteins, the N-terminal portion of MLL is preserved, while the C-terminal part is replaced by a portion of another protein, leading to novel oncogenic functionalities.1−4 A key concept in the current model of leukemic transformation by MLL fusions is the aberrant maintenance of a stem cell-like state through persistent expression of genes such as HOXA9 and MEIS1. At least a subset of MLL fusion proteins, including those with the frequently observed fusion partners AF4, AF9, and AF10, depend on DOT1L as a cofactor for driving expression of these critical genes.1−4 DOT1L is the only known histone 3 lysine 79 (H3K79) methyltransferase, and the H3K79me2 mark is broadly associated with active transcription.5 In mouse models of acute lymphoblastic or myeloid leukemia caused by MLL-AF4 or MLL-AF9, aberrantly increased H3K79me2 was detected at target genes of those fusion proteins.6,7 Genetic inactivation of DOT1L led to downregulation of target genes and loss of the leukemic phenotype with minimal effects on global gene expression.7 Accordingly, the DOT1L inhibitor EPZ004777 demonstrated selective activity against MLL-rearranged leukemia models.8 A related, further optimized DOT1L inhibitor named pinometostat (EPZ-5676) was subsequently shown to produce robust antitumor activity against an MV4-11 xenograft model in rats.9 This S-adenosyl methionine (SAM) competitive agent, which carries the adenosyl portion of the SAM cofactor, is not suitable for oral absorption and needs to be administered as a continuous infusion in order to achieve sufficient exposure and sustained target inhibition. Pinometostat is also the only DOT1L inhibitor candidate that has been investigated in clinical trials to date, albeit with limited success.10
We applied several approaches to discover new DOT1L inhibitors that overcome the PK limitations of SAM-related compounds,11−13 including high throughput screening (HTS) with a coupled luminescence-based readout for detection of the methyltransferase product SAH that yielded compound 1 (Table 1), a single micromolar hit (IC50 SPA DOT1L = 8.5 μM). In a similar manner as described for a fragment-based screening hit,13 compound 1 is not binding in the SAM binding pocket but in an adjacent induced pocket formed by engaging the SAM pocket lid loop (Figure 1). Despite no overlap with the SAM cofactor binding pocket, compound 1 and SAM have mutually exclusive binding, leading to an apparent SAM competitive inhibition for this new series.
Table 1. DOT1L Inhibitors 1 to 11.

| compound | R1 | R2 | R3 | X | IC50 SPA DOT1L [μM] |
|---|---|---|---|---|---|
| 1 | –NO2 | phenyl | phenyl | –NH2 | 8.5b |
| 2a | –NO2 | 3-chlorophenyl/2-pyridyl | 2-pyridyl/3-chlorophenyl | –Me | 4.6b |
| 3 | pyrimidin-2-ylamino | 2-pyridyl | 3-chlorophenyl | –Me | 0.27b |
| 4 | pyrimidin-2-ylamino | 3-chlorphenyl | 2-pyridyl | –Me | 22.8b |
| 5 | pyrimidin-2-ylamino | 2-pyridyl | 3-chloro-2-fluorophenyl | –Me | 0.097b |
| 6 | (5-carbamoylpyrimidin-2-yl)amino | 2-pyridyl | 3-chloro-2-fluorophenyl | –Me | 0.0039b |
| 0.0021c | |||||
| 7 | pyrimidin-2-ylamino | 3-fluoro-2-pyridyl | 3-chloro-2-fluorophenyl | –Me | 0.016b |
| 8 | (4,6-dimethoxy-1,3,5-triazin-2-yl)amino | 3-fluoro-2-pyridyl | 3-chloro-2-fluorophenyl | –Me | 0.00059c |
| 9 | (4,6-dimethoxy-1,3,5-triazin-2-yl)amino | 3-chloro-2-pyridyl | 2,2-difluorobenz[d][1,3]dioxole-4-yl | –Me | 0.00019c |
| 10 | (4-methoxy-6-(piperazin-1-yl)-1,3,5-triazin-2-yl)amino | 3-chloro-2-pyridyl | 2,2-difluorobenz[d][1,3]dioxole-4-yl | –Me | 0.00011c |
| 11 | (4-amino-6-methoxy-1,3,5-triazin-2-yl)amino | 3-chloro-2-pyridyl | 2,2-difluorobenz[d][1,3]dioxole-4-yl | –NH2 | 0.00017c |
Figure 1.
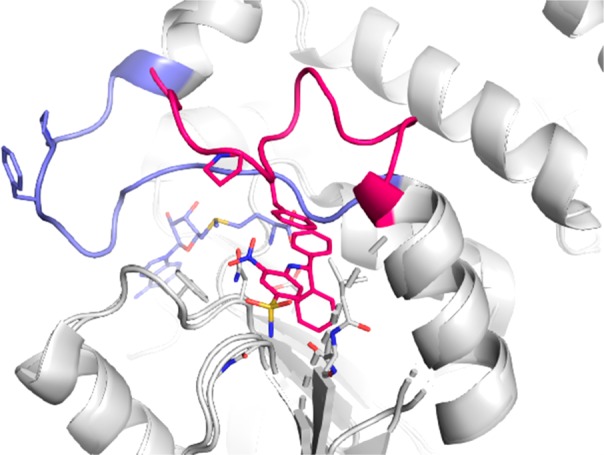
Overlay of X-ray crystal structures of Dot1L with SAM (pdb 1nw3) and 1 (undisclosed structure). Cartoon representation of Dot1L (gray) with the flexible loop 126–140 shown in light blue for SAM and red for 1; stick models of ligands 1 (red) and SAM (blue). The residues Phe129 and Pro130 that form the roof of the induced pocket are shown as sticks.
The sulfonamide group of our ligand is a key contributor to the potency of the ligand by forming three direct hydrogen-bonds with DOT1L, but at the same time it brings a large contribution to its polar surface area and a desolvation penalty upon binding. We envisioned replacing the sulfonamide by the more hydrophobic methylsulfone. The acceptor capability of such compounds is unchanged while the donors are now polarized C–H. Despite being weaker hydrogen-bond donors, they are also paying less energetic contribution to their desolvation. We observed in general a loss of around 3-fold in potency for sulfonamide to methylsulfone match pairs. In order to favor the binding conformation and to desymmetrize the benzhydrylamino moiety, we introduced a nitrogen at the position 2 of one of the phenyl groups to allow an intramolecular hydrogen-bond interaction. Further, we decorated the second phenyl at position 3 with a chloro group in compound 2 (IC50 SPA DOT1L = 4.6 μM) to map the extension of this pocket on one side of the phenyl.
One undesired feature of compound 2 was still the nitro group. It was replaced with a 2-pyrimidinylamino moiety that interacted more efficiently with Asn214 by forming a bidentate hydrogen-bond interaction and inserted between Pro130 and Phe243. At the same time, it opened a new growth vector. The more active (S)-enantiomer compound 3 had submicromolar activity (IC50 SPA DOT1L = 270 nM) and, as one would expect based on the binding mode of this series, the (R)-enantiomer was inactive (compound 4, IC50 SPA DOT1L = 22.8 μM). A key stabilizing interaction of the lid loop rearrangement is the π–π stacking of Phe131 with the central phenyl ring of compound 3. The ligand is engaging three backbone hydrogen-bond interactions with Ser311: one pseudo H-bond with a polarized C–H as well as a dual donor and acceptor interaction with the methylsulfone group. The methylsulfone is also donor for the Ser269 backbone carbonyl. The chlorophenyl is filling a hydrophobic pocket (Leu143, Met147, Val267, Ser269, Tyr311) and the pyridyl a hydrophobic channel (Phe131, Tyr136, S140, Val169) partitioned by Val144, Phe239, and Asn241 (Figure 2).
Figure 2.
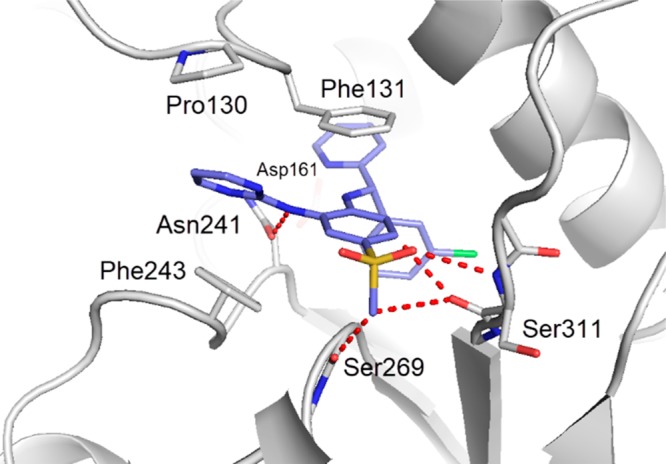
Detailed interactions of 3 with Dot1L (pdb 6TE6). Amino acid side chains engaging in key interactions with the ligand are illustrated as sticks, and polar contacts are highlighted as dotted red lines. The binding mode of 3 is driven by the stacking of aromatic groups and three key polar contacts with Asn241, Ser311 and Ser269. The Asp161 does not engage the ligand. Compared to 1, the pyrimidine moiety of 3 increases the stacking contacts with Phe243 and Phe131-Pro130, and the intramolecular hydrogen bond of the pyridine with the central aniline preorganizes the bound conformation.
One concern with compound 3 was the high clearance measured in rat liver microsomes (Clint = 553 μL × min–1 × mg–1). Adding to the 3-chlorophenyl moiety a fluoro substituent at position 2 led to compound 5 with a 2-fold improvement of potency (IC50 SPA DOT1L = 97 nM) and significant reduction of intrinsic clearance (Clint = 50 μL × min–1 × mg–1), possibly by steric shielding of the benzylic metabolic weak spot and/or an electronic effect.
A further improvement in potency was achieved by introducing an aminocarbonyl substituent at position 4 of the pyrimidine (compound 6). In addition to its electron withdrawing effect, this group extended the stacking capability of the pyrimidine with Pro130 and Phe243 (IC50 SPA DOT1L = 2.1 nM). This low nanomolar inhibitor of DOT1L was tested in cellular assays11 to assess the ability to inhibit the dimethylation of H3K79 in HeLa cells (ED50 H3K79me2 Elisa = 300 nM) and HOXA9 gene expression in Molm-13 cells (ED50 HOXA9 RGA = 4500 nM). The compound showed a suitable PK profile in mouse and rat with an oral bioavailability of 26% and 36%, respectively, at 3 mg/kg (Tables s1 and s2). At a maximal dose of 280 mg/kg p.o., the blood exposure was above 10 μM for 8 h. Treating mice bearing MV4-11 subcutaneous xenograft tumors for 23 days at a dose of 200 mg/kg twice daily lead to no tumor growth inhibition. While global H3K79 dimethylation levels in tumor were reduced by 50%, no impact on expression of the MLL-AF4 target genes HOXA9 and MEIS1 was observed. We concluded from this experiment that we needed to improve the exposure/potency ratio significantly to reach efficacy after oral dosing.
An increase in potency was achieved by introducing a halogen such as fluorine at position 3 of the pyridyl group (compound 7, IC50 SPA DOT1L = 16 nM). Replacement of the pyrimidine with the more electron deficient triazine to allow extension of the stacking capability by adjunction of two methoxy groups provided a subnanomolar inhibitior in our biochemical assay (compound 8, IC50 SPA DOT1L = 0.59 nM) that translated in potent cell activity (ED50 H3K79me2 Elisa = 43 nM). Further fine-tuning of the van der Waals contacts by replacing the 3-fluoropyridyl group with a 3-chloropyridyl group and the 2-fluoro-3-chlorophenyl group with a 2,2-difluorobenzo[d][1,3]dioxole-4-yl group provided compound 9 (IC50 SPA DOT1L = 0.19 nM, ED50 H3K79me2 Elisa = 12 nM, and ED50 HOXA9 RGA = 170 nM). While both compounds 8 and 9 showed an excellent blood exposure after a single dose, multiple dosing uncovered an issue of cytochrome P450 (Cyp450) 3A4 isoform induction leading to reduced exposure over time (e.g., compound 8 at 100 mg/kg: AUC0–12 = 216 h × μM at day 1 vs AUC0–12 = 47 h × μM at day 20, Figure 3). Induction was confirmed by rodent and human Cyp450 hepatocyte functional assays.
Figure 3.
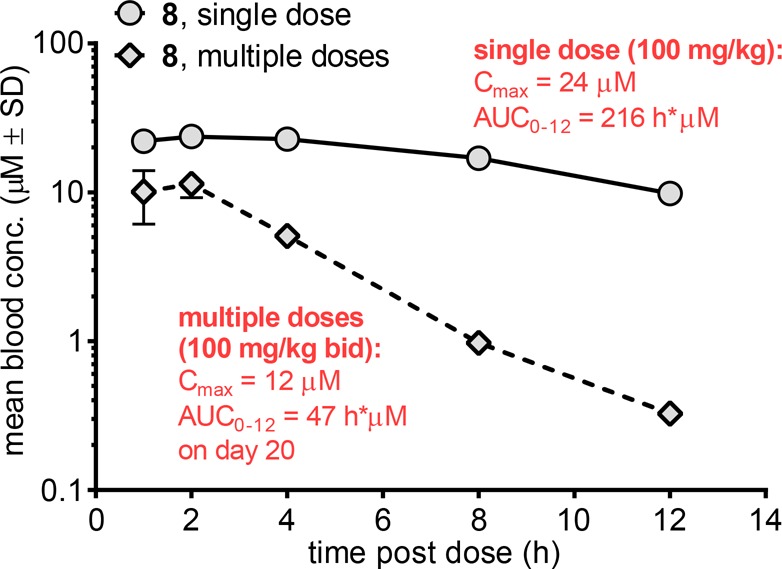
Mouse PK single vs multiple doses of 8 indicative of a cytochrome P450 induction.
Compound 9 was modified by replacing one methoxy group with an ionizable piperazine that conferred a logD (pH 7.4) decrease superior to 1.4 and a further increase in potency (compound 10, IC50 SPA DOT1L = 0.11 nM, ED50 H3K79me2 Elisa = 1.7 nM and ED50 HOXA9 RGA = 33 nM; logDpH 7.4 = 3.3). Cocrystallization of compound 10 showed that the binding mode is similar to the one of compound 3 with optimized van der Waals contacts and extended stacking between Phe243 and Pro130. The 2,2-difluorobenzo[d][1,3]dioxole-4-yl group is shapewise nicely complementary to the hydrophobic pocket. However, the hydrogen-bond pattern with Asn241 was altered in comparison to compound 3 with only the NH donor at adequate distance to form an efficient hydrogen-bond interaction with the Asn241 carbonyl (Figure 4 A). Compound 10 was shown to interact with DOT1L by SPR with slow kinetics (t1/2 > 5 h). Selectivity testing for compound 10 against a limited panel of histone methyl transferases showed no inhibition below 5 μM (Table S4).
Figure 4.
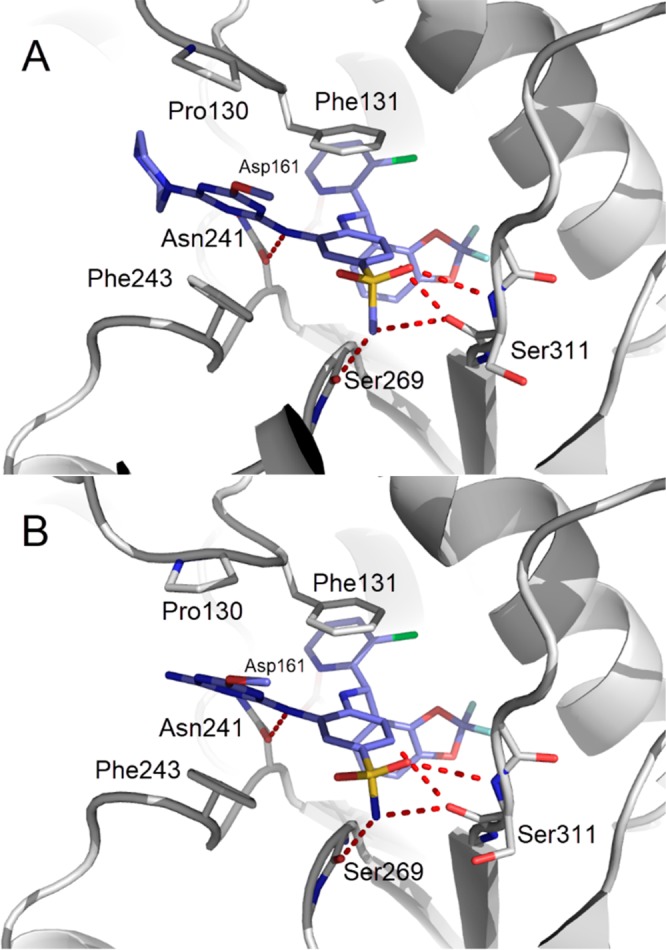
(A) Detailed interactions of 10 with Dot1L (pdb 6TEL). Amino acid side chains engaging in key interactions with the ligand are illustrated as sticks, polar contacts are highlighted as dotted red lines. Compared to 3, the piperazine triazine moiety of 10 increases the stacking contacts with Phe243 and Phe129-Pro130 and the hydrophobic pocket of the benzhydrylamino moiety is filled more efficiently. (B) Detailed interactions of 11 with Dot1L (pdb 6TEN). Amino acid side chains engaging in key interactions with the ligand are illustrated as sticks, and polar contacts are highlighted as dotted red lines. Compared to 10, the amino methoxy triazine moiety of 11 makes efficient stacking contacts with Phe243 and Phe129-Pro130 and the reintroduction of the sulfonamide as hydrogen bond donor shortens the ligand distance with Ser269 and Ser311 carbonyl groups.
Compound 10 was formulated as solution (Solutol SH15/saline 10/90%) and could be administered at 300 mg/kg allowing to reach 4 to 8 μM in blood over 24 h after a single administration and no loss of exposure upon multiple daily administrations, but rather the expected accumulation (Figure s1). Unfortunately, this compound was not tolerated at such a high dose by tumor xenograft bearing mice, and at a 6-fold reduced dose, the tumor growth as well as the HOXA9 reporter gene mRNA were reduced only by less than half as compared to control animals.
CYP450 3A4 isoform induction of compound 9 could also be mitigated by restoring the initial sulfonamide instead of the methylsulfonyl group (Figure 4B) and by changing one methoxy group for an amino group (compound 11, IC50 SPA DOT1L = 0.17 nM, ED50 H3K79me2 Elisa = 2.9 nM and ED50 HOXA9 RGA = 30 nM; log DpH 7.4 = 3.5). Similarly to compound 10, the logD was decreased as compared to compound 9. However, with a substantially increased PSA and hydrogen bond donor number, the compound lost its oral bioavailability and had to be administered subcutaneously as a solution (20% Kolliphor HS 15/80% saline by volume). At a dose of 50 mg/kg twice daily, exposure was maintained after 7 days. Mice bearing subcutaneous MV4-11 tumor xenografts were treated for 20 days at 75 mg/kg once or twice daily by subcutaneous injection. While once daily administration did not lead to tumor growth inhibition, 73% growth inhibition was measured in the twice daily treated group (Figure 5A) with only temporary body weight loss in the beginning of treatment (Figure 5B). While both treatment regimens resulted in very strong inhibition of global H3K79 dimethylation level in tumor (Figure 5C), the efficacious regimen was superior in reducing the mRNA expression of the target genes HOXA9 (25% in qd regimen vs 53% in bid regimen) and MEIS1 (53% vs 74%) (Figure 5D)
Figure 5.
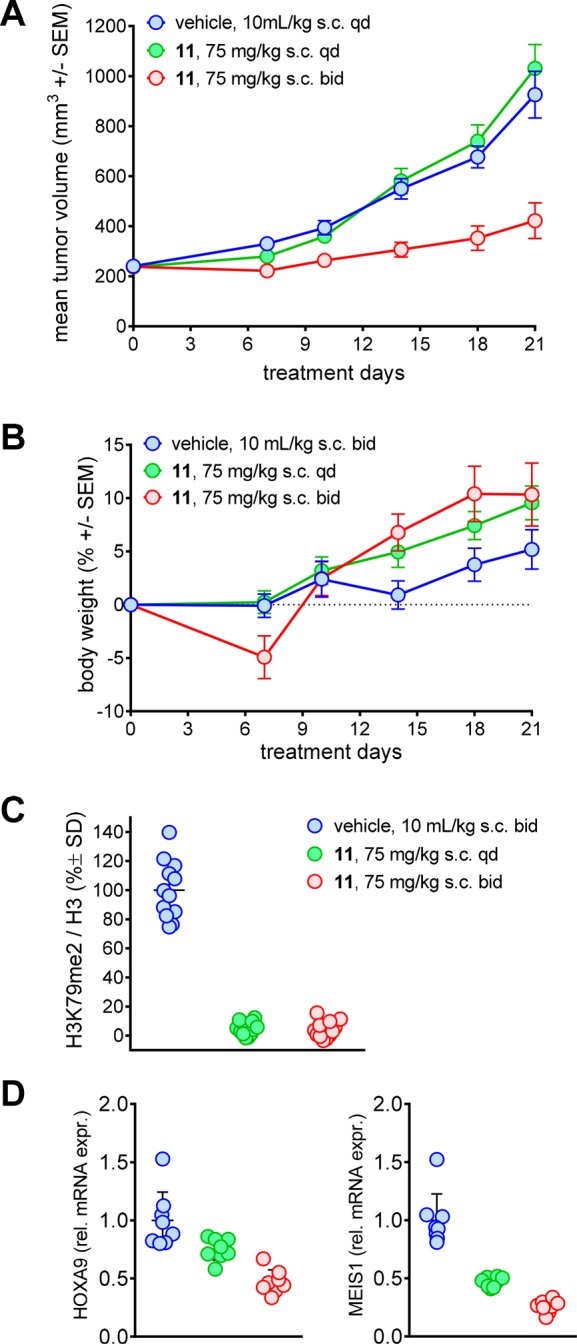
(A) Volume of MV4-11 xenograft tumors upon treatment with 11 or vehicle control. SEM, standard error of the mean; s.c., subcutaneous injection; qd, once daily; bid, twice daily. (B) Body weight of xenograft-bearing NOD-SCID mice. (C) Global H3K79me2 normalized by total H3, measured by ELISA. All values as percent of vehicle mean. SD, standard deviation. (D) Relative mRNA expression of HOXA9 or MEIS1 determined by qPCR. All values as fold vehicle mean.
Compound 11 was synthesized starting with the condensation of the commercially available 2,2-difluorobenzo[d][1,3]dioxole-4-carbaldehyde with (R)-2-methylpropane-2-sulfinamide catalyzed by tetraethoxytitanium (Scheme 1). Commercially available 2-bromo-3-chloro-pyridine was reacted with isopropylmagnesium bromide to generate the Grignard reagent to stereoselectively14 engage with intermediate 12 to yield after careful flash chromatography the desired diastereoisomer 13. After acid-catalyzed deprotection, amine 14 was reacted in an aromatic nucleophilic substitution with the commercially available 4-fluoro-3-nitrobenzenesulfonamide. Reduction of the nitro group of intermediate 15 generated the aniline used to selectively displace the first chloro substituent of the commercially available dichloro-methoxytriazine to provide intermediate 16 that was submitted to a final aromatic nucleophilic substitution with ammonia to yield compound 11.
Scheme 1. Synthetic Route to Compounds 11.

Reagents and conditions: (a) (R)-2-methylpropane-2-sulfinamide, Ti(OEt)4, THF, 45 °C, 2 h, yield 92%; (b) (i) 2-bromo-3-chloropyridine, iPrMgBr, THF, −55 °C to rt, 2 h; (ii) to 12, −50 °C to rt, toluene, 1 h, yield 71%; (c) 2 M HCl, MeOH/dioxane, rt, 1 h, yield 85%; (d) 4-fluoro-3-nitrobenzenesulfonamide, DMA, 50 °C, 6 h, yield 97%; (e) (i) 1.1 bar H2, Ra–Ni, MeOH/THF, rt, 15 h; (ii) 2,4-dichloro-6-methoxy-1,3,5-triazine, 2,6-lutidine, THF, 0 °C, 0.5 h then rt, 5 h, yield quantitative; (f) 7 M NH3, MeOH, rt, 4.25 h, yield 52%.
In conclusion, we progressed toward the aspired goal of making DOT1L inhibitors with improved rodent PK properties compared to EPZ-5676.15 Further optimization is still required to generate a viable clinical candidate from the series described here. The limited efficacy obtained with compound 11 illustrates the difficulty to achieve a sustained and very profound level of DOT1L inhibiton in vivo needed in order to effectively suppress MLL-fusion tumor growth. When administering EPZ-5676 subcutaneously at or slightly beyond the maximally tolerated dose in the same tumor model, no antitumor efficacy was obtained (data not shown).
Subcutaneous administration of compound 11 was adequately delivering a steady concentration of inhibitor in mice that allowed exploring a maximally tolerated dose that led to deep and sustained target inhibition. However, even this level of DOT1L inhibition was not able to fully control growth of the MV4-11 tumor model. The apparent disconnect between substantial reduction of global H3K79 dimethylation and less marked downstream pharmacodynamic responses, namely target gene suppression and leukemic cell death or differentiation, underlines the need for extreme target inhibition to achieve the desired functional consequences. With the better understanding of MLL-rearranged leukemia, alternative treatment strategies are emerging.16 Even if more potent DOT1L inhibitors can be discovered, we expect that combination treatment will be needed in order to fully exploit the potential of DOT1L inhibition in mixed lineage leukemia and achieve substantial clinical benefit in a large proportion of patients.
Acknowledgments
The authors thank Flavia Adler, Céline Be, Alexandra Buhles, Patrick Graff, Aude Izaac, Julia Klopp, Elke Koch, Corinne Marx, Kerstin Pollehn, Paul Westwood, and Aurelie Winterhalter for excellent technical assistance, and Anton Kessler, Martin Klumpp and Kim Twesten for HTS assay.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00452.
PK compound 10 Figure S1, selectivity HMT panel Table S4, summary of reported activities, HTS assay principle, and experimental procedures (PDF)
The authors declare the following competing financial interest(s): Authors are shareholders of Novartis and/or employees of Novartis.
Notes
PyMol was used for structural visualization and figure preparation.17
Supplementary Material
References
- Slany R. K. The molecular biology of mixed lineage leukemia. Haematologica 2009, 94, 984–993. 10.3324/haematol.2008.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntean A. G.; Hess J. L. The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol.: Mech. Dis. 2012, 7, 283–301. 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A. J.; Bradner J.; Armstrong S. A. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012, 33, 563–570. 10.1016/j.it.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov A. V.; Hoshii T.; Armstrong S. A.. Mixed-Lineage Leukemia Fusions and Chromatin in Leukemia. Cold Spring Harbor Perspect. Med. 2017, 7, a026658. 10.1101/cshperspect.a026658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood K.; Tellier M.; Murphy S.. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. 10.3390/biom8010011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov A. V.; Feng Z.; Lemieux M. E.; Faber J.; Vempati S.; Sinha A. U.; Xia X.; Jesneck J.; Bracken A. P.; Silverman L. B.; Kutok J. L.; Kung A. L.; Armstrong S. A. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14, 355–368. 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt K. M.; Zhu N.; Sinha A. U.; Vempati S.; Faber J.; Krivtsov A. V.; Feng Z.; Punt N.; Daigle A.; Bullinger L.; Pollock R. M.; Richon V. M.; Kung A. L.; Armstrong S. A. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Majer C. R.; Sneeringer C. J.; Song J.; Johnston L. D.; Scott M. P.; Smith J. J.; Xiao Y.; Jin L.; Kuntz K. W.; Chesworth R.; Moyer M. P.; Bernt K. M.; Tseng J. C.; Kung A. L.; Armstrong S. A.; Copeland R. A.; Richon V. M.; Pollock R. M. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Basavapathruni A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Raimondi A.; Scott M. P.; Waters N. J.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein E. M.; Garcia-Manero G.; Rizzieri D. A.; Tibes R.; Berdeja J. G.; Savona M. R.; Jongen-Lavrenic M.; Altman J. K.; Thomson B.; Blakemore S. J.; Daigle S. R.; Waters N. J.; Suttle A. B.; Clawson A.; Pollock R.; Krivtsov A.; Armstrong S. A.; DiMartino J.; Hedrick E.; Lowenberg B.; Tallman M. S. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. 10.1182/blood-2017-12-818948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Zhu H.; Stauffer F.; Caravatti G.; Vollmer S.; Machauer R.; Holzer P.; Mobitz H.; Scheufler C.; Klumpp M.; Tiedt R.; Beyer K. S.; Calkins K.; Guthy D.; Kiffe M.; Zhang J.; Gaul C. Discovery of Novel Dot1L Inhibitors through a Structure-Based Fragmentation Approach. ACS Med. Chem. Lett. 2016, 7, 735–740. 10.1021/acsmedchemlett.6b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobitz H.; Machauer R.; Holzer P.; Vaupel A.; Stauffer F.; Ragot C.; Caravatti G.; Scheufler C.; Fernandez C.; Hommel U.; Tiedt R.; Beyer K. S.; Chen C.; Zhu H.; Gaul C. Discovery of Potent, Selective, and Structurally Novel Dot1L Inhibitors by a Fragment Linking Approach. ACS Med. Chem. Lett. 2017, 8, 338–343. 10.1021/acsmedchemlett.6b00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C.; Mobitz H.; Gaul C.; Ragot C.; Be C.; Fernandez C.; Beyer K. S.; Tiedt R.; Stauffer F. Optimization of a Fragment-Based Screening Hit toward Potent DOT1L Inhibitors Interacting in an Induced Binding Pocket. ACS Med. Chem. Lett. 2016, 7, 730–734. 10.1021/acsmedchemlett.6b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plobeck N.; Powell D. Asymmetric synthesis of diarylmethylamines by diastereoselective addition of organometallic reagents to chiral N-tert-butanesulfinimines: switchover of diastereofacial selectivity. Tetrahedron: Asymmetry 2002, 13, 303–310. 10.1016/S0957-4166(02)00099-X. [DOI] [Google Scholar]
- Basavapathruni A.; Olhava E. J.; Daigle S. R.; Therkelsen C. A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Faimondi A.; Porter Scott M.; Dovletoglou A.; Richon V. M.; Pollock R. M.; Copeland R. A.; Moyer M. P.; Chesworth R.; Pearson P. G.; Waters N. J. Nonclinical pharmacokinetics and metabolism of EPZ-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm. Drug Dispos. 2014, 35, 237–252. 10.1002/bdd.1889. [DOI] [PubMed] [Google Scholar]
- Steinhilber D.; Marschalek R. How to effectively treat acute leukemia patients bearing MLL-rearrangements. Biochem. Pharmacol. 2018, 147, 183–190. 10.1016/j.bcp.2017.09.007. [DOI] [PubMed] [Google Scholar]
- PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.