

Abstract
Background
The X-linked form of Charcot-Marie-Tooth disease type 1 (CMTX1) is an inherited peripheral neuropathy that arises in patients with mutations in the gap-junction beta-1 gene (GJB1).
Case presentation
Three young male patients from Southern China with pes cavus experienced multiple episodes of transient central nervous system (CNS) dysfunction. Three patients all had reversible posterior leukoencephalopathy as detected by brain diffusion-weighted magnetic resonance imaging (MRI-DWI). Nerve conduction velocity (NCV) showed sensorimotor polyneuropathy with mixed demyelinating and axonal features. Genetic testing indicated a c.425G > A (p.Arg142Glu) or c.563 C > T (p.Thr188Ile) or c.103G > C (p.Val35Leu) mutation in GJB1. The unique feature of this report is the identification of two novel mutations: c.563 C > T and sc.103G > C of the GJB1 gene detected in two families respectively. Another unique feature is that peripheral neuropathy symptoms in the three patients were insidious and found at the onset of CNS symptoms.
Conclusions
Posterior leukoencephalopathy is involved in CMTX1 patients. The white matter changes in MRI of CMTX1 patients are reversible and recover later than CNS symptoms.
Keywords: X-linked form of Charcot-Marie-tooth disease type 1, Gap-junction beta-1 gene, Reversible posterior leukoencephalopathy
Background
The X-linked form of Charcot-Marie-Tooth disease type 1 (CMTX1) is an inherited peripheral neuropathy that arises in patients with mutations in the gap-junction beta-1 gene (GJB1). GJB1 encodes the transmembrane channel protein, connexin 32 (Cx32). Cx32 has been found in myelinating Schwann cells, oligodendrocytes, and astrocytes; it is believed to develop intracellular channels between adjacent myelin loops to form a pathway for small molecules or ions across the myelin sheath [1].
Here, we describe three patients from three families from South China with CMTX1 who experienced multiple episodes of transient central nervous system (CNS) dysfunction associated with reversible posterior leukoencephalopathy; this was confirmed by family history, brain magnetic resonance images (MRI), nerve conduction velocity (NCV), and GJB1 mutations. Two novel mutations of GJB1 gene were detected in our patients.
Case presentation
Family 1
The proband, a 17-year-old male, was admitted to our hospital on July 25, 2018 after experiencing acute onset of profound dysarthria, chorea-choreiform movements, and confusion. His abnormal movements and confusion resolved over the course of several hours, but slurred speech remained. In the following two days, he experienced two episodes of similar symptoms and recovered after a few hours, but no triggering factors were identified.
A review of the patient’s past records revealed that he had experienced an episode of weakness in all four limbs at the age of 12. The episode was of sudden onset and resolved completely over the course of three hours without special treatment. He was evaluated at once with head CT scanning and no abnormalities were found in the first episode.
His physical examination was notable for his pes cavus when he arrived in our department. A detailed pedigree of other family members revealed that the patient’s mother, maternal grandfather, three maternal aunts, and other relatives also had pes cavus deformities, although none reported sudden onset of weakness or dysarthria episodes.
His neurological examination revealed the atrophy of his distal lower extremities, mild weakness of ankle dorsi- and plantar-flexion, absent deep tendon reflexes in the lower extremities, and negative bilateral Babinski signs.
Polymerase chain reaction (PCR) for Coxsackievirus IgG was positive in the serum but negative for Coxsackievirus IgM. The rest of the routine serum analyses were within the normal range. A serum lactic acid exercise test was applied to exclude mitochondrial encephalomyopathy. Serum lactic acid before exercise was 3.49 mmol/L (normal value: 0.63~2.44 mmol/L), 3.73 mmol/L after exercise, and 6.75 mmol/L 30 min after exercise. Cell count, protein, glucose, and chloride levels were normal in the cerebrospinal fluid (CSF). CSF and the serum were negative for antibodies against AMPAR1 and AMPAR2, NMDAR, GABABR, LGI1, and Caspr2. A wide range of abnormalities in slow waves was found on the electroencephalograms (EEG). A brain diffusion-weighted magnetic resonance imaging (DWI) obtained on July 24, 2018 showed hyperintensities in the splenium of the corpus callosum and posterior subcortical white matter (Fig. 1-a). MR angiography (MRA) and MR spectroscopy (MRS) were normal. Two weeks later, a second MRI performed on August 6, 2018 showed only minor white matter lesions in the splenium of the corpus callosum and posterior periventricular areas. MRS was normal (Fig. 1-b). NCV data demonstrated a moderate to severe sensorimotor polyneuropathy with mixed demyelinating and axonal features at both upper and lower extremities. Genetic testing showed a c.425G > A (p.Arg142Glu) hemizygous point mutation in GJB1. His mother, older maternal aunt and her daughter, as well as a younger maternal aunt, who all had pes cavus also had the same point mutations in GJB1 (Fig. 2). This mutation c.425G > A (p.Arg142Glu) in the GJB1 gene was classified as “Likely pathogenic”according to the criteria of the American College of Medical Genetics and Genomics (ACMG) standards and guidelines. CMTX1 coexistence with reversible posterior leukoencephalopathy was thus diagnosed by his family history, brain MRI, NCV data, and GJB1 mutation.
Fig. 1.

Brain magnetic resonance images of patient 1. a: MRI on 24 July 2018: Diffuse white matter lesions in posterior subcortical areas and the splenium of the corpus callosum. MRA was normal. b: MRI on 6 August 2018: the demyelinating changes of white matter had largely disappeared. MRS was normal
Fig. 2.

Pedigree of Family 1. The proband, his mother, older maternal aunt and her daughter as well as younger maternal aunt who have pes cavus deformities have a c.425G > A (p.Arg142Glu) hemizygous point mutation in the GJB1 gene
Intravenous methylprednisolone (500 mg/day for 3 days) followed by oral prednisolone (1 mg/kg/day) was administered because acute demyelinative encephalitis was diagnosed at arrival. Prednisolone was tapered when reversible posterior leukoencephalopathy was confirmed. The patient’s slurred speech improved without episode onset of chorea-choreiform movements and confusion when he was discharged on August 7, 2018. He had recovered to his baseline and symptoms of peripheral neuropathy remained at a 3-month outpatient follow-up.
Family 2
The proband, a 15-year-old male, presented with acute onset of dysarthria, weakness, and numbness in all four limbs on February 16, 2018. In 2 h, his symptoms had improved but he experienced another two episodes with similar symptoms and recovered after a few hours on the next day. The initial brain MRI DWI on February 17, 2018 showed hyperintensities in both posterior periventricular areas and subcortical white matter of occipital and parietal lobe (Fig. 3-a); MRA and MRS were normal. A possible diagnosis of adrenoleukodystrophy was made when he was admitted to our hospital on March 3, 2018. His physical examination was normal except for pes cavus and diminished deep tendon reflexes in all extremities. The patient’s mother, maternal grandfather, aunt and her daughter also had pes cavus (Fig. 4). His routine serum and CSF analyses were within normal range. The serum of very-long-chain fatty acids, adrenal cortical hormones, and adrenal gland CT with enhancement were checked to exclude adrenoleukodystrophy and all were normal. The findings in EEGs were unremarkable. NCV indicated slight axonal sensorimotor neuropathy at lower extremities. Eighteen days later, a second MRI performed on March 6, 2018 showed normal findings (Fig. 3-b). Whole-exome sequencing of this patient showed a c.563 C > T (p.Thr188Ile) hemizygous point mutation in GJB1. This mutation c.563 C > T (p.Thr188Ile) in the GJB1 gene was classified as “Uncertain”according to the criteria of the ACMG standards and guidelines. It has not been described previously based on the Human Gene Mutation Database (HGMD). Unfortunately, his family members refused genetic testing. The typical clinical presentations and continuous MRI findings as well as this variation not identified in 500 control subjects supported the hypothesis that the only mutation c.563 C > T (p.Thr188Ile) with whole-exome sequencing may be pathogenic.
Fig. 3.
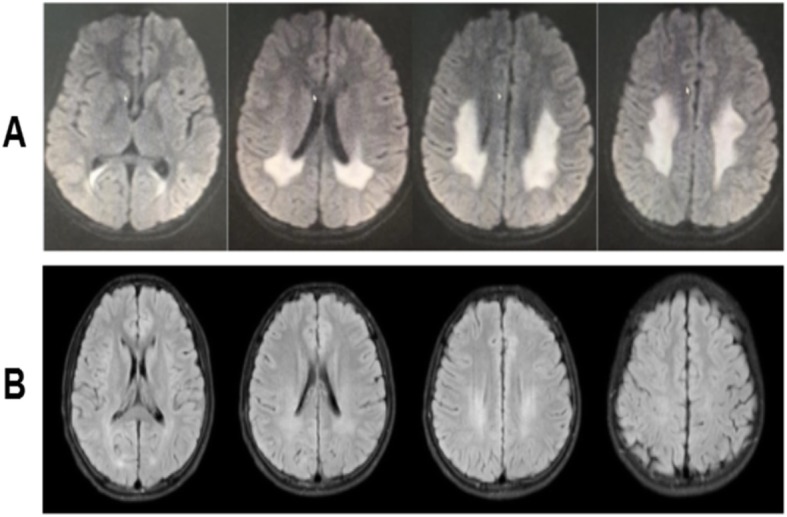
Brain MRI of patient 2. a: MRI on 17 February 2018 indicated white matter lesions in bilateral posterior ventricular areas; b: White matter lesions nearly complete resolution on 6 March 2018. MRS was normal
Fig. 4.
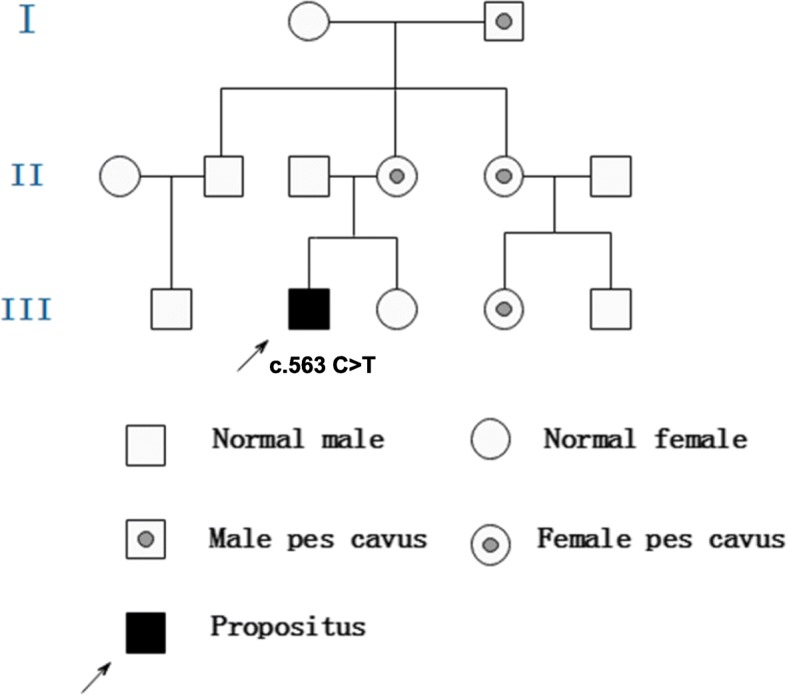
Pedigree of Family 2. The proband has a c.563 C > T (p.Thr188Ile) hemizygous point mutation in the GJB1 gene
He had recovered to his baseline without episodes of dysarthria, weakness, and numbness with no special treatment when he was discharged on March 13, 2018. He presented normal at a 3-month outpatient follow-up.
Family 3
The proband, an 18-year-old male, presented with acute onset of left arm weakness on January 24, 2019. Within 20 h, his symptoms had gradually improved, but he experienced new symptoms as dizziness, weakness and numbness in all four limbs, difficulties in raising his head and opening his mouth, dysarthria, and dysphagia with normal CT scanning on 11 AM, January 25, 2019 and recovered after treatment in a local hospital in a few hours. He had another two episodes with similar symptoms and recovered in a few hours on January 25 and 26. The brain MRI DWI on January 26, 2019 indicated hyperintensities in both posterior periventricular areas (Fig. 5-a). He was admitted to our hospital with diagnosis of adrenoleukodystrophy on January 29, 2019. He fell from a 2-m height in the middle of December, 2018 and was treated in the hospital with diagnosis of abdominal injury, spleen and left kidney contusion, and a bilateral lumbar transverse process fracture. His physical examination was normal except for diminished deep tendon reflexes in all extremities.
Fig. 5.

Brain MRI of patient 3. a: MRI on January 26, 2019 showed white matter lesions in bilateral posterior ventricular areas and the splenium of the corpus callosum; b: MRI on February 11, 2019 showed the abnormal signal almost complete resolution; c: MRI on February 26, 2019 showed no obvious abnormality
His neurological examination revealed pes cavus, horizontal nystagmus on both eyes, atrophy in his hands and distal lower extremities, normal muscle strength, and absent deep tendon reflexes in all extremities. The finger-nose test and heel–knee test showed mild dysmetria on right limbs when closing eyes. The Romberg sign was positive when closing eyes. NCV findings were suggestive of sensorimotor polyneuropathy at both upper and lower extremities. Genetic testing showed a c.103G > C (p.Val35Leu) hemizygous point mutation in GJB1. His mother, younger maternal aunt and grandfather’s brother who had pes cavus also had the same point mutations in GJB1 (Fig. 6). The pathogenicity of c.103G > C (p.Val35Leu) mutation in the GJB1 gene was classified as “Uncertain”according to the criteria of the ACMG standards and guidelines. Based on the typical clinical and electrophysiological presentations, mutation on the X chromosome identified in the proband and his mother, but not in his father which matches the X-linked recessive inheritance disease pattern, as well as this mutation not found in 500 control subjects, the c.103G > C (p.Val35Leu) mutation may be pathogenic even though it has not been described previously based on the HGMD.
Fig. 6.

Pedigree of Family 3. The proband, his mother, yonger maternal aunt and maternal grandfather who have pes cavus deformities have a c.103G > C (p.Val35Leu) hemizygous point mutation in the GJB1 gene
He had recovered to his baseline without special treatment when he was discharged on January 31, 2019. He presented a normal brain MRI DWI within 2 weeks (on February 11, 2019, Fig. 5-b) and within one month (February 26, 2019, Fig. 5-C) during outpatient follow-up.
Electrophysiological characteristics in these three male probands were shown in Tables 1 and 2. Motor nerve conduction velocities (MNCV) and sensory nerve conduction velocities (SNCV) were severely reduced in lower limbs as well as upper limbs in case 1 and case 3; however, MNCV and SNCV in case 2 were almost within normal range. Markedly low amplitude of sensory and motor responses was found at both peroneal nerves in case 1 and case 3. Long latency was detected in most motor nerves in the three cases.
Table 1.
Electrophysiological findings in lower limbs of three probands with CMTX
| Case1 | Case2 | Case3 | ||
|---|---|---|---|---|
| Peroneal. L | ||||
| Conduction velocity | M | 25 m/s(>40) | 39 m/s(>40) | 30 m/s(>40) |
| S | NA | 50 m/s(>40) | NA | |
| Amplitude | M | 0.1 mV(>5) | 2.2 mV(>5) | 0.3 mV(>5) |
| S | NR | 4.2 μV(>5) | NR | |
| Latency | M | 5.4 ms(<3.2) | 4.6 ms(<3.2) | 4.9 ms(<3.2) |
| Peroneal. R | ||||
| Conduction velocity | M | NA | 48 m/s(>40) | NA |
| S | NA | 49 m/s(>40) | NA | |
| Amplitude | M | NR | 1.6 mV(>5) | NR |
| S | NR | 3.5 μV(>5) | NR | |
| Latency | M | NR | 4.2 ms(<3.2) | NR |
| Tibial. L | ||||
| Conduction velocity | M | 35 m/s(>40) | 45 m/s(>40) | 24 m/s(>40) |
| S | NA | 39 m/s(>40) | NA | |
| Amplitude | M | 0.5 mV(>5) | 6.0 mV(>5) | 0.3 mV(>5) |
| S | NR | 4.4 μV(>5) | NR | |
| Latency | M | 3.9 ms(<3.2) | 4.4 ms(<3.2) | 10.1 ms(<3.2) |
| Tibial. R | ||||
| Conduction velocity | M | 31 m/s(>40) | 49 m/s(>40) | 23 m/s(>40) |
| S | 33 m/s(>40) | 41 m/s(>40) | NA | |
| Amplitude | M | 0.3 mV(>5) | 6.6 mV(>5) | 0.5 mV(>5) |
| S | 3.1 μV(>5) | 4.0 μV(>5) | NR | |
| Latency | M | 6.3 ms(<3.2) | 5.8 ms(<3.2) | 10.1 ms(<3.2) |
M Motor nerve, S Sensory nerve, NA Not applicable, NR Not recordable, (): normal values
Table 2.
Electrophysiological findings in upper limbs of three probands with CMTX
| Case1 | Case2 | Case3 | ||
|---|---|---|---|---|
| Median. L | ||||
| Conduction velocity | M | 37 m/s(>45) | 58 m/s(>45) | 35 m/s(>45) |
| S | 38 m/s(>45) | 50 m/s(>45) | NA | |
| Amplitude | M | 9.6 mV(>5) | 5.1 mV(>5) | 2.9 mV(>5) |
| S | 2.0 μV(>15) | 9.9 μV(>15) | NR | |
| Latency | M | 4.4 ms(<3) | 3.6 ms(<3) | 5.8 ms(<3) |
| Median. R | ||||
| Conduction velocity | M | 27 m/s(>45) | 53 m/s(>45) | 35 m/s(>45) |
| S | 41 m/s(>45) | 44 m/s(>45) | NA | |
| Amplitude | M | 4.2 mV(>5) | 9.4 mV(>5) | 2.1 mV(>5) |
| S | 2.9 μV(>15) | 19.8 μV(>15) | NR | |
| Latency | M | 4.5 ms(<3) | 3.8 ms(<3) | 6.8 ms(<3) |
| Ulnar. L | ||||
| Conduction velocity | M | 37 m/s(>45) | 60 m/s(>45) | 36 m/s(>45) |
| S | NA | 52 m/s(>45) | NA | |
| Amplitude | M | 4.6 mV(>5) | 5.4 mV(>5) | 3.9 mV(>5) |
| S | NR | 7.9 μV(>5) | NR | |
| Latency | M | 3.6 ms(<3) | 2.4 ms(<3) | 5.6 ms(<3) |
| Ulnar. R | ||||
| VConduction velocity | M | 36 m/s(>45) | 57 m/s(>45) | 30 m/s(>45) |
| S | 36 m/s(>45) | 48 m/s(>45) | NA | |
| Amplitude | M | 2.7 mV(>5) | 5.7 mV(>5) | 4.0 mV(>5) |
| S | 5.7 μV(>5) | 7.4 μV(>5) | NR | |
| Latency | M | 3.5 ms(<3) | 2.8 ms(<3) | 5.2 ms(<3) |
M Motor nerve, S Sensory nerve, NA Not applicable, NR Not recordable, (): normal values
Discussion
To date, over 30 cases of white matter lesion involvement in patients with Charcot-Marie-Tooth disease and 22 GJB1 gene mutations have been described [2–18]. Most these cases, as well as our cases, have similar clinical features: 1. Young males with onset age at 10–20 years; however, a female patient has been reported in previous literature. Males were more severely clinically affected and had slower MNCVs than females [3]; 2. Patients and their maternal female relatives having pes cavus deformities with X-linked dominant inheritance; 3. Experiences of recurrent and transient episodes of CNS symptoms which recover after a few hours or days. There are also diffuse hyperintense lesions in the periventricular areas and corpus callosum as well as deep cerebral white matter with a posterior predominance found in T2WI or DWI. These signal abnormalities largely disappeared in a few weeks or months; 4. NCV data show mixed demyelinating and axonal sensorimotor neuropathy; 5. Genetic testing identifies a hemizygous point mutation in GJB1; 6. Acute fulminant CNS dysfunction typically triggered by conditions of systemic inflammation and metabolic stress, such as febrile illness, returning from high altitudes, intense exercise, hyperventilation, and concussion or trauma were found in some cases [18–20]. Positive Coxsackievirus IgG and negative IgM in the serum indicate past and not a recent Coxsackievirus infection in our case 1. This fact indicates viral infection may not be one of the triggering factors in case 1; trauma and following surgeries may trigger CNS lesions in our case 3; 7. Good outcome of CNS lesions in most cases.
Connexin32 is expressed by not only Schwann cells in peripheral nerves, but also by myelinating oligodendrocytes and astrocytes in the central nervous system [1]. Interruption of the gap junction-mediated coupling between oligodendrocytes and astrocytes likely causes an inability of these cells to properly regulate ion communication and fluid exchange, which may explain the restricted diffusion seen on the MRI of the patient with GJB1 gene mutations [21]. The possible mechanisms underlying reversible posterior leukoencephalopathy are myelin splitting and intra-myelin edema, with compression of the extracellular spaces. This is because reduction of apparent diffusion coefficient values (MRI-ADC) in white matter might reverse after a few months and cytotoxic edema usually lasts less than 2 weeks [22].
The interesting and unique feature of this present report is the identification of two novel mutations in GJB1, which were detected in Family 2: c.563 C > T (p.Thr188Ile) and Family 3: c.103G > C (p.Val35Leu) according to the HGMD. The phenotypes present in the three probands in current study are similar but severity is different. The proband in case 1 and 3 with more severe symptoms compared with case 2. There may be due to their different genotypes [23].
Another unique and interesting feature of this present report is that the peripheral neuropathy in our patients was insidious and only found at onset of CNS symptoms. This is a common reason for initial misdiagnosis. The initial diagnosis of patient 1 was mitochondrial encephalomyopathy according to his CNS symptoms, brain MRI, and elevated serum lactic acid level after exercise. In the case of patient 2, extensive investigations were performed to exclude adrenoleukodystrophy.
The time point relation between CNS symptoms and lesions in the MRI is also interesting. In patient 1, the second MRI in twelve days showed that only minor white matter lesions remained when his CNS symptoms diminished after 10 days. The second MRI for patient 2 was normal when his CNS symptoms relived in eighteen days. For patient 3, the second and third MRI after two weeks and one month were all normal. A closer MRI scanning may be helpful to further explore this relationship.
The limitation of this study is that the three cases lack nerve biopsy data because the patients refused invasive examinations.
In conclusion, 3 CMTX1 cases with recurrent episodes of a reversible posterior leukoencephalopathy with a c.425G > A (p.Arg142Glu) or c.563 C > T (p.Thr188Ile) or c.103G > C (p.Val35Leu) mutation in the GJB1 gene were presented in this report. The unique features of this study are the identification of two novel mutations: c.563 C > T and c.103G > C of the GJB1 gene, insidious peripheral neuropathy symptoms in the three propands, and the reversible white matter changes in MRI recovering later than CNS symptoms.
Acknowledgments
We gratefully thank the participants and their families.
Abbreviations
- CMTX1
X-linked form of Charcot-Marie-Tooth disease type 1
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- DWI
Diffusion-weighted magnetic resonance imaging
- EEG
Electroencephalograms
- GJB1
Gap-junction beta-1
- MRI
Magnetic resonance image
- NCV
Nerve conduction velocity
Authors’ contributions
YLL, JLL, DBC, YW, LHM and WH were all directly involved in the clinical management of this case. YW was a major contributor in writing the manuscript under the guidance of WH. All authors read and approved the final manuscript.
Funding
Data collection, analysis, and interpretation of the study were supported by grants from the Natural Science Foundation of Guangxi Province (CN) (2017GXNSFAA198042).
Availability of data and materials
Data generated during this study are included in this published article.
Ethics approval and consent to participate
This study was approved by the institutional review board of First Affiliated Hospital, Guangxi Medical University. Written informed consent was obtained from the patients or patient’s parent to participate.
Consent for publication
Written informed consent was obtained from the patients or patient’s parent for publication of this paper and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH. Connexin mutations in X-linked Charcot-Marie-tooth disease. Science. 1993;262(5142):2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 2.Panas M, Karadimas C, Avramopoulos D, Vassilopoulos D. Central nervous system involvement in four patients with Charcot-Marie-tooth disease with connexin 32 extracellular mutations. J Neurol Neurosurg Psychiatry. 1998;65(6):947–948. doi: 10.1136/jnnp.65.6.947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee MJ, Nelson I, Houlden H, Sweeney MG, Hilton-Jones D, Blake J, Wood NW, Reilly MM. Six novel connexin32 (GJB1) mutations in X-linked Charcot-Marie-tooth disease. J Neurol Neurosurg Psychiatry. 2002;73(3):304–306. doi: 10.1136/jnnp.73.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isoardo G, Di Vito N, Nobile M, Benetton G, Fassio F. X-linked Charcot-Marie-tooth disease and progressive-relapsing central demyelinating disease. Neurology. 2005;65(10):1672–1673. doi: 10.1212/01.wnl.0000186032.06791.94. [DOI] [PubMed] [Google Scholar]
- 5.Parman Y, Ciftci F, Poyraz M, Halefoglu AM, Oge AE, Eraksoy M, Saruhan-Direskeneli G, Deymeer F, Battaloglu E. X-linked Charcot-Marie-tooth disease and multiple sclerosis. J Neurol. 2007;254(7):953–955. doi: 10.1007/s00415-006-0324-7. [DOI] [PubMed] [Google Scholar]
- 6.Fusco C, Frattini D, Pisani F, Spaggiari F, Ferlini A, Della Giustina E. Coexistent central and peripheral nervous system involvement in a Charcot-Marie-tooth syndrome X-linked patient. J Child Neurol. 2010;25(6):759–763. doi: 10.1177/0883073809344119. [DOI] [PubMed] [Google Scholar]
- 7.Anand G, Maheshwari N, Roberts D, Padeniya A, Hamilton-Ayers M, van der Knaap M, Fratter C, Jayawant S. X-linked hereditary motor sensory neuropathy (type 1) presenting with a stroke-like episode. Dev Med Child Neurol. 2010;52(7):677–679. doi: 10.1111/j.1469-8749.2010.03674.x. [DOI] [PubMed] [Google Scholar]
- 8.Sato K, Kubo S, Fujii H, Okamoto M, Takahashi K, Takamatsu K, Tanaka A, Kuriyama M. Diffusion tensor imaging and magnetic resonance spectroscopy of transient cerebral white matter lesions in X-linked Charcot-Marie-tooth disease. J Neurol Sci. 2012;316(1–2):178–180. doi: 10.1016/j.jns.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 9.Karadima G, Koutsis G, Raftopoulou M, Floroskufi P, Karletidi KM, Panas M. Four novel connexin 32 mutations in X-linked Charcot-Marie-tooth disease. Phenotypic variability and central nervous system involvement. J Neurol Sci. 2014;341(1–2):158–161. doi: 10.1016/j.jns.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Xie Y, Zhu X, Wang H, Li Y, Li J. Transient, recurrent, white matter lesions in x-linked Charcot-Marie-tooth disease with novel mutation of gap junction protein beta 1 gene in China: a case report. BMC Neurol. 2014;14:156. doi: 10.1186/s12883-014-0156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu N, Said S, Sabat S, Wicklund M, Stahl MC. Recurrent episodes of stroke-like symptoms in a patient with Charcot-Marie-tooth neuropathy X type 1. Case Rep Neurol. 2015;7(3):247–252. doi: 10.1159/000442410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koutsis G, Karadima G, Floroskoufi P, Raftopoulou M, Panas M. Relapsing remitting multiple sclerosis in x-linked charcot-marie-tooth disease with central nervous system involvement. Case Rep Neurol Med. 2015;2015:841897. doi: 10.1155/2015/841897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JK, Han SA, Kim SJ. X-linked Charcot-Marie-tooth disease with GJB1 mutation presenting as acute disseminated encephalomyelitis-like illness: a case report. Medicine. 2017;96(49):e9176. doi: 10.1097/MD.0000000000009176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicholson PD, Pulst SM. Centrally involved X-linked Charcot-Marie-tooth disease presenting as a stroke-mimic. Neurol Genet. 2017;3(1):e128. doi: 10.1212/NXG.0000000000000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu YY, Lyu H, Jin SQ, Zuo YH, Liu J, Wang ZX, Zhang W, Yuan Y. Clinical and genetic features of Chinese X-linked Charcot-Marie-tooth type 1 disease. Chin Med J. 2017;130(9):1049–1054. doi: 10.4103/0366-6999.204925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sagnelli A, Piscosquito G, Chiapparini L, Ciano C, Salsano E, Saveri P, Milani M, Taroni F, Pareyson D. X-linked Charcot-Marie-tooth type 1: stroke-like presentation of a novel GJB1 mutation. J Peripher Nerv Syst. 2014;19(2):183–186. doi: 10.1111/jns5.12070. [DOI] [PubMed] [Google Scholar]
- 17.Hu G, Zhang L, Zhang M, Yang C, Nie X, Xiang F, Chen L, Dong Z, Yu S. Novel gap junction protein beta-1 gene mutation associated with a stroke-like syndrome and central nervous system involvement in patients with X-linked Charcot-Marie-tooth type 1: a case report and literature review. Clin Neurol Neurosurg. 2019;180:68–73. doi: 10.1016/j.clineuro.2019.03.018. [DOI] [PubMed] [Google Scholar]
- 18.Hanemann CO, Bergmann C, Senderek J, Zerres K, Sperfeld AD. Transient, recurrent, white matter lesions in X-linked Charcot-Marie-tooth disease with novel connexin 32 mutation. Arch Neurol. 2003;60(4):605–609. doi: 10.1001/archneur.60.4.605. [DOI] [PubMed] [Google Scholar]
- 19.Taylor RA, Simon EM, Marks HG, Scherer SS. The CNS phenotype of X-linked Charcot-Marie-tooth disease: more than a peripheral problem. Neurology. 2003;61(11):1475–1478. doi: 10.1212/01.WNL.0000095960.48964.25. [DOI] [PubMed] [Google Scholar]
- 20.Paulson HL, Garbern JY, Hoban TF, Krajewski KM, Lewis RA, Fischbeck KH, Grossman RI, Lenkinski R, Kamholz JA, Shy ME. Transient central nervous system white matter abnormality in X-linked Charcot-Marie-tooth disease. Ann Neurol. 2002;52(4):429–434. doi: 10.1002/ana.10305. [DOI] [PubMed] [Google Scholar]
- 21.Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23(13):5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Citton V, Burlina A, Baracchini C, Gallucci M, Catalucci A, Dal Pos S, Burlina A, Manara R. Apparent diffusion coefficient restriction in the white matter: going beyond acute brain territorial ischemia. Insights Imaging. 2012;3(2):155–164. doi: 10.1007/s13244-011-0114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao ZH, Chen ZT, Zhou RL, Wang YZ. A Chinese pedigree with a novel mutation in GJB1 gene and a rare variation in DHTKD1 gene for diverse CharcotMarieTooth diseases. Mol Med Rep. 2019;19(5):4484–4490. doi: 10.3892/mmr.2019.10058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data generated during this study are included in this published article.