

Pulmonary arterial hypertension (PAH) is a life‐threatening disease with poor prognosis. As a subtype of pulmonary hypertension, PAH results from various etiologies. Accordingly, it is mainly divided into idiopathic PAH, heritable PAH, drug‐ and toxin‐induced PAH, and PAH associated with connective tissue diseases; portal hypertension; HIV infection; congenital heart disease; and schistosomiasis.1 PAH is characterized by vascular lumen narrowing attributable to pulmonary vascular remodeling, which leads to increased pulmonary arterial pressure.2, 3 In detail, excessive and persistent pulmonary vasoconstriction results from various factors, stimulating increased pulmonary arterial pressure.4 Furthermore, irreversible vascular remodeling, including pulmonary artery endothelial cell (PAEC) dysfunction, abnormal proliferation and apoptosis resistance of pulmonary arterial smooth muscle cells (PASMCs), extravagant fibroblast proliferation and activation, and immune dysfunction are the main driving factors in PAH progression.3, 5 Together with structural remodeling, pulmonary vascular dysfunction aggravates PAH progression and eventually leads to heart failure and death.
The pathophysiological mechanisms of PAH are complex, involving immunological abnormality and inflammation.6 Recently, it was demonstrated that T cells play a critical role in pulmonary vascular remodeling and the development of PAH.7 Tamosiuniene et al8 suggested that athymic nude rats lacking mature T cells developed more severe pulmonary hypertension than immune‐reconstituted rats, which indicated that T lymphocytes attenuate early inflammation following Sugen 5416–induced vascular injury in a rat model and subsequently protect against the development of PAH.
However, the T‐lymphocyte subsets, which include helper T cells (Th cells), cytotoxic T cells, and regulatory T cells (Tregs), play different roles in PAH. Among the helper T‐cell subtypes, exploration has shown that Th1 and Th17 cells widely cause the inflammatory and autoimmune responses in PAH by producing interleukin‐6, interleukin‐2, interleukin‐21, interferon‐γ, and tumor necrosis factor‐α.9, 10 It has been directly demonstrated that Th17 cells promote the course of PAH in hypoxia‐induced rats via the secretion of interleukin‐17A, while inhibition of Th17 cells significantly attenuated the increased pressure and remodeling response to chronic hypoxia.11 In addition, it has been verified that Th2 cells exacerbate PAH progression. Inhibition of Th2 cells by chemoattractant receptor homologous molecule expressed on Th2 cells could suppress interleukin‐4 and interleukin‐13 secretion to reduce inflammation and thereby alleviate the elevated right ventricular systolic pressure and pulmonary vascular remodeling in rodent PAH models.12 Moreover, Austin et al13 found that patients with idiopathic PAH and heritable PAH had abnormally increased cytotoxic T cells in the peripheral lung tissue, and identified that cytotoxic T cells promote the development of PAH via its potent cytolytic activity to trigger the local inflammatory response. In contrast, Tregs were recently found to contribute to immunosuppressive progress and further ameliorate PAH by regulating pulmonary vascular wall inflammation and inhibiting pulmonary artery remodeling.8
The Treg population and the subtype ratio of T lymphocytes can be considered a critical biomarker of PAH prognosis. Under interleukin‐12, interleukin‐4, interleukin‐6, interleukin‐10, and transforming growth factor‐β (TGF‐β) stimulation, T lymphocytes could differentiate into different subsets including Th1, Th2, Th17, and Tregs, with relative balanced quantities in normal physiological homeostasis.14 Conversely, disproportion of these subsets, especially reduced Tregs and increased Th17 cells, also described as Treg/Th17 ratio imbalance, may lead to significant perturbations of the immune response, thereby affecting the natural progression of PAH. Furthermore, disproportionate Treg/Th17 ratios have been reported in various PAH, especially in idiopathic PAH, heritable PAH, and PAH associated with connective tissue diseases, which has been identified as positively correlated with PAH severity and prognosis.15, 16
This interesting finding of an imbalanced Treg/Th17 ratio in PAH may be a result of epigenetic modification of the gene promoters. The reduced Tregs may be attributable to increased methylation levels of forkhead box protein 3, a core transcription factor for maintaining Treg quantity and function, thereby reducing forkhead box protein 3 expression and impairing Treg levels in PAH.17 Similarly, the increased Th17 cells are attributable to suppressed methylation levels in the RAR‐related orphan receptor C gene, which represents elevated RAR‐related orphan receptor γt mRNA levels and Th17 expression.18 In addition, Tregs have been found to suppress the numbers and function of other subtypes of T lymphocytes, including Th1, Th2, and Th17, via direct cell‐cell interactions or producing immunoregulatory factors TGF‐β and interleukin‐10.19 However, some studies have also observed increased Tregs in PAH.20 It has been speculated that a possible explanation is related to reactive Treg elevation in the peripheral blood, which is a result of attempting to suppress peripheral proinflammatory activity.13 Although Treg levels in PAH remain controversial, their immunosuppressive function protects against the pathological process of PAH.
Recently, it has been found that Tregs are essential for regulating the course of PAH. Experimental animal models have verified that athymic rats with reduced Treg numbers and activity are more susceptible to severe PAH than wild rats with normal Treg number and function.21 Female athymic rats presented with more serious pulmonary inflammation and augmented right ventricular (RV) fibrosis than their male counterparts after treatment with Sugen 5416 and hypoxia.22 Mechanistically, Treg immune reconstitution protected against PAH development in the present study by increasing expression of the cardiopulmonary vasoprotective factors cyclooxygenase 2, prostacyclin I2 synthase, programmed cell death 1 ligand 1, heme oxygenase 1, prostacyclin, and nitric oxide to exert vasodilative effects.22 This interesting result on PAH severity appears to be sex dependent, and this might be related to the pleiotropic effects of estrogen on inflammation. In abnormal immune homeostasis, the effects of estrogen on Tregs show both immune‐suppressive and immune‐stimulatory effects, and the latter account for a primary role in PAH pathological processes, which leads to a more severe PAH course in female than male patients.23, 24 Therefore, maintaining Treg number and function is important for protecting against PAH, especially in women.
Tregs play a protective role in multiple pathological processes of PAH.8, 25 Tregs can regulate PAH initiation and progression by secreting positive cytokines, interacting directly or indirectly with other immune cells to ameliorate PAEC injury, regulate PASMC proliferation and apoptosis, control fibroblast proliferation and activation, and maintain immune homeostasis (Figures 1 and 2). Accordingly, we systematically summarized the advanced findings of Treg in the reduction of PAEC impairment, inhibition of PASMC abnormal proliferation, repressing inappropriate fibroblast activation, and balancing immune dysfunction to illustrate the regulatory function of Tregs in PAH courses, with the aim of identifying potential therapeutic targets in PAH.
Figure 1.
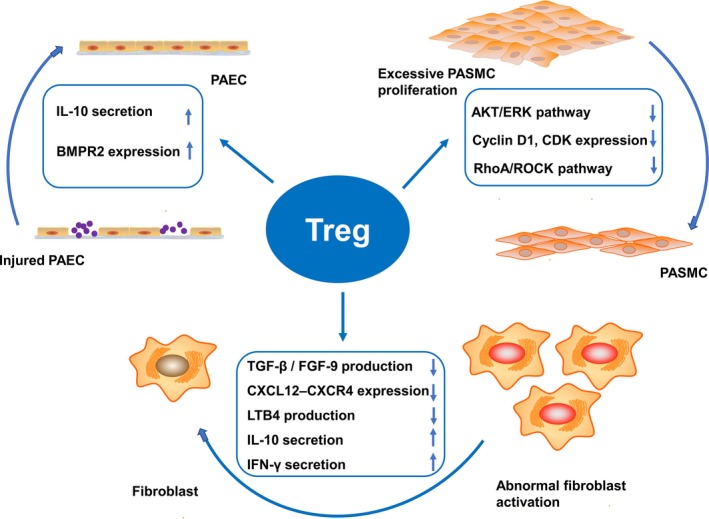
Underlying mechanisms of Tregs in PAH. Tregs repair injured PAECs and endothelium integrity by secreting interleukin‐10 and upregulating BMPR2 expression. Tregs inhibit excessive PASMC proliferation and apoptosis resistance by suppressing the Akt and ERK pathway, reducing cyclin D1 and CDK expression, and restraining the RhoA/ROCK pathway. Tregs regulate abnormal fibroblast proliferation and activation by suppressing TGF‐β and FGF‐9 production, inhibiting the CXCL12–CXCR4 axis, suppressing leukotriene B4 production, and directly secreting interleukin‐10 and interferon‐γ. BMPR2 indicates bone morphogenetic protein receptor type 2; CDK, cyclin‐dependent kinase; CXCL12–CXCR4, chemokine (C‐X‐C motif) ligand 12—CXC receptor 4; ERK, extracellular signal‐regulated kinase; FGF‐9, fibroblast growth factor‐9; LTB4, leukotriene B4; PAEC, pulmonary arterial endothelial cell; PASMC, pulmonary arterial smooth muscle cell; TGF‐β, transforming growth factor‐β.
Figure 2.
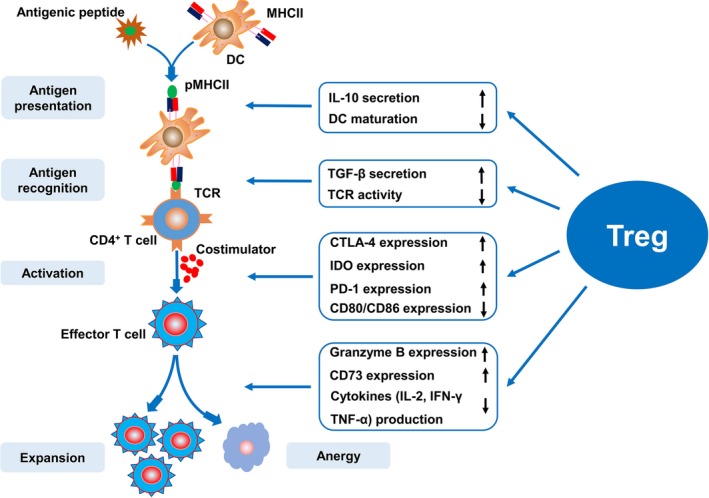
Mechanisms of Treg inhibition of excessive immune response. Tregs are involved in antigen presentation and recognition, as well as T‐lymphocyte activation and expansion. Tregs inhibit antigen presentation by inducing interleukin‐10 expression and inhibiting DC maturation. Tregs suppress antigen recognition by secreting TGF‐β and inhibiting TCR activity. Tregs suppress T‐lymphocyte activation by upregulating the costimulatory molecules CTLA‐4 and reducing CD80 and CD86 expression, upregulating IDO expression indirectly as well as expression of the negative costimulatory signal PD‐1. Tregs control T‐lymphocyte proliferation and differentiation by upregulating granzyme B and CD73 expression, as well as hindering the production of various cytokines such as interleukin‐2, interferon‐γ, and TNF‐α. CTLA‐4 indicates cytotoxic T lymphocyte associated with antigen‐4; DCs, dendritic cells; IDO, indoleamine 2,3‐dioxygenase; MHCII, major histocompatibility complex class II; PD‐1, programmed death 1; pMHCII, peptide–major histocompatibility complex class II complexes; TCR, self‐reactive T‐cell receptors; TGF‐β, transforming growth factor‐β; TNF‐α, tumor necrosis factor‐α.
Tregs Ameliorate PAEC Injury
In addition to acting as a physical barrier, PAECs can also regulate vasoconstriction and vasodilation by synthesizing and transporting a series of vasoactive substances.26 Under the stress of inflammation and stimulation, endothelial dysfunction may lead to decreased production of vasodilators such as nitric oxide and prostacyclin, as well as persistent overexpression of vasoconstrictors such as endothelin‐1.27 This results in increased pulmonary arterial pressure attributable to excessive pulmonary arterial contraction, and eventually accelerates the progression of PAH.28
Investigations have provided evidence that, in PAH, the pulmonary arterial endothelium changes mainly include endothelial injury, endothelial cell proliferation, and invasion of the intima by myofibroblast‐like cells, which is also related to endothelial‐to‐mesenchymal transition.29 Generally, the pulmonary endothelium is a critical local source of several key mediators for vascular remodeling, including growth factors (fibroblast growth factor‐2, angiotensin II), vasoactive peptides (nitric oxide, prostacyclin), cytokines (interleukin‐1, interleukin‐6), and adipokines (leptin).30, 31 PAEC dysfunction could stimulate the production of vasoconstrictors and inflammatory factors to promote cell growth and inflammation—in particular, regulating excessive PASMC proliferation.32
Accordingly, pulmonary artery endothelial injury is an important driving factor of PAH. Xiao et al33 revealed that monocrotaline triggered endothelial damage and finally induced PAH by inducing the aggregation and activation of the calcium‐sensing receptor in PAEC. The calcium‐sensing receptor structure is rich in histidine, tryptophan, and cysteine, which can all bind easily to monocrotaline and further damage the pulmonary arterial endothelium.33 Furthermore, BMPR2 (bone morphogenetic protein receptor type 2) is mainly secreted by PAECs, which feeds back to PAECs and subsequently prevents their proliferation and apoptosis.34 Hong et al35 observed that BMPR2 conditional knockout mice had significantly higher median right ventricular systolic pressure than healthy control mice, indicating that injured PAECs are predisposed to PAH because of reduced BMPR2 secretion.
Treg dysfunction interacts with PAEC injury and thereby aggravates PAH progression. A recent study showed that abnormal Treg function or quantity could lead to impaired PAEC anti‐inflammatory function, which is an important factor in PAH formation.7 Furthermore, Huertas et al36 proved that in patients with idiopathic PAH, PAEC dysfunction may downregulate the function of peripheral circulating Tregs by synthesizing leptin, thereby failing to exert anti‐inflammatory and immunomodulatory effects to improve PAH. Therefore, the abnormal Tregs and PAECs could interact with each other and aggravate the course of PAH.
Endothelial‐to‐mesenchymal transition is a biological process in which the endothelial phenotype of PAECs is progressively changed into a mesenchymal or myofibroblastic phenotype.37 A recent study demonstrated that BMPR2 levels inhibited the chromatin architectural factor high‐mobility group AT‐hook 1 and thereby impeded endothelial‐to‐mesenchymal transition to protect against PAH progression.38, 39 Intriguingly, it has been determined that Tregs may attenuate perivascular inflammation and endothelial cell apoptosis by upregulating BMPR2 expression.35 Therefore, Tregs may inhibit endothelial‐to‐mesenchymal transition by upregulating BMPR2 expression and subsequently reduce high‐mobility group AT‐hook 1 in PAECs.
Moreover, Tregs help maintain normal PAEC function by interacting with other cytokines such as interleukin‐10. Tregs protect against pulmonary vascular endothelial injury and eventually ameliorate PAH by secreting interleukin‐10, which is involved in regulating vascular endothelial function by enhancing endothelial nitric oxide synthase phosphorylation and attenuating oxidation by inhibiting nicotinamide adenine dinucleotide phosphate oxidase activity, ultimately achieving the purpose of relaxing the pulmonary arterioles.40 In summary, Tregs can ameliorate the course of PAH by repairing injured PAECs and endothelium integrity.
Tregs Regulate PASMC Proliferation and Apoptosis
Smooth muscle cells function to contract blood vessels and regulate vascular tone, blood pressure, and blood flow distribution. Under normal circumstances, PASMCs are static and differentiated, exhibiting low proliferation and synthetic activity.41 However, PASMCs become proliferative and apoptosis resistant and have enhanced synthesis and excellent plasticity under pathological conditions such as anoxia and inflammation, which contributes to the development of vascular remodeling.42 The PASMCs of patients with PAH exhibit cancer‐like characteristics, which refers to excessive proliferation and apoptosis resistance.3 Others have also defined these cells as smooth muscle (SM)‐like cells.43
There are several sources for SM‐like cells. The PASMCs in PAH have been considered as being derived from resident PASMCs of the medial layer that migrate to the intima and further proliferate and transform into SM‐like cells.44 Moreover, progenitor cells such as pulmonary pericytes are a potential source of SM‐like cells that could contribute to pulmonary vascular remodeling and lead to PAH.31 Moreover, recent studies have demonstrated that resident endothelial cells within the intima transition to a mesenchymal or SM‐like phenotype, which is involved in the pathogenesis of PAH.38, 45 Furthermore, increased SM‐like cells may be a critical cause of PAH pathogenesis.46 Therefore, inhibiting the transition of SM‐like cells may have potential application in targeted therapy of PAH.47
However, different from the biological effects of PAECs, Tregs inhibit PASMC proliferation and defer the PASMC cycle mainly by increasing anti‐inflammatory cytokine levels to suppress the inflammatory response in PAH. Tregs protect against hypoxia‐induced PAH by suppressing PASMC proliferation.48 In vivo experiments have shown that, in hypoxic conditions, mice injected with purified Tregs had significant reduction of the increased right ventricular systolic pressure and proinflammatory cytokine expression than mice without purified Treg treatment. In an in vitro study, human PASMCs cultured and incubated with Tregs in hypoxic conditions had significantly reduced optical density value compared with the control group when assayed using the 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5 diphenyl tetrazolium bromide method. Mechanistically, the anti‐inflammatory factor interleukin‐10 was increased, and proinflammatory factors, including interleukin‐6, interleukin‐1β, and monocyte chemoattractant protein‐1 were markedly reduced in Treg‐treated human PASMCs as compared with the control group. This suggests that Tregs play a critical role in ameliorating PAH by inhibiting PASMC proliferation and acting against inflammation.48 Recently, it was reported that increased Tregs ameliorated atherosclerosis by suppressing vascular smooth muscle cell proliferation.49
Previous studies have reported the partial mechanisms of Treg regulation of PASMC proliferation. Chu et al48 have shown that Tregs reduce PASMC proliferation and PAH progression by suppressing Akt and extracellular signal–regulated kinase, triggering cell cascade responses, including cell growth, proliferation, survival, and motility.48, 50 They also proved that Tregs reduce pulmonary vascular remodeling by reducing cyclin D1 and cyclin‐dependent kinase expression, as cyclin D1 and cyclin‐dependent kinase promote PASMC proliferation by facilitating G1–S transition in cells.48
As mentioned above, Treg numbers in the peripheral blood are significantly reduced and are accompanied by increased Th17 cells in rats treated with hypoxia.51 A recent study verified that inhibition of the RhoA/Rhoa kinase pathway could reverse the abnormal Treg/Th17 ratio in a hypoxic PAH rat model. Moreover, correcting the disproportionate ratio suppressed the excessive PASMC growth and hypertrophy, and thereby ameliorated PAH progression.51 This finding suggests that Tregs may improve PAH by inhibiting the RhoA/Rhoa kinase signaling pathway. Therefore, Tregs may be a novel target for inhibiting PASMC proliferation in improving the course of PAH.
Tregs Regulate Fibroblast Proliferation and Activation
Fibrosis occurs as a result of sustained injury such as hypoxia, high‐perfusion flow, or alkaloid treatment, which causes fibroblast recruitment, activation, and proliferation and the production of high amounts of collagen and extracellular matrix proteins.52 Various types of cells, especially fibroblast and myofibroblast expansion and differentiation, are mainly implicated in the fibrotic process.53 In the course of PAH, fibroblasts are improperly activated and expanded. Subsequently, collagen and extracellular matrix proteins are synthesized and deposited in the lung, pulmonary artery (especially the pulmonary artery adventitia), and right ventricle, which is important for driving the course of PAH.54
Previous studies have demonstrated that Tregs have a vital role in antifibrosis by inhibiting excessive activation of fibroblasts and the immune response.55, 56, 57 Although there is some discrepancy in the net effects of Tregs on the fibrotic processes, Tregs play an important role in regulating fibroblast proliferation and activation, thereby controlling fibrosis of the lung, pulmonary artery, and right ventricle, and consequently ameliorating PAH progression.
Tregs Regulate Fibroblasts in the Lung
Generally, Tregs antagonize pulmonary fibrosis by inhibiting pulmonary fibroblasts. Tregs exert suppressive effects on collagen accumulation and fibroblast recruitment by suppressing TGF‐β1 and fibroblast growth factor‐9 in a TGF‐β1–induced pulmonary fibrosis model.55, 56 Moreover, Garibaldi et al57 found that Tregs resolved fibroblast proliferation in a lipopolysaccharide‐induced acute lung injury model by reducing fibrocyte recruitment and inhibiting the chemokine (C‐X‐C motif) ligand 12 (CXCL12)–CXCR4 axis. This suggests that Tregs exert antifibrotic effects on pulmonary fibrosis by suppressing fibroblast recruitment and proliferation.
However, several studies have reported that Tregs have profibrotic properties in silicosis‐ or bleomycin‐induced lung fibrosis experimental models.58, 59 Lo Re et al58 indicated that Tregs contribute to pulmonary fibrosis by stimulating fibroblasts through the secretion of platelet‐derived growth factor‐B in silicosis‐induced lung fibrosis. Xiong et al59 reported that Tregs contribute to fibrosis progression in a bleomycin‐induced mouse model by promoting the effects of Th17 and CD8+ T cells, while inhibiting interferon‐γ expression.
The possible explanations for the disparity of these findings may depend on the different lung fibrosis models, which implicate different underlying pathophysiological mechanisms.59 More interestingly, it was recently reported that Tregs seem to be detrimental in the early stages of pulmonary fibrosis in bleomycin‐induced idiopathic pulmonary fibrosis mice, but are protective in the later stages.60 This suggests that Tregs exert different effects on the PAH pathological stages, which may be an important reason for this deviation.
Tregs Attenuate Excessive Proliferation and Activation of Pulmonary Artery Adventitial Fibroblasts
Tregs are involved in the improvement of pulmonary artery fibrosis, especially pulmonary artery adventitial fibrosis. In recent years, the concept of the “outside‐in” mechanism has been increasingly supported by scholars, and mainly refers to adventitial fibroblasts serving as a source of pathologic stimuli permeating the vascular wall.61 Pulmonary artery adventitial fibroblasts (PAAFs) are activated by a variety of pathways that result in excessive proliferative, migratory, fibrotic, and inflammatory activities and thereby promote PAH progression.62 Moreover, a variety of factors regulate PAAFs, thereby affecting the progression of pulmonary artery fibrosis.
Tregs exert suppressive effects on PAAF proliferation. In general, Treg activation can increase macrophage responses to promote injury resolution. In the absence of Treg regulation, macrophages are excessively activated and recruited in a pathological state, which not only directly induces PAAF proliferation, but also enhances this effect by promoting leukotriene B4 production that further stimulates the function of PAAF, ultimately accelerating PAH progression.63, 64 Therefore, Tregs play an important role in inhibiting PAAF proliferation and attenuating adventitial fibrosis in PAH.
Tregs Regulate Fibroblasts in Right Ventricle
In the pathological processes of PAH, chronic RV pressure overload increases wall stress and promotes ventricular remodeling, including “adaptive remodeling,” which is characterized by concentric RV hypertrophy and preserved systolic and diastolic function; “maladaptive remodeling” is recognized by eccentric remodeling, RV dilatation, reduced RV function, and early deterioration into RV failure.65 RV fibrosis is the hallmark of maladaptive remodeling, which is a result of RV pressure overload after pulmonary arterial pressure is elevated.65 Furthermore, adaptive RV remodeling is associated with a lower risk of clinical deterioration compared with maladaptive RV remodeling in patients with PAH.66 Therefore, RV fibrosis and function help to assess the prognosis of patients with PAH.
Tregs have been proven to play an indispensable role in regulating cardiac fibroblasts to control myocardial fibrosis and maintain cardiac function. Cao et al67 demonstrated the protective effect of Tregs on myocardial fibrosis via the secretion of the immune‐suppressive cytokine interleukin‐10, suggesting that Tregs potentially control ventricular remodeling and myocardial fibrosis in a coxsackievirus B3–induced mouse myocardial fibrosis model. Consistently, Ramjee et al68 also found that promoting Treg recruitment and activation was conducive to alleviating myocardial fibrosis and heart failure in a mouse model, which is related to the Treg chemokine interferon‐γ reducing profibrotic cytokine secretion and mediating fibroblast migration, thereby inhibiting myocardial fibroblast proliferation and activation. Although the direct function of Tregs in RV failure secondary to PAH has not been identified, its potent antifibrotic properties mean that it may be a promising target for RV protection in PAH.
However, the function of Tregs in regulating cardiac fibrosis is also disputed, and some studies have reported that Tregs promote cardiac fibroblast proliferation and survival, aggravating myocardial fibrosis and chronic heart failure.69, 70 Bansal et al69 reported that Treg depletion resulted in a significant reduction in collagen I and collagen III expression and in the inhibition of interstitial fibrosis in ischemic cardiomyopathy mice, which indicated the cardiac profibrotic effects of Tregs.69 A recent study further reported that in patients with late‐stage heart failure, Tregs could secrete TGF‐β in an autocrine or paracrine manner and then stimulate cardiac fibroblast proliferation, thereby facilitating the progression of heart failure.70 Overall, proper fibrosis in the early stage after cardiac injury may contribute to repair of the lesions, while excessive fibrosis results in cardiac remodeling and dysfunction. The contradictory findings of Tregs in myocardial fibrosis may be related to this process and still require further exploration.
Tregs Inhibit Excessive Immune Response
Immune dysfunction is a critical factor in pulmonary perivascular inflammation and the pathological progresses of PAH.71 Previous studies have suggested that sustaining Treg function to control the aberrant immune response and maintain immune homeostasis is of great significance for overcoming PAH progression.72, 73 Furthermore, recent studies have shown that Tregs suppress the immune response by suppressing various immune‐associated cells, including CD4+ and CD8+ T cells, natural killer cells, and antigen‐presenting cells (APCs, mainly involving dendritic cells [DCs], macrophages, B cells). Further work has shown that Tregs mediate APC, natural killer cell, and CD8+ T‐cell cytolysis by upregulating granzyme and perforin expression.74 Although Treg‐mediated suppression is related to several kinds of immune cells, the mechanisms of Treg suppression of T‐lymphocyte–mediated immune responses are the most widely studied.
Treg immunosuppression of the processes of T‐lymphocyte–mediated immune response are mainly implicated in the following stages. First, Tregs can interfere with the formation of peptide–major histocompatibility complex class II complexes and can hamper APC presentation of peptide–major histocompatibility complex class II (MHCII) complexes to T lymphocytes. Second, Tregs control the immune response by inhibiting the antigen recognition process. Finally, Tregs suppress T‐lymphocyte activation and expansion (Figure 2).
Tregs induce significant interleukin‐10 expression, and interleukin‐10 induces immune suppression primarily by inhibiting MHCII expression, which is essential for APC antigen presentation to T cells.75 In addition, conventional DCs are a primary APC subset that mediate Treg immunosuppression.76 Tregs hinder antigen presenting and suppress the immune response by obstructing DC maturation.77 Accordingly, Tregs can inhibit APC antigen presentation to T lymphocytes by reducing MHCII expression and DC maturation.
Next, antigen recognition is associated with the specific binding of T‐cell receptors (TCRs) to the APC‐presented peptide‐MHCII, which promotes the generation of costimulatory molecules. Accordingly, the costimulatory signals generated by the interaction among costimulatory molecules are critical for activating and expanding specific T lymphocytes. However, Kim et al72 verified that Tregs suppress the immune response by preventing the antigen recognition process via the inhibition of self‐reactive TCR activity on T lymphocytes. Moreover, TGF‐β interferes with TCRs and further suppresses the costimulatory receptor signaling pathways by inhibiting Tec kinase activation, suppressing calcium influx elicited by TCR/CD28 stimulation in CD4+ T cells.78 Furthermore, Tregs might protect against PAH by secreting TGF‐β.79 Therefore, we assume that Tregs play an immunosuppressive role and further protect against PAH by blocking antigen recognition attributable to the increased expression of TGF‐β.
After combining with MHCII on APCs, these specific T lymphocytes will be activated and undergo further expansion. Accordingly, T‐cell activation requires not only antigenic stimulation signals (the first signal) provided by peptide‐MHCII, but also costimulatory signals (the second signal) delivered by costimulatory molecules to TCRs.80 According to this model, T lymphocytes that receive only antigen‐specific TCR stimulation in the absence of costimulatory molecules will be rendered unresponsive (anergic) to antigenic challenge. Intriguingly, the costimulatory pathways can provide positive costimulatory signals that promote T‐cell activation as well as negative costimulatory signals that inhibit T‐cell responses.81 Maeda et al82 suggested that Tregs suppressed responder T‐cell activation by downregulating the expression of the costimulatory molecules CD80 and CD86 on APCs, which decreased costimulatory signal production. Moreover, cytotoxic T‐lymphocyte–associated protein 4, highly expressed on Treg surfaces and which interacts with CD80 and CD86, may induce DCs to upregulate indoleamine 2,3‐dioxygenase 1 expression, leading to increased negative costimulatory signals and subsequently damaging responder T‐cell activation, inhibiting T‐cell responses and exerting immunosuppressive effects.83 Programmed cell death 1 is a negative costimulatory signal that delivers inhibitory signals and prevents effector T‐cell responses. Tregs suppress T‐lymphocyte activation and expansion by upregulating and maintaining programmed cell death 1 expression.80
Finally, after T‐lymphocyte activation, it is also necessary to promote their proliferation and differentiation under the action of cytokines such as interleukin‐2, interferon‐γ, and tumor necrosis factor‐α. Responder T cells under Treg‐mediated suppression remain hypoproliferative and produce low amounts of cytokines, which can be immunologically defined as “anergy.”82 Gondek et al84 also suggested that Tregs exert their immunosuppressive effects by upregulating granzyme B expression, which induces apoptosis in responder T cells, weakening the immune response. More recently, Smyth et al85 demonstrated that Treg‐derived exosomes inhibit T‐cell proliferation and further prevent the aberrant immune response by expressing CD73, which converts extracellular adenosine‐5‐monophosphate to adenosine, describing a new mechanism of Treg inhibition of immune response. Taken together, these studies demonstrate that Tregs exert immunosuppressive effects at various stages of the immune response, providing solid evidence of Tregs correcting the immune dysfunction in PAH.
Tregs as a Therapeutic Target in PAH
Well‐documented studies have identified Tregs as a promising therapeutic target, and their clinical application has been tested for several diseases.86, 87 Clinical data have shown that both exogenous Treg transplantation and induction of intrinsic Treg activation are effective for preventing and treating several dominant self‐tolerance and autoimmune diseases.87, 88, 89 Although there are no definitive data on the efficiency and safety of transplanted exogenous Tregs in patients with PAH yet, it is a promising approach for Treg clinical application, given the multiple effects of Treg induction in PAH.
At present, studies on exogenous Treg transplantation are relatively more feasible in the field of disease treatment. Recently, Bluestone et al90 conducted a phase 1 trial to assess the safety of adoptively transferred Tregs for immunotherapy. Fourteen adult subjects with type 1 diabetes mellitus were infused ex vivo with expanded autologous polyclonal Tregs via the peripheral intravenous route. The authors reported that, after isolation and expansion in the new hosts, the transferred Tregs were long lived, with up to 25% of the peak level remaining in the circulation at 1 year after transfer. That study showed transient increases in Tregs in the recipients, who retained a broad Treg phenotype long term. Moreover, there were no infusion reactions or cell therapy–related adverse events.90 This indicates that autologous Treg transplantation may be a safe and relatively effective treatment for autoimmune disease. However, the main challenges to successfully implementing exogenous Treg transplantation therapy are the effective production of high‐quality Tregs and balancing the yield and purity of the products.91 Further studies are required to identify more effective methods for Treg application in clinical therapy.
Recently, it was found that inducing internal Treg expansion is a promising means of increasing endogenous Treg activity. CD28 superagonists are highly effective for treating rodent models of autoimmunity because of the potent capability for inducing Treg expansion and increasing Treg activity.92 A number of studies have also demonstrated significantly therapeutic effects of interleukin‐2 in patients with various autoimmune diseases by inducing endogenous Treg expansion.93, 94 In addition, a recent study reported that physical exercise induces increased endogenous Tregs, which contributes to ameliorating pulmonary allergic inflammation.95 These studies suggest that there may be unexplored, more concise and efficient methods for inducing Treg expansion.
It has also been reported that correcting the abnormal Treg metabolism could prolong Treg survival duration under pathological conditions. Approaches to improving Treg metabolism may be also a promising strategy for Treg‐targeted therapy of PAH.96 Liver kinase B1, a potential regulator of cellular homeostasis in PAH courses, is believed to mediate Treg metabolism and maintain immunological homoeostasis by regulating multiple metabolic pathways.97, 98 Liver kinase B1 has been found to be essential for Treg survival and function. Treg‐specific deletion of the Lkb1 gene in mice causes serious mitochondrial metabolism dysfunction and alters cellular metabolism pathways, leading to loss of Treg number and function and causing early‐onset autoimmune disorder; conversely, liver kinase B1 gain of function significantly reverses this tendency.97, 98 This finding suggests that approaches to normalizing Treg metabolism and thereby extending the life span of Tregs may be another effective method for treating PAH.
Conclusions
A large amount of experimental and preclinical evidence shows that Tregs play a critical regulatory role in both the initiation and development stages of PAH pathogenesis.48, 99 Reduced Treg number and impaired Treg function caused by innate or environmental elements may aggravate the processes of PAH. Otherwise, recovering abnormal Tregs may protect against PAH by improving PAEC injury, PASMC proliferation and apoptosis, fibroblast proliferation and activation, and immune dysfunction. However, restrictions on using Tregs for clinical treatment remain. Clearly, Tregs might be a promising therapeutic target in PAH. At the same time, it might also improve our understanding of the immunomodulatory mechanisms involved in PAH.
Disclosures
None.
(J Am Heart Assoc. 2019;8:e014201 DOI: 10.1161/JAHA.119.014201.)
References
- 1. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–D41. [DOI] [PubMed] [Google Scholar]
- 2. Montani D, Lau EM, Dorfmuller P, Girerd B, Jais X, Savale L, Perros F, Nossent E, Garcia G, Parent F, Fadel E, Soubrier F, Sitbon O, Simonneau G, Humbert M. Pulmonary veno‐occlusive disease. Eur Respir J. 2016;47:1518–1534. [DOI] [PubMed] [Google Scholar]
- 3. Thompson AAR, Lawrie A. Targeting vascular remodeling to treat pulmonary arterial hypertension. Trends Mol Med. 2017;23:31–45. [DOI] [PubMed] [Google Scholar]
- 4. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nicolls MR, Taraseviciene‐Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26:1110–1118. [DOI] [PubMed] [Google Scholar]
- 7. Voelkel NF, Tamosiuniene R, Nicolls MR. Challenges and opportunities in treating inflammation associated with pulmonary hypertension. Expert Rev Cardiovasc Ther. 2016;14:939–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res. 2011;109:867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan CC, Caspi RR. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin‐6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–244, 28p following 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maston LD, Jones DT, Giermakowska W, Howard TA, Cannon JL, Wang W, Wei Y, Xuan W, Resta TC, Gonzalez Bosc LV. Central role of T helper 17 cells in chronic hypoxia‐induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2017;312:L609–L624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen G, Zuo S, Tang J, Zuo C, Jia D, Liu Q, Liu G, Zhu Q, Wang Y, Zhang J, Shen Y, Chen D, Yuan P, Qin Z, Ruan C, Ye J, Wang XJ, Zhou Y, Gao P, Zhang P, Liu J, Jing ZC, Lu A, Yu Y. Inhibition of CRTH2‐mediated Th2 activation attenuates pulmonary hypertension in mice. J Exp Med. 2018;215:2175–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Austin ED, Rock MT, Mosse CA, Vnencak‐Jones CL, Yoder SM, Robbins IM, Loyd JE, Meyrick BO. T lymphocyte subset abnormalities in the blood and lung in pulmonary arterial hypertension. Respir Med. 2010;104:454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, Chen W, Zhang C, Zhang Y. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol. 2016;13:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huertas A, Phan C, Bordenave J, Tu L, Thuillet R, Le Hiress M, Avouac J, Tamura Y, Allanore Y, Jovan R, Sitbon O, Guignabert C, Humbert M. Regulatory T cell dysfunction in idiopathic, heritable and connective tissue‐associated pulmonary arterial hypertension. Chest. 2016;149:1482–1493. [DOI] [PubMed] [Google Scholar]
- 16. Gaowa S, Zhou W, Yu L, Zhou X, Liao K, Yang K, Lu Z, Jiang H, Chen X. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediators Inflamm. 2014;2014:247372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hew KM, Walker AI, Kohli A, Garcia M, Syed A, McDonald‐Hyman C, Noth EM, Mann JK, Pratt B, Balmes J, Hammond SK, Eisen EA, Nadeau KC. Childhood exposure to ambient polycyclic aromatic hydrocarbons is linked to epigenetic modifications and impaired systemic immunity in T cells. Clin Exp Allergy. 2015;45:238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang D, Huang S, Yuan X, Liang J, Xu R, Yao G, Feng X, Sun L. The regulation of the Treg/Th17 balance by mesenchymal stem cells in human systemic lupus erythematosus. Cell Mol Immunol. 2017;14:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saito S, Nakashima A, Shima T, Ito M. Th1/Th2/Th17 and regulatory T‐cell paradigm in pregnancy. Am J Reprod Immunol. 2010;63:601–610. [DOI] [PubMed] [Google Scholar]
- 20. Galati D, De Martino M, Trotta A, Rea G, Bruzzese D, Cicchitto G, Stanziola AA, Napolitano M, Sanduzzi A, Bocchino M. Peripheral depletion of NK cells and imbalance of the Treg/Th17 axis in idiopathic pulmonary fibrosis patients. Cytokine. 2014;66:119–126. [DOI] [PubMed] [Google Scholar]
- 21. Nicolls MR, Mizuno S, Taraseviciene‐Stewart L, Farkas L, Drake JI, Al Husseini A, Gomez‐Arroyo JG, Voelkel NF, Bogaard HJ. New models of pulmonary hypertension based on VEGF receptor blockade‐induced endothelial cell apoptosis. Pulm Circ. 2012;2:434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tamosiuniene R, Manouvakhova O, Mesange P, Saito T, Qian J, Sanyal M, Lin YC, Nguyen LP, Luria A, Tu AB, Sante JM, Rabinovitch M, Fitzgerald DJ, Graham BB, Habtezion A, Voelkel NF, Aurelian L, Nicolls MR. Dominant role for regulatory T cells in protecting females against pulmonary hypertension. Circ Res. 2018;122:1689–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cohen‐Solal JF, Jeganathan V, Grimaldi CM, Peeva E, Diamond B. Sex hormones and SLE: influencing the fate of autoreactive B cells. Curr Top Microbiol Immunol. 2006;305:67–88. [DOI] [PubMed] [Google Scholar]
- 24. Nicolls MR, Voelkel NF. The roles of immunity in the prevention and evolution of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2017;195:1292–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perros F, Ranchoux B, Izikki M, Bentebbal S, Happe C, Antigny F, Jourdon P, Dorfmuller P, Lecerf F, Fadel E, Simonneau G, Humbert M, Bogaard HJ, Eddahibi S. Nebivolol for improving endothelial dysfunction, pulmonary vascular remodeling, and right heart function in pulmonary hypertension. J Am Coll Cardiol. 2015;65:668–680. [DOI] [PubMed] [Google Scholar]
- 27. Li H, Lu W, Cai WW, Wang PJ, Zhang N, Yu CP, Wang DL, Liu BC, Sun W. Telmisartan attenuates monocrotaline‐induced pulmonary artery endothelial dysfunction through a PPAR gamma‐dependent PI3K/Akt/eNOS pathway. Pulm Pharmacol Ther. 2014;28:17–24. [DOI] [PubMed] [Google Scholar]
- 28. Nie X, Tan J, Dai Y, Liu Y, Zou J, Sun J, Ye S, Shen C, Fan L, Chen J, Bian JS. CCL5 deficiency rescues pulmonary vascular dysfunction, and reverses pulmonary hypertension via caveolin‐1‐dependent BMPR2 activation. J Mol Cell Cardiol. 2018;116:41–56. [DOI] [PubMed] [Google Scholar]
- 29. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8:443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guignabert C, Tu L, Girerd B, Ricard N, Huertas A, Montani D, Humbert M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: importance of endothelial communication. Chest. 2015;147:529–537. [DOI] [PubMed] [Google Scholar]
- 31. Ricard N, Tu L, Le Hiress M, Huertas A, Phan C, Thuillet R, Sattler C, Fadel E, Seferian A, Montani D, Dorfmuller P, Humbert M, Guignabert C. Increased pericyte coverage mediated by endothelial‐derived fibroblast growth factor‐2 and interleukin‐6 is a source of smooth muscle‐like cells in pulmonary hypertension. Circulation. 2014;129:1586–1597. [DOI] [PubMed] [Google Scholar]
- 32. Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xiao R, Su Y, Feng T, Sun M, Liu B, Zhang J, Lu Y, Li J, Wang T, Zhu L, Hu Q. Monocrotaline induces endothelial injury and pulmonary hypertension by targeting the extracellular calcium‐sensing receptor. J Am Heart Assoc. 2017;6:e004865 DOI: 10.1161/JAHA.116.004865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Diebold I, Hennigs JK, Miyagawa K, Li CG, Nickel NP, Kaschwich M, Cao A, Wang L, Reddy S, Chen PI, Nakahira K, Alcazar MA, Hopper RK, Ji L, Feldman BJ, Rabinovitch M. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015;21:596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, Li E, Raizada MK, Bloch KD, Oh SP. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118:722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huertas A, Tu L, Gambaryan N, Girerd B, Perros F, Montani D, Fabre D, Fadel E, Eddahibi S, Cohen‐Kaminsky S, Guignabert C, Humbert M. Leptin and regulatory T‐lymphocytes in idiopathic pulmonary arterial hypertension. Eur Respir J. 2012;40:895–904. [DOI] [PubMed] [Google Scholar]
- 37. Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, Holmes AM. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol. 2015;185:1850–1858. [DOI] [PubMed] [Google Scholar]
- 38. Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial‐to‐mesenchymal transition via HMGA1 and its target slug. Circulation. 2016;133:1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ranchoux B, Antigny F, Rucker‐Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmuller P, Remy S, Lecerf F, Plante S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen‐Kaminsky S, Perros F. Endothelial‐to‐mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. [DOI] [PubMed] [Google Scholar]
- 40. Tamosiuniene R, Nicolls MR. Regulatory T cells and pulmonary hypertension. Trends Cardiovasc Med. 2011;21:166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bourgeois A, Lambert C, Habbout K, Ranchoux B, Paquet‐Marceau S, Trinh I, Breuils‐Bonnet S, Paradis R, Nadeau V, Paulin R, Provencher S, Bonnet S, Boucherat O. FOXM1 promotes pulmonary artery smooth muscle cell expansion in pulmonary arterial hypertension. J Mol Med (Berl). 2018;96:223–235. [DOI] [PubMed] [Google Scholar]
- 42. Hong Z, Chen KH, DasGupta A, Potus F, Dunham‐Snary K, Bonnet S, Tian L, Fu J, Breuils‐Bonnet S, Provencher S, Wu D, Mewburn J, Ormiston ML, Archer SL. MicroRNA‐138 and microRNA‐25 down‐regulate mitochondrial calcium uniporter, causing the pulmonary arterial hypertension cancer phenotype. Am J Respir Crit Care Med. 2017;195:515–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen B, Calvert AE, Meng X, Nelin LD. Pharmacologic agents elevating cAMP prevent arginase II expression and proliferation of pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2012;47:218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coll‐Bonfill N, Musri MM, Ivo V, Barbera JA, Tura‐Ceide O. Transdifferentiation of endothelial cells to smooth muscle cells play an important role in vascular remodelling. Am J Stem Cells. 2015;4:13–21. [PMC free article] [PubMed] [Google Scholar]
- 45. Qiao L, Nishimura T, Shi L, Sessions D, Thrasher A, Trudell JR, Berry GJ, Pearl RG, Kao PN. Endothelial fate mapping in mice with pulmonary hypertension. Circulation. 2014;129:692–703. [DOI] [PubMed] [Google Scholar]
- 46. Sahara M, Sata M, Morita T, Nakamura K, Hirata Y, Nagai R. Diverse contribution of bone marrow‐derived cells to vascular remodeling associated with pulmonary arterial hypertension and arterial neointimal formation. Circulation. 2007;115:509–517. [DOI] [PubMed] [Google Scholar]
- 47. Huang S, Zhu X, Huang W, He Y, Pang L, Lan X, Shui X, Chen Y, Chen C, Lei W. Quercetin inhibits pulmonary arterial endothelial cell transdifferentiation possibly by Akt and Erk1/2 pathways. Biomed Res Int. 2017;2017:6147294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chu Y, Xiangli X, Xiao W. Regulatory T cells protect against hypoxia‐induced pulmonary arterial hypertension in mice. Mol Med Rep. 2015;11:3181–3187. [DOI] [PubMed] [Google Scholar]
- 49. Ji Q, Meng K, Yu K, Huang S, Huang Y, Min X, Zhong Y, Wu B, Liu Y, Nie S, Zhang J, Zhou Y, Zeng Q. Exogenous interleukin 37 ameliorates atherosclerosis via inducing the Treg response in ApoE‐deficient mice. Sci Rep. 2017;7:3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Orlandi A, Bochaton‐Piallat ML, Gabbiani G, Spagnoli LG. Aging, smooth muscle cells and vascular pathobiology: implications for atherosclerosis. Atherosclerosis. 2006;188:221–230. [DOI] [PubMed] [Google Scholar]
- 51. Li C, Liu PP, Tang DD, Song R, Zhang YQ, Lei S, Wu SJ. Targeting the RhoA‐ROCK pathway to regulate T‐cell homeostasis in hypoxia‐induced pulmonary arterial hypertension. Pulm Pharmacol Ther. 2018;50:111–122. [DOI] [PubMed] [Google Scholar]
- 52. Baicu CF, Li J, Zhang Y, Kasiganesan H, Cooper G IV, Zile MR, Bradshaw AD. Time course of right ventricular pressure‐overload induced myocardial fibrosis: relationship to changes in fibroblast postsynthetic procollagen processing. Am J Physiol Heart Circ Physiol. 2012;303:H1128–H1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28:23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peng X, Moore MW, Peng H, Sun H, Gan Y, Homer RJ, Herzog EL. CD4+CD25+FoxP3+ regulatory Tregs inhibit fibrocyte recruitment and fibrosis via suppression of FGF‐9 production in the TGF‐beta1 exposed murine lung. Front Pharmacol. 2014;5:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. MacDonald KP, Blazar BR, Hill GR. Cytokine mediators of chronic graft‐versus‐host disease. J Clin Invest. 2017;127:2452–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garibaldi BT, D'Alessio FR, Mock JR, Files DC, Chau E, Eto Y, Drummond MB, Aggarwal NR, Sidhaye V, King LS. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol. 2013;48:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lo Re S, Lecocq M, Uwambayinema F, Yakoub Y, Delos M, Demoulin JB, Lucas S, Sparwasser T, Renauld JC, Lison D, Huaux F. Platelet‐derived growth factor‐producing CD4+ Foxp3+ regulatory T lymphocytes promote lung fibrosis. Am J Respir Crit Care Med. 2011;184:1270–1281. [DOI] [PubMed] [Google Scholar]
- 59. Xiong S, Guo R, Yang Z, Xu L, Du L, Li R, Xiao F, Wang Q, Zhu M, Pan X. Treg depletion attenuates irradiation‐induced pulmonary fibrosis by reducing fibrocyte accumulation, inducing Th17 response, and shifting IFN‐γ, IL‐12/IL‐4, IL‐5 balance. Immunobiology. 2015;220:1284‐1291. [DOI] [PubMed] [Google Scholar]
- 60. Boveda‐Ruiz D, D'Alessandro‐Gabazza CN, Toda M, Takagi T, Naito M, Matsushima Y, Matsumoto T, Kobayashi T, Gil‐Bernabe P, Chelakkot‐Govindalayathil AL, Miyake Y, Yasukawa A, Morser J, Taguchi O, Gabazza EC. Differential role of regulatory T cells in early and late stages of pulmonary fibrosis. Immunobiology. 2013;218:245–254. [DOI] [PubMed] [Google Scholar]
- 61. El Kasmi KC, Pugliese SC, Riddle SR, Poth JM, Anderson AL, Frid MG, Li M, Pullamsetti SS, Savai R, Nagel MA, Fini MA, Graham BB, Tuder RM, Friedman JE, Eltzschig HK, Sokol RJ, Stenmark KR. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol. 2014;193:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stenmark KR, Frid MG, Yeager M, Li M, Riddle S, McKinsey T, El Kasmi KC. Targeting the adventitial microenvironment in pulmonary hypertension: a potential approach to therapy that considers epigenetic change. Pulm Circ. 2012;2:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Qian J, Tian W, Jiang X, Tamosiuniene R, Sung YK, Shuffle EM, Tu AB, Valenzuela A, Jiang S, Zamanian RT, Fiorentino DF, Voelkel NF, Peters‐Golden M, Stenmark KR, Chung L, Rabinovitch M, Nicolls MR. Leukotriene B4 activates pulmonary artery adventitial fibroblasts in pulmonary hypertension. Hypertension. 2015;66:1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, Strassheim D, Meyrick B, Yeager ME, Flockton AR, McKeon BA, Lemon DD, Horn TR, Anwar A, Barajas C, Stenmark KR. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol. 2011;187:2711–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van de Veerdonk MC, Bogaard HJ, Voelkel NF. The right ventricle and pulmonary hypertension. Heart Fail Rev. 2016;21:259–271. [DOI] [PubMed] [Google Scholar]
- 66. Prins KW, Duval S, Markowitz J, Pritzker M, Thenappan T. Chronic use of PAH‐specific therapy in World Health Organization Group III Pulmonary Hypertension: a systematic review and meta‐analysis. Pulm Circ. 2017;7:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cao Y, Xu W, Xiong S. Adoptive transfer of regulatory T cells protects against Coxsackievirus B3‐induced cardiac fibrosis. PLoS One. 2013;8:e74955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ramjee V, Li D, Manderfield LJ, Liu F, Engleka KA, Aghajanian H, Rodell CB, Lu W, Ho V, Wang T, Li L, Singh A, Cibi DM, Burdick JA, Singh MK, Jain R, Epstein JA. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J Clin Invest. 2017;127:899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD. Dysfunctional and proinflammatory regulatory T‐lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation. 2019;139:206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shao PP, Liu CJ, Xu Q, Zhang B, Li SH, Wu Y, Sun Z, Cheng LF. Eplerenone reverses cardiac fibrosis via the suppression of Tregs by inhibition of Kv1.3 channel. Front Physiol. 2018;9:899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li C, Liu P, Song R, Zhang Y, Lei S, Wu S. Immune cells and autoantibodies in pulmonary arterial hypertension. Acta Biochim Biophys Sin (Shanghai). 2017;49:1047–1057. [DOI] [PubMed] [Google Scholar]
- 72. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. [DOI] [PubMed] [Google Scholar]
- 73. Yuan X, Cheng G, Malek TR. The importance of regulatory T‐cell heterogeneity in maintaining self‐tolerance. Immunol Rev. 2014;259:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica‐Worms DR, Ley TJ. Granzyme B and perforin are important for regulatory T cell‐mediated suppression of tumor clearance. Immunity. 2007;27:635–646. [DOI] [PubMed] [Google Scholar]
- 75. Sziksz E, Pap D, Lippai R, Beres NJ, Fekete A, Szabo AJ, Vannay A. Fibrosis related inflammatory mediators: role of the IL‐10 cytokine family. Mediators Inflamm. 2015;2015:764641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Perry JSA, Lio CJ, Kau AL, Nutsch K, Yang Z, Gordon JI, Murphy KM, Hsieh CS. Distinct contributions of Aire and antigen‐presenting‐cell subsets to the generation of self‐tolerance in the thymus. Immunity. 2014;41:414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, Lafaille JJ, Dustin ML. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med. 2006;203:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chen CH, Seguin‐Devaux C, Burke NA, Oriss TB, Watkins SC, Clipstone N, Ray A. Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J Exp Med. 2003;197:1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Farkas L, Kolb MRJ. A switch in TGF‐beta signaling explains contradictory findings in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2018;197:157–159. [DOI] [PubMed] [Google Scholar]
- 80. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD‐L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Francisco LM, Sage PT, Sharpe AH. The PD‐1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Maeda Y, Nishikawa H, Sugiyama D, Ha D, Hamaguchi M, Saito T, Nishioka M, Wing JB, Adeegbe D, Katayama I, Sakaguchi S. Detection of self‐reactive CD8(+) T cells with an anergic phenotype in healthy individuals. Science. 2014;346:1536–1540. [DOI] [PubMed] [Google Scholar]
- 83. Wang Y, Liu H, McKenzie G, Witting PK, Stasch JP, Hahn M, Changsirivathanathamrong D, Wu BJ, Ball HJ, Thomas SR, Kapoor V, Celermajer DS, Mellor AL, Keaney JF Jr, Hunt NH, Stocker R. Kynurenine is an endothelium‐derived relaxing factor produced during inflammation. Nat Med. 2010;16:279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact‐mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B‐dependent, perforin‐independent mechanism. J Immunol. 2005;174:1783–1786. [DOI] [PubMed] [Google Scholar]
- 85. Smyth LA, Ratnasothy K, Tsang JY, Boardman D, Warley A, Lechler R, Lombardi G. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol. 2013;43:2430–2440. [DOI] [PubMed] [Google Scholar]
- 86. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Blazar BR, MacDonald KPA, Hill GR. Immune regulatory cell infusion for graft‐versus‐host disease prevention and therapy. Blood. 2018;131:2651–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ratnasothy K, Jacob J, Tung S, Boardman D, Lechler RI, Sanchez‐Fueyo A, Martinez‐Llordella M, Lombardi G. IL‐2 therapy preferentially expands adoptively transferred donor‐specific Tregs improving skin allograft survival. Am J Transplant. 2019;19:2092–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Canavan JB, Scotta C, Vossenkamper A, Goldberg R, Elder MJ, Shoval I, Marks E, Stolarczyk E, Lo JW, Powell N, Fazekasova H, Irving PM, Sanderson JD, Howard JK, Yagel S, Afzali B, MacDonald TT, Hernandez‐Fuentes MP, Shpigel NY, Lombardi G, Lord GM. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn's disease. Gut. 2016;65:584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7:315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tang Q, Bluestone JA. Regulatory T‐cell therapy in transplantation: moving to the clinic. Cold Spring Harb Perspect Med. 2013;3:a015552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tabares P, Berr S, Romer PS, Chuvpilo S, Matskevich AA, Tyrsin D, Fedotov Y, Einsele H, Tony HP, Hunig T. Human regulatory T cells are selectively activated by low‐dose application of the CD28 superagonist TGN1412/TAB08. Eur J Immunol. 2014;44:1225–1236. [DOI] [PubMed] [Google Scholar]
- 93. Xiao J, Yu K, Li M, Xiong C, Wei Y, Zeng Q. The IL‐2/anti‐IL‐2 complex attenuates cardiac ischaemia‐reperfusion injury through expansion of regulatory T cells. Cell Physiol Biochem. 2017;44:1810–1827. [DOI] [PubMed] [Google Scholar]
- 94. Whitehouse G, Gray E, Mastoridis S, Merritt E, Kodela E, Yang JHM, Danger R, Mairal M, Christakoudi S, Lozano JJ, Macdougall IC, Tree TIM, Sanchez‐Fueyo A, Martinez‐Llordella M. IL‐2 therapy restores regulatory T‐cell dysfunction induced by calcineurin inhibitors. Proc Natl Acad Sci USA. 2017;114:7083–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fernandes P, de Mendonca Oliveira L, Bruggemann TR, Sato MN, Olivo CR, Arantes‐Costa FM. Physical exercise induces immunoregulation of TREG, M2, and pDCs in a lung allergic inflammation model. Front Immunol. 2019;10:854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia‐Vazquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL, Pelletier J, Piccirillo CA, Krawczyk CM, Divangahi M, Jones RG. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42:41–54. [DOI] [PubMed] [Google Scholar]
- 97. Yang K, Blanco DB, Neale G, Vogel P, Avila J, Clish CB, Wu C, Shrestha S, Rankin S, Long L, Kc A, Chi H. Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signalling. Nature. 2017;548:602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. He N, Fan W, Henriquez B, Yu RT, Atkins AR, Liddle C, Zheng Y, Downes M, Evans RM. Metabolic control of regulatory T cell (Treg) survival and function by Lkb1. Proc Natl Acad Sci USA. 2017;114:12542–12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhu R, Chen L, Xiong Y, Wang N, Xie X, Hong Y, Meng Z. An upregulation of CD8(+)CD25(+)Foxp3(+) T cells with suppressive function through interleukin 2 pathway in pulmonary arterial hypertension. Exp Cell Res. 2017;358:182–187. [DOI] [PubMed] [Google Scholar]