

Abstract
Background
Uric acid (UA) is a plasmatic antioxidant that has possible effects on blood pressure. The effects of UA on endothelial function are unclear. We hypothesize that endothelial function is not impaired unless significant UA depletion is achieved through selective xanthine oxidase inhibition with febuxostat and recombinant uricase (rasburicase).
Methods and Results
Microvascular hyperemia, induced by iontophoresis of acetylcholine and sodium nitroprusside, and heating‐induced local hyperemia after iontophoresis of saline and a specific nitric oxide synthase inhibitor were assessed by laser Doppler imaging. Blood pressure and renin‐angiotensin system markers were measured, and arterial stiffness was assessed. CRP (C‐reactive protein), allantoin, chlorotyrosine/tyrosine ratio, homocitrulline/lysine ratio, myeloperoxidase activity, malondialdehyde, and interleukin‐8 were used to characterize inflammation and oxidative stress. Seventeen young healthy men were enrolled in a randomized, double‐blind, placebo‐controlled, 3‐way crossover study. The 3 compared conditions were placebo, febuxostat alone, and febuxostat together with rasburicase. The allantoin (μmol/L)/UA (μmol/L) ratio differed between sessions (P<0.0001). During the febuxostat‐rasburicase session, heating‐induced hyperemia became altered in the presence of nitric oxide synthase inhibition; and systolic blood pressure, angiotensin II, and myeloperoxidase activity decreased (P≤0.03 versus febuxostat). The aldosterone concentration decreased in the febuxostat‐rasburicase group (P=0.01). Malondialdehyde increased when UA concentration decreased (both P<0.01 for febuxostat and febuxostat‐rasburicase versus placebo). Other parameters remained unchanged.
Conclusions
A large and short‐term decrease in UA in humans alters heat‐induced endothelium‐dependent microvascular vasodilation, slightly reduces systolic blood pressure through renin‐angiotensin system activity reduction, and markedly reduces myeloperoxidase activity when compared with moderate UA reduction. A moderate or severe hypouricemia leads to an increase in lipid peroxidation through loss of antioxidant capacity of plasma.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT03395977.
Keywords: febuxostat, nitric oxide synthase, rasburicase, renin‐angiotensin‐aldosterone system
Subject Categories: Physiology, Oxidant Stress, Endothelium/Vascular Type/Nitric Oxide
Clinical Perspective
What Is New?
Uric acid (UA) is a well‐known antioxidant in the plasma but may also be a deleterious oxidant product when its concentration increases.
We studied and compared the effects of moderate and extreme reduction in UA concentration on microvascular function and oxidative stress. Microvascular function was assessed by laser Doppler imaging.
We explored the physiological characteristics between UA, blood pressure, and the renin‐angiotensin system by measuring different components of the renin‐angiotensin system; and we developed a human model of severe hypouricemia, which is more reliable than using animal models where purine metabolism is completely different from humans.
What Are the Clinical Implications?
In normal men, unfavorable effects of extreme UA reduction on heating‐induced hyperemia are only seen when nitric oxide synthase is inhibited, and microvascular effects of UA might thus become more apparent in older or diseased subjects with a dysfunctional endothelium.
In this study, UA withdrawal also suggests a positive link between uricemia, systolic blood pressure, and angiotensin II.
Last, myeloperoxidase activity decreased while lipid peroxidation increased; thus, the effects of UA suppression on oxidative stress markers in humans are complex and nonuniform.
Introduction
The role of uric acid (UA) in cardiovascular disease remains controversial; however, some consider it a real cardiovascular risk factor.1 A J‐shaped relationship between UA concentration and cardiovascular events has been described in hypertensive patients (PIUMA [Progetto Ipertensione Umbria Monitoraggio Ambulatoriale] study), suggesting that hyperuricemia and hypouricemia are both associated with an increased cardiovascular risk.2, 3 UA represents >50% of the total antioxidant plasmatic capacity.4, 5 Among all cardiovascular diseases, the strongest association is with hypertension, where UA seems to play a role in renal renin‐angiotensin system (RAS) activation.4, 6, 7 Impaired flow‐mediated dilation was observed in uric acid transporter 1 (URAT1) homozygote‐mutated Japanese subjects with a UA concentration <47.6 μmol/L (<0.8 mg/dL).8, 9 UA alone could not account for this condition because of the feedback effect of UA on xanthine oxidoreductase (XOR) activity. Indeed, in addition to the reduction in antioxidant capacity, a reduction in UA concentration will supposedly lead to an increase in XOR activity and, thus, reactive oxygen species (ROS) production.10, 11, 12, 13 In contrast, inhibition of xanthine oxidase (XO) with allopurinol improved endothelial function in patients with heart failure compared with probenecid, a URAT1 blocker.14 Similarly, despite a 61% reduction in UA concentration, rasburicase alone failed to improve endothelial function in patients with type 2 diabetes mellitus.15 In general, lower UA levels may alter plasma antioxidant capacity. As a result of the inhibitory feedback of UA on XOR activity, however,10, 11 URAT1 mutation, probenecid, and rasburicase may also enhance XOR activity and ROS production. The latter is not seen with allopurinol, which inhibits XOR. These indirect comparisons suggest differential effects of XOR‐mediated ROS production and UA‐related plasma antioxidant properties on endothelial function. The restoration of endothelial function by UA infusion in patients with type 1 diabetes mellitus and smokers, as assessed by venous occlusion plethysmography, provides further evidence that UA is necessary for endothelial balance.16
The XO isoform of the XOR enzyme is involved in purine metabolism and production of ROS.17 XO, along with nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and decoupled endothelial nitric oxide (NO) synthase (NOS), is a major contributor to ROS production.18 Endothelial dysfunction is the result of impaired NO bioavailability, resulting from the imbalance of antioxidant and oxidant factors (ROS).
More potent and better tolerated therapies may allow patients to experience lower levels of UA than previously. There is growing concern that low levels of UA may prove deleterious. The recent CARES (Cardiovascular Safety of Febuxostat and Allopurinol in Patients with Gout and Cardiovascular Morbidities) trial showed that more cardiovascular deaths occurred in patients treated with febuxostat compared with those treated with allopurinol.19 A larger proportion of febuxostat‐treated patients reached a UA concentration <300 μmol/L (<5 mg/dL). This trial, however, had several methodological pitfalls. Our study aims to provide a timely, in‐depth insight of the cardiovascular consequences of acute hypouricemia. We believe that studying the effects of different levels of UA reduction is of importance because of lack of data about the threshold to initiate urate‐lowering therapies,1 especially as UA has been suggested to increase the risk of hypertension in younger subjects.4
Therefore, in this randomized, double‐blind, placebo‐controlled, 3‐way crossover study, febuxostat (a highly specific nonpurinergic XOR inhibitor) was used to prevent ROS production related to UA production. Concomitantly, recombinant uricase (rasburicase) was given to achieve extremely and unprecedented low levels of UA.
This study is, to the best of our knowledge, the first to achieve an acute hypouricemia with the aim to determine the effects of UA on microvascular function, independent of XOR, and to compare the effects of acute moderate and severe hypouricemia. We hypothesized that microvascular function would not become impaired until extremely low UA concentrations were achieved. Microvascular function was assessed by acetylcholine iontophoresis induced hyperemia as well as by local heating after N‐nitro‐L‐arginine methyl ester (L‐NAME; a specific NOS inhibitor) iontophoresis. Sodium nitroprusside (SNP) iontophoresis induced hyperemia was used to differentiate endothelium‐dependent and endothelium‐independent responses. Arterial stiffness, CRP (C‐reactive protein), allantoin, chlorotyrosine/tyrosine ratio, homocitrulline/lysine ratio, myeloperoxidase activity, malondialdehyde, and interleukin‐8 characterized arterial function, inflammation, and oxidative stress during the different experimental sessions.
Methods
The data that support the findings of this study are available from the corresponding author on request.
Population and Design
The protocol was reviewed and approved by Erasme Hospital's ethics committee (reference P2017/296; NCT03395977). The study was conducted in accordance with the principles outlined in the Declaration of Helsinki. Written informed consent was obtained from all participants.
Between January and June 2018, 22 healthy male subjects with a normal physical examination were recruited at the Erasme Hospital (Brussels, Belgium). Female subjects were not included in the study because of the uricosuric effect of estrogens in premenopausal women.20 Smokers and alcohol consumers were excluded, and no long‐term medication (even vitamin supplementation) was allowed. Patients with glucose‐6‐phosphate dehydrogenase deficiency were excluded.
Participants were enrolled in a randomized, double‐blind, placebo‐controlled, 3‐way (A, B, and C) crossover study with a 10‐day washout period between experimental sessions. Sessions were as follow: A, placebo given orally, intravenous saline; B, febuxostat given orally, intravenous saline; and C, febuxostat given orally, intravenous rasburicase (Figure 1).
Figure 1.
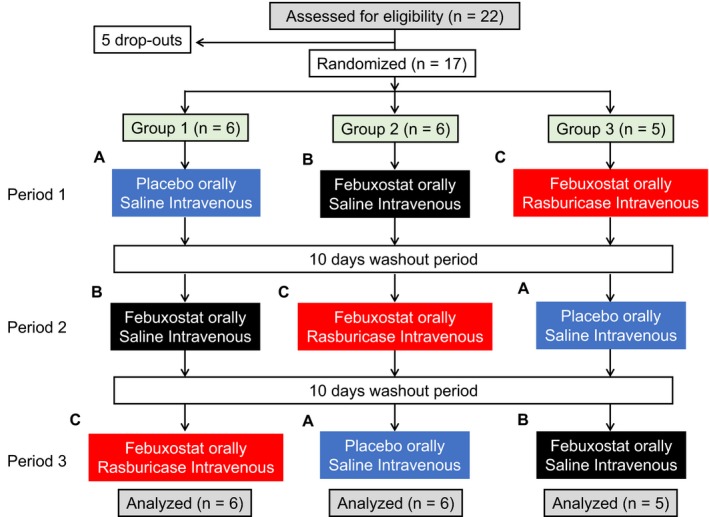
Flowchart diagram. Twenty‐two healthy volunteers were enrolled, but 5 did not complete the study. Seventeen participants were randomized to 3 groups. Each group was composed of 3 treatments separated by a 10‐day washout period. Treatments were placebo and intravenous saline (A), febuxostat given orally and intravenous saline (B), and febuxostat given orally and intravenous rasburicase (C).
For each session, participants took oral pills for 3 days (placebo or febuxostat, 240 mg/d). On the third day, patients were infused with 0.9% saline solution or 3 mg of rasburicase for 30 minutes. Pills (placebo or febuxostat packaged in identical white capsules) and infusions were prepared by a pharmacist in accordance with the randomization protocol (3 treatment orders were established: ABC, BCA, or CAB). Order sequences were determined before inclusion and allocated to participants after enrollment. Volunteers and investigators were blinded to the medications administered during the study. The highest tested dosage of febuxostat was administered to ensure XOR would be completely blocked.21 A 24‐hour washout period between rasburicase or saline infusion and measurements was observed to achieve the lowest concentration of UA, as previously described.15 Investigations (microvascular function, hemodynamic parameters, and arterial stiffness) were performed on day 4 in a quiet room maintained at 22°C±2°C. These measurements were performed at the same time of day for all subjects.
Blood samples were collected at recruitment and on the fourth day of each session. Venous samples for UA measurement were transported on ice to inhibit potential rasburicase activity. Blood analyses were performed in our hospital directly after collection. Serum and plasma samples (after centrifugation at 4000g for 15 minutes at 20°C) were isolated and stored at −80°C in our biobank (BE_BERA1; Biobanque Hôpital Erasme–Université Libre de Bruxelles; BE_NBWB1; Biothèque Wallonie Bruxelles; BBMRI‐ERIC) until analysis.
Drugs
Lactose given orally and 0.9% saline solution administered intravenously were used as placebo. The active oral and intravenous drugs included 240 mg of febuxostat (Adenuric; Menarini, Florence, Italy) and 3 mg of rasburicase (Fasturtec; Sanofi, Paris, France) reconstituted in 50 mL sterile 0.9% saline solution, respectively.
Acetylcholine, SNP, L‐NAME, and saline solution were used for iontophoresis, as previously described.22, 23 Acetylcholine‐ and SNP‐induced hyperemias were used to assess the endothelium‐dependent and endothelium‐independent vasomotor response, respectively. Heating‐induced hyperemias after L‐NAME and saline iontophoresis were also used to assess the endothelial vasomotor response. L‐NAME facilitated the analysis of the non–NO‐mediated vasodilation response to heating. This noninvasive technique is central to evaluating peripheral endothelial function.24
Primary Outcome
Endothelial function
Microcirculatory vasomotor function was assessed by a laser Doppler Imager (Moor Instruments Ltd, Axminster, UK) that measured blood flow in a skin surface area of 3.8 cm², as previously described.22 Twelve scans were obtained for each test; the first 2 scans corresponded to baseline cutaneous flow. Thirty minutes before the measurements being taken, 5% Emla cream (lidocaine, 2.5%, and prilocaine, 2.5%; AstraZeneca, London, UK) was applied to the anterior surface of both forearms to prevent nonspecific vasodilatation induced by the electric current. We performed acetylcholine and SNP hyperemias, where molecules were administered percutaneously using iontophoresis, on one forearm. On the other, the skin was heated to 44°C using dedicated skin heater electrodes and a temperature monitor (SH02; Moor Instruments Ltd) after L‐NAME and saline iontophoresis.22 Data were expressed in perfusion units. The heating response is biphasic and depends on the endothelial system, adrenergic nerves, and sensory nerves.25 The first phase is mainly mediated by transient axon reflex vasodilatation and less by NO. The second (plateau) phase is mainly related to NO release and the endothelium.25 Therefore, we only compared the late‐phase response between sessions in terms of the skin response to heat, as in our previous studies.22
Secondary Outcomes
Biological measures
A venous blood sample was used to collect biological data (hematologic, renal, and hepatic functions, lipid profile, and glycemia). Homocitrulline/lysine and 3‐chlorotyrosine/tyrosine ratios, allantoin, interleukin‐8, myeloperoxidase activity, and malondialdehyde were used to assess inflammation and oxidative stress. 3‐Chlorotyrosine is a specific product of myeloperoxidase activity, and the formation of homocitrulline may be catalyzed by myeloperoxidase. Oxidation of the amino acid residues tyrosine and lysine leads to formation of 3‐chlorotyrosine and homocitrulline, respectively. These products were measured by acid hydrolysis, derivatization, and liquid chromatography–tandem mass spectrometry (Data S1 provides further information).26 Allantoin is a marker of oxygen free radical load in humans not receiving rasburicase and results from a nonenzymatic reaction between UA and ROS. Allantoin was also measured by liquid chromatography–tandem mass spectrometry (Data S1). Myeloperoxidase is a major oxidative enzyme, and its activity was measured by the Specific Immunological Extraction Followed by Enzymatic Detection (SIEFED) method.27 Interleukin‐8 is a well‐known marker of inflammation and was measured by ELISA (BD Biosciences). Malondialdehyde, an end product formed through the degradation of certain lipid peroxidation products, was detected using the thiobarbituric acid reactive substances technique, as previously described.28
ELISA kits were used to measure arachidonic and epoxyeicosatrienoic acids (AA and EET, respectively; MyBiosource). EET, a product of AA, is implicated in the non–NO‐mediated vasodilation response to heating, most apparent when NOS is inhibited by L‐NAME.29
The activity of the RAS was evaluated in a post hoc analysis secondary to the effects of UA concentration on blood pressure (BP). To fully understand this system, we measured angiotensin II, renin concentration, plasma renin activity, aldosterone, and angiotensin‐converting enzyme using different ELISA kits (angiotensin‐converting enzyme and aldosterone: Abcam, UK; renin concentration: Creative Diagnostics; plasma renin activity: Alpco; and angiotensin II: Merck, Germany).
Hemodynamic parameters and arterial stiffness
BP (mobile sphygmomanometer; Welch Allyn; Hill‐Rom, Batesville, IN), heart rate, and saturation of peripheral oxygen were measured for each session (Capnostream 20; Oridion; Medtronic, Minneapolis, MN). Carotid‐femoral pulse wave velocity, and radial augmentation index (AIx) and heart rate‐corrected augmentation index (AIx@75) were used to assess the aortic stiffness and were measured through carotid, femoral and radial artery applanation tonometry, respectively. Pulse wave velocity and augmentation index were assessed noninvasively using a fully automated device (SphygmoCor; Atcor Medical, Sydney, NSW, Australia), as previously described.30
Adverse effects
Participants were advised that gastrointestinal and other less common adverse effects could occur. All participants were requested to report diarrhea, abdominal pain, headache, nausea, fatigue, hot spells, or any other complaints to the principal investigator.
Statistical Analysis
Variables were expressed as mean±SEM when normally distributed; otherwise, they were expressed as median (quartile 1; quartile 3). Normality was assessed using the Shapiro‐Wilk test. The data analysis for the crossover design was performed using a global linear model for repeated measures (mixed ANOVA). The 3 conditions (placebo, febuxostat, and febuxostat‐rasburicase) were used as within‐subjects factor, and the order sequence of the sessions (ABC, BCA, or CAB) was used as between‐subjects factor to assess the carryover effect. The absence of carryover effect was assumed when no statistical significance was found for the order sequences. Data that were not normally distributed were analyzed using the nonparametric Friedman test for repeated measures. Binary variables, such as adverse effects, were analyzed using a χ2 test. Bonferroni correction was applied for multiple comparisons. P<0.05 was considered statistically significant. All statistical analyses were performed using SPSS, version 22.0 (IBM Corporation, Armonk, NY). N=17 for all measures, except for EET and AA determination, as a result of aberrant values, where N=16. Placebo‐corrected values for the febuxostat and febuxostat‐rasburicase groups were calculated by the subtraction of placebo value to febuxostat and febuxostat‐rasburicase values, respectively. They were analyzed using a paired t test when statistical differences were found in variables related to microvascular function, BP, or RAS. Pearson correlation test was used to assess the relationship between variables. For heat‐induced hyperemia after L‐NAME iontophoresis, a period by period (ie, every 2.5 minutes) comparison was performed between the 3 groups and between the 2 groups when data were expressed as placebo‐corrected values. Sample size was computed with G3*Power, version 3.1.9.2 (Kiel University, Germany). The effect size from a previous study in our laboratory22 enabled us to estimate a total sample size of 12 for an α error of 5% and a power of 80%. Twenty‐two subjects were enrolled because the rigorous study protocol made us anticipate that 40% of the participants might not complete the study in the worst‐case scenario.
Results
Five participants left the study because of time constraints; their data were not included in the present article. Seventeen subjects were randomized to all 3 treatment sequences (Figure 1).
Baseline Characteristics of the Participants
UA concentration in our population was within the normal range (Table 1).
Table 1.
Characteristics of the Study Participants at Baseline
| Characteristics | Value |
|---|---|
| Age, ya | 23 (22; 25) |
| BMI, kg/m2 a | 23.9 (22.9; 26.0) |
| Platelets, ×10³/μL | 224.1±13.6 |
| G6PD, U/g of hemoglobin | 10.0±0.3 |
| CRP, nmol/La | 5.2 (4.8; 12.3) |
| Urea, mmol/L | 5.0±0.2 |
| Creatinine, μmol/La | 88.4 (79.6; 92.8) |
| Urea/creatinine ratio | 59.8±3.2 |
| Sodium, mmol/La | 142.0 (140.5; 142.5) |
| Potassium, mmol/L | 3.9±0.0 |
| Chloride, mmol/L | 101.3±0.3 |
| Uric acid, μmol/L | 334.2±14.7 |
| Calcium, mmol/L | 2.4±0.0 |
| Phosphorus, mmol/L | 1.0±0.0 |
| Bilirubin, μmol/La | 10.8 (8.6; 13.2) |
| ALP, nkat/La | 1190.0 (1037.0; 1215.5) |
| GGT, nkat/La | 283.4 (200.0; 416.8) |
| ALT, nkat/L | 457.8±58.9 |
| AST, nkat/L | 356.9±20.9 |
| LDH, nkat/L | 2621.5±86.4 |
| Cholesterol, mmol/L | 4.3±0.2 |
| Triglyceride, mmol/La | 0.9 (0.6; 1.3) |
| HDL, mmol/La | 1.3 (1.3; 1.5) |
| LDL, mmol/L | 2.4±0.2 |
| Albumin, μmol/L | 70.5±1.0 |
| Glucose, mmol/L | 4.7±0.1 |
Data are given as mean±SEM or median (quartile 1; quartile 3). ALP indicates alkaline phosphatase; ALT, alanine transaminase; AST, aspartate transaminase; BMI, body mass index; CRP, C‐reactive protein; G6PD, glucose‐6‐phosphate dehydrogenase; GGT, γ‐glutamyl transpeptidase; HDL, high‐density lipoprotein; LDH, spolactate dehydrogenase; LDL, low‐density lipoprotein.
Not normally distributed.
Biological Analyses
The plasmatic concentration of UA decreased in participants treated with febuxostat and/or febuxostat‐rasburicase (P<0.0001) (Table 2). Plasmatic concentrations of urea, creatinine and the urea/creatinine ratio differed between sessions (overall P=0.04; P=0.015 and P=0.045, respectively). No difference was found between other biological variables. There was no carryover effect.
Table 2.
Biological, Adverse Effects and Hemodynamic Parameters
| Measures | Placebo (n=17) | Febuxostat (n=17) | Febuxostat‐Rasburicase (n=17) | P Value | |||
|---|---|---|---|---|---|---|---|
| ANOVA | Placebo vs Febuxostat | Placebo vs Febuxostat‐Rasburicase | Febuxostat vs Febuxostat‐Rasburicase | ||||
| Biological | |||||||
| Uric acid, μmol/L | 321.2±13.6 | 126.7±10.1 | 18.2±1.7 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| Sodium, mmol/La | 142.0 (141.0; 142.0) | 142.0 (141.0; 142.0) | 141.0 (141.0; 142.3) | 1.0 | ··· | ··· | ··· |
| Potassium, mmol/L | 3.9±0.1 | 3.8±0.1 | 3.8±0.1 | 0.5 | ··· | ··· | ··· |
| Chloride, mmol/L | 101.1±0.2 | 100.6±0.3 | 101.2±0.4 | 0.3 | ··· | ··· | ··· |
| Urea, mmol/L | 4.6±0.2 | 4.8±0.2 | 4.4±0.2 | 0.04 | 0.2 | 0.9 | 0.1 |
| Creatinine, μmol/La | 79.6 (70.7; 88.4) | 88.4 (79.6; 88.4) | 88.4 (79.6; 88.4) | 0.015 | 0.09 | 0.04 | 1.0 |
| Urea/creatinine ratio | 56.6±2.7 | 56.7±2.2 | 52.0±2.8 | 0.045 | 1.0 | 0.2 | 0.2 |
| Platelets, ×10³/mm³ | 230.5±11.9 | 231.7±11.2 | 228.1±12.7 | 0.9 | ··· | ··· | ··· |
| CRP, nmol/La | 4.8 (4.8; 11.9) | 5.9 (4.8; 9.5) | 4.8 (4.8; 18.1) | 0.5 | ··· | ··· | ··· |
| Adverse effects, n (%) | |||||||
| Any | 6 (35.3) | 12 (70.6) | 15 (88.2) | 0.004 | 0.12 | 0.003 | 0.6 |
| Diarrhea | 0 (0.0) | 4 (23.5) | 3 (17.6) | 0.1 | ··· | ··· | ··· |
| Abdominal pain | 1 (5.9) | 5 (29.4) | 4 (23.5) | 0.2 | ··· | ··· | ··· |
| Headache | 3 (17.6) | 4 (23.5) | 8 (47.1) | 0.1 | ··· | ··· | ··· |
| Nausea | 0 (0.0) | 3 (17.6) | 3 (17.6) | 0.2 | ··· | ··· | ··· |
| Fatigue | 0 (0.0) | 2 (11.8) | 3 (17.6) | 0.2 | ··· | ··· | ··· |
| Hot spell | 1 (5.9) | 3 (17.6) | 5 (29.4) | 0.2 | ··· | ··· | ··· |
| Other | 1 (5.9) | 0 (0.0) | 1 (5.9) | 0.4 | ··· | ··· | ··· |
| Hemodynamic parameters | |||||||
| PWV, m/s | 5.3±0.1 | 5.5±0.1 | 5.3±0.2 | 0.2 | ··· | ··· | ··· |
| AIx, % | 0.6±2.6 | 0.4±1.8 | 1.5±1.8 | 0.9 | ··· | ··· | ··· |
| AIx@75, % | −7.0±2.4 | −6.2±1.8 | −6.1±1.9 | 0.9 | ··· | ··· | ··· |
| Systolic BP, mm Hg | 109.4±2.5 | 111.2±1.6 | 106.5±2.1 | 0.023 | 0.9 | 0.4 | 0.01 |
| Diastolic BP, mm Hg | 69.2±1.9 | 69.6±1.5 | 67.2±1.7 | 0.5 | ··· | ··· | ··· |
| HR, /min | 61.7±1.3 | 63.6±1.0 | 62.9±1.6 | 0.3 | ··· | ··· | ··· |
| SpO2, % | 97.7±0.5 | 97.7±0.3 | 97.5±0.4 | 0.4 | ··· | ··· | ··· |
Data are given as mean±SEM or median (quartile 1; quartile 3). AIx and AIx@75 indicate augmentation index and heart rate‐corrected augmentation index; BP, blood pressure; CRP, C‐reactive protein; HR, heart rate; PWV, pulse wave velocity; SpO2, saturation of peripheral oxygen.
Not normally distributed.
Endothelial Function
Acetylcholine‐ and SNP‐induced hyperemias were similar between groups. Overall area under the curve was 4336.0±233.8, 4312.5±203.8, and 4387.2±258.3 perfusion units and 3709.7±248.9, 3657.0±236.4, and 3534.2±331.7 perfusion units for the placebo, febuxostat, and febuxostat‐rasburicase groups, respectively (P=0.89 and P=0.58, respectively) (Figure 2A and 2B).
Figure 2.
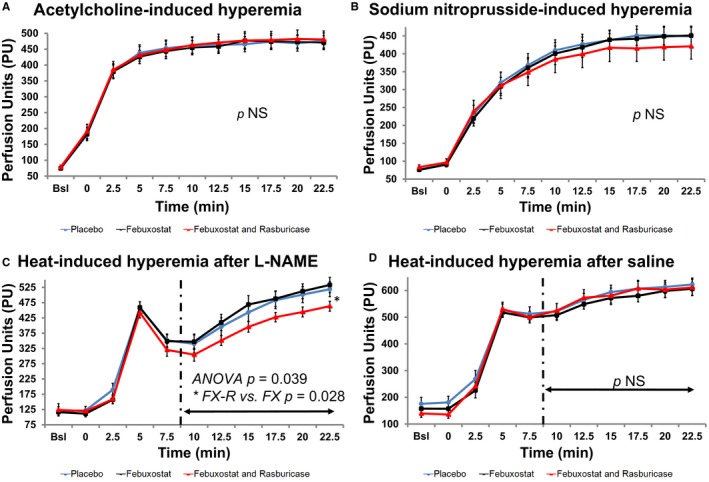
Endothelium function assessment. A and B, Acetylcholine‐ and sodium nitroprusside (SNP)–induced hyperemias were similar in both groups (placebo, febuxostat, and febuxostat and rasburicase). C and D, Heating‐induced hyperemia after pretreatment with N‐nitro‐L‐arginine methyl ester (L‐NAME; C) was impaired in the febuxostat‐rasburicase group compared with the febuxostat group. The vasodilation response was similar between groups after pretreatment with saline (D). NS indicates not significant; PU, perfusion unit.
Late‐phase area under the curve was 2682.7±160.1, 2759.3±147.7, and 2386.4±99.1 perfusion units after pretreatment by L‐NAME iontophoresis for the placebo, febuxostat, and febuxostat‐rasburicase groups, respectively (overall P=0.039; febuxostat versus febuxostat‐rasburicase P=0.028) (Figures 2C and 3). No difference was found between groups in terms of heating‐induced hyperemia when pretreated with saline iontophoresis (P=0.52) (Figure 2D). Carryover effects were not observed.
Figure 3.
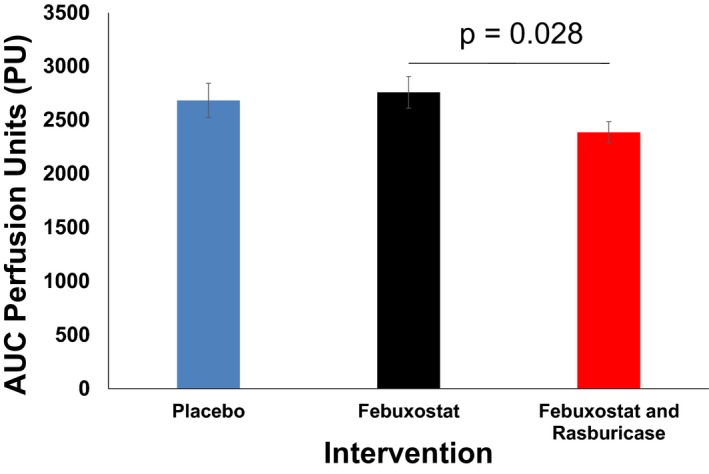
Histogram of late‐phase areas under curves (AUCs) of heating‐induced skin vasodilatation after N‐nitro‐L‐arginine methyl ester (L‐NAME) iontophoresis in the 3 groups (placebo, febuxostat, and febuxostat and rasburicase). Vasodilation response was impaired in the febuxostat‐rasburicase group compared with the febuxostat group.
The analysis of the placebo‐corrected changes between the febuxostat and febuxostat‐rasburicase groups yielded similar results (Data S1).
Markers of Oxidative Stress
Allantoin concentration decreased with febuxostat and, as expected, increased significantly with febuxostat‐rasburicase (P<0.0001) (Table 3). Myeloperoxidase activity decreased when the UA concentration was extremely low, and malondialdehyde increased when the UA concentration decreased (both overall P=0.001). No difference was found with regard to the 3‐chlorotyrosine/tyrosine and homocitrulline/lysine ratios. Carryover effects were not present. Interleukin‐8 concentration was under the sensitivity threshold (<1 pg/mL) for all subjects except one (Data S1).
Table 3.
Markers of Oxidative Stress and RAS
| Measures | Placebo (n=17) | Febuxostat (n=17) | Febuxostat‐Rasburicase (n=17) | P Value | |||
|---|---|---|---|---|---|---|---|
| ANOVA | Placebo vs Febuxostat | Placebo vs Febuxostat‐Rasburicase | Febuxostat vs Febuxostat‐Rasburicase | ||||
| Allantoin, μmol/L | 0.8±0.1 | 0.4±0.0 | 24.9±1.6 | <0.0001 | 0.003 | <0.0001 | <0.0001 |
| Chlorotyrosine/tyrosine ratio, ×10−5 a | 1.5 (0.3; 5.5) | 1.7 (0.5; 6.7) | 2.0 (0.4; 7.0) | 0.9 | ··· | ··· | ··· |
| Homocitrulline/lysine ratio, ×10−5 | 55.9±2.6 | 54.7±2.6 | 55.3±2.7 | 0.5 | ··· | ··· | ··· |
| Myeloperoxidase activity, mU/mLa | 1.2 (1.0; 1.6) | 1.7 (0.8; 1.9) | 0.5 (0.4; 0.8) | 0.001 | 0.3 | 0.027 | 0.03 |
| Malondialdehyde, μmol/La | 0.3 (0.3; 0.4) | 0.5 (0.3; 0.6) | 0.5 (0.4; 0.7) | 0.001 | 0.001 | 0.006 | 1.0 |
| PRA, ng/mL per ha | 1.1 (1.0; 1.7) | 1.7 (1.1; 2.4) | 1.2 (0.9; 2.0) | 0.2 | ··· | ··· | ··· |
| Renin concentration, pg/mLa | 18.1 (13.0; 21.9) | 22.3 (16.1; 25.5) | 17.6 (12.3; 32.6) | 0.6 | ··· | ··· | ··· |
| Aldosterone, pg/mLa | 52.8 (39.9; 70.3) | 66.8 (29.0; 90.0) | 35.0 (27.0; 54.0) | 0.01 | 1.0 | 0.1 | 0.08 |
| Aldosterone/PRA ratio | 55.3±18.7 | 45.0±7.5 | 37.3±5.7 | 0.048 | 0.5 | 0.049 | 0.9 |
| Aldosterone/renin ratioa | 3.1 (2.5; 4.8) | 2.7 (1.5; 4.3) | 2.3 (1.2; 3.5) | 0.08 | ··· | ··· | ··· |
| Angiotensin II, pg/mLa | 1.8 (1.4; 2.4) | 1.9 (1.4; 2.6) | 1.7 (1.3; 2.3) | 0.007 | 1.0 | 0.3 | 0.02 |
| ACE, pg/mL | 93.8±7.9 | 94.3±7.8 | 88.0±9.3 | 0.4 | ··· | ··· | ··· |
Data are given as mean±SEM or median (quartile 1; quartile 3). ACE indicates angiotensin‐converting enzyme; PRA, plasma renin activity; RAS, renin‐angiotensin system.
Not normally distributed.
Adverse Effects
In the placebo, febuxostat, and febuxostat‐rasburicase groups, 35.3%, 68.8%, and 88.9% of participants, respectively, presented with at least one adverse effect (P=0.004) (Table 2). There was no difference between groups when adverse effects were analyzed separately.
Hemodynamic Parameters
Arterial stiffness, assessed by the pulse wave velocity and augmentation index, did not change between groups. Systolic BP was lowest in the febuxostat‐rasburicase group (overall P=0.02; febuxostat versus febuxostat‐rasburicase P=0.01) (Table 2). There was no difference in diastolic BP, heart rate, and saturation of peripheral oxygen between groups. Carryover effects were not evident.
AA and EET Pathways (Post Hoc Analyses)
The concentration of EET was 44.8 (36.6; 65.0), 55.7 (38.4; 75.2), and 45.8 (39.9; 70.6) pg/mL in the placebo, febuxostat, and febuxostat‐rasburicase sessions, respectively (overall P=0.003; placebo versus febuxostat P=0.015) (Table 4). AA concentration and the EET/AA ratio did not differ between groups.
Table 4.
AA and EET Pathways
| Measures | Placebo (n=16) | Febuxostat (n=16) | Febuxostat‐Rasburicase (n=16) | P Value | |||
|---|---|---|---|---|---|---|---|
| ANOVA | Placebo vs Febuxostat | Placebo vs Febuxostat‐Rasburicase | Febuxostat vs Febuxostat‐Rasburicase | ||||
| EET, pg/mLa | 44.8 (36.6; 65.0) | 55.7 (38.4; 75.2) | 45.8 (39.9; 70.6) | 0.003 | 0.015 | 1.0 | 0.15 |
| AA, ng/mL | 10 657.3±956.5 | 12 235.2±1235.7 | 11 689.0±877.8 | 0.2 | ··· | ··· | ··· |
| EET/AA, ×10−6 a | 4.7 (2.9; 7.1) | 4.4 (2.6; 7.0) | 4.8 (2.6; 6.6) | 0.8 | ··· | ··· | ··· |
Data are given as mean±SEM or median (quartile 1; quartile 3). AA indicates arachidonic acid; EET, epoxyeicosatrienoic acid.
Not normally distributed.
RAS (Post Hoc Analyses)
The concentration of angiotensin‐converting enzyme, renin, and plasma renin activity did not differ between sessions. On the contrary, the concentration of angiotensin II and aldosterone decreased with UA reduction (overall P=0.007; febuxostat versus febuxostat‐rasburicase P=0.02 for angiotensin II and overall P<0.01 for aldosterone) (Table 3).
Analyses of placebo‐corrected changes revealed that an additional UA reduction of 108.5 μmol/L decreased systolic BP by 4.7 mm Hg, angiotensin II by 0.24 pg/mL, and aldosterone by 21.4 pg/mL in the febuxostat‐rasburicase group compared with febuxostat alone (all P<0.05; Data S1 provide detailed values). A positive correlation between angiotensin II and aldosterone concentrations was found when we analyzed the febuxostat‐rasburicase changes corrected for placebo (Pearson test; P<0.01). No other correlations with BP or UA were found, nor in the febuxostat changes corrected for placebo.
Discussion
To the best of our knowledge, this is the first study on severe and short‐term experimentally induced hypouricemia in healthy men. Furthermore, this is the first trial to study and compare the effects of short‐term moderate and extreme reduction in UA concentration on microvascular function, oxidative stress, inflammation, and hemodynamic parameters. We used 2 different drugs to achieve an experimental condition to enable us to unravel the role of UA and XOR on the cardiovascular system. The main strengths of our study include the following: (1) its randomized, placebo‐controlled, double‐blind, 3‐way crossover design; (2) effects of marked reduction in UA were assessed in the absence of interference of ROS production by XO; and (3) microvascular function was assessed by different means, providing an insight into acetylcholine, SNP, NOS dependent and independent pathways.
The major new findings of our study include the following: (1) low UA concentrations do not impair NO‐mediated endothelial function in healthy volunteers; (2) UA is involved in non–NO‐mediated (endothelium‐derived hyperpolarizing factor [EDHF]) pathway vasodilation response to local heating; (3) low UA concentrations are accompanied by a slight reduction in systolic BP through reduction in RAS activity, especially angiotensin II and aldosterone; (4) myeloperoxidase activity is greatly reduced when UA reached its lowest concentration; and (5) lipid peroxidation increases when UA decreases.
UA and Endothelial Function in the Absence of NOS Inhibition
A reduction in UA as a result of XOR inhibition or, even more strikingly, during the addition of rasburicase did not impair NO‐mediated endothelial function, as assessed by acetylcholine iontophoresis. Similar results were found with SNP iontophoresis. ROS production during UA metabolism is dependent on the predominant XOR isoform. XO uses oxygen as an electron acceptor and is, thus, able to produce ROS.4 XO inhibition by allopurinol improved endothelial function in patients with heart failure compared with probenecid, a URAT1 blocker.14 Probenecid or other therapies that lower UA and do not block XOR, such as rasburicase, may enhance XOR activity and ROS production because of loss of UA inhibitory feedback on XOR activity. In our study, we expected to see impaired markers of endothelial function with the lowest UA concentrations. This is because of the J‐shaped relationship between UA and cardiovascular events, as reported in the PIUMA study,2 impaired flow‐mediated dilation observed in URAT1 mutated subjects,8 and concerns of an increase in mortality in the CARES trial.19 On the contrary, extremely low concentrations of UA did not alter acetylcholine‐ and SNP‐induced vasodilatation, arterial stiffness, inflammation, and markers of oxidative stress. The fact that we investigated young and healthy subjects may have played a role in these findings. Indeed, hypertensive patients in the PIUMA study were older (mean age, 51 years), whereas patients with high cardiovascular risk and gout were enrolled in the CARES trial. Dysfunctional endothelial NOS and XO isoform predominance may render such patients2, 19 more sensitive to plasma antioxidant capacity reduction. This is different from healthy subjects, in whom xanthine dehydrogenase is likely the main XOR isoform and does not produce ROS during purine metabolism. The fact that interleukin‐8 concentration was almost undetectable in our volunteers also supports this hypothesis. The absence of impairment in acetylcholine‐induced hyperemia in the febuxostat‐rasburicase group could be explained by the myeloperoxidase reduction. Indeed, myeloperoxidase is known to modulate the NO pathway in the vasculature, and myeloperoxidase‐deficient mice aortic rings do not express a reduction in acetylcholine‐induced relaxation in an acute inflammation model.31 This suggests the reduction in myeloperoxidase could falsely improve the endothelial function through decline in NO consumption. Last, the parameters in our study were investigated under different experimental conditions as the 3 aforementioned trials that studied the effects of persistent UA reduction (months to years).2, 8, 19
UA and Endothelial Function in the Presence of NOS Inhibition
A main finding of our study is the impairment of the non–NO‐mediated late thermal vasodilation response in healthy humans by short‐term UA reduction to extremely low values by a febuxostat‐rasburicase combination. During heat‐induced hyperemia, the placebo and febuxostat groups did not differ in the presence of L‐NAME. Also, the response to acetylcholine did not differ between the placebo, febuxostat, and febuxostat‐rasburicase groups. Thus, there is no reason to believe that UA levels, per se, affected the inhibition of NOS activity by L‐NAME in our study. This non–NO‐mediated response is thought to be played by the EDHF pathway. The EDHF pathway does not seem to play an important role when NOS is active. Conversely, the EDHF pathway plays a larger “backup” role in vasodilatation when NOS is inhibited or decoupled.32, 33 The skin response to local warming is 60% mediated by NO and 40% mediated by EDHF.29 Two different EDHF pathways have been described.34 The “classic pathway” involves hyperpolarization of the endothelial cell secondary to an increase in intracellular calcium, which leads to K+ channel activation. Smooth cell hyperpolarization occurs by K+ channel activation as a result of K+ release from the endothelial cell or by means of electrical transmission through gap junctions. The “nonclassic pathway” does not require hyperpolarization of the endothelial cell. Rather, factors produced by the endothelial cell (H2O2, NO, prostaglandin, and EET) act on K+ channels or transporters of the smooth muscle cell to induce hyperpolarization and cell relaxation.34 It has recently been reported that EET accounts for 50% of the EDHF response when skin is exposed to local heating.29 This might explain our findings because UA interferes with EET metabolism. Indeed, EET is a product of AA,29 and UA is known to increase AA metabolism.35, 36 The EET/AA ratio remains unchanged between groups. The relationship between EET and UA concentration, however, does not appear linear. A small reduction in UA concentration by febuxostat alone increased the EET concentration in our study (P=0.015). This might prove beneficial with regard to the vasodilation and anti‐inflammatory activity of EET.37 This effect, however, disappeared when UA concentration became extremely low (P=1.0). The relationship between the concentration of UA and EET might thus follow a reverse U‐shaped relationship. This type of association has already been described for creatinine,38 and a J‐ or U‐shaped relationship is described between UA concentration and the rate of cardiovascular events in hypertensive patients.2 Among the 4 isomers of EET (5.6, 8.9, 11.12, and 14.15 EET),39 only 11.12 and 14.15 EET were detectable by the ELISA kit used in this study. EETs act as vasodilators, anti‐inflammatory agents, and cell protectors40 and are rapidly inactivated by epoxide hydrolase.39 The detection of only 2 of 4 EET isomers, EET synthesis confined to endothelial cells,41 and its rapid metabolism by hydrolase can explain the results of this study. UA could also act on other components of the EDHF pathway not assessed here.
UA, Allantoin, and Other Markers of Oxidative Stress
Through imidazoline I‐1 receptors, intravenous allantoin has short‐term and only transient antihypertensive properties in animals and at a dose of 0.5 mg/kg.42 Allantoin in humans is a marker of oxidative stress without known active cardiovascular effects and results exclusively from the nonenzymatic interaction between plasmatic UA and oxidants.13 Thus, the lower concentration of allantoin in the febuxostat group (who did not receive rasburicase) could be explained by a reduction in UA concentration. This reduction in allantoin concentration in the febuxostat group is unlikely caused by a reduction in ROS load, as myeloperoxidase activity was similar in the placebo and febuxostat groups.
The elevated concentration of allantoin in the febuxostat‐rasburicase group reveals that UA was almost entirely degraded by rasburicase. Endothelial assessment took place 24 hours after the administration of rasburicase when a large amount of allantoin was excreted in urine. We believe the observed effects are not attributable to allantoin. There has been no effect of allantoin on endothelial function described thus far.
Myeloperoxidase activity decreased by 2‐fold in the febuxostat‐rasburicase group. UA concentration is associated with myeloperoxidase concentration and activity in patients with gout.43 We hypothesize that extreme reductions in UA concentration reduce the release of myeloperoxidase from neutrophils. This could ultimately result in a lower chlorotyrosine concentration, which was not observed in our study.
Conversely, we observed an increase in malondialdehyde when UA decreased in the febuxostat and febuxostat‐rasburicase groups. This increase in lipid peroxidation is consistent with reduced plasma antioxidant capacity, resulting from lower UA levels. This mechanism has been proposed by other in vitro studies44, 45, 46, 47 and suspected after short‐term UA infusion in acute ischemic stroke,48 whereas long‐term use of XOR inhibitors reduced malondialdehyde in patients with heart failure and diabetes mellitus when oxidative stress was supposedly high.49, 50, 51
This is different from the reduction in myeloperoxidase activity we observed in the febuxostat‐rasburicase group, suggestive of reduced intracellular oxidative stress, with myeloperoxidase being an oxidant product stored in neutrophils. A reduction in myeloperoxidase activity could have beneficial effects in reducing oxidative stress and does not seem to increase the risk of infection.31 These complex relationships between UA and oxidative stress support the oxidant‐antioxidant paradox of UA.52
UA and BP, Urea, and Creatinine
Systolic BP decreased by 4.7 mm Hg in the febuxostat‐rasburicase compared with febuxostat sessions. As previously mentioned, UA plays a role in hypertension physiological characteristics through the activation of RAS in kidneys.4 Angiotensin II was reduced by 0.24 pg/mL and aldosterone by 21.4 pg/mL in febuxostat‐rasburicase compared with febuxostat sessions in accordance with the underlying physiological characteristics.4 A positive correlation was found between the aldosterone and angiotensin II concentrations but not with BP levels or UA concentrations. The changes in the concentration of angiotensin II were consistent but modest, as would be expected in healthy subjects with a normal RAS activity and BP. Given the similar concentration of renin between groups, extremely low UA concentrations may have reduced the release of angiotensinogen from the liver, likely because of cytokine changes.53 An increase in angiotensinogen and angiotensin II in rat smooth muscle cells54 and mouse preadipocyte cells55 incubated with UA supports this hypothesis. The positive correlation between UA and urinary angiotensinogen in adolescents with primary hypertension,56 as well as with plasmatic angiotensinogen in obese hypertensive patients,57 also supports this hypothesis.
Short‐term effects on urea and creatinine were not expected in our study. Diarrhea was more prevalent during febuxostat and febuxostat‐rasburicase sessions; however, the urea/creatinine ratios remained within normal range (40–100) and did not differ between the experimental sessions. In addition, heart rate did not differ between the groups. Dehydration is, thus, unlikely to explain our findings, but intrarenal effects of hypouricemia may have played a role. A U‐shaped relationship between UA concentration and intrarenal hemodynamic parameters has been described in healthy subjects and is similar to the relationship with cardiovascular events.38 Mild hypouricemia and hyperuricemia are both associated with indirect markers of increased afferent arteriolar resistance.38 Effects of UA on kidneys may also involve the RAS.38 Whether these findings also apply to the acute UA reductions seen in our study is unclear. Even with a significant difference, the effect on renal parameters is small and values remained within normal ranges.
Other Limitations
The novel findings in our study pertain only to young male healthy adults. Finally, the time lapse between the interventions and biological measures in our study was possibly too short to demonstrate an effect on proteins, resulting in reduced sensitivity of our measures.
Conclusions
A large and short‐term decrease in UA in humans alters heat‐induced endothelium‐dependent microvascular vasodilation, slightly reduces systolic BP through reduction in RAS activity, and markedly reduces myeloperoxidase activity when compared with a moderate UA reduction. Moderate or severe hypouricemia leads to an increase in lipid peroxidation through loss of antioxidant capacity of plasma. Our study highlights the mechanism underlying the relationship between UA and microvascular function, maintenance of BP through RAS activation, and the complex pro‐oxidant/oxidant role of UA with its pro‐oxidant intracellular and antioxidant plasmatic roles.
Perspectives
Further studies are urgently needed to determine the effects of aging, endothelial dysfunction, and increased oxidative stress on our observations. This is important as such patients could be more affected by reductions in plasma antioxidant capacity. Moreover, the EDHF pathway is likely to have a larger role in vasodilation in a dysfunctional endothelium and may, thus, result in even larger impairments when patients at risk of cardiovascular disease become hypouricemic.
Sources of Funding
Benjamin De Becker is a Research Fellow—Fonds National de la Recherche Scientifique (Belgium). Funding received for this work: grants from the “Fonds National de la Recherche Scientifique”, the “Docteur et Madame René Tagnon” fund from the King Baudouin Foundation, the “Fonds pour la Chirurgie Cardiaque”, and the “Fonds Erasme” from the Erasme Hospital–Université Libre de Bruxelles (Brussels, Belgium).
The Analytical Platform of the Faculty of Pharmacy (Université Libre de Bruxelles) is supported by Fonds National de la Recherche Scientifique (Belgium) and ULB Platform.
Disclosures
Pr van de Borne's employer received honoraria for lecturing/advisory boards from Amgen, Bayer, Boehgingher‐Ingelheim, Daïchi‐Sankyo, Idorsia, Menarini, Novo Nordisk, and Sanofi. The remaining authors have no disclosures to report.
Supporting information
Data S1.
Acknowledgments
The authors express their gratitude to the “Fonds National de la Recherche Scientifique” (Belgium), the “Docteur et Madame René Tagnon” Fund (Belgium), the “Fonds pour la Chirurgie Cardiaque” (Belgium), and the “Fonds Erasme” from the Erasme Hospital–Université Libre de Bruxelles (Brussels, Belgium). The authors thank all the participants for their participation.
(J Am Heart Assoc. 2019;8:e013130 DOI: 10.1161/JAHA.119.013130.)
References
- 1. Borghi C, Rosei EA, Bardin T, Dawson J, Dominiczak A, Kielstein JT, Manolis AJ, Perez‐Ruiz F, Mancia G. Serum uric acid and the risk of cardiovascular and renal disease. J Hypertens. 2015;33:1729–1741. [DOI] [PubMed] [Google Scholar]
- 2. Verdecchia P, Schillaci G, Reboldi G, Santeusanio F, Porcellati C, Brunetti P. Relation between serum uric acid and risk of cardiovascular disease in essential hypertension: the PIUMA study. Hypertension. 2000;36:1072–1078. [DOI] [PubMed] [Google Scholar]
- 3. Kuo C‐F, See L‐C, Yu K‐H, Chou I‐J, Chiou M‐J, Luo S‐F. Significance of serum uric acid levels on the risk of all‐cause and cardiovascular mortality. Rheumatology (Oxford). 2013;52:127–134. [DOI] [PubMed] [Google Scholar]
- 4. De Becker B, Borghi C, Burnier M, van de Borne P. Uric acid and hypertension: a focused review and practical recommendations. J Hypertens. 2019;37:878–883. [DOI] [PubMed] [Google Scholar]
- 5. Johnson RJ, Sautin YY, Oliver WJ, Roncal C, Mu W, Gabriela Sanchez‐Lozada L, Rodriguez‐Iturbe B, Nakagawa T, Benner SA. Lessons from comparative physiology: could uric acid represent a physiologic alarm signal gone awry in western society? J Comp Physiol B. 2009;179:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grayson PC, Young Kim S, Lavalley M, Choi HK. Hyperuricemia and incident hypertension: a systematic review and meta‐analysis. Arthritis Care Res. 2011;63:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Feig DI. Serum uric acid and the risk of hypertension and chronic kidney disease. Curr Opin Rheumatol. 2014;26:176–185. [DOI] [PubMed] [Google Scholar]
- 8. Sugihara S, Hisatome I, Kuwabara M, Niwa K, Maharani N, Kato M, Ogino K, Hamada T, Ninomiya H, Higashi Y, Ichida K, Yamamoto K. Depletion of uric acid due to SLC22A12 (URAT1) loss‐of‐function mutation causes endothelial dysfunction in hypouricemia. Circ J. 2015;79:1125–1132. [DOI] [PubMed] [Google Scholar]
- 9. Iso T, Kurabayashi M. Extremely low levels of serum uric acid are associated with endothelial dysfunction in humans. Circ J. 2015;79:978–980. [DOI] [PubMed] [Google Scholar]
- 10. Radi R, Tan S, Prodanov E, Evans RA, Parks DA. Inhibition of xanthine oxidase by uric acid and its influence on superoxide radical production. Biochim Biophys Acta. 1992;1122:178–182. [DOI] [PubMed] [Google Scholar]
- 11. Tan S, Radi R, Gaudier F, Evans RA, Rivera A, Kirk KA, Parks DA. Physiologic levels of uric acid inhibit xanthine oxidase in human plasma. Pediatr Res. 1993;34:303–307. [DOI] [PubMed] [Google Scholar]
- 12. Becker BF, Reinholz N, Ozcelik T, Leipert B, Gerlach E. Uric‐acid as radical scavenger and antioxidant in the heart. Pflugers Arch. 1989;415:127–135. [DOI] [PubMed] [Google Scholar]
- 13. Becker BF. Towards the physiological function of uric acid. Free Radic Biol Med. 1993;14:615–631. [DOI] [PubMed] [Google Scholar]
- 14. George J, Carr E, Davies J, Belch JJF, Struthers A. High‐dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation. 2006;114:2508–2516. [DOI] [PubMed] [Google Scholar]
- 15. Waring WS, McKnight JA, Webb DJ, Maxwell SRJ. Lowering serum urate does not improve endothelial function in patients with type 2 diabetes. Diabetologia. 2007;50:2572–2579. [DOI] [PubMed] [Google Scholar]
- 16. Waring WS, Mcknight JA, Webb DJ, Maxwell SRJ. Uric acid restores endothelial function in patients with type 1 diabetes and regular smokers. Diabetes. 2006;55:3127–3132. [DOI] [PubMed] [Google Scholar]
- 17. George J, Struthers A. Role of urate, xanthine oxidase and the effects of allopurinol in vascular oxidative stress. Vasc Health Risk Manag. 2009;5:265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. [DOI] [PubMed] [Google Scholar]
- 19. White WB, Saag KG, Becker MA, Borer JS, Gorelick PB, Whelton A, Hunt B, Castillo M, Gunawardhana L. Cardiovascular safety of febuxostat or allopurinol in patients with gout. N Engl J Med. 2018;378:1200–1210. [DOI] [PubMed] [Google Scholar]
- 20. Gaffo AL, Jacobs DR, Sijtsma F, Lewis CE, Mikuls TR, Saag KG. Serum urate association with hypertension in young adults: analysis from the Coronary Artery Risk Development in Young Adults cohort. Ann Rheum Dis. 2013;72:1321–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kamel B, Graham GG, Williams KM, Pile KD, Day RO. Clinical pharmacokinetics and pharmacodynamics of febuxostat. Clin Pharmacokinet. 2017;56:459–475. [DOI] [PubMed] [Google Scholar]
- 22. Wauters A, Dreyfuss C, Pochet S, Hendrick P, Berkenboom G, Van De Borne P, Argacha JF. Acute exposure to diesel exhaust impairs nitric oxide‐mediated endothelial vasomotor function by increasing endothelial oxidative stress. Hypertension. 2013;62:352–358. [DOI] [PubMed] [Google Scholar]
- 23. Dreyfuss C, Wauters A, Adamopoulos D, Pochet S, Azarkan M, Berkenboom G, Van De Borne P, Argacha JF. L‐NAME iontophoresis: a tool to assess NO‐mediated vasoreactivity during thermal hyperemic vasodilation in humans. J Cardiovasc Pharmacol. 2013;61:361–368. [DOI] [PubMed] [Google Scholar]
- 24. Kubli S, Waeber B, Dalle‐Ave A, Feihl F. Reproducibility of laser Doppler imaging of skin blood flow as a tool to assess endothelial function. J Cardiovasc Pharmacol. 2000;36:640–648. [DOI] [PubMed] [Google Scholar]
- 25. Johnson JM, Kellogg DL. Local thermal control of the human cutaneous circulation. J Appl Physiol. 2010;109:1229–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Delporte C, Franck T, Noyon C, Dufour D, Rousseau A, Madhoun P, Desmet JM, Serteyn D, Raes M, Nortier J, Vanhaeverbeek M, Moguilevsky N, Nève J, Vanhamme L, Van Antwerpen P, Zouaoui Boudjeltia K. Simultaneous measurement of protein‐bound 3‐chlorotyrosine and homocitrulline by LC‐MS/MS after hydrolysis assisted by microwave: application to the study of myeloperoxidase activity during hemodialysis. Talanta. 2012;99:603–609. [DOI] [PubMed] [Google Scholar]
- 27. Franck T, Minguet G, Delporte C, Derochette S, Boudjeltia KZ, Van Antwerpen P, Gach O, Deby‐dupont G, Mouithys‐mickalad A, Serteyn D. An immunological method to combine the measurement of active and total myeloperoxidase on the same biological fluid and its application in finding inhibitors which interact directly with the enzyme. Free Radic Res. 2015;49:790–799. [DOI] [PubMed] [Google Scholar]
- 28. Wang Z, Forceville X, Van Antwerpen P, Piagnerelli M, Ahishakiye D, Macours P, De Backer D, Neve J, Vincent J. A large‐bolus injection, but not continuous infusion of sodium selenite improves outcome in peritonitis. Shock. 2009;32:140–146. [DOI] [PubMed] [Google Scholar]
- 29. Brunt VE, Minson CT. KCa channels and epoxyeicosatrienoic acids: major contributors to thermal hyperaemia in human skin. J Physiol. 2012;590:3523–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adamopoulos D, Argacha J‐F, Gujic M, Preumont N, Degaute J‐P, van de Borne P. Acute effects of nicotine on arterial stiffness and wave reflection in healthy young non‐smokers. Clin Exp Pharmacol Physiol. 2009;36:784–789. [DOI] [PubMed] [Google Scholar]
- 31. Eiserich JP, Baldus S, Brennan M, Ma W, Zhang C, Tousson A, Castro L, Lusis AJ, Nauseef WM, White CR, Freeman BA. Myetoperoxidase, a leukocyte‐derived vascular NO oxidase. Science. 2002;296:2391–2394. [DOI] [PubMed] [Google Scholar]
- 32. Brunt VE, Fujii N, Minson CT. Endothelial‐derived hyperpolarization contributes to acetylcholine‐mediated vasodilation in human skin in a dose‐dependent manner. J Appl Physiol. 2015;119:1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Félétou M. The Endothelium Part 2: EDHF‐Mediated Responses “The Classical Pathway.” San Rafael (CA): Morgan & Claypool Life Sciences Publishers Series INTEGRATED; 2011. [PubMed] [Google Scholar]
- 34. Edwards G, Félétou M, Weston AH. Endothelium‐derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch. 2010;459:863–879. [DOI] [PubMed] [Google Scholar]
- 35. Deby C, Deby‐Dupont G, Noël FX, Lavergne L. In vitro and in vivo arachidonic acid conversions into biologically active derivatives are enhanced by uric acid. Biochem Pharmacol. 1981;30:2243–2249. [DOI] [PubMed] [Google Scholar]
- 36. El Ridi R, Tallima H. Physiological functions and pathogenic potential of uric acid: a review. J Adv Res. 2017;8:487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fleming I. The factor in EDHF: cytochrome P450 derived lipid mediators and vascular signaling. Vascul Pharmacol. 2016;86:31–40. [DOI] [PubMed] [Google Scholar]
- 38. Uedono H, Tsuda A, Ishimura E, Nakatani S, Kurajoh M, Mori K, Uchiada J, Emoto M, Nakatani T, Inaba M. U‐shaped relationship between serum uric acid levels and intrarenal hemodynamic parameters. Am J Physiol Ren Physiol. 2017;312:F992–F997. [DOI] [PubMed] [Google Scholar]
- 39. Jia G, Durante W, Sowers JR. Endothelium‐derived hyperpolarizing factors: a potential therapeutic target for vascular dysfunction in obesity and insulin resistance. Diabetes. 2016;65:2118–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oni‐orisan A, Edin ML, Lee JA, Wells MA, Christensen ES, Vendrov KC, Lih FB, Tomer KB, Bai X, Taylor JM, Stouffer GA, Zeldin DC, Lee CR. Cytochrome P450‐derived epoxyeicosatrienoic acids and coronary artery disease in humans: a targeted metabolomics study. J Lipid Res. 2016;57:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ellinsworth DC, Sandow SL, Shukla N, Liu Y, Jeremy JY, Gutterman DD. Endothelium‐derived hyperpolarization and coronary vasodilation: diverse and integrated roles of epoxyeicosatrienoic acids, hydrogen peroxide and gap junctions. Microcirculation. 2017;23:15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen MF, Tsai JT, Chen LJ, Wu TP, Yang JJ, Te Yin L, Yang YL, Chiang TA, Lu HL, Wu MC. Antihypertensive action of allantoin in animals. Biomed Res Int. 2014;2014:690135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stamp LK, Turner R, Khalilova IS, Zhang M, Drake J, Forbes LV, Kettle AJ. Myeloperoxidase and oxidation of uric acid in gout: implications for the clinical consequences of hyperuricaemia. Rheumatology (Oxford). 2014;53:1958–1965. [DOI] [PubMed] [Google Scholar]
- 44. Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant‐ and radical‐caused aging and cancer: a hypothesis. Proc Natl Acad Sci USA. 1981;78:6858–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishida Y. Inhibition of lipid peroxidation by methylated analogues of uric acid. J Pharm Pharmacol. 1991;43:885–887. [DOI] [PubMed] [Google Scholar]
- 46. Stinefelt B, Leonard SS, Blemings KP, Shi X, Klandorf H. Free radical scavenging, DNA protection, and inhibition of lipid peroxidation mediated by uric acid. Ann Clin Lab Sci. 2005;35:37–45. [PubMed] [Google Scholar]
- 47. Muraoka S, Miura T. Inhibition by uric acid of free radicals that damage biological molecules. Pharmacol Toxicol. 2003;93:284–289. [DOI] [PubMed] [Google Scholar]
- 48. Amaro S, Soy D, Obach V, Cervera A, Planas AM, Chamorro A. A pilot study of dual treatment with recombinant tissue plasminogen activator and uric acid in acute ischemic stroke. Stroke. 2007;38:2173–2175. [DOI] [PubMed] [Google Scholar]
- 49. Farquharson CAJ, Butler R, Hill A, Belch JJF, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation. 2002;106:221–226. [DOI] [PubMed] [Google Scholar]
- 50. Butler R, Morris AD, Belch JJF, Hill A, Struthers AD. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension. 2000;35:746–751. [DOI] [PubMed] [Google Scholar]
- 51. Desco M‐C, Asensi M, Ma R, Pallardo FV, Sastre J. Xanthine oxidase is involved in free radical production in type 1 diabetes. Diabetes. 2001;51:1118–1124. [DOI] [PubMed] [Google Scholar]
- 52. Sautin YY, Johnson RJ. Uric acid: the oxidant‐antioxidant paradox. Nucleosides Nucleotides Nucleic Acids. 2008;27:608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Satou R, Penrose H, Navar LG. Inflammation as a regulator of the renin‐angiotensin system and blood pressure. Curr Hypertens Rep. 2018;20:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Corry DB, Eslami P, Yamamoto K, Nyby MD, Makino H, Tuck ML. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin‐angiotensin system. J Hypertens. 2008;26:269–275. [DOI] [PubMed] [Google Scholar]
- 55. Wu B, Hao Y, Shi J, Geng N, Li T, Chen Y, Sun Z, Zheng L, Li H, Li N, Zhang X, Sun Y. Association between xanthine dehydrogenase tag single nucleotide polymorphisms and essential hypertension. Mol Med Rep. 2015;12:5685–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuroczycka‐saniutycz E, Wasilewska A. Urinary angiotensinogen as a marker of intrarenal angiotensin II activity in adolescents with primary hypertension. Pediatr Nephrol. 2013;28:1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang J, Zhang Y, Deng W, Chen B. Elevated serum uric acid is associated with angiotensinogen in obese patients with untreated hypertension. J Clin Hypertens. 2014;16:569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.