

Abstract
Background
Alexander disease is caused by dominantly acting mutations in glial fibrillary acidic protein (GFAP), the major intermediate filament of astrocytes in the central nervous system.
Main body
In addition to the sequence variants that represent the origin of disease, GFAP accumulation also takes place, together leading to a gain-of-function that has sometimes been referred to as “GFAP toxicity.” Whether the nature of GFAP toxicity in patients, who have mixtures of both mutant and normal protein, is the same as that produced by simple GFAP excess, is not yet clear.
Conclusion
The implications of these questions for the design of effective treatments are discussed.
Keywords: GFAP, Astrocyte, Antisense oligonucleotides
Background
Alexander disease, once a minor disorder known primarily to neuropathologists and the small number of neurologists who followed the leukodystrophies, has now gained increased attention as the first well-documented example of a primary disease of astrocytes. Since the first case report by W. Stewart Alexander [1], its recognition as a distinct entity evolved over time (prolonged due to the rarity of the disease). By the late 1960s, the condition had been recognized in both children and adults, although its origin remained a mystery. A genetic basis was presumed, but not identified. The breakthrough came from the world of mouse genetics. In studies originally designed to study the role of intermediate filaments in reactive astrocytosis (or “gliosis”), transgenic mouse models were designed to force over-expression of the major astrocyte intermediate filament, GFAP. The surprising result was that astrocytes in these mice formed the hallmark protein aggregates of Alexander disease—Rosenthal fibers [2]. Mice engineered to have the highest levels of GFAP died within weeks after birth. Although these mice did not have a leukodystrophy, the link between GFAP and Rosenthal fibers provided sufficient rationale to pursue GFAP as a candidate gene for the disease [3]. We and others quickly found that nearly all Alexander disease patients carried heterozygous missense mutations in the coding region of GFAP, and that such mutations could account for all forms of the disease [4, 5].
The dominant nature of the GFAP variants, coupled with the minimal phenotype associated with complete GFAP deficiency as illustrated in mouse knockouts, supports the hypothesis that Alexander disease is a gain-of-function disease [6, 7]. Indeed, no null variants have ever been found in human patients. That the hallmark aggregate, Rosenthal fibers, could be induced by forced over-expression, and that this by itself could be lethal (in the mouse), also led to the idea of GFAP “toxicity” (due to GFAP excess) as a unifying hypothesis to explain how mutations cause disease. However, focusing only on GFAP excess is a mistake. The goal of this brief review is to stress the broader concept that the initiating event in disease pathogenesis must be mutant GFAP, and that this occurs prior to any change in levels. Any subsequent rise in total GFAP would then act to exacerbate the disease process.
Main text
GFAP levels are indeed elevated in Alexander disease, and one important question is why? Given the significant tissue damage that often exists, along with the expected reactive response by astrocytes, it is no surprise that GFAP levels rise. In a series of six patients, Walker et al. [8] showed increased levels of protein that at least roughly corresponded to the severity of disease (as defined by age of onset). Increased levels of GFAP mRNA had previously been documented for two patients by Hageman et al. [9], and so one can assume that increased synthesis is at least one mechanisms contributing to the overall change in levels. Mouse models engineered to carry a disease-associated variant in their endogenous Gfap gene also display increased levels of both mRNA and protein [10]. Using luciferase reporter lines of mice that serve as indirect monitors of the murine Gfap promoter, Jany et al. [11] found that mutant mice dramatically increase promoter activity during the second postnatal week, and in the absence of any significant pathology (Fig. 1). Hence, we believe this change in GFAP expression reflects an early and spontaneous alteration of astrocyte function, equivalent to activation of other downstream stress pathways, which in this particular instance unfortunately upregulates expression of the very protein that is causative for disease. A recent study using a transgenic mouse expressing the human R239H variant suggests that abnormal calcium signaling may be a key factor contributing to the upregulation of GFAP [12].
Fig. 1.
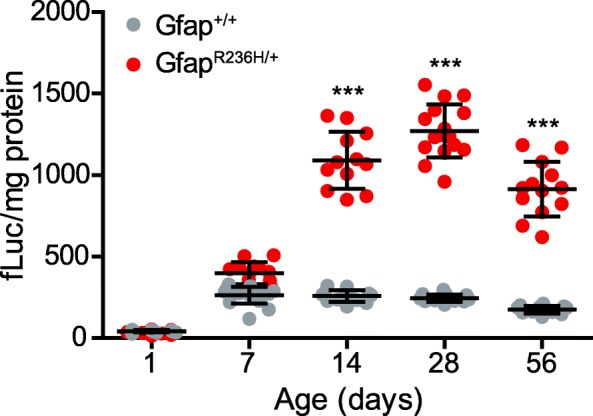
Increase in activity of the Gfap promoter during early postnatal development of a mouse model of Alexander disease. Promoter activity (monitored through expression of a Gfap-luciferase reporter) in mutants rises above that in wild-type mice between postnatal days 7 to 14 and remains elevated through at least 8 weeks of age. Figure used with permission [11]
In theory, impaired degradation could also contribute to the accumulation of GFAP protein, but the evidence for this is less certain. Early results suggested that mutant GFAPs interfered with function of the proteasome [13]. Furthermore, the small heat shock protein, αB-crystallin, which had previously been shown to be protective in mouse models of the disease [14], could relieve the block on the proteasome through binding to small oligomers of GFAP [15]. However, other evidence indicated that autophagy might be increased [16], so that the net effect on overall degradation rate remained unclear. Recently, we investigated the turnover rate of GFAP directly in mouse models, by introducing amino acids containing heavy nitrogen via the diet and following the conversion of the total GFAP population from the light to heavy forms of nitrogen using mass spectrometry [17]. To our surprise, the turnover rate of GFAP in the mutant mice was roughly twice as fast as in control mice, indicating that degradation must have increased, at least in the adult animals that were the focus of this study.
The simplest explanation of these results is that an early event in pathogenesis is an increase in synthesis, followed by an undetermined lag period after which degradation also increases (Fig. 2). If degradation had increased immediately to match the change in synthesis, no change in protein levels would occur. It is the lag in the compensatory response that allows protein levels to rise, eventually reaching a new but higher equilibrium. What initiates the change in synthesis is not yet known, but presumably reflects activation of one or more cellular stress pathways by the initial production of even small amounts of mutant protein. One way or another, GFAP levels are elevated in Alexander disease. This phenomenon begins in the astrocyte, but is also evident in the cerebrospinal fluid (CSF) of most patients and even in blood of some [19]. The release of detectable levels of GFAP into body fluids that are more accessible for biopsy is a feature that may prove useful for following the response to experimental therapies.
Fig. 2.
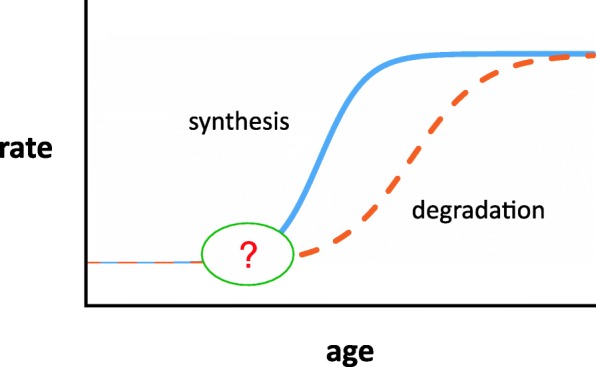
Proposed model for changes in rates of synthesis and degradation of GFAP caused by the presence of mutant protein. Initially rates of synthesis and degradation are equal, with stable levels of protein. A change occurs which increases synthesis, but there is a lag period before degradation increases, during which the imbalance results in increased levels of protein. Eventually, synthesis and degradation reach a new equilibrium, but maintaining a higher level of total GFAP. Figure used with permission [18]
The question of whether GFAP elevation by itself causes all of the downstream effects (i.e., microglial activation, neuronal and oligodendroglial dysfunction) is unresolved. Clearly some aspects of the disease are replicated simply by increased levels of normal protein, such as formation of Rosenthal fibers and activation of multiple stress response pathways. However, no patients have yet been identified with disease caused by excess GFAP of normal sequence, such as might be produced by GFAP duplications [20]. We believe, but do not yet have proof, that the threshold for toxicity is lower when mutant protein is involved. It is interesting that, in the mouse at least, GFAP is not uniformly elevated in all regions of the CNS. Indeed, the cervical spinal cord, a site that is consistently affected in human Alexander disease, shows only modest increase in Gfap mRNA and even a decrease in the level of protein [11]. This occurs despite the clear presence of Rosenthal fibers in this location. Whether the same anomaly occurs in human spinal cord has not yet been investigated.
Recently, two publications have appeared that report the generation and characterization of induced pluripotent stem cells (iPSC) derived from patients with Alexander disease [21, 22]. In both cases, astrocytes differentiated from these iPS cells form Rosenthal-like fibers and acquire many distinct abnormalities compared with isogenic controls in which the GFAP variant was corrected to the normal sequence. Although Li et al. [21] do not comment on whether GFAP levels differed between the mutant and control cell lines, Jones et al. [22] assert many phenotypic changes that take place in the absence of evident change in GFAP.
In the disease setting, where patients are heterozygous, the GFAP pool consists of both normal and mutant protein, and it would be very useful to be able to distinguish one from the other. However, antibodies capable of distinguishing the two (which usually differ in only a single amino acid) exist for just one variant—the Arg416Trp mutation. In this case, Perng et al. [23] showed that both proteins were present within the Rosenthal fibers of a human patient carrying this mutation. Using a cell-free assay designed to study assembly of individual monomers into mature 10-nm intermediate filaments, they could show that having just 25% mutant protein was sufficient to cause aggregation.
In another patient, reported by Flint et al. [24], an unusual splice site mutation resulted in an in-frame deletion of exon 4, predicting synthesis of a protein missing 54 amino acids (207–260 of the normal 432) in the rod domain. For this patient, brain mRNA was available for analysis, which revealed that only 8% of the GFAP mRNA derived from the mutant allele. Using a cell culture model for studying filament assembly, they found that the mutant protein could disrupt polymerization even at very low levels, or 2.5% of the total.
Since so little is understood about the normal functions of GFAP, it is difficult to discuss in precise terms exactly how Alexander disease-associated mutations might cause gain or loss of any particular functions. The arguments reviewed above support the idea that increasing total levels replicate some key aspects of disease, and if mutant proteins acquire new and toxic properties, this qualifies as a different type of gain-of-function. Dominant negative mutations, which are genetically dominant but produce phenotypes that often resemble the complete deficiency state modeled by mouse knockouts, are well-known causes of other disorders. But Alexander disease bears little resemblance to the minimal phenotype observed in mouse knockouts of GFAP. With the present state of knowledge about GFAP and Alexander disease, we must accept the possibility that disease reflects a combination of both gain and loss of different functions, although we would argue that ultimately it is the gain-of-function that dominates.
Conclusion
What implications do these findings have for design of potential treatments? Several approaches have been proposed in the past, targeting different downstream effects of mutant protein [25], but the most straightforward idea is that of reducing or eliminating production of the protein that initiates the disease process—GFAP. The number of known disease-causing variants already exceeds 100, a seemingly insurmountable number for a strategy of allele-specific suppression. At present, the most feasible means for reducing GFAP is generalized suppression, involving reduction of both mutant and normal protein. The rationale for GFAP suppression remains the same whether one starts from a baseline of apparently normal levels (with some being mutant) or the elevated levels seen in most patients or regions. Previous attempts to identify suppressors of GFAP expression through screens of known drugs or compounds suffered from modest or inconsistent effects (clomipramine— [26]), lack of in vivo data (curcumin— [27]), or unacceptable side effects (lithium— [28]).
A dramatic advance on the therapeutic front is our recently reported finding that antisense oligonucleotides (ASOs) are a remarkably effective means for suppressing GFAP expression, and can even reverse established pathology [29]. The effects of single intracerebroventricular injections of such ASOs become manifest within weeks after injection and persist for several months. Rosenthal fibers disappear, and several downstream markers of activated astrocytes and/or microglia return close to normal levels.
The degree to which astrocytes are completely normalized by ASO suppression remains to be seen. Nevertheless, these findings have generated considerable interest in the clinical community and offer the first real promise of a therapeutic worth testing in a formal clinical trial. ASO approaches for neurological diseases are already approved or in advanced stages of clinical development for other conditions, such as spinal muscular atrophy, Huntington’s disease, and amyotrophic lateral sclerosis [30]. For Alexander disease, application of the ASO approach to treatment will require better understanding of how closely the GFAP levels in CSF and blood reflect those in the brain and spinal cord, so that each individual’s response to treatment can be assessed in the least invasive way possible. In addition, it is important to recognize that human patients typically have more extensive pathology than any of the animal models to date, and the degree of rescue that is achievable in the clinical setting will only be learned through experience, and may require the adoption of secondary forms of treatment that complement the reduction or elimination of toxic GFAP.
Acknowledgements
Not applicable
Abbreviations
- ASO
Antisense oligonucleotide
- CSF
Cerebrospinal fluid
- GFAP
Glial fibrillary acidic protein
- iPSC
Induced pluripotent stem cells
Author’s contribution
The author read and approved the final manuscript.
Funding
The work from the author’s laboratory that is described in this review was supported by grants from the National Institutes of Health (HD-076892), the United Leukodystrophy Foundation, the Juanma Fund, the Palamaro Fund, and the Jelte Rijkaart Fund, and in part by a grant from the National Institutes of Health to the Waisman Center (HD-090256).
Availability of data and materials
Not applicable
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The author declares that he has no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949;72:373–381. doi: 10.1093/brain/72.3.373. [DOI] [PubMed] [Google Scholar]
- 2.Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998;152:391–398. [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez D, Gauthier F, Bertini E, Bugiani M, Brenner M, N’guyen S, et al. Infantile Alexander disease: spectrum of GFAP mutations and genotype-phenotype correlation. Am J Hum Genet. 2001;69:1134–1140. doi: 10.1086/323799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li R, Johnson AB, Salomons G, Goldman JE, Naidu S, Quinlan R, et al. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol. 2005;57:310–326. doi: 10.1002/ana.20406. [DOI] [PubMed] [Google Scholar]
- 6.Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE. Alexander disease. J Neurosci. 2012;32:5017–5023. doi: 10.1523/JNEUROSCI.5384-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li R, Messing A, Goldman JE, Brenner M. GFAP mutations in Alexander disease. Int J Dev Neurosci. 2002;20:259–268. doi: 10.1016/S0736-5748(02)00019-9. [DOI] [PubMed] [Google Scholar]
- 8.Walker AK, LaPash Daniels CM, Goldman JE, Trojanowski JQ, Lee VM-Y, Messing A. Astrocytic TDP-43 pathology in Alexander disease. J Neurosci. 2014;34:6448–6458. doi: 10.1523/JNEUROSCI.0248-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagemann TL, Gaeta SA, Smith MA, Johnson DA, Johnson JA, Messing A. Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum Mol Genet. 2005;14:2443–2458. doi: 10.1093/hmg/ddi248. [DOI] [PubMed] [Google Scholar]
- 10.Hagemann TL, Connor JX, Messing A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J Neurosci. 2006;26:11162–11173. doi: 10.1523/JNEUROSCI.3260-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jany PL, Hagemann TL, Messing A. GFAP expression as an indicator of disease severity in mouse models of Alexander disease. ASN Neuro. 2013;5:e00109. doi: 10.1042/AN20130003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saito K, Shigetomi E, Yasuda R, Sato R, Nakano M, Tashiro K, et al. Aberrant astrocyte Ca2+ signals “AxCa signals” exacerbate pathological alterations in an alexander disease model. Glia. 2018;66:1053–1067. doi: 10.1002/glia.23300. [DOI] [PubMed] [Google Scholar]
- 13.Tang G, Xu Z, Goldman JE. Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem. 2006;281:38634–38643. doi: 10.1074/jbc.M604942200. [DOI] [PubMed] [Google Scholar]
- 14.Hagemann TL, Boelens W, Wawrousek E, Messing A. Suppression of GFAP toxicity by αB-crystallin in mouse models of Alexander disease. Hum Mol Genet. 2009;18:1190–1199. doi: 10.1093/hmg/ddp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) inhibit the proteasome system in Alexander disease astrocytes, and the small heat shock protein, αB-crystallin, reverses the inhibition. J Biol Chem. 2010;285:10527–10537. doi: 10.1074/jbc.M109.067975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang G, Yue Z, Talloczy Z, Hagemann T, Cho W, Messing A, et al. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17:1540–1555. doi: 10.1093/hmg/ddn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moody LR, Barrett-Wilt GA, Sussman MR, Messing A. Glial fibrillary acidic protein exhibits altered turnover kinetics in a mouse model of Alexander disease. J Biol Chem. 2017;292:5814–5824. doi: 10.1074/jbc.M116.772020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Messing A. Alexander disease: a guide for patients and families: Morgan & Claypool Publishers; 2018. 106 p.
- 19.Jany PL, Agosta GE, Benko WS, Eickhoff JC, Keller SR, Köehler W, et al. Cerebrospinal fluid and blood levels of GFAP in Alexander disease. eNeuro. 2015;2:e0080–15.2015. doi: 10.1523/ENEURO.0080-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira MC, Dorboz I, Rodriguez D, Boespflug-Tanguy O. Screening for GFAP rearrangements in a cohort of Alexander disease and undetermined leukoencephalopathy patients. Eur J Med Genet. 2015;58:466–470. doi: 10.1016/j.ejmg.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Li L, Tian E, Chen X, Chao J, Klein J, Qu Q, et al. GFAP mutations in astrocytes impair oligodendrocyte progenitor proliferation and myelination in an hiPSC model of Alexander disease. Cell Stem Cell. 2018;23:239–251. doi: 10.1016/j.stem.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones JR, Kong L, Hanna MG, 4th, Hoffmann B, Krencik R, Bradley R, et al. Mutations in GFAP disrupt the distribution and function of organelles in human astrocytes. Cell Reports. 2018;25:947–958. doi: 10.1016/j.celrep.2018.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perng MD, Su M, Wen SF, Li R, Gibbon T, Prescott AR, et al. The Alexander disease-causing GFAP mutant, R416W, accumulates into Rosenthal fibers by a pathway involving filament aggregation and the association of αB-crystallin and HSP27. Am J Hum Genet. 2006;79:197–213. doi: 10.1086/504411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flint D, Li R, Webster LS, Naidu S, Kolodny E, Percy A, et al. Splice site, frameshift and chimeric GFAP mutations in Alexander disease. Hum Mutat. 2012;11:1141–1148. doi: 10.1002/humu.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Messing A, Daniels CM, Hagemann TL. Strategies for treatment in Alexander disease. Neurotherapeutics. 2010;7:507–515. doi: 10.1016/j.nurt.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho W, Brenner M, Peters N, Messing A. Drug screening to identify suppressors of GFAP expression. Hum Mol Genet. 2010;19:3169–3178. doi: 10.1093/hmg/ddq227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachetti T, Di Zanni E, Balbi P, Ravazzolo R, Sechi G, Ceccherini I. Beneficial effects of curcumin on GFAP filament organization and down-regulation of GFAP expression in an in vitro model of Alexander disease. Exp Cell Res. 2012;318:1844–1854. doi: 10.1016/j.yexcr.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 28.LaPash Daniels CM, Paffenroth E, Austin EV, Glebov K, Lewis D, Walter J, et al. Lithium decreases glial fibrillary acidic protein in a mouse model of Alexander disease. PLoS ONE. 2015;10:e0138132. doi: 10.1371/journal.pone.0138132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagemann TL, Powers B, Mazur C, Kim A, Wheeler S, Hung G, et al. Antisense suppression of GFAP as a treatment for Alexander disease. Ann Neurol. 2018;83:27–39. doi: 10.1002/ana.25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoch KM, Miller TM. Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron. 2017;94:1056–1070. doi: 10.1016/j.neuron.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable